

Abstract
Recruitment of effector T cells to inflamed peripheral tissues is regulated by chemokines and their receptors, but the factors regulating recruitment to tumors remain largely undefined. Ionizing radiation (IR) therapy is a common treatment modality for breast and other cancers. Used as a cytocidal agent for proliferating cancer cells, IR in combination with immunotherapy has been shown to promote immune-mediated tumor destruction in pre-clinical studies. Here we demonstrate that IR markedly enhanced the secretion by mouse and human breast cancer cells of CXCL16, a chemokine that binds to CXCR6 on Th1 and activated CD8 effector T cells, and plays an important role in their recruitment to sites of inflammation. Employing a poorly immunogenic mouse model of breast cancer, we found that irradiation increased the migration of CD8+CXCR6+ activated T cells to tumors in vitro and in vivo. CXCR6-deficient mice showed reduced infiltration of tumors by activated CD8 T cells and impaired tumor regression following treatment with local IR to the tumor and antibodies blocking the negative regulator of T cell activation CTLA-4.
These results provide the first evidence that IR can induce the secretion by cancer cells of pro-inflammatory chemotactic factors that recruit anti-tumor effector T cells. The ability of IR to convert tumors into “inflamed” peripheral tissues could be exploited to overcome obstacles at the effector phase of the anti-tumor immune response and improve the therapeutic efficacy of immunotherapy.
Keywords: Tumor immunity, Cell trafficking, Chemokines, T Cells, Cytotoxic
INTRODUCTION
The molecular identification of tumor-associated antigens has provided many potential targets for the development of therapeutic cancer vaccines (1). Much research in the field of immunotherapy (IT)4 is focused on optimizing vaccination strategies to boost pre-existing and/or prime de novo anti-tumor responses. However, successful vaccination as measured by expansion of vaccine-specific T cells does not necessarily translate into clinical tumor responses (2), emphasizing the need to overcome obstacles also at the effector phase of the anti-tumor immune response (3).
Although in IT the therapeutic agent is a cell (i.e., an anti-tumor T cell), the requirements for IT to be successful are the same as for other anti-tumor agents: the agent must be effective in the microenvironment of the tumor, and it must reach the target cells in optimal quantities (4). The inability to meet one or both of these requirements is often responsible for the ineffective destruction of established vascularized solid tumors by tumor-specific CTL (5–8).
Chemokines and their receptors play a crucial role in T cell recruitment to different tissues, and regulate both homeostatic and inflammation-dependent homing of T cells (9). The chemokine receptor(s) involved in recruiting effector T cells to a specific site are recognized as stimulus and organ-dependent, but little is known about the receptors involved in recruitment of effector T cells to tumors (3). The chemokine receptor CXCR6 is expressed at very low levels on naive T cells and is up-regulated upon activation under Th1-polarizing conditions (10). Importantly, CXCR6 is expressed at high levels by CD8 T cells with cytotoxic effector function (11, 12), and has been implicated in the recruitment of these cells to inflamed tissues (13–15).
Cancer cells produce several chemokines, largely to recruit leukocytes that promote tolerance and immune escape, and to aid tumor growth by enhanced angiogenesis (16, 17).However, tumor cells engineered to express pro-inflammatory chemokines such as IP-10/CXCL10 and RANTES/CCL5 have been shown to recruit lymphocytes that can reject the tumors (18, 19).
Evidence is accumulating that ionizing radiation (IR) therapy, a treatment modality routinely employed to kill cancer cells, can modulate the expression of several receptors and cytokines by cancer cells and tumor stroma, resulting in modifications of the tumor microenvironment that can be exploited to enhance the effects of IT (reviewed in references 20 and 21). Some of the IR-induced modifications appear to facilitate T cell recruitment to tumors, in part, by promoting normalization of the vasculature and/or by up-regulating the expression of endothelial adhesion molecules (22–24). However, specific mechanisms by which IR regulates trafficking of T cells to solid tumors remain largely undefined.
We have employed the 4T1 preclinical model of metastatic breast cancer to test the therapeutic potential of local IR combined with IT (25). When injected into syngeneic mice, 4T1 cells form highly angiogenic, metastatic, and poorly immunogenic tumors, hence recapitulating many of the characteristics of aggressive human breast cancer (26–28). Treatment of mice with established (Day 13) 4T1 tumors with local IR and CTLA-4 blockade in combination but not as single modalities induced CD8-mediated anti-tumor responses inhibiting metastases and inducing regression of the primary irradiated tumors (25). Here we show that most of the CD8 T cells infiltrating 4T1 tumors following the combination treatment were CXCR6+. Tumor irradiation increased recruitment of tumor-specific activated CD8 cells and markedly enhanced the expression and release of CXCR6 ligand, the pro-inflammatory chemokine CXCL16, by 4T1 cells. CXCR6-deficient mice showed reduced treatment-induced CD8 cell infiltration in tumors, and reduced tumor inhibition. Taken together, these data indicate that IR has the ability to induce chemokines involved in recruitment of effector T cells, effectively converting tumors into “inflamed” peripheral tissues that are rendered susceptible to the effector phase of the anti-tumor immune response.
MATERIALS AND METHODS
Mice
Six to eight week old BALB/c mice were purchased from Taconic Animal Laboratory (Germantown, NY). CXCR6+/gfp and CXCR6gfp/gfp mice have been described (11), and were provided in the BALB/c background by Dan Littman (New York University School of Medicine, NY). BALB/c CL4-TCR mice transgenic for an influenza virus influenza hemagglutinin (HA) H2-Kd-restricted epitope (29) were purchased from Jackson Laboratory (Bar Harbor, ME). All experiments were approved by the Institutional Animal Care and Use Committee of New York University.
Cells and antibodies
4T1, 4T07, 66cl4, 168FARN, and 67NR BALB/c mouse-derived mammary carcinoma cell lines (26), and 4T1-HA derivatives (30), were grown in DMEM medium (Invitrogen Corporation, Carlsbad, CA) supplemented with 2mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 2.5 × 10−5 M 2-mercapthoethanol, and 10% FBS (Gemini Bio-Products Woodland, CA) (complete medium). Human cell lines MCF10A, CRL2324, CRL1902, and HTB-20 were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and were cultured as recommended by ATCC. Mouse Baf-3 cells were transduced with control vector MSCV-IRES-GFP or MSCV-CXCR6-IRES-GFP expressing mouse CXCR6, selected by sorting for GFP+ cells, and cultured in the presence of 10 ng/ml IL-3.
Anti-CTLA-4 hamster mAb 9H10 was purified as previously described (31). CXCL16-Fc fusion protein (10) was a gift of M. Matloubian. Goat anti-mouse CXCL16 and isotype control were purchased from R&D System, (Minneapolis, MN). FITC-Donkey anti-goat IgG was from Jackson ImmunoResearch (West Grove, PA). PE-Cy5-CD8, PE-Cy5-CD3, PE-CD4, PE-CD8, PE-CD69, PE-CD62L, and PE-Thy1.1 were from BD PharMingen, (San Diego, CA).
In vitro CD8 T cell activation and expansion
CD8 T cells were purified from spleen single cell suspensions with CD8α (Ly-2) MACS beads (Miltenyi Biotec, Auburn, CA). Cells were cultured in 24-well tissue culture plates coated with 5μg/ml anti-CD3 (145-2C11 clone, eBioScience, San Diego, CA) and 5μg/ml anti-CD28 (37.51, BD Pharmingen, San Diego, CA) for 48 h, followed by culture in fresh RPMI 1640 medium supplemented with 2mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 50nM 2-mercapthoethanol, 10% FBS (T cell medium) and 50μU/ml human rIL-2 (provided by the National Cancer Institute BRB Preclinical Repository) (10). After 8 to 10 days of expansion, activated CD8 cells were used for phenotype analysis, chemotaxis assays, or adoptive transfer. For chemotaxis assays and adoptive transfer, CD8 cells from WT mice were labeled with 5μM or 10μM CFSE (Molecular Probe, Eugene, OR), respectively. IFNγ production by CL4 CD8 cells was measured in supernatants by ELISA (Diaclone Tepnel, Lifecodes Corp. Stamford, CT) after 20 h culture with irradiated (100 Gy) 4T1 or 4T1-HA cells at E/T ratio of 10 in duplicate wells.
Chemotaxis assays
Chemotaxis assays were performed as described (32) using 5-μm transwells for Baf-3 cells and 3-μm transwells for T cells (Corning Costar Corp., Corning, NY). Briefly, 3×105 BaF3 or 5×105 T cells were added to the upper chamber in 100μl of DMEM 1% BSA (chemotaxis buffer). The lower chamber contained 600μl of chemotaxis buffer and mouse rCXCL16 (Peprotech, Rocky Hill, NJ) or 1× 105 4T1 cells untreated or irradiated with 12 Gy and pre-seeded in the bottom of 24-well plate. The adherent tumor cells were washed twice with PBS and changed to 600μl of chemotactic buffer 16 hours before the assay. For neutralization of CXCL16 activity, 1μg/ml anti-mouse CXCL16 or control Ab was added to the lower chamber 30 min beforehand. After 4 to 6 h incubation at 37°C transmigrated GFP+ or CFSE+ cells were counted by flow cytometry. An equal number of 5μm polysterene beads (Polysciences, PA) was added to each sample prior to analysis to correct for variability in volume aspirated by the cytometer.
Tumor challenge and treatment
Mice were injected s.c. in the right flank with 5×104 4T1 cells in 0.1 ml of DMEM medium without additives on Day 0. Perpendicular tumor diameters were measured with a Vernier caliper, and tumor volumes were calculated as length × width2 × 0.52. Treatment was started on Day 13 when tumors reached the average diameter of 5 mm (approximately 65 mm3 in volume). Radiation was administered to a field including the tumor with <5 mm margins using a 60CO radiation source by two fractions of 12 Gy each on Days 13 and 14 as previously described (25). Control hamster IgG and 9H10 were given i.p. at 200 μg at 1, 4 and 7 days after IR.
Analysis of TIL
Tumors were dissected carefully removing surrounding normal tissue, minced into ~1 mm pieces, and digested with collagenase D (400U/ml) for 25 minutes at 37 °C in a shaker. Aliquots of 106 tumor-derived cells were stained at 4°C with various mAbs and analyzed using a FACScan flow cytometer and FlowJo version 6.4.4 (Tree Star, Ashland, OR).
Adoptive transfer
WT mice bearing 4T1-HA tumors of ~5 mm average diameter were injected i.v. with 20×106 CFSE-labeled in vitro activated CL4 CD8 T cells which express the congenic marker Thy1.1. After 24 h the number of adoptively transferred T cells infiltrating the spleen and tumors was determined as described (33). Briefly, to obtain sufficient cell numbers for analysis tumors from 4 mice per group were pooled and digested with collagenase D as described above. Obtained cell suspensions were stained with PE-Thy1.1 and PE-Cy5-CD8, and analyzed by flow cytometry. An equal number of 5μm polysterene beads (Polysciences, PA) was added to each sample prior to analysis to estimate the total number of CD8+Thy1.1+ cells.
Immunohistochemistry
4T1 tumors were harvested 48 h after IR or mock treatment, fixed for 1 h at 4°C in 4% paraformaldehyde followed by overnight incubation in 30% sucrose, and frozen in OCT medium. 7μm sections were incubated with 0.3% H2O2 to quench endogenous peroxidase activity and stained overnight at 4°C with 1μg/ml polyclonal goat anti-mouse CXCL16 or control Ab followed by FITC-Donkey anti-goat IgG. Subsequently, slides were incubated with peroxidase conjugated anti-FITC Ab (Roche, Indianapolis, IN), visualized with 3,3-diaminobenzidine (DAB Substrate Kit, BD Pharmingen, San Diego, CA), and counterstained with hematoxylin.
RT-PCR and real-time PCR
Total RNA isolated from carcinoma cells with TRIZOL (Invitrogen, CA) was subjected to RT-PCR using specific primers for mouse CXCL16 (forward: 5′ – GCT TTG GAC CCT TGT CTC TTG C – 3′; reverse: 5′ – GTG CTG AGT GCT CTG ACT ATG TGC – 3′); or human CXCL16 (forward: 5′ – GGG GGC AGT CAC CGC AGT CCT– 3′; reverse: 5′ – ATT AGC CGG GTG TGG TGG TGA GCA– 3′).
Real time PCR was performed using SYBR Green Quantitative RT-PCR Kit (SIGMA, MO) and Light Cycler (Roche, IN). The following primers were used: mouse CXCL16 (forward: 5′ – CCT TGT CTC TGG CGT TCT TCC – 3′; reverse: 5 – TCC AAA GTA CCC TGC GGT ATC – 3′); mouse ADAM10 (forward: 5 – AGC AAC ATC TGG GGA CAA AC – 3′; reverse: 5′-TGG CCA GAT TCA ACA AAA CA – 3′); mouse ADAM17 (forward: 5′ – GTA CGT CGA TGC AGA GCA AA – 3′; reverse: 5′ – AAA CCA GAA CAG ACC CAA CG – 3′). The estimated amount of the gene of interest was normalized to the amount of eIF4GII.
Soluble CXCL16 measurement
Mouse and human carcinoma cells were plated at 1× 104 cells/well in duplicate wells of a 96-well plate 48 h after radiation or mock treatment. The medium was changed to DMEM containing 2mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 1% FBS. Some wells contained 200 ng/ml PMA and/or 20 μM BB94 (Batimastat). Released CXCL16 was measured in supernatants after 4 h by ELISA (RayBiotech, Norcross, GA).
CXCL16 knockdown
Lentiviral mediated knockdown of murine CXCL16 was achieved by insertion of a 58-bp DNA duplex oligo containing a specific 21-bp target region directed against the 3′UTR of the CXCL16 gene, via EcoR1-AgeI cloning sites, immediately downstream of a U6, DNA-polIII promoter in the pLKO.1.puro vector, which ultimately generates a shRNA specifically disrupting generation of target protein (sh-CXCL16). A control vector with a 58-bp non-silencing sequence (sh-NS) was also created. Knockdowns were produced by transfection of the 293GP packaging line with the specific pLKO.1 vector, plus pCI-VSV-G and pCMVΔ 8.2R′ to generate active virions. 4T1 cells transduced with control (sh-NS-4T1) and silencing (sh-CXCL16-4T1) were selected in puromycin at 1.5 μg/ml.
Statistical methods
Tumor volume data was acquired longitudinally from WT and CXCR6gfp/gfp mice. Random coefficients regression was used to model the square root of tumor volume as a function of genotype, treatment and elapsed time from treatment onset. The square root of volume was modeled since the temporal change in this measure was well approximated as linear thereby allowing a straightforward assessment of the interaction between treatment and genotype in terms of their effects on tumor growth. The covariance structure was modeled by assuming observations to be correlated only when obtained from the same animal and by allowing the error variance to differ across animal groups. All reported p values are two-sided and were declared significant at the 5% level. Statistical computations were carried out using SAS for Windows, version 9.0 (SAS Institute, Cary, NC).
RESULTS
Local IR increases recruitment of tumor-specific activated CD8 T cells to the tumor
We have previously shown in the mouse 4T1 carcinoma model that local IR used in combination with CTLA-4 blockade promotes immune-mediated tumor destruction (25). To test whether radiation-induced changes in the tumor microenvironment may facilitate recruitment of tumor-specific effector T cells, mice were injected with 4T1-HA cells expressing the reporter antigen HA. HA-specific CD8 T cells were obtained by in vitro activation of CD8 T cells purified from the spleen of CL4 TCR transgenic mice. Following activation with plate-bound anti-CD3 and anti-CD28 mAb and culture in IL-2, the majority of these cells expressed the activation marker CD69, and the chemokine receptor CXCR6, as detected by staining with CXCL16-Fc fusion protein (10), a characteristic of T-cytotoxic 1 (Tc1) effector cells (12) (Figure 1A). Consistent with this, activated CL4 cells exhibited antigen-specific effector functions as demonstrated by γIFN production in response to 4T1-HA, but not 4T1 cells (Figure 1B). Next, activated CL4 T cells were CFSE-labeled before i.v. injection into mice bearing 4T1-HA s.c. tumors 48 h after tumor irradiation or mock treatment. Mice were sacrificed 24 h after CL4 T cell transfer, and spleen and tumors harvested. The number of adoptively transferred T cells infiltrating the spleen and tumors was determined by staining with Thy1.1 and CD8, and flow cytometry. As assessed by CFSE dilution, the transferred CD8 T cells did not proliferate within this time (Figure 1C). Whereas the number of adoptively transferred cells present in the spleen of irradiated and non-irradiated mice was similar (mean 3.2×106 and 3.6 ×106 cells/mouse respectively), irradiated tumors contained almost 2-fold more CD8 T cells (Figure 1D). These results are consistent with previous observations that radiation can enhance T cell infiltration of tumors (22, 23), and suggest that CD8 T cells of Tc-1 type are responsive to changes produced by tumor irradiation.
Figure 1. Irradiation of 4T1 tumors enhances recruitment of adoptively transferred tumor-specific CD8 T cells.
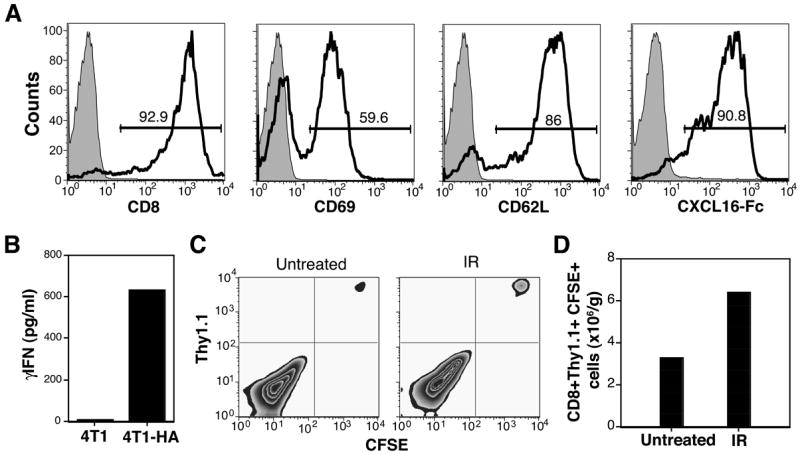
WT mice were injected with 4T1-HA cells. Local IR or mock treatment (Untreated) was delivered to the tumors in two 12 Gy fractions at 24 h interval. 48 h after last IR mice received adoptive transfer of CFSE-labeled in vitro activated CL4 T cells. (A) Phenotype of activated CL4 T cells. Shaded histograms are cells stained with control antibody. Numbers indicate the percentage of cells in the gate. (B) Activated CL4 T cells produced IFNγ in response to 4T1-HA ut not parental 4T1 cells. (C, D) Tumors were harvested 24 h after adoptive transfer, and obtained single cell suspensions stained with PE-Cy5-anti-CD8 and PE-anti-Thy1.1 mAbs to identify the transferred T cells. (C) T cells recovered from the tumors did not proliferate within this time as indicated by lack of CFSE dilution. (D) Recruitment of T cells to tumors was enhanced by radiation. Data are from 4 mice of each group. Errors bars are absent because pooling of tumors within each group was necessary to count the CD8 T cells. Results are representative of two experiments.
The majority of CD8 TIL in 4T1 tumor-bearing mice treated with IR and CTLA-4 blockade expresses CXCR6
The anti-tumor response elicited by treatment with the combination of IR and CTLA-4 blockade in 4T1 tumor-bearing mice is mediated by CD8 T cells (25). To test whether CD8 cells that infiltrate regressing tumors following treatment express CXCR6, we injected 4T1 cells into heterozygous GFP-knockin in the CXCR6 locus (CXCR6+/gfp) mice. In these mice CD8 cells become strongly GFP+ when they up-regulate CXCR6 expression (11). Mice were left untreated or treated with IR delivered exclusively to the tumor and CTLA-4 blocking mAb 9H10 (Figure 2A), and tumor and tumor draining lymph nodes (TDLN) were analyzed at Day 26 when the initial IR-mediated tumor growth delay is followed by an immune-mediated regression phase (Figure 2B). H&E-stained tumor sections showed increased tumor infiltrating lymphocytes (TIL) in treated tumors, many of which were GFP+ by fluorescence microscopy analysis (Figure 2C). Flow cytometric analysis of lymphocytes within dissociated tumors showed that 70–80% and 45–50% of GFP+ TIL were CD8 cells in treated and untreated mice, respectively (not shown). The majority of CD8 cells in tumors, but not in TDLN of treated mice, expressed high levels of CXCR6 (Figure 2D). The CD8+CXCR6+ cells within the tumors were negative for the lymphoid homing receptor CD62L and positive for the activation marker CD69, whereas in TDLN this subset was less activated (Figure 2E). Some of CD8+CXCR6+ cells in TDLN retained high expression of CD62L, although the percentage was lower than in CD8+CXCR6− cells (27% versus 88%, respectively, data not shown), as previously reported (12). Overall, these results suggest that CD8 T cells that home to 4T1 tumors following treatment with IR and CTLA-4 blockade are Tc-1 effectors expressing high levels of CXCR6. In TDLN the CD8+CXCR6+ cells may represent an early effector population (34).
Figure 2. CD8 TIL in regressing 4T1 tumors from CXCR6+/gfp mice treated with local IR and CTLA-4 blockade express CXCR6.

(A) Treatment schedule. (B) Growth of representative tumors from untreated (black squares) and IR+9H10-treated (red diamonds) mice. (C, D) CXCR6+ (as detected by GFP-positivity) TIL in treated tumors. Each tumor was bisected and half was processed for microscopic evaluation whereas the other half was used for flow cytometric analysis. For the latter, tumors from mice in the same group were pooled. (C) Sequential sections of representative tumors from untreated or treated (IR+9H10) mice were stained with H&E (upper panels) or analyzed by fluorescence microscopy for the presence of GFP+ cells (lower panels). In tumors from treated mice residual tumor cells (yellow arrow) were admixed with TIL (green arrow), and many of the TIL were GFP+. Bar, 50 μm. (D, E) Flow cytometric analysis of lymphocytes within dissociated tumors and TD-LN of treated mice. (D) Cells were stained with PE-Cy5-anti-CD8α and gated on CD8+ cells. Numbers indicate the percentage of cells in each gate. (E) Bars show the percentage of CD8+GFP+ cells from tumors (red) and TDLN (black) expressing CD69 or CD62L. Results are from 4 to 8 mice per group, and are representative of 3 experiments.
Radiation induces up-regulation of CXCL16 expression and release by 4T1 tumor cells
Migration of CXCR6+ effector T cells to peripheral sites of inflammation has been shown to be dependent, in part, on up-regulation of CXCR6 ligand, the chemokine CXCL16, in the target tissue (13, 14, 35). To determine whether CXCL16 is expressed in vivo in 4T1 tumors, immunohistochemistry for CXCL16 was performed 48 h after tumor irradiation or mock treatment. In mock-treated 4T1 tumors weak CXCL16 immunoreactivity was detectable in some vessels, with a faint staining in tumor cells (Figure 3A). In contrast, irradiated tumors showed more intense staining of vessels, as well as strong staining in the majority of tumor cells. To confirm expression of CXCL16 by the carcinoma cells, in vitro cultured 4T1 cells were tested by RT-PCR, which showed a clear positive band (not shown, see also Figure 6A).
Figure 3. Release of CXCL16 by 4T1 cells is mediated by a MPase and enhanced by radiation.
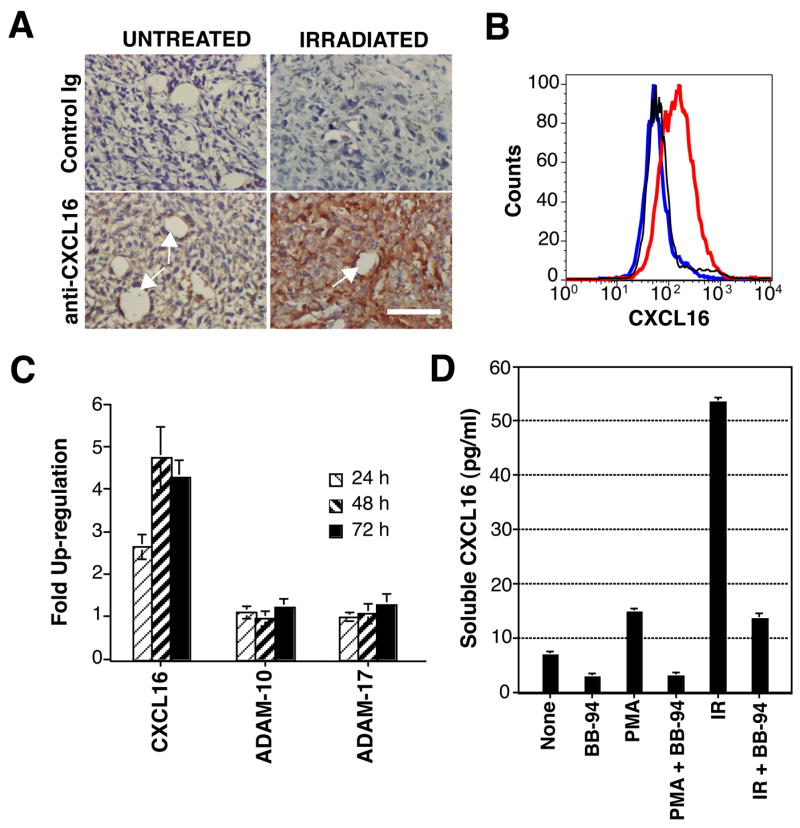
(A) Up-regulation of CXCL16 by local IR of 4T1 tumors in vivo. Tumors were harvested 48 h after IR (IRRADIATED) or mock treatment (UNTREATED) and stained with anti-CXCL16 or control Ig to detect expression of CXCL16. White arrows indicate blood vessels. Bar, 50 μm. (B) MPase inhibition leads to accumulation of CXCL16 molecules on the surface of 4T1 cells. Cells were incubated for 2 h at 37°C in the presence (red line) or absence (blue line) of BB-94 (20 μM), and stained with anti-CXCL16, or with control Ig (black line), followed by FITC-Donkey anti-Goat Ab. (C) Real time RT-PCR measurement of the expression of CXCL16, ADAM-10 and ADAM-17 in 4T1 cells at different times post-radiation. Samples were normalized to eIF4G II, and expression on untreated cells was assigned a relative value of 1.0. Data are expressed as the mean ±SD (n=3). (D) Soluble CXCL16 was measured by ELISA in supernatants of untreated or irradiated (12 Gy, IR) 4T1 cells cultured for 4 h in the presence or absence of MPase activators/inhibitors, as indicated. Data are expressed as the mean ±SD (n=3).
Figure 6. Comparison between WT and CXCR6gfp/gfp mice with established 4T1 carcinoma in the response to treatment with local IR and CTLA-4 blockade.
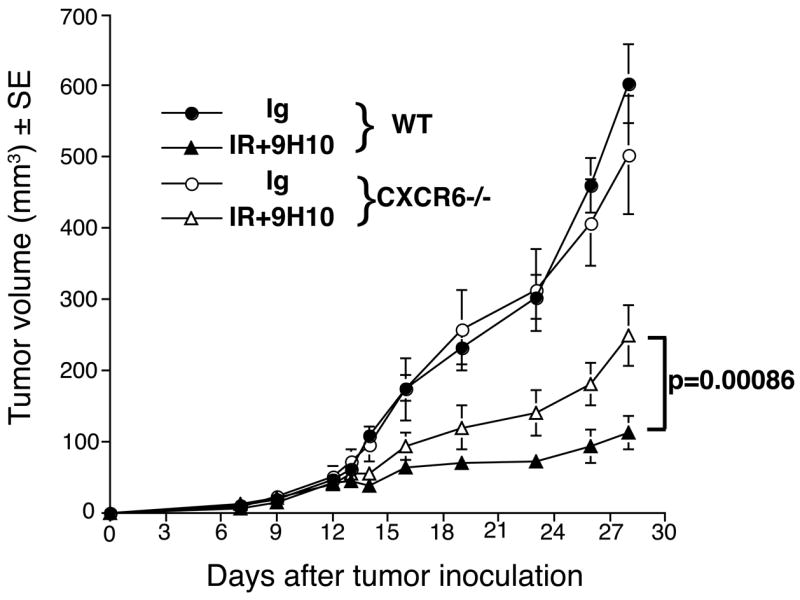
Treatment was started on Day 12 post-s.c. inoculation of 4T1 cells in the flank. RT was delivered in two fractions of 12 Gy to the s.c. tumors on Day 12 and 13. Ab were given i.p. 1, 4 and 7 days post-IR. Tumor volume is shown as the mean ± SE in each treatment group up to Day 28 when all animals were alive. Tumor growth was not significantly different (p=0.9) in WT (full circles, n=8) and CXCR6gfp/gfp (empty circles, n=7) mice receiving the control IgG. In contrast, CXCR6gfp/gfp mice receiving IR+9H10 (empty triangles, n=7) had a significantly (p=0.00086) higher tumor volume than WT mice receiving IR+9H10 (full triangles, n=7). Tumor volume differences between RT+9H10 and control (IgG) mice were statistically significant (p<0.0001) within each genotype.
CXCL16 is expressed as a transmembrane molecule, and is shed in soluble form from the cell surface by the disintegrin-like metalloproteinase (MPase) ADAM10 (36, 37). Therefore, we tested 4T1 cells for expression of surface CXCL16 by immunofluorescence staining and flow cytometry. Surface expression of CXCL16 on 4T1 cells was detected only when its MPase-mediated cleavage was inhibited (Figure 3B), indicating that CXCL16 is rapidly shed in soluble form from these cells.
Next, to investigate the effects of IR on CXCL16 expression, real time RT-PCR was performed at different times after in vitro irradiation of 4T1 cells. Results indicated an over fourfold induction of CXCL16 mRNA peaking at 48 h post-IR (Figure 3C). In contrast, mRNA levels of the MPases ADAM-10 and ADAM-17, implicated in constitutive and PMA-induced cleavage of CXCL16, respectively, (36–38), were unchanged. Measurement of released CXCL16 in 4T1 cell supernatants showed that, as expected, activation of the MPase by PMA enhanced CXCL16 release, whereas incubation with the specific MPase inhibitor BB-94 reduced it (Figure 3D). Remarkably, release of CXCL16 was markedly increased following irradiation of 4T1 cells. The radiation-induced release was inhibited by BB-94. This indicates that the MPase mediated the CXCL16 release.
IR-induced CXCL16 release attracts CXCR6+ cells to the 4T1 tumor cells
Next, chemotaxis assays were performed to determine the ability of CXCL16 released by 4T1 cells to induce the migration of CXCR6+ cells. Baf-3 cells transduced to express the CXCR6 receptor (CXCR6+Baf) migrated in a dose-dependent manner towards rCXCL16 (Figure 4A). Remarkably, the migration of CXCR6+Baf, but not Baf-3 cells, towards irradiated 4T1 cells was increased by almost 8-fold compared to non-irradiated 4T1 cells (Figure 4B), indicating that the enhanced production of CXCL16 by 4T1 cells following IR is functionally relevant. To determine the requirement for CXCL16 for chemotaxis induction, 4T1 cells with selective CXCL16 gene knockdown were generated by transduction of cells with lentivirus short hairpin RNA (shRNA) expression vectors targeting CXCL16 (shCXCL16). Effective knockdown of >90% was detected by RT-PCR and Western Blotting (data not shown) and shCXCL16-4T1 cells showed markedly reduced ability to release soluble CXCL16 and to induce chemotaxis of CXCR6+Baf cells (Figure 4C and D).
Figure 4. CXCL16/CXCR6 interactions regulate the migration of CD8 activated T cells to irradiated 4T1 cancer cells in vitro.
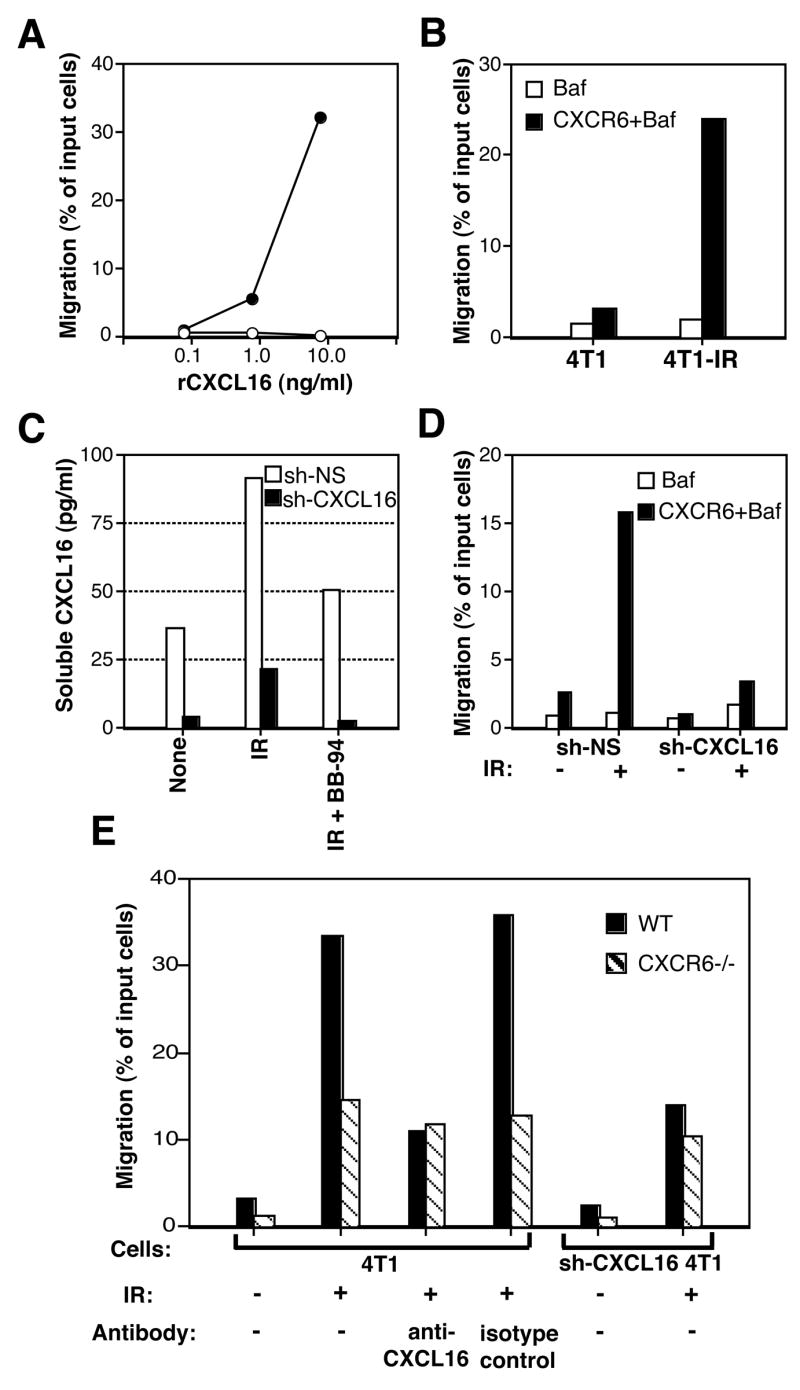
Results of migration assays are presented as the percentage of input cells migrating to the lower chamber of a transwell filter. (A) Response of Baf-3 (empty circles) and CXCR6+Baf-3 (full circles) cells to various concentrations of rCXCL16. (B) Migration of Baf (white bars) and CXCR6+Baf-3 (black bars) cells towards untreated (4T1) or irradiated with 12 Gy 48 h earlier (4T1-IR) 4T1 cells. (C) 4T1 cells transduced with control (sh-NS, white bars) and silencing (sh-CXCL16, black bars) retroviral vectors were left untreated (None) or irradiated (IR). CXCL16 release was tested in supernatants after 4 h culture in the presence or absence of BB-94. (D) Migration of Baf (white bars) and CXCR6+Baf-3 (black bars) to 4T1 cells transduced with sh-NS and sh-CXCL16. (E) Migration of in vitro activated CD8 T cells from WT (black bars) and CXCR6gfp/gfp (CXCR6−/−, shaded bars) mice to 4T1 and sh-CXCL16-4T1 cells, untreated (−) or irradiated with 12 Gy 48 hours earlier (+). In some wells 1 μg/ml of anti-CXCL16 or isotype control Ab was added, as indicated. Data are representative of two experiments.
To determine if migration of activated CD8 cells to 4T1 cells is enhanced by radiation, CD8 cells were purified from the spleen of naïve WT and CXCR6gfp/gfp (CXCR6−/−) mice. Cells were activated in vitro with plate-bound anti-CD3 and anti-CD28 mAb and culture in IL-2, and expression of CXCR6 was confirmed by staining with CXCL16-Fc fusion protein (see Figure 1A), and found to be >90% on cells from WT mice, whereas a comparable percentage of CXCR6−/− cells was GFP+. Migration of CD8 cells of both genotypes towards untreated 4T1 cells was minimal, although higher for WT cells (~3% versus 1%) (Figure 4E). Migration of WT CD8 cells towards irradiated 4T1 cells was increased by over 10-fold. This increased migration was markedly reduced by incubation with CXCL16 blocking antibody and by CXCL16 knockdown (Figure 4E). Interestingly, although the migration of CXCR6−/− CD8 cells towards irradiated 4T1 cells was also increased, it was markedly lower as compared to WT cells. Blocking CXCL16 reduced the migration of WT CD8 cells to the level observed for CXCR6−/−CD8 cells. Taken together, these data indicate that CXCR6/CXCL16 interactions drive, in large part, the migration of CD8 cells towards irradiated 4T1 carcinoma cells, while additional chemotactic factors may be induced by radiation and contribute to increased T cell migration.
Efficient tumor infiltration by effector CD8 cells requires expression of a functional CXCR6 receptor
To determine whether in vivo CXCR6/CXCL16 interactions can regulate the infiltration of 4T1 tumors by effector CD8 cells elicited by treatment with IR and CTLA-4 blockade, CXCR6+/gfp and CXCR6gfp/gfp mice were injected with 4T1 cells and treated as shown in Figure 2A. The percentage of the CD4 and CD8 T cell subsets in TDLN of CXCR6+/gfp and CXCR6gfp/gfp mice was similar (data not shown). In contrast, the absolute number of CD8, but not CD4, TIL was markedly lower in treated CXCR6gfp/gfp as compared to CXCR6+/gfp mice (Figure 5A). The majority of CD8 TIL was positive for GFP and for the activation marker CD69 in both CXCR6+/gfp and CXCR6gfp/gfp mice (Figure 5B), suggesting that CD8 T cells in CXCR6gfp/gfp mice are not defective in the ability to become activated. In mice of both genotypes, the majority of TIL expressing GFP were CD8+ cells (80 and 63% in CXCR6+/gfp and CXCR6gfp/gfp mice, respectively) with only a small percentage of CD4+ cells (13 and 23% in CXCR6+/gfp and CXCR6gfp/gfp mice, respectively).
Figure 5. CXCR6-deficient mice cannot increase CD8 TIL following treatment with IR and CTLA-4 blockade.

CXCR6+/gfp (white bars) and CXCR6gfp/gfp (black bars) mice were injected s.c. with 4T1 cells and left untreated or treated as described in figure 2A. On Day 26 tumors and TDLN were collected and obtained single cell suspensions stained with PE-Cy5-anti-CD3, and PE-anti-CD8 or anti-CD4 mAbs, followed by flow cytometry analysis. The lymphocyte gate was set based on the scattered plots in TDLN. (A) The percentage of cells in the lymphocyte gate positive for CD3, CD3 and CD8 (CD8), CD3 and CD4 (CD4), and GFP was multiplied for the percentage of cells in the lymphocyte gate and for the total number of viable cells isolated from the tumors, and divided for the tumor weight to obtain the number of cells per mg of tumor. (B) TIL isolated from treated mice were gated on CD8+ cells and analyzed for the expression of GFP and CD69, as indicated. Numbers are the percentage of cells in each quadrant. (C) For each marker, the percentage of positive cells in the lymphocyte gate in tumors isolated from treated mice was divided by the percentage of cells isolated from untreated mice. A ratio of one (line) indicates no change, > 1 indicates an increase in treated tumors, and < 1 indicates a decrease in treated tumors. Data are the mean of 8 mice of each genotype per treatment group. Errors bars are absent because pooling of tumors within each group was necessary to count the T cells. Results are representative of two experiments.
To determine whether similar changes in TIL are induced by treatment in CXCR6+/gfp and CXCR6gfp/gfp mice, the percentage of TIL positive for each marker in treated versus untreated mice of each genotype was compared. As shown in Figure 5C, in treated mice of both genotypes, there was a reduction in CD4 TIL. In contrast, the treatment-induced increase in CD8 TIL observed in CXCR6+/gfp was not detected in CXCR6gfp/gfp mice, suggesting that a functional CXCR6 receptor is required for accumulation of effector CTL in irradiated tumors.
Impaired tumor inhibition in CXCR6-deficient mice treated with IR and CTLA-4 blockade
To determine whether the reduced tumor infiltration by CD8 effectors in CXCR6gfp/gfp mice reflected in reduced treatment-induced tumor inhibition, WT and CXCR6gfp/gfp mice were injected with 4T1 cells and left untreated or treated with IR and CTLA-4 blockade starting on Day 12 post-injection. There was no significant difference in tumor growth (p=0.9) between WT and CXCR6gfp/gfp mice receiving control Ig (Figure 6). Treatment with IR and 9H10 significantly reduced the rate of tumor growth among animals of both genotypes (p<0.0001). However, tumor growth in CXCR6gfp/gfp mice receiving IR+9H10 was significantly higher than in WT mice (p=0.00086). The ability of treatment to cause tumor growth inhibition was significantly weaker (p=0.017) in CXCR6gfp/gfp as compared to WT mice.
Overall, theses data indicate that CXCR6/CXCL16 interactions regulate the recruitment of 9H10-activated CD8 cells to irradiated tumors, and the overall tumor inhibition.
Expression of CXCL16 by mouse and human breast cancer cells is common
To determine whether the expression of CXCL16 and its response to radiation are common among epithelial malignancies of the breast, four additional mouse mammary carcinoma cell lines of differing metastatic potential (26) were tested by RT-PCR. In addition to the very aggressive and highly metastatic 4T1 cells, CXCL16 was found to be expressed by the non-metastatic 67NR cells and by the weakly metastatic 4T07 cells (Figure 7A). 67NR and 4T07 cells responded to irradiation similarly to 4T1 cells, by up-regulating the production of soluble CXCL16 (Figure 7B).
Figure 7. Expression of CXCL16 is common in mouse and human breast cancer lines.
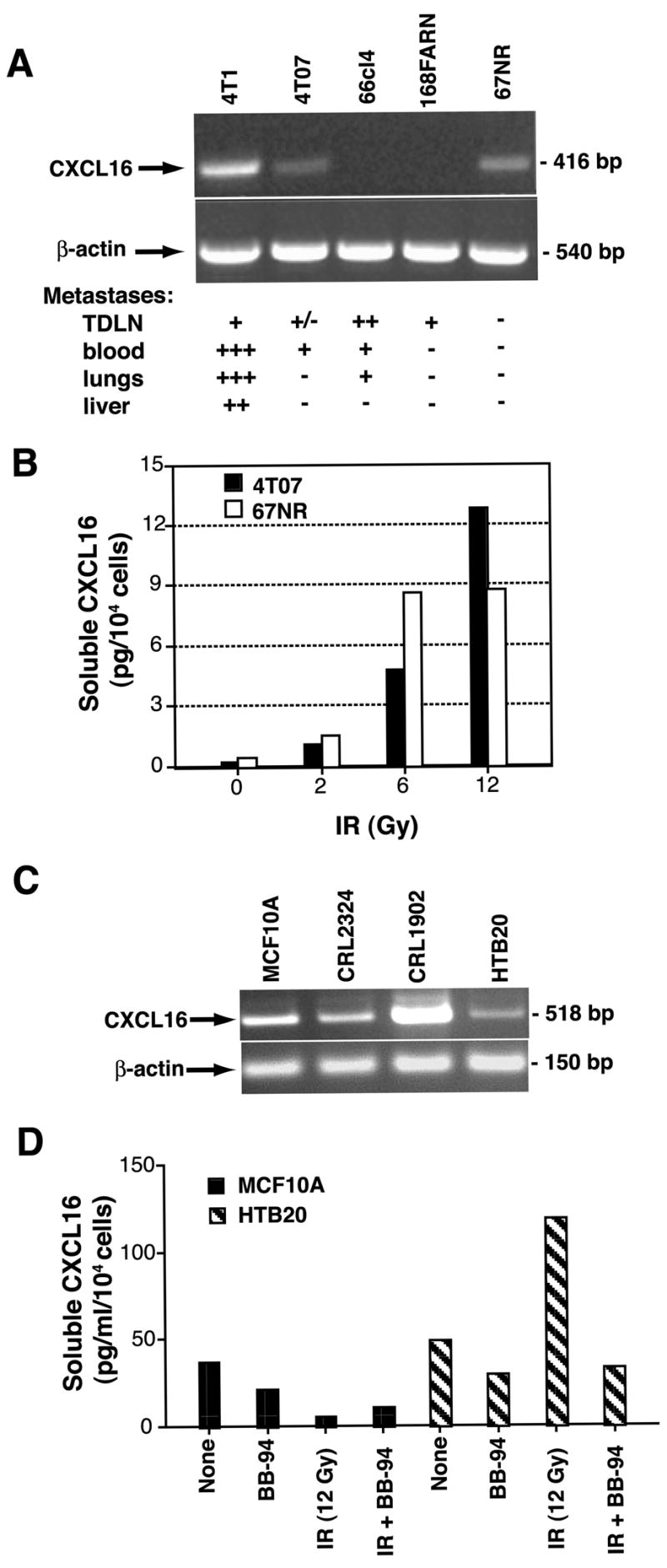
(A) CXCL16 was expressed in 3 out of 5 mouse mammary carcinoma lines of differing metastatic potential (26) tested by RT-PCR. (B) IR enhanced in a dose-dependent fashion the release of soluble CXCL16 by the non-metastatic 67NR and the metastatic 4T07 cells. (C) CXCL16 was expressed in 4 human cell lines derived from primary breast specimens of benign breast (MCF 10A) or invasive breast cancer (CRL-2324, CRL-1902, and HTB-20) tested by RT-PCR. (D) Soluble CXCL16 was measured by ELISA in supernatants of untreated or irradiated (12 Gy, IR) MCF 10A and HTB-20 cells cultured for 6 h in the presence or absence of MPase inhibitor BB-94, as indicated. Radiation increased the release of CXCL16 by the HTB-20 tumor cells but not by the epithelial MCF 10A cells derived from benign breast tissue. Data are the mean of duplicate wells and are representative of two experiments.
Next, four human breast epithelial cell lines were tested and found to be positive for expression of CXCL16 by RT-PCR (Figure 7C). The MCF10A cells were derived by cloning of spontaneously immortalized epithelial cells from a patient with fibrocystic disease and resemble normal ductal epithelium (39), whereas the other three lines were derived from primary invasive breast cancers (40). Both MCF10A and HTB20 showed MPase-mediated release of soluble CXCL16, but only HTB20 responded to IR with increased release (Figure 7D). Overall, these data suggest that expression of CXCL16 by breast epithelial cells is common, and the response to radiation may be influenced by neoplastic transformation.
DISCUSSION
In this study, we show for the first time that mouse and human breast cancer cells express the chemokine CXCL16, and that IR strikingly up-regulates its expression and release. CXCL16, the only known ligand for CXCR6, is one of only two chemokines that are expressed as transmembrane molecules (10). The transmembrane form can mediate adhesion to CXCR6+ cells, as well as function as a scavenger receptor for oxidized low density lipoproteins, phosphatidylserine, bacteria and dextran sulfate (37, 41). The chemokine domain of CXCL16 is cleaved from the cell surface by the activity of the MPase ADAM10 (36). Although we cannot completely exclude that the transmembrane form plays some role as a receptor on 4T1 cells, analysis of cell surface expression showed that CXCL16 was undetectable unless the MPase activity was inhibited (Figure 3B), suggesting that it does not accumulate on the surface of the cancer cells in significant quantities.
Soluble CXCL16 has been shown to induce strong chemotaxis of activated CD8 T cells, which express high levels of CXCR6, and to a lesser degree of activated CD4 cells, which have lower levels of the receptor (10). Consistent with this, our data showed that irradiated 4T1 tumor cells induced strong, CXCL16-dependent chemotaxis of activated CD8 cells in vitro (Figure 4E), and that local tumor irradiation in vivo enhanced the recruitment of tumor-specific CXCR6+ CD8 cells (Figure 1). Importantly, comparison between CXCR6+/gfp and CXCR6gfp/gfp mice indicated that CXCR6 expression by CD8 cells is required for efficient infiltration of 4T1 tumors (Figure 5). Since endogenous CD8 cell activation was triggered in these mice by treatment with IR and CTLA-4 blockade, it cannot be excluded that CXCR6-deficient T cells were impaired in their ability to become activated. However, this is not very likely since TIL from CXCR6+/gfp and CXCR6gfp/gfp mice expressed similar levels of the activation marker CD69 (Figure 5B), and CXCR6-deficiency has been shown to decrease homing to inflamed organs, but not CD8 cell activation in other experimental systems (14).
CXCL16 is known to be expressed by dendritic cells, macrophages, and some endothelia, and to be up-regulated during inflammation in different tissues and organs (13, 14, 35, 42). In a recent report Hojo et al., have shown that human colon carcinoma cells also express CXCL16 (43). Importantly, patients with tumors expressing higher levels of CXCL16 had increased TIL, and a significantly better prognosis than patients with low CXCL16 expression in the tumor (43), suggesting that high expression of CXCL16 enhances the effectiveness of immune-mediated tumor control by improving recruitment of anti-tumor T cells. Our data presented here provide support for this hypothesis by showing that the increased CXCL16 expression induced by IR in breast carcinoma cells enhanced recruitment of anti-tumor CD8 cells, and that in mice deficient in CXCR6 expression tumor inhibition was less efficient than in WT mice.
It remains to be determined if malignancies other than breast and colon cancer express CXCL16 or other chemokines capable of recruiting Th1 and Tc-1 cells. The ability of radiation to enhance, albeit to a lesser extent, the migration of CXCR6-deficient CD8 cells towards 4T1 cancer cells (Figure 4E) suggests that additional chemokines are induced by IR. The observation that pre-operative IR correlates with enhanced infiltration of oral squamous cell carcinoma by activated CD8 cells (44) supports the hypothesis that induction of T cell chemotactic factors may be a relatively common component of the stress response that follows radiation exposure of neoplastic cells. Interestingly, MCF10A cells, which are derived from normal breast epithelium, did not increase CXCL16 release in response to IR (Figure 7D). However, it remains to be determined if this differential response is a characteristic of non-neoplastic cells. Up-regulation of some pro-inflammatory chemokines has been reported in normal lung and skin after IR at doses that cause inflammation and fibrosis in susceptible mice strains (45, 46). However, it is unclear if this is a primary effect of IR, or it is secondary to other inflammatory stimuli, and which cell type is responsible for chemokine production.
It is intriguing that CXCL16 has been reported to stimulate proliferation, chemotaxis, and tube formation in human umbilical vein endothelial cells, suggesting that it may act as an angiogenic factor (47). Since increased secretion of pro-angiogenic factors is a major part of the response of neoplastic cells to radiation (48), it is possible that CXCL16 plays a dual role in tumor growth by contributing to angiogenesis while at the same time attracting anti-tumor T cells. Improved understanding of the molecular changes that are induced by IR in tumors opens the possibility of exploiting the tumor response to radiation with a timely immunotherapeutic intervention that, by enhancing anti-tumor T cell activation, will contribute to immune-mediated tumor destruction.
The molecular mechanism(s) responsible for up-regulation of CXCL16 following IR of breast cancer cells are presently undefined. A possible candidate is the PI3K/Akt signaling pathway, which is activated by IR in tumor and endothelial cells (49). This pathway promotes survival of tumor cells, and has been recently linked to induction of CXCL16 in a mouse model of mammary tumorigenesis (50).
IR is a standard treatment modality in oncology, and can now be very precisely targeted to tumors (51) minimizing immunosuppressive side effects, an advantage to systemic chemotherapy. Recently, the possibility of combining local IR with IT has been explored by us and others in pre-clinical and some clinical studies (reviewed in references 20 and 21). IR has been shown to induce changes in both, the cancer and tumor stromal cells that survive, that promote their recognition by anti-tumor CD8 cells (52–56). Here we provide evidence for another mechanism that enhances immune-mediated tumor destruction, the IR-induced up-regulation of a chemokine recruiting activated T cells to the tumor. We propose that IR should be further considered as an easily translatable strategy to overcome immune barriers at the effector phase of the anti-tumor immune response (57).
Acknowledgments
We thank Dan Littman for CXCR6gfp/gfp mice, Mehrdad Matloubian for the CXCR6-Fc construct, James P. Allison for 9H10 antibody, Fred Miller for mouse breast cancer cell lines, Ivan Borrello for 4T1-HA cells, and the personnel of NYU Cancer Institute Flow Cytometry facility for expert assistance. We are grateful to William H. McBride for critical reading of the manuscript.
Footnotes
This work was supported by NIH R01 CA113851, Research Scholar award RSG-05-145-01-LIB from the American Cancer Society, and by a grant from The Chemotherapy Foundation, (to S. Demaria). Additional support was provided by NIH R01AI55037 (to M.L. Dustin) and Cancer Research Institute post-doctoral fellowship (to T.O. Cameron). NYU Cancer Institute is supported by NIH 5P30CA016087-27.
Non Standard Abbreviations: HA, hemagglutinin; IR, ionizing radiation therapy; IT, immunotherapy; MPase, metalloproteinase; Tc1, T-cytotoxic 1; TDLN, tumor-draining lymph nodes; TIL, tumor infiltrating lymphocytes; shRNA, short hairpin RNA.
DISCLOSURES. The authors have no financial conflict of interests.
Publisher's Disclaimer: This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institute of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.
Contributor Information
Satoko Matsumura, Department of Pathology, New York University School of Medicine, 550 First Avenue, New York, NY, 10016.
Baomei Wang, Department of Pathology, New York University School of Medicine, 550 First Avenue, New York, NY, 10016.
Noriko Kawashima, Department of Pathology, New York University School of Medicine, 550 First Avenue, New York, NY, 10016.
Steve Braunstein, Department of Microbiology, New York University School of Medicine, 550 First Avenue, New York, NY, 10016.
Michelle Badura, Department of Microbiology, New York University School of Medicine, 550 First Avenue, New York, NY, 10016.
Thomas O. Cameron, Department of Pathology, New York University School of Medicine, 550 First Avenue, New York, NY, 10016, and the Program in Molecular Pathogenesis, The Helen L. and Martin S. Kimmel Center for Biology and Medicine at the Skirball Institute for Biomolecular Medicine, 540 First Avenue, NY 10016
James S. Babb, Department of Radiology, New York University School of Medicine, 550 First Avenue, New York, NY, 10016
Robert J. Schneider, Department of Microbiology, New York University School of Medicine, 550 First Avenue, New York, NY, 10016
Silvia C. Formenti, Department of Radiation Oncology, New York University School of Medicine, 550 First Avenue, New York, NY, 10016
Michael L. Dustin, Department of Pathology, New York University School of Medicine, 550 First Avenue, New York, NY, 10016, and the Program in Molecular Pathogenesis, The Helen L. and Martin S. Kimmel Center for Biology and Medicine at the Skirball Institute for Biomolecular Medicine, 540 First Avenue, NY 10016
Sandra Demaria, Department of Pathology, New York University School of Medicine, 550 First Avenue, New York, NY, 10016.
References
- 1.Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer. 2005;5:615–625. doi: 10.1038/nrc1669. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg SA, Sherry RM, Morton KE, Scharfman WJ, Yang JC, Topalian SL, Royal RE, Kammula U, Restifo NP, Hughes MS, Schwartzentruber D, Berman DM, Schwarz SL, Ngo LT, Mavroukakis SA, White DE, Steinberg SM. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol. 2005;175:6169–6176. doi: 10.4049/jimmunol.175.9.6169. [DOI] [PubMed] [Google Scholar]
- 3.Gajewski TF, Meng Y, Blank C, Brown I, Kacha A, Kline J, Harlin H. Immune resistance orchestrated by the tumor microenvironment. Immunol Rev. 2006;213:131–145. doi: 10.1111/j.1600-065X.2006.00442.x. [DOI] [PubMed] [Google Scholar]
- 4.Jain RK. Transport of molecules, particles, and cells in solid tumors. Annu Rev Biomed Eng. 1999;1:241–263. doi: 10.1146/annurev.bioeng.1.1.241. [DOI] [PubMed] [Google Scholar]
- 5.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, Heimann DM, Klebanoff CA, Yu Z, Hwang LN, Feigenbaum L, Kruisbeek AM, Rosenberg SA, Restifo NP. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carlos TM. Leukocyte recruitment at sites of tumor: dissonant orchestration. J Leukoc Biol. 2001;70:171–184. [PubMed] [Google Scholar]
- 7.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frey AB, Monu N. Effector-phase tolerance: another mechanism of how cancer escapes antitumor immune response. J Leukoc Biol. 2006;79:652–662. doi: 10.1189/jlb.1105628. [DOI] [PubMed] [Google Scholar]
- 9.Kunkel EJ, Butcher EC. Chemokines and the tissue-specific migration of lymphocytes. Immunity. 2002;16:1–4. doi: 10.1016/s1074-7613(01)00261-8. [DOI] [PubMed] [Google Scholar]
- 10.Matloubian M, David A, Engel S, Ryan JE, Cyster JG. A transmembrane CXC chemokine is a ligand for HIV-coreceptor Bonzo. Nat Immunol. 2000;1:298–304. doi: 10.1038/79738. [DOI] [PubMed] [Google Scholar]
- 11.Unutmaz D, Xiang W, Sunshine MJ, Campbell J, Butcher E, Littman DR. The primate lentiviral receptor Bonzo/STRL33 is coordinately regulated with CCR5 and its expression pattern is conserved between human and mouse. J Immunol. 2000;165:3284–3292. doi: 10.4049/jimmunol.165.6.3284. [DOI] [PubMed] [Google Scholar]
- 12.Kim CH, Kunkel EJ, Boisvert J, Johnston B, Campbell JJ, Genovese MC, Greenberg HB, Butcher EC. Bonzo/CXCR6 expression defines type 1-polarized T-cell subsets with extralymphoid tissue homing potential. J Clin Invest. 2001;107:595–601. doi: 10.1172/JCI11902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamauchi R, Tanaka M, Kume N, Minami M, Kawamoto T, Togi K, Shimaoka T, Takahashi S, Yamaguchi J, Nishina T, Kitaichi M, Komeda M, Manabe T, Yonehara S, Kita T. Upregulation of SR-PSOX/CXCL16 and recruitment of CD8+ T cells in cardiac valves during inflammatory valvular heart disease. Arterioscler Thromb Vasc Biol. 2004;24:282–287. doi: 10.1161/01.ATV.0000114565.42679.c6. [DOI] [PubMed] [Google Scholar]
- 14.Sato T, Thorlacius H, Johnston B, Staton TL, Xiang W, Littman DR, Butcher EC. Role for CXCR6 in recruitment of activated CD8+ lymphocytes to inflamed liver. J Immunol. 2005;174:277–283. doi: 10.4049/jimmunol.174.1.277. [DOI] [PubMed] [Google Scholar]
- 15.Nanki T, Shimaoka T, Hayashida K, Taniguchi K, Yonehara S, Miyasaka N. Pathogenic role of the CXCL16-CXCR6 pathway in rheumatoid arthritis. Arthritis Rheum. 2005;52:3004–3014. doi: 10.1002/art.21301. [DOI] [PubMed] [Google Scholar]
- 16.Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- 17.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 18.Luster AD, Leder P. IP-10, a -C-X-C- chemokine, elicits a potent thymus-dependent antitumor response in vivo. J Exp Med. 1993;178:1057–1065. doi: 10.1084/jem.178.3.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mule JJ, Custer M, Averbook B, Yang JC, Weber JS, Goeddel DV, Rosenberg SA, Schall TJ. RANTES secretion by gene-modified tumor cells results in loss of tumorigenicity in vivo: role of immune cell subpopulations. Hum Gene Ther. 1996;7:1545–1553. doi: 10.1089/hum.1996.7.13-1545. [DOI] [PubMed] [Google Scholar]
- 20.Demaria S, Bhardwaj N, McBride WH, Formenti SC. Combining radiotherapy and immunotherapy: a revived partnership. Int J Radiat Oncol Biol Phys. 2005;63:655–666. doi: 10.1016/j.ijrobp.2005.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Demaria S, Formenti SC. Sensors of ionizing radiation effects on the immunological microenvironment of cancer. Int J Radiat Biol. 2007;83:1–7. doi: 10.1080/09553000701481816. [DOI] [PubMed] [Google Scholar]
- 22.Ganss R, Ryschich E, Klar E, Arnold B, Hammerling GJ. Combination of T-cell therapy and trigger of inflammation induces remodeling of the vasculature and tumor eradication. Cancer Res. 2002;62:1462–1470. [PubMed] [Google Scholar]
- 23.Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol. 2005;174:7516–7523. doi: 10.4049/jimmunol.174.12.7516. [DOI] [PubMed] [Google Scholar]
- 24.Hallahan D, Kuchibhotla J, Wyble C. Cell adhesion molecules mediate radiation-induced leukocyte adhesion to the vascular endothelium. Cancer Res. 1996;56:5150–5155. [PubMed] [Google Scholar]
- 25.Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, Formenti SC. Immune-mediated inhibition of metastases following treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res. 2005;11:728–734. [PubMed] [Google Scholar]
- 26.Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992;52:1399–1405. [PubMed] [Google Scholar]
- 27.Lin P, Buxton JA, Acheson A, Radziejewski C, Maisonpierre PC, Yancopoulos GD, Channon KM, Hale LP, Dewhirst MW, George SE, Peters KG. Antiangiogenic gene therapy targeting the endothelium-specific receptor tyrosine kinase Tie2. Proc Natl Acad Sci U S A. 1998;95:8829–8834. doi: 10.1073/pnas.95.15.8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 29.Morgan DJ, Liblau R, Scott B, Fleck S, McDevitt HO, Sarvetnick N, Lo D, Sherman LA. CD8(+) T cell-mediated spontaneous diabetes in neonatal mice. J Immunol. 1996;157:978–983. [PubMed] [Google Scholar]
- 30.Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, Dolcetti L, Bronte V, Borrello I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. 2006;203:2691–2702. doi: 10.1084/jem.20061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ngo VN, Tang HL, Cyster JG. Epstein-Barr virus-induced molecule 1 ligand chemokine is expressed by dendritic cells in lymphoid tissues and strongly attracts naive T cells and activated B cells. J Exp Med. 1998;188:181–191. doi: 10.1084/jem.188.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petersen CC, Petersen MS, Agger R, Hokland ME. Accumulation in tumor tissue of adoptively transferred T cells: A comparison between intravenous and intraperitoneal injection. J Immunother. 2006;29:241–249. doi: 10.1097/01.cji.0000203078.97493.c3. [DOI] [PubMed] [Google Scholar]
- 34.Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, Finkelstein SE, Theoret MR, Rosenberg SA, Restifo NP. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. 2005;115:1616–1626. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van der Voort R, van Lieshout AW, Toonen LW, Slöetjes AW, van den Berg WB, Figdor CG, Radstake TR, Adema GJ. Elevated CXCL16 expression by synovial macrophages recruits memory T cells into rheumatoid joints. Arthritis Rheum. 2005;52:1381–1391. doi: 10.1002/art.21004. [DOI] [PubMed] [Google Scholar]
- 36.Abel S, Hundhausen C, Mentlein R, Schulte A, Berkhout TA, Broadway N, Hartmann D, Sedlacek R, Dietrich S, Muetze B, Schuster B, Kallen KJ, Saftig P, Rose-John S, Ludwig A. The transmembrane CXC-chemokine ligand 16 is induced by IFN-gamma and TNF-alpha and shed by the activity of the disintegrin-like metalloproteinase ADAM10. J Immunol. 2004;172:6362–6372. doi: 10.4049/jimmunol.172.10.6362. [DOI] [PubMed] [Google Scholar]
- 37.Shimaoka T, Nakayama T, Kume N, Takahashi S, Yamaguchi J, Minami M, Hayashida K, Kita T, Ohsumi J, Yoshie O, Yonehara S. Cutting edge: SR-PSOX/CXC chemokine ligand 16 mediates bacterial phagocytosis by APCs through its chemokine domain. J Immunol. 2003;171:1647–1651. doi: 10.4049/jimmunol.171.4.1647. [DOI] [PubMed] [Google Scholar]
- 38.Ludwig A, Schulte A, Schnack C, Hundhausen C, Reiss K, Brodway N, Held-Feindt J, Mentlein R. Enhanced expression and shedding of the transmembrane chemokine CXCL16 by reactive astrocytes and glioma cells. J Neurochem. 2005;93:1293–1303. doi: 10.1111/j.1471-4159.2005.03123.x. [DOI] [PubMed] [Google Scholar]
- 39.Soule HD, Maloney TM, Wolman SR, Peterson WDJ, Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50:6075–6086. [PubMed] [Google Scholar]
- 40.Zimonjic DB, Keck-Waggoner CL, Yuan BZ, Kraus MH, Popescu NC. Profile of genetic alterations and tumorigenicity of human breast cancer cells. Int J Oncol. 2000;16:221–230. doi: 10.3892/ijo.16.2.221. [DOI] [PubMed] [Google Scholar]
- 41.Shimaoka T, Kume N, Minami M, Hayashida K, Kataoka H, Kita T, Yonehara S. Molecular cloning of a novel scavenger receptor for oxidized low density lipoprotein, SR-PSOX, on macrophages. J Biol Chem. 2000;275:40663–40666. doi: 10.1074/jbc.C000761200. [DOI] [PubMed] [Google Scholar]
- 42.Geissmann F, Cameron TO, Sidobre S, Manlongat N, Kronenberg M, Briskin MJ, Dustin ML, Littman DR. Intravascular immune surveillance by CXCR6+ NKT cells patrolling liver sinusoids. PLoS Biol. 2005;3:e113. doi: 10.1371/journal.pbio.0030113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hojo S, Koizumi K, Tsuneyama K, Arita Y, Cui Z, Shinohara K, Minami T, Hashimoto I, Nakayama T, Sakurai H, Takano Y, Yoshie O, Tsukada K, Saiki I. High-level expression of chemokine CXCL16 by tumor cells correlates with a good prognosis and increased tumor-infiltrating lymphocytes in colorectal cancer. Cancer Res. 2007;67:4725–4731. doi: 10.1158/0008-5472.CAN-06-3424. [DOI] [PubMed] [Google Scholar]
- 44.Suwa T, Saio M, Umemura N, Yamashita T, Toida M, Shibata T, Takami T. Preoperative radiotherapy contributes to induction of proliferative activity of CD8+ tumor-infiltrating T-cells in oral squamous cell carcinoma. Oncol Rep. 2006;15:757–763. [PubMed] [Google Scholar]
- 45.Johnston CJ, Williams JP, Okunieff P, Finkelstein JN. Radiation-induced pulmonary fibrosis: examination of chemokine and chemokine receptor families. Radiat Res. 2002;157:256–265. doi: 10.1667/0033-7587(2002)157[0256:ripfeo]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 46.Liang L, Hu D, Liu W, Williams JP, Okunieff P, Ding I. Celecoxib reduces skin damage after radiation: selective reduction of chemokine and receptor mRNA expression in irradiated skin but not in irradiated mammary tumor. Am J Clin Oncol. 2003;26:S114–121. doi: 10.1097/01.COC.0000074149.95710.40. [DOI] [PubMed] [Google Scholar]
- 47.Zhuge X, Murayama T, Arai H, Yamauchi R, Tanaka M, Shimaoka T, Yonehara S, Kume N, Yokode M, Kita T. CXCL16 is a novel angiogenic factor for human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2005;331:1295–1300. doi: 10.1016/j.bbrc.2005.03.200. [DOI] [PubMed] [Google Scholar]
- 48.Moeller BJ, Dewhirst MW. Raising the bar: how HIF-1 helps determine tumor radiosensitivity. Cell Cycle. 2004;3:1107–1110. [PubMed] [Google Scholar]
- 49.Zingg D, Riesterer O, Fabbro D, Glanzmann C, Bodis S, Pruschy M. Differential activation of the phosphatidylinositol 3′-kinase/Akt survival pathway by ionizing radiation in tumor and primary endothelial cells. Cancer Res. 2004;64:5398–5406. doi: 10.1158/0008-5472.CAN-03-3369. [DOI] [PubMed] [Google Scholar]
- 50.Ju X, Katiyar S, Wang C, Liu M, Jiao X, Li S, Zhou J, Turner J, Lisanti MP, Russell RG, Mueller SC, Ojeifo J, Chen WS, Hay N, Pestell RG. Akt1 governs breast cancer progression in vivo. Proc Natl Acad Sci U S A. 2007;104:7438–7443. doi: 10.1073/pnas.0605874104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verellen D, Ridder MD, Linthout N, Tournel K, Soete G, Storme G. Innovations in image-guided radiotherapy. Nat Rev Cancer. 2007;7:949–960. doi: 10.1038/nrc2288. [DOI] [PubMed] [Google Scholar]
- 52.Chakraborty M, Abrams SI, Coleman CN, Camphausen K, Schlom J, Hodge JW. External beam radiation of tumors alters phenotype of tumor cells to render them susceptible to vaccine-mediated T-cell killing. Cancer Res. 2004;64:4328–4337. doi: 10.1158/0008-5472.CAN-04-0073. [DOI] [PubMed] [Google Scholar]
- 53.Garnett CT, Palena C, Chakarborty M, Tsang KY, Schlom J, Hodge JW. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res. 2004;64:7985–7994. doi: 10.1158/0008-5472.CAN-04-1525. [DOI] [PubMed] [Google Scholar]
- 54.Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, Camphausen K, Luiten RM, de Ru AH, Neijssen J, Griekspoor A, Mesman E, Verreck FA, Spits H, Schlom J, van Veelen P, Neefjes JJ. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203:1259–1271. doi: 10.1084/jem.20052494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Newcomb EW, Demaria S, Lukyanov Y, Shao Y, Schnee T, Kawashima N, Lan L, Dewyngaert JK, Zagzag D, McBride WH, Formenti SC. The combination of ionizing radiation and peripheral vaccination produces long-term survival of mice bearing established invasive GL261 gliomas. Clin Cancer Res. 2006;12:4730–4737. doi: 10.1158/1078-0432.CCR-06-0593. [DOI] [PubMed] [Google Scholar]
- 56.Zhang B, Bowerman NA, Salama JK, Schmidt H, Spiotto MT, Schietinger A, Yu P, Fu YX, Weichselbaum RR, Rowley DA, Kranz DM, Schreiber H. Induced sensitization of tumor stroma leads to eradication of established cancer by T cells. J Exp Med. 2007;204:49–55. doi: 10.1084/jem.20062056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gajewski TF. Failure at the effector phase: immune barriers at the level of the melanoma tumor microenvironment. Clin Cancer Res. 2007;13:5256–5261. doi: 10.1158/1078-0432.CCR-07-0892. [DOI] [PubMed] [Google Scholar]