

Abstract
Glutamate receptor-interacting protein 1 (GRIP1) and GRIP2 are closely related proteins that bind GluR2-containing AMPA receptors and couple them to structural and signaling complexes in neurons. Cerebellar long-term synaptic depression (LTD) is a model system of synaptic plasticity that is expressed by persistent internalization of GluR2-containing AMPA receptors. Here, we show that genetic deletion of both GRIP1 and GRIP2 blocks LTD expression in primary cultures of mouse cerebellar neurons but that single deletion of either isoform allows LTD to occur. In GRIP1/2 double knock-out Purkinje cells, LTD can be fully rescued by a plasmid-driving expression of GRIP1 and partially rescued by a GRIP2 plasmid. These results indicate that the GRIP family comprises an essential molecular component for cerebellar LTD.
Keywords: glutamate, AMPA receptor, plasticity, motor learning, cerebellum, dendrite
Introduction
Long-term synaptic depression (LTD) of the parallel-fiber–Purkinje-cell synapse (commonly referred to as “cerebellar LTD”) is a well defined cellular model of information storage that has been hypothesized to underlie certain forms of motor learning (Ito, 2001). It is triggered by brief postsynaptic activation of a metabotropic glutamate receptor 1 (mGluR1)/PKC cascade (Linden and Connor, 1991) and is expressed by clathrin-mediated endocytosis of GluR2-containing AMPA receptors (Matsuda et al., 2000; Wang and Linden, 2000). In recent years, some of the molecular mechanisms linking PKC activation to AMPA receptor internalization have been revealed. The C-terminal tail of GluR2 contains a sequence (IESVKI) that functions as both a PKC phosphorylation site (at Ser880) and a type II postsynaptic density-95/Discs large/zona occludens-1 (PDZ) ligand. In the dephospho-Ser880 state, GluR2 preferentially binds a particular PDZ domain, PDZ5, of the multi-PDZ domain proteins glutamate receptor-interacting protein 1 (GRIP1) and GRIP2 (GRIP2 is also called ABP) (Dong et al., 1997, 1999; Srivastava et al., 1998). However, when GluR2 Ser880 is phosphorylated by PKC, the affinity of GRIP1/2 for GluR2 is dramatically reduced, and this allows for binding of another PDZ domain protein, protein interacting with C kinase 1 (PICK1), to this site (Matsuda et al., 1999; Chung et al., 2000). LTD expression is blocked by PKC inhibitors (Linden and Connor, 1991) or by peptides that inhibit PICK1–GluR2 interaction (Xia et al., 2000). In addition, mutant mice lacking the PKCα isoform (Leitges et al., 2004), or PICK1 (Steinberg et al., 2006), do not express LTD. Moreover, mutant mice that have knock-in mutations that delete the GluR2 C-terminal PDZ ligand entirely (Steinberg et al., 2006), or make the GluR2 Ser880 unphosphorylatable by PKC (Chung et al., 2003; Steinberg et al., 2006), do not express LTD. What has not been clear in this model is whether GRIP1 and/or GRIP2 have an explicit role in cerebellar LTD expression. Here, Purkinje cells derived from mutant mice lacking GRIP1 and/or GRIP2 have been used together with biolistic transfection of plasmids encoding wild-type (wt) or mutant GRIPs to test the hypothesis that GRIP1/2 are required for expression of cerebellar LTD.
Materials and Methods
Generation of GRIP1 conditional knock-out mice.
A targeting vector containing two exons encoding the first PDZ domain of GRIP1 was constructed and introduced into embryonic stem (ES) cells by the homologous recombination technique, using neomycin resistance gene (neor) cassette for a positive selection and thymidine kinase (data not shown) for a negative selection (Fig. 1A). The neor cassette was flanked by flippase recombinase target (FRT) sequences at both ends and inserted into the intron between the two PDZ1-encoding exons. Two loxP sites were inserted before the first exon and after FRT-flanked neor cassette so that the introduction of Cre recombinase resulted in the deletion of the first PDZ1-encoding exon and a frame shift causing an immediate nonsense mutation in the entire GRIP1 transcript. Correctly targeted ES cells containing the neor cassette were injected into C57BL/6 blastocysts. Chimeric mice carrying the mutant allele were bred to C57BL/6 mice to generate heterozygous mice [GRIP1(flox) neor (+)/wt]. Heterozygous mice were then bred to transgenic mice carrying flippase to delete the neor cassette via FRT-flippase system [GRIP1(flox) neor (−)/wt] and then intercrossed to produce the GRIP1(flox/flox) conditional knock-out (KO) homozygous mice. The success of the procedures was confirmed by PCR (data not shown) and Southern blot analysis (Fig. 1B). The GRIP2 conventional knock-out mice were generated as described previously (Takamiya et al., 2004), and the double-mutant mice GRIP1(flox/flox) and GRIP2(−/−) were generated by intercrossing these two lines.
Figure 1.

Mutant mice harboring mutations in GRIP1 and GRIP2. A, Schematic representation of the GRIP1 conditional targeting strategy. The targeted allele of GRIP1 after homologous recombination is shown at the top, with two exons (white boxes) encoding the first PDZ domain. The neor cassette (black box; PGK-neo bpA), flanked by FRT sequences (gray boxes), was inserted between the two exons. Two loxP sites (arrowheads) were inserted before the first PDZ1 exon and immediately after the neor cassette; thus, expression of Cre recombinase would result in deletion of the first PDZ1 exon and cause a frameshift mutation that introduces an immediate stop codon in the GRIP1 transcript. The neor cassette was deleted by crossing the mutant mice with flippase transgenic mice. Two EcoRI restriction sites and the region for the probe for Southern blot analysis are also shown. B, Southern blot analysis of GRIP1 conditional knock-out mice. The genomic DNA was digested by EcoRI and detected with the probe illustrated in A. Wild-type mice gave a fragment of 3.6 kb, whereas the neor(+) mice extend the size of the fragment to 5.7 kb. When the neor cassette is deleted, the probed fragment is decreased to 3.8 kb. Lane 1: wt/wt, 3.6 kb; lane 2: heterozyogotes with the targeting vector, GRIP1(flox)neor (+)/wt, 5.7/3.6 kb; lane 3: heterozygotes after neor deletion, GRIP1(flox/+)neor (−)/wt, 3.8/3.6 kb. C, Cre lentivirus-mediated GRIP1 deletion in mouse cortical neurons. Cortical cultures were prepared from GRIP1(flox/flox) and GRIP2(−/−) mice and infected with EGFP or EGFP-IRES-Cre lentiviruses. Four, seven, and ten days after infection, neurons were harvested to detect GRIP1 expression level. Western blots of GRIP1 using anti-GRIP1 antibody (left) and GluR2 (right) are shown. D, GRIP1 immunocytochemistry after Cre-lentivirus infection. GRIP1(flox/flox) and GRIP2(−/−) cortical neurons were infected with EGFP-IRES-Cre or EGFP lentiviruses for 7 d and fixed and stained with anti-GRIP1 monoclonal antibody (left). Differential interference contrast (DIC) images of the same neurons are shown on the right.
Preparation of mouse embryonic cerebellar cultures, biolistic transfection, voltage-clamp recording, and Ca imaging were performed as described previously (Leitges et al., 2004; Steinberg et al., 2006). Please see supplemental Methods, available at www.jneurosci.org as supplemental material, for details and exemplar Ca images.
Results
To directly examine the roles of GRIP1 and GRIP2 in cerebellar LTD, GRIP1 and GRIP2 knock-out mice were used. GRIP2 knock-out mice are viable (Takamiya et al., 2004); however, because GRIP1 conventional knock-out mice have an early embryonic lethal phenotype (Bladt et al., 2002; Takamiya et al., 2004), a Cre-loxP system was used to generate GRIP1 conditional knock-out mice. Although we have not been able to achieve efficient cleavage of the GRIP1 conditional allele in vivo, we have successfully knocked out the gene using lentiviral expression of Cre recombinase in cultured neurons from the mutant mice. To achieve conditional knock-out of GRIP1 in the cultured neurons, lentiviruses expressing Cre recombinase (EGFP-IRES-Cre) or an EGFP control were introduced into young cortical cultures at 3 d in vitro (DIV) prepared from GRIP1(flox/flox) and GRIP2(−/−) double-mutant mice to confirm the efficiency of the Cre-loxP deletion system. Four days after EGFP-IRES-Cre lentivirus infection, the expression of GRIP1 protein began to decrease, as shown by Western blot analysis using a specific anti-GRIP1 polyclonal antibody recognizing the C terminus of GRIP1 (Fig. 1C). Seven to ten days after EGFP-IRES-Cre virus infection, GRIP1 protein could no longer be detected. In contrast, neurons infected with control EGFP lentivirus showed strong GRIP1 expression. To further verify the effect of Cre lentivirus-mediated knock-out of GRIP1, immunocytochemistry using an anti-GRIP1 monoclonal antibody recognizing the PDZ domain 7 region was performed on cortical cultures prepared from GRIP1(flox/flox) and GRIP2(−/−) double-mutant mice. Immunoreactivity for GRIP1 completely disappeared 7 d after EGFP-IRES-Cre lentiviral infection (the efficiency and time course of GRIP1 deletion using other Cre lentiviruses were similar to that of the Cre-EGFP lentivirus (Fig. 1D). [The efficiency and time course of GRIP1 deletion using other Cre lentiviruses were similar to that of the Cre-EGFP lentivirus (data not shown).] Because it took 7 d to completely eliminate GRIP1 protein in our experimental paradigm, lentiviruses were usually applied to neurons at 2 DIV, and Purkinje neurons were used 7–10 d later for analysis.
Cerebellar LTD was examined in the cultured Purkinje cells using whole-cell voltage-clamp recordings and Ca imaging. Purkinje cells were held at −70 mV, and iontophoresis was used to deliver test pulses of glutamate (30–100 ms duration; 0.05 Hz). After a stable baseline recording period, repeated glutamate/depolarization pairing was used to induce LTD, after which test pulses were resumed for an additional 45 min monitoring period (Fig. 2A). Two different control treatments were used. In one, Purkinje cells derived from floxed GRIP1 mice were infected with lentivirus expressing the marker EGFP (control 1). These cells showed robust LTD (55 ± 8.6% of baseline at t = 45 min; n = 9 cells; mean ± SEM), as did another control group in which wild-type cells were infected with bicistronic lentiviral Cre (EGFP-IRES-Cre) and EGFP (control 2, 47 ± 9.3% of baseline at t = 45 min; n = 8). When knock-out of both GRIP1 and GRIP2 was achieved (in GRIP2 KO × double-floxed GRIP1 Purkinje cells infected with EGFP-IRES-Cre lentiviral, GRIP1/2 DKO), LTD was completely abolished (GRIP1/2 DKO, 106 ± 8.0% of baseline at t = 45 min; n = 8). However, single knock-out of either GRIP1 (50 ± 6.8% of baseline at t = 45 min; n = 7) or GRIP2 (56 ± 7.5% of baseline at t = 45 min; n = 7) had no effect on the amplitude or time course of LTD.
Figure 2.
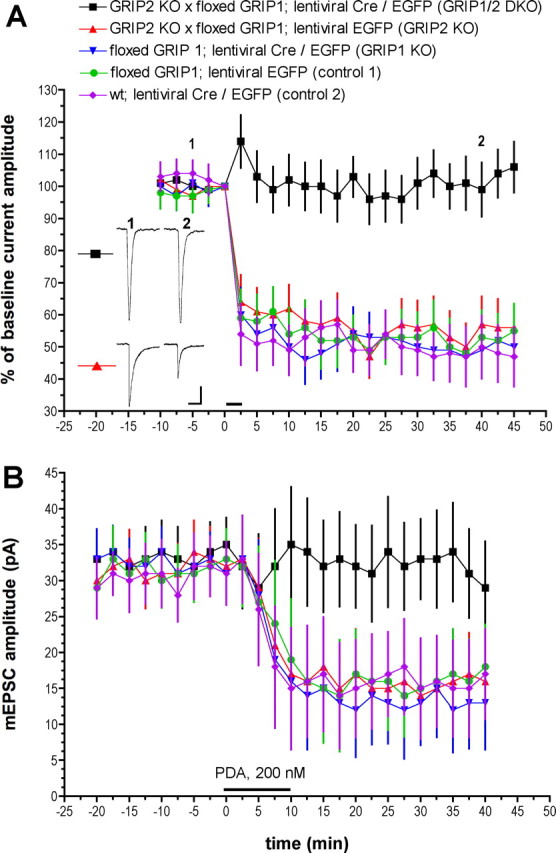
Cerebellar LTD is abolished in GRIP1/2 double knock-out Purkinje cells. A, LTD induction by glutamate/depolarization pairing. Baseline responses were evoked by glutamate pulses delivered to Purkinje cell dendrites by microiontophoresis at 0.05 Hz. After a baseline recording period, LTD was induced by pairing a single glutamate pulse with a 1-s-long depolarization from −70 to 0 mV, repeated six times at the test pulse frequency. This is indicated on the graph by a horizontal bar at t = 0 min. Exemplar traces are single, unaveraged responses recording at the time points indicated on the graph. Calibration: 50 pA, 1 s. GRIP1/2 DKO, n = 8; GRIP2 KO, n = 7; GRIP1 KO, n = 7; control 1, n = 9; control 2, n = 8. Error bars indicate SEM here and in Figure 3. B, Chemical LTD of mEPSCs produced by bath application of the PKC activator PDA. PDA (200 nm) was applied from t = 0 to10 min, as indicated by the horizontal bar. mEPSCs were measured and are expressed here as the mean ± SEM of raw amplitudes (not normalized values). n = 12 cells per group.
GRIP1/2 DKO, GRIP1 KO, and GRIP2 KO Purkinje cells all displayed normal input resistance (Rinput) and miniature EPSC (mEPSC) kinetics, arguing against generalized electrophysiological disruption by these treatments (supplemental Table 1, available at www.jneurosci.org as supplemental material). In addition, it is unlikely that GRIP1/2 deletion, either doubly or singly, blocked the initial signals necessary for LTD induction as Ca imaging experiments revealed that both depolarization-evoked and mGluR1 agonist- [DHPG (3,4-dihydroxyphenylglycine)] evoked dendritic Ca transients were normal (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). To assess baseline synaptic transmission, mEPSCs were measured. Manipulations of GRIP1 and GRIP2, either singly or in combination, did not affect baseline mEPSC amplitude, arguing against occlusion as a mechanism for LTD blockade in GRIP1/2 DKO Purkinje cells (Fig. 2B). It has been shown previously that brief bath application of the PKC-activating phorbol ester phorbol-12,13-diacetate (PDA) can produce a chemical form of LTD that mutually occludes LTD induced by glutamate/depolarization pairing (Linden and Connor, 1991; Chung et al., 2003). Robust chemical LTD was evoked in control 1 (mEPSC amplitude, 29 ± 3.7 pA at t = −20 min; 18 ± 5.9 pA at t = 40 min; n = 12 cells), control 2 (29 ± 4.3 pA at t = −20 min; 17 ± 6.3 pA at t = 40 min; n = 12 cells), GRIP1 KO (33 ± 4.2 pA at t = −20 min; 13 ± 6.5 pA at t = 40 min; n = 12 cells), and GRIP2 KO (30 ± 3.8 pA at t = −20 min; 16 ± 5.7 pA at t = 40 min; n = 12 cells) Purkinje cells. However, chemical LTD was completely blocked in GRIP1/2 DKO Purkinje cells (33 ± 4.2 pA at t = −20 min; 29 ± 6.5 pA at t = 40 min; n = 12 cells). Together with the pairing-induced LTD experiments, these findings suggest that a GRIP family member is necessary for LTD induction, but that either GRIP1 or GRIP2 will suffice.
As a further test of this model, GRIP1/2 DKO Purkinje cells were transfected with a plasmid encoding either wild-type GRIP1 or GRIP2 to rescue the KOs. In both cases, these transfections included a second plasmid coated on the same biolistic particle for driving expression of dsRed as a marker. In the GRIP1/2 DKO background, GRIP1 overexpression produced a complete rescue of both pairing-induced LTD (Fig. 3) (46 ± 8.4% of baseline at t = 45 min; n = 7) and chemical LTD (33 ± 3.4 pA at t = −20 min; 14 ± 6.3 pA at t = 40 min; n = 12 cells). However, overexpression of a mutant form of GRIP1 in which PDZ5 had been mutated to interfere with GluR2 binding (K577A/K578A; binding data not shown) completely failed to rescue either pairing-induced LTD (107 ± 7.0% of baseline at t = 45 min; n = 8) or chemical LTD (33 ± 4.1 pA at t = −20 min; 30 ± 7.5 pA at t = 40 min; n = 12 cells). Similar results were found when this experiment was repeated with double the concentration of GRIP1 PDZ5 mutant plasmid on each particle (data not shown in Fig. 3) (pairing-induced LTD, 100 ± 7.3% of baseline at t = 45 min, n = 6; chemical LTD, 34 ± 4.6 pA at t = −20 min; 33 ± 6.2 pA at t = 40 min, n = 12 cells).
Figure 3.

Transfection with plasmids driving expression of either GRIP1 or GRIP2 restores cerebellar LTD in GRIP1/2 double knock-out Purkinje cells. A, LTD induction by glutamate/depolarization pairing. Calibration: 50 pA, 1 s. GRIP1/2 DKO, GRIP1 plasmid, n = 7; GRIP1/2 DKO, GRIP2 plasmid, n = 8; GRIP1/2 DKO, GRIP1 PDZ5 mutant plasmid, n = 8. B, Chemical LTD of mEPSCs. n = 12 cells/group.
Interestingly, overexpression of GRIP2 in GRIP1/2 DKO Purkinje cells produced a partial rescue of both pairing-induced LTD (75 ± 6.2% of baseline at t = 45 min; n = 8) and chemical LTD (29 ± 4.1 pA at t = −20 min; 23 ± 5.8 pA at t = 40 min; n = 12 cells) that was intermediate between the effect of wild-type GRIP1 and the inactive PDZ5 mutant GRIP1 control. It is not clear why GRIP2 overexpression produced a partial rescue of LTD, whereas GRIP1 overexpression resulted in a complete rescue. Perhaps the expression levels of GRIP1 and GRIP2 from these plasmids in Purkinje cells are different. However, doubling the concentration of GRIP2 plasmid coated onto each particle failed to produce complete LTD rescue (data not shown in Fig. 3) (pairing-induced LTD, 70 ± 6.9% of baseline at t = 45 min, n = 5; chemical LTD: 31 ± 4.0 pA at t = −20 min; 22 ± 6.0 pA at t = 40 min; n = 12). It is possible that the expression levels of GRIP1 and GRIP2 might be similar, but for some reason GRIP2 is not as efficient in the rescue of LTD. Unfortunately, GRIP transgene expression levels are not straightforward to measure biochemically, because Purkinje cells only comprise ∼2% of the cells in the culture, and the transfection efficiency of the gene gun is low, typically yielding only 10–20 transfected Purkinje cells per dish.
Discussion
The main conclusion of this work is that a GRIP family member is necessary for cerebellar LTD expression. Although single knock-out experiments indicate that either GRIP1 or GRIP2 will suffice for LTD expression, rescues in the double knock-out background showed complete rescue with GRIP1 and partial rescue with GRIP2, possibly indicating a preferential role for GRIP1. The complete blockade of LTD expression in GRIP1/2 DKO Purkinje cells is not a result of side effects of this manipulation on the initial signals necessary for LTD induction (mGluR1 function, voltage-sensitive Ca channel function), nor is it a consequence of manipulating basal synaptic strength (as indexed by mEPSC amplitude) or general electrophysiological viability (as indexed by mEPSC kinetics and Rinput). The observation that PDZ5-mutant GRIP1 fails to rescue LTD in GRIP1/2 DKO Purkinje cells argues that GRIP–GluR2 binding is a necessary step in the expression of LTD.
Previous work in cerebellar Purkinje cells has shown that internal application of a peptide that interferes with both GRIP and PICK1 interactions with GluR2 blocked expression of LTD (Xia et al., 2000). In hippocampal pyramidal neurons, this peptide also blocked LTD induction and could reverse previous LTD (Daw et al., 2000). It is likely that other domains and functions of GRIP are necessary for LTD expression. It has been suggested that the linker II domain of GRIP binds the single PDZ domain of activated PICK1 dimers and thereby recruits them to GluR2-containing AMPA receptors to initiate their endocytosis in LTD expression (Lu and Ziff, 2005). GRIP has also been reported to bind other signaling molecules in the neurons such as GRASP1 (GRIP-associated protein 1) (Ye et al., 2000), ephrin B receptors (Hoogenraad et al., 2005), liprin-α (Wyszynski et al., 2002), and matrix metalloproteinase 5 (Monea et al., 2006). It will be useful to determine whether any of these GRIP interactors also function in cerebellar LTD.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants R37 MH51106 (D.J.L.) and RO1 NS036715 (R.L.H.). D.J.L. and R.L.H. were supported by a Silvio Conte Center for Neuroscience Research Grant from the NIH. We thank Roland Bock and Devorah Vanness for excellent technical assistance and Yilin Yu for preparing mice for culture.
References
- Bladt F, Tafuri A, Gelkop S, Langille L, Pawson T. Epidermolysis bullosa and embryonic lethality in mice lacking the multi-PDZ domain protein GRIP1. Proc Natl Acad Sci USA. 2002;99:6816–6821. doi: 10.1073/pnas.092130099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HJ, Xia J, Scannevin RH, Zhang X, Huganir RL. Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. J Neurosci. 2000;20:7258–7267. doi: 10.1523/JNEUROSCI.20-19-07258.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HJ, Steinberg J, Huganir RL, Linden DJ. Requirement of AMPA receptor GluR2 phosphorylation for cerebellar long-term depression. Science. 2003;300:1751–1755. doi: 10.1126/science.1082915. [DOI] [PubMed] [Google Scholar]
- Daw MI, Chittajallu R, Bortolotto ZA, Dev KK, Duprat F, Henley JM, Collingridge GL, Isaac JT. PDZ proteins interacting with C-terminal GluR2/3 are involved in a PKC-dependent regulation of AMPA receptors at hippocampal synapses. Neuron. 2000;28:873–886. doi: 10.1016/s0896-6273(00)00160-4. [DOI] [PubMed] [Google Scholar]
- Dong H, O'Brien RJ, Fung ET, Lanahan AA, Worley PF, Huganir RL. GRIP: a synaptic PDZ domain-containing protein that interacts with AMPA receptors. Nature. 1997;386:279–284. doi: 10.1038/386279a0. [DOI] [PubMed] [Google Scholar]
- Dong H, Zhang P, Song I, Petralia RS, Liao D, Huganir RL. Characterization of the glutamate receptor-interacting proteins GRIP1 and GRIP2. J Neurosci. 1999;19:6930–6941. doi: 10.1523/JNEUROSCI.19-16-06930.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenraad CC, Milstein AD, Ethell IM, Henkemeyer M, Sheng M. GRIP1 controls dendrite morphogenesis by regulating EphB receptor trafficking. Nat Neurosci. 2005;8:906–915. doi: 10.1038/nn1487. [DOI] [PubMed] [Google Scholar]
- Ito M. Cerebellar long-term depression: characterization, signal transduction, and functional roles. Physiol Rev. 2001;81:1143–1195. doi: 10.1152/physrev.2001.81.3.1143. [DOI] [PubMed] [Google Scholar]
- Leitges M, Kovac J, Plomann M, Linden DJ. A unique PDZ ligand in PKCalpha confers induction of cerebellar long-term synaptic depression. Neuron. 2004;44:585–594. doi: 10.1016/j.neuron.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Linden DJ, Connor JA. Participation of postsynaptic PKC in cerebellar long-term depression in culture. Science. 1991;254:1656–1659. doi: 10.1126/science.1721243. [DOI] [PubMed] [Google Scholar]
- Lu W, Ziff EB. PICK1 interacts with ABP/GRIP to regulate AMPA receptor trafficking. Neuron. 2005;47:407–421. doi: 10.1016/j.neuron.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Mikawa S, Hirai H. Phosphorylation of serine-880 in GluR2 by protein kinase C prevents its C terminus from binding with glutamate receptor-interacting protein. J Neurochem. 1999;73:1765–1768. doi: 10.1046/j.1471-4159.1999.731765.x. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Launey T, Mikawa S, Hirai H. Disruption of AMPA receptor GluR2 clusters following long-term depression induction in cerebellar Purkinje neurons. EMBO J. 2000;19:2765–2774. doi: 10.1093/emboj/19.12.2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monea S, Jordan BA, Srivastava S, DeSouza S, Ziff EB. Membrane localization of membrane type 5 matrix metalloproteinase by AMPA receptor binding protein and cleavage of cadherins. J Neurosci. 2006;26:2300–2312. doi: 10.1523/JNEUROSCI.3521-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Osten P, Vilim FS, Khatri L, Inman G, States B, Daly C, DeSouza S, Abagyan R, Valtschanoff JG, Weinberg RJ, Ziff EB. Novel anchorage of GluR2/3 to the postsynaptic density by the AMPA receptor-binding protein ABP. Neuron. 1998;21:581–591. doi: 10.1016/s0896-6273(00)80568-1. [DOI] [PubMed] [Google Scholar]
- Steinberg JP, Takamiya K, Shen Y, Xia J, Rubio ME, Yu S, Jin W, Thomas GM, Linden DJ, Huganir RL. Targeted in vivo mutations of the AMPA receptor subunit GluR2 and its interacting protein PICK1 eliminate cerebellar long-term depression. Neuron. 2006;49:845–860. doi: 10.1016/j.neuron.2006.02.025. [DOI] [PubMed] [Google Scholar]
- Takamiya K, Kostourou V, Adams S, Jadeja S, Chalepakis G, Scambler PJ, Huganir RL, Adams RH. A direct functional link between the multi-PDZ domain protein GRIP1 and the Fraser syndrome protein Fras1. Nat Genet. 2004;36:172–177. doi: 10.1038/ng1292. [DOI] [PubMed] [Google Scholar]
- Wang YT, Linden DJ. Expression of cerebellar long-term depression requires postsynaptic clathrin-mediated endocytosis. Neuron. 2000;25:635–647. doi: 10.1016/s0896-6273(00)81066-1. [DOI] [PubMed] [Google Scholar]
- Wyszynski M, Kim E, Dunah AW, Passafaro M, Valtschanoff JG, Serra-Pagès C, Streuli M, Weinberg RJ, Sheng M. Interaction between GRIP and liprin-alpha/SYD2 is required for AMPA receptor targeting. Neuron. 2002;34:39–52. doi: 10.1016/s0896-6273(02)00640-2. [DOI] [PubMed] [Google Scholar]
- Xia J, Chung HJ, Wihler C, Huganir RL, Linden DJ. Cerebellar long-term depression requires PKC-regulated interactions between GluR2/3 and PDZ domain-containing proteins. Neuron. 2000;28:499–510. doi: 10.1016/s0896-6273(00)00128-8. [DOI] [PubMed] [Google Scholar]
- Ye B, Liao D, Zhang X, Zhang P, Dong H, Huganir RL. GRASP-1: a neuronal RasGEF associated with the AMPA receptor/GRIP complex. Neuron. 2000;26:603–617. doi: 10.1016/s0896-6273(00)81198-8. [DOI] [PubMed] [Google Scholar]