

Abstract
The bile salt export pump (Bsep) represents the major bile salt transport system at the canalicular membrane of hepatocytes. When examined in model cell lines, genetic mutations in the BSEP gene impair its targeting and transport function, contributing to the pathogenesis of PFIC II. PFIC II mutations are known to lead to a deficiency of BSEP in human hepatocytes, suggesting that PFIC II mutants are unstable and degraded in the cell. To investigate this further, we have characterized the impact of several PFIC II mutations on the processing and stability of rat Bsep. G238V, D482G, G982R, R1153C and R1286Q all retain Bsep to the endoplasmic reticulum (ER) to different extents. Except for R1153C, the PFIC II mutants are degraded with varying half-lives. G238V and D482G are partially misfolded and can be stabilized by low temperature and glycerol. The proteasome provides the major degradation pathway for the PFIC II mutants, while the lysosome also contributes to the degradation of D482G. The PFIC II mutants appear to be more heavily ubiquitinated compared with the wild-type (wt) Bsep, and their ubiquitination is increased by the proteasome inhibitors. Overexpression of several E3 ubiquitin ligases, which are involved in ER-associated degradation (ERAD), lead to the decrease of both mutant and wt Bsep. Gene knockdown studies revealed that the ERAD E3s Rma1 and TEB4 contribute to the degradation of G238V, while HRD1 contributes to the degradation of a mutant lacking the lumenal glycosylation domain (ΔGly). Furthermore we present evidence that G982R weakly associates with various components of the ER quality control system. These data together demonstrate that the PFIC II mutants and ΔGly are degraded by the ERAD pathway.
Bile secretion is mediated by several ATP binding cassette (ABC) transporters located in the canalicular membrane of hepatocytes (for reviews, see [1, 2]). Among these ABC transporters, the bile salt export pump (BSEP/Bsep, ABCB11/ABCb11) represents the primary if not the sole transport system for the canalicular excretion of bile salts [3, 4]. Bile secretory failure results in cholestasis. Progressive familial intrahepatic cholestasis (PFIC) of infancy represents a group of inherited cholestatic diseases that are classified into three subtypes. One of these subtypes, PFIC II, is associated with mutations in the BSEP gene [4, 5]. PFIC I and PFIC III are caused by mutations in the genes encoding FIC I (ATP8B1) and MDR3 (ABCB4), respectively [6, 7]. A large number of premature termination, missense and frame shift mutations in the BSEP gene have been identified from PFIC II patients. These mutations impair the canalicular location or the transport function (or both) of Bsep [8-13].
When different variants of Bsep were examined in model cell lines, several missense mutations reduced the expression level of the Bsep protein [8-10, 13]. Since the mRNA transcript levels of the mutant Bsep are not affected [9, 10, 13], these data raise the possibility that these mutations may cause the Bsep protein to become unstable and degraded. This assumption is consistent with an earlier immunohistochemistry study that BSEP was not detected in many PFIC II patients [5]. The mechanism by which these mutations cause Bsep to become unstable and degraded is not known. In the present study, we have examined the impact of a number of missense PFIC II mutations on processing and stability of rat Bsep.
In several genetic diseases like cystic fibrosis, ubiquitin and the proteasome have been implicated in the degradation of aberrant ABC transporters [14]. The most common mutation in cystic fibrosis patients is cystic fibrosis transmembrane conductance regulator (CFTR) ΔF508, which causes the mutant CFTR protein, instead of being expressed at the cell surface, to be completely degraded at the ER by the proteasome, a multisubunit ATP-dependent protease in the cytosol. Inhibition of proteasomes also stabilized Bsep G238V, E297G and D482G when examined in Madin-Darby canine kidney (MDCK) cells and human embryonic kidney (HEK) cells [8, 10, 13]. These findings suggest that the proteasome plays a major role in the degradation of these BSEP mutants in PFIC II patients. In this paper, we assessed the contribution of the proteasome and lysosome to the degradation of the Bsep PFIC II mutants.
Ubiquitination involves three key enzymes, namely an ubiquitin-activating enzyme (E1), an ubiquitin-conjugating enzyme (E2) that transiently carries the activated ubiquitin molecule, and an ubiquitin ligase (E3) that recognizes specific substrates and transfers the activated ubiquitin from the E2 to the substrate. Among these three enzymes, E3s are believed to be responsible for the high efficiency and exquisite selectivity of ubiquitination reactions [15].
The ER is the major site where newly synthesized membrane proteins such as Bsep enter the secretory pathway and fold. Only proteins that are correctly folded and assembled are usually able to leave the ER. Most misfolded or unassembled proteins are retained in the ER and subsequently degraded in the cytosol by a process known as the ER-associated protein degradation (ERAD). During the ERAD process, an aberrant membrane protein is first recognized and ubiquitinated by a specific E3, exported out of the ER by the retro-translocation machinery and finally degraded by the proteasomes [16]. In this paper, we demonstrated that several Bsep PFIC II mutants are degraded by the ERAD pathway. We analyzed the role of known or suspected ERAD E3s in the degradation of the PFIC II mutants and probed the interaction between Bsep mutants and other components of the ER quality control system.
Materials and Methods
Chemicals, Cells and Antibodies
All the chemicals and reagents were purchased from Sigma, Merck or J.T. Baker unless otherwise stated. HEK 293 cells were from American Type Culture Collection (Manassas, VA). The following antibodies were from commercial sources: the green fluorescent protein (GFP), ubiquitin, calnexin and Hsc70 (Santa Cruz), FLAG (Sigma) and hemagglutinin (HA, Covance). The following antibodies were generous gifts from various labs: HRD1 and TEB4 (Emmanuel Wiertz), Rma1 (Ze'ev Ronai), CHIP (Cam Patterson), p97 (Rasmus Hartmann-Petersen), HERP (Koichi Kokame) and derlin-1 (Yihong Ye).
Plasmid construction
The construction and characterization of pEGFP-Bsep, a full-length cDNA encoding rat Bsep N-terminally fused with GFP and the PFIC II mutants of pEGFP-Bsep have been described previously [8]. We have used rat Bsep as a model protein because the rat Bsep is highly homologous to the human orthologue with an 82.3% identity at the amino acid level [3, 4]. pFLAG-Bsep was created using pEGFP-Bsep as a template by restriction digestion and polymerase chain reaction (PCR). The fusion construct (pFLAG-Bsep) encodes a chimeric protein, FLAG-Bsep starting with FLAG tag, followed by a 17 amino-acid linker and the Bsep coding sequence minus the initiation codon ATG. For the construction of the PFIC II mutants of pFLAG-Bsep, site-directed mutagenesis was performed using the Quikchange PCR mutagenesis kit (Stratagene, La Jolla, CA). The following missense mutants were studied in this work, G238V, D482G, G982R, R1153C and R1286Q. The localization of these mutations is shown in a topology model of rat Bsep (Fig.1). A deletion mutant ΔGly was also created by PCR such that it lacks the amino acids 109-127 of Bsep, a domain containing the lumenal glycosylation motifs of Bsep. The cDNAs encoding HRD1, Rma1 and CHIP were gifts from Drs. Yuval Reiss, Ze'ev Ronai and Cam Patterson, respectively. A N-terminal myc fusion construct of CHIP was created by PCR. The cDNA encoding TEB4 has been described previously [17]. The constructs generated in this study were all confirmed by DNA sequencing at the W.M. Keck facility of Yale University.
Fig.1. The locations of PFIC II mutations and ΔGly in Bsep.

The positions of G238V, D482G, G982R, R1153C and R1286Q are indicated by star signs in a predicted topology model of rat Bsep. The Walker A and B regions are shown in boxes and a number of potential glycosylation sites are indicated by the branch structures in the first extracellular loop of Bsep. ΔGly represents a mutation that lacks the potential glycosylation sites. The orientation of the plasma membrane is indicated.
Cell culture and transfection
HEK 293 cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). For transient transfection, confluent HEK 293 cells were transfected using LipofectAmine (Invitrogen, Carlsbad, CA) as described by the manufacturer for 24 or 48 hrs. To knockdown TEB4, Rma1 and CHIP, the cells were transfected with the following small interfering RNAs (siRNAs) for 48 hrs: TEB4 (TTAAGAGTGTGCTGCCTAA), Rma1 (GCGACCTTCGAATGTAATA) and CHIP (GGAGCAGGGCAATCGTCTG). A short hairpin RNA (shRNA) construct was generated for HRD1 against the sequence GGTGTTTGGCTTTGAGTAT. For drug treatment, MG132 (Calbiochem, San Diego, CA) 20μg/ml was added to the culture medium to inhibit proteasome for time indicated in each experiment. To inhibit lysosome, 15mM ammonium chloride, 10μg/ml leupeptin and 10μg/ml pepstatin was added to the medium for up to 5 hrs. For cycloheximide chase, 50μg/ml cycloheximide was added to the culture for time indicated in each experiment. After transfection, the cell monolayers were directly solubilized in the RIPA buffer (50mM Tris-HCl, pH 7.4, 150mM NaCl, 1mM EDTA) containing 1% NP-40 and 0.5% deoxycholate on ice for 30 minutes. The lysates were centrifuged at 12,500 g for 5 minutes. The PNGase F (New England Biolabs, Ipswich, MA) treatment was performed as described by the manufacturer.
Metabolic labeling and immunoprecipitation
HEK293 cells were transfected with FLAG-Bsep and were starved of methionine in methionine-free DMEM (Invitrogen, Carlsbad, CA) for 1 hr at 37 °C and metabolically labeled for 20 minutes with Tran35S-label (100 μCi per well; 1,200 Ci mmol−1; MP Biomedicals, Solon, OH). Cells were washed twice with 1 ml of phosphate buffered saline (PBS) and lysed immediately or supplemented with methionine and cycloheximide and incubated in DMEM for a chase period. Cell extracts for denaturing immunoprecipitations were prepared in RIPA buffer containing 1% NP-40, 0.5% deoxycholate and 0.1% SDS. Cell extracts for non-denaturing immunoprecipitations were prepared in RIPA buffer containing 0.3% NP-40. Before initiation of immunoprecipitation reactions, all lysates were precleared with Protein G agarose for 1hr at 4 °C, and proteins were immunoprecipitated using Protein A- and G-agarose beads. Dried polyacrylamide gels were analyzed using phosphor imaging.
Electrophoresis and immunoblotting
Cell lysates from HEK 293 cells were analyzed by SDS-PAGE [18]. The separated polypeptides were electrotransferred to PVDF membranes (Bio-Rad, Hercules, CA) and then subject to Western blotting using a standard protocol. The immuoreactive proteins were visualized using an enhanced chemiluminescence (ECL) kit (Amersham, Piscataway, NJ) or alkaline phosphatase.
Fluorescent microscopy
After transfection, HEK 293 cells grown on coverslips were fixed with paraformaldehyde for 20 mins at room temperature. Coverslips were mounted on glass microscope slides using VectaShield (Vector Labs, Burlingame, CA). Cells were viewed on a Zeiss LSM 510 confocal microscope (Zeiss, Thornwood, NJ). Digital images were saved and analyzed with Adobe PhotoShop.
Results
Expression of Bsep PFIC II mutants in HEK 293 cells
To examine the expression level and modification of the rat Bsep PFIC II mutants, we first expressed the PFIC II mutants of GFP-Bsep along with the wild-type (wt) GFP-Bsep in HEK cells. As can be seen in Fig. 2A, the majority of wt Bsep was detected as a 190 kDa protein, consistent with previous data with MDCK cells [8]. A minority of wt Bsep was detected as a 170 kDa protein, which presumably represents the core-glycosylated form. This was confirmed by de-glycosylation with PNGase F (Fig. 2B). In contrast, the PFIC II mutants were mostly detected as core-glycosylated proteins, except G238V and D482G, for which a fraction of 190 kDa mature glycosylated form was also observed. Also included for comparison was ΔGly, a mutant construct that lacks the lumenal glycosylation domain of Bsep. ΔGly was expressed exclusively as the nonglycosylated form with mobility faster than the core-glycosylated GFP-Bsep. The core-glycosylation of the PFIC II mutants is consistent with the intracellular sequestration pattern previously reported [8] and suggests that these mutants may not traffic beyond the ER.
Fig.2. Expression of PFIC II mutants and ΔGly in HEK 293 cells.


(A) The HEK 293 cells were transiently transfected with wt GFP-Bsep, PFIC II mutants and ΔGly. The lysates (100 μg protein each lane) were separated by SDS-PAGE and GFP-Bsep was detected by immunoblotting using an antibody against GFP. β-actin was also probed to indicate the equal loading of lysates. ECL was used to visualize the protein of interest. 190 kDa and 170 kDa indicate the mature and immature glycosylated GFP-Bsep. (B) The cell lysate of HEK cells transfected with wt GFP-Bsep was digested with PNGase F. (C) mRNAs were isolated from the HEK cells 24 hrs after transfection. Real-time quantitative PCR was performed and data represent triplicate measurements for each sample. Bsep gene expression was normalized to the expression of β-actin.
To examine the predicted ER localization of PFIC II mutants, we transfected HEK cells with GFP-tagged Bseps and analyzed their co-localization with an ER marker, calnexin (Supplement Fig.1). As it can be seen, there is substantial co-localization between GFP-Bsep and calnexin in mutants that are exclusively core-glycosylated or nonglycosylated, i.e. G982R, R1153C, R1286Q and ΔGly. For G238V and D482G, which are partially core-glycosylated, there is a partial co-localization between GFP-Bsep and calnexin. The wt GFP-Bsep is primarily expressed at the periphery (cell surface) of the HEK cells, in which case there is virtually no co-localization between GFP-Bsep and calnexin. These data together demonstrate that the PFIC II mutants and ΔGly are either exclusively or partially localized to the ER.
Besides different glycosylation pattern, PFIC II mutants were also noticeably expressed at lower levels compared with the wt protein, with D482G, G982R and ΔGly showing the lowest expression (Fig.2A). Because the PFIC II mutations could potentially affect the transcription of Bsep, we next examined the expression of the Bsep mRNA using quantitative PCR. Bsep expression was normalized against the expression of β-actin. As it can be seen in Fig.2C, the mRNA levels were comparable between the wt and mutant Bseps, suggesting that the transcription of the PFIC II mutants are not affected. Thus the low expression levels of the PFIC II mutant proteins suggest that they may be subject to rapid degradation.
The stability of the PFIC II mutants
We next examined the stability of the GFP-tagged PFIC II mutants by adding cycloheximide to block further protein synthesis and following the amount of protein remaining by anti-GFP immunoblotting. As can be seen in Fig.3A, for wt GFP-Bsep, the core-glycosylated form disappears with the chase, while the mature glycosylated form increases concomitantly, indicating that the core-glycosylated form can be converted to the mature form. In G238V, the core-glycosylated form disappears while the mature form is not significantly increased, indicating that the core-glycosylated form is degraded over the chase period. For D482G, about 30% of the core-glycosylated protein was degraded, while a significant fraction was converted to the mature glycosylated form, indicating that this mutant is probably only weakly misfolded and a portion of correctly folded D482G can traffic through the secretory pathway. G982R, R1286Q and ΔGly were degraded over time as indicated by the decrease in the core-glycosylated form (the only form detected), whereas R1153C was relatively stable. These data were confirmed by pulse-chase studies (Fig.3B-D). As can be seen in Fig.3B, about 60% of wt FLAG-Bsep is converted from a 140 kDa immature glycosylated form to a 160 kDa mature glycosylated protein over the 120 min chase and 40% of the 140 kDa protein remains. In contrast, G982R and R1286Q are degraded with a half-life of around 1 hr (Fig.3C). R1153C is relatively stable. For G238V and D482G, respectively, only about 20% and 40% of the 140 kDa protein is converted to the 160 kDa protein, while about 50% of the 140 kDa protein is degraded over 120 min chase (Fig. 3D). These data suggest that the core-glycosylated G238V and D482G have a half-life of around 2 hr, which is consistent with the results from the cycloheximide chase studies, which used chase times up to 4 hrs (Fig. 3A). The higher conversion percentage for D482G is also consistent with the data in Fig.3A.
Fig.3. The stability of PFIC II mutants and ΔGly in HEK 293 cells.




(A) HEK 293 cells were transfected with wt GFP-Bsep, PFIC II mutants and ΔGly. Cycloheximide was added to the cell culture 24 hrs after transfection and the cells were harvested at time zero, 1.5 hr and 4 hr of the cycloheximide chase. GFP-Bsep in each cell lysate was analyzed by immunoblotting using an antibody against GFP. β-actin was also probed to indicate the equal loading of lysates. Alkaline phosphatase was used to visualize the protein of interest. The amount of GFP-Bsep at each time point was quantified using densitometry, normalized against that of β-actin and expressed as percentage of the total GFP-Bsep at time zero. Each data point represents mean ± SE of triplicate determination. The HEK 293 cells were transfected with wt FLAG-Bsep (B) and the PFIC II mutants (C and D). The HEK cells were labeled with S35-methionine and FLAG-Bsep was immunoprecipitated. 160 kDa and 140 kDa indicate the mature and immature glycosylated FLAG-Bsep. Each data point represents mean ± SE of triplicate determination.
The effect of low temperature and chemical chaperone on G238V and D482G
The data in Fig.3 suggest that a portion of D482G and G238V can be converted to the mature glycosylated form over time during biosynthesis. We next asked if conditions such as low temperature or chemical chaperones can stabilize D482G and G238V, as these conditions favor protein folding and have been shown to stabilize the misfolded mutant protein CFTR ΔF508 during its biogenesis [19]. Both incubation at low temperature (30°C) and addition of glycerol increases the peripheral, cell surface expression of GFP-tagged D482G and G238V in HEK cells (Fig.4A). This is confirmed by a higher percentage of mature glycosylated form in D482G and G238V in HEK cells under these conditions (Fig.4B). These data together confirm the notion that a fraction of correctly folded D482G and G238V can traffic through secretory pathway and this fraction is increased under the conditions that favor protein folding.
Fig.4. Low temperature and glycerol stabilize G238V and D482G.


(A) The HEK 293 cells were transfected with wt GFP-Bsep, G238V and D482G. Twenty-four hours after transfection, the cells were incubated at 37°C for 24 hrs (control), 30°C for 24 hrs or treated with 10% glycerol for 24 hrs. GFP-Bsep was visualized by fluorescence microscopy. All the images were taken with the same microscopic setting. Bar=10 μm. (B) The lysates of the HEK 293 cells under these conditions were immunoblotted for GFP-Bsep and β-actin. ECL was used to visualize the protein of interest.
The contribution of the proteasome and lysosome to PFIC II mutant turnover
Since the proteasome and lysosome provide two of the major degradation mechanisms in the cell, we next examined the contribution of these two pathways to the stability of G238V, D482G and G982R. Treatment with either MG132, a specific proteasome inhibitor or the combination of ammonium chloride, leupeptin and pepstatin, which inhibits lysosomal proteolysis, had little or no impact on wt GFP-Bsep degradation (Fig. 5A). In contrast, treatment with MG132 significantly stabilized the 170 kDa core-glycosylated form of G982R, D482G and G238V. These data suggest that the proteasome is a major pathway for degradation of the immature glycosylated forms of these mutant proteins. In contrast, the combination of ammonium chloride, leupeptin and pepstatin only moderately increases the mature glycosylated form of D482G, while not affecting the expression of G238V or G982R. These data suggest that lysosome is not the major degradation pathway for these mutants. It is possible that D482G became prone to lysosome degradation when it traffics to a later stage of secretory pathway. It is also worth noting that the 190 kDa mature glycosylated form of D482G also increases by the treatment of MG132 compared with the control condition. This may reflect a situation that when the proteasome function is inhibited, the stabilized immature glycosylated D482G can traffic beyond the ER and is thus converted to the 190 kDa mature glycosylated form. The stabilization of G982R by the proteasome inhibitor was confirmed by pulse-chase studies (Fig.5C), whereas the expression of wt Bsep is little affected by MG132 (Fig.5B).
Fig.5. Proteasome is the major pathway to degrade PFIC II mutants.


(A) The HEK 293 cells were transfected with the wt GFP-Bsep, G238V, D482G, and G982R. Cycloheximide was added to the cell culture 24 hrs after transfection and the cells were harvested at indicated time points. GFP-Bsep in each cell lysate was analyzed by immunoblotting using an antibody against GFP. β-actin was also probed to indicate the equal loading of lysates. To inhibit proteasome, 20μg/ml MG132 was added to the culture medium during the cycloheximide chase. Equal amount of DMSO, a solvent used to dissolve MG132, was added to the control medium. To inhibit lysosome, 15mM ammonium chloride, 10μg/ml leupeptin and 10μg/ml pepstatin was added to the medium. ECL was used to visualize the protein of interest. The HEK 293 cells were transfected with wt FLAG-Bsep (B) and G982R (C). The cells were incubated with medium with DMSO (mock) or with 20μg/ml MG132 during the chase period. FLAG-Bsep was immunoprecipitated. Each data point represents mean ± SE of triplicate determination.
Ubiquitination of PFIC II mutants
Because the proteins subjected to the proteasome pathway are covalently modified by ubiquitin, we next examined the ubiquitination of the PFIC II mutants of Bsep. The wt and mutant FLAG-Bsep was co-expressed with HA-ubiquitin (HA-Ub) in HEK cells. The FLAG-Bsep was immunoprecipitated and probed for FLAG-Bsep and HA-Ub (Fig.6). We also compared the ubiquitination in the presence and absence of MG132, the specific proteasome inhibitor. As expected, MG132, which inhibits proteasome function, significantly increases the ubiquitination of both the wt and mutant Bseps (Fig.6). However the increase in ubiquitination is moderate for wt Bsep, compared with the mutant proteins G238V, D482G, G982R and ΔGly. This is more obvious if one normalizes the expression level of the mutant protein and the wt Bsep. Notably, R1286Q is more heavily ubiquitinated than wt Bsep even in the absence of MG132. A similar finding was seen for the endogenous ubiquitination of R1286Q in cells not co-expressing HA-Ub (Supplement Fig.2). These data together suggest that the PFIC II mutants and ΔGly are more heavily ubiquitinated than wt Bsep. More experiments will be needed to establish that this is the key difference underlying the differences in their degradation rates.
Fig.6. The ubiquitination of PFIC II mutants and ΔGly in the HEK 293 cells.

The HEK 293 cells were co-transfected with FLAG-Bsep and HA-Ubiquitin (HA-Ub) for 24 hrs. The cells were then incubated with medium with DMSO or treated with MG132 20μg/ml for 6 hrs. Untransfected HEK cells were used as a negative control (mock). FLAG-Bsep was immunoprecipitated, and the immunoprecipitate was immunoblotted with an anti-FLAG or an anti-HA antibody. ECL was used to visualize the protein of interest. FLAG-Bsep and polyubiquitinated Bsep (Ubn-Bsep) are indicated. IP, immunoprecipitate. IB, immunoblot.
The effect of the ERAD E3s
Taken together, the data presented above demonstrate that the short-lived PFIC II mutants and ΔGly are retained in the ER and are likely to be degraded by the ubiquitin/proteasome pathway. Thus the PFIC II mutants and ΔGly are subject to the ER-associated protein degradation (ERAD). Inhibition of a specific ubiquitination enzyme of the ERAD pathway should stabilize a Bsep mutant, whereas over-expression of a specific ERAD ubiquitination enzyme should accelerate its degradation. To examine if the ERAD E3s affect the expression of the Bsep mutants, we co-expressed the ERAD E3s HRD1, TEB4, Rma1 or CHIP with the wt Bsep, G238V and ΔGly proteins. Increased levels of all the E3s resulted in decreased amounts of G238V and ΔGly compared to the mutant expressed alone (Fig. 7A). However, a similar pattern was also seen with the wt Bsep and other PFIC mutants (data not shown), suggesting that these overexpression effects might be relatively nonspecific. For at least one of the E3s, Rma1, overexpression was associated with increased ubiquitination of wt FLAG-tagged Bsep (Fig. 7B). Enhanced ubiquitination of G238V was seen with the other ERAD E3s (data not shown). These data suggest that the overexpressed ERAD E3s can ubiquitinate Bsep proteins and target them for degradation, but they do not show the substrate specificity expected from the differences in protein half-lives observed among the wt and mutant Bseps.
Fig.7. The co-expression of the ERAD E3 increases the ubiquitination of Bsep and decreases its level.

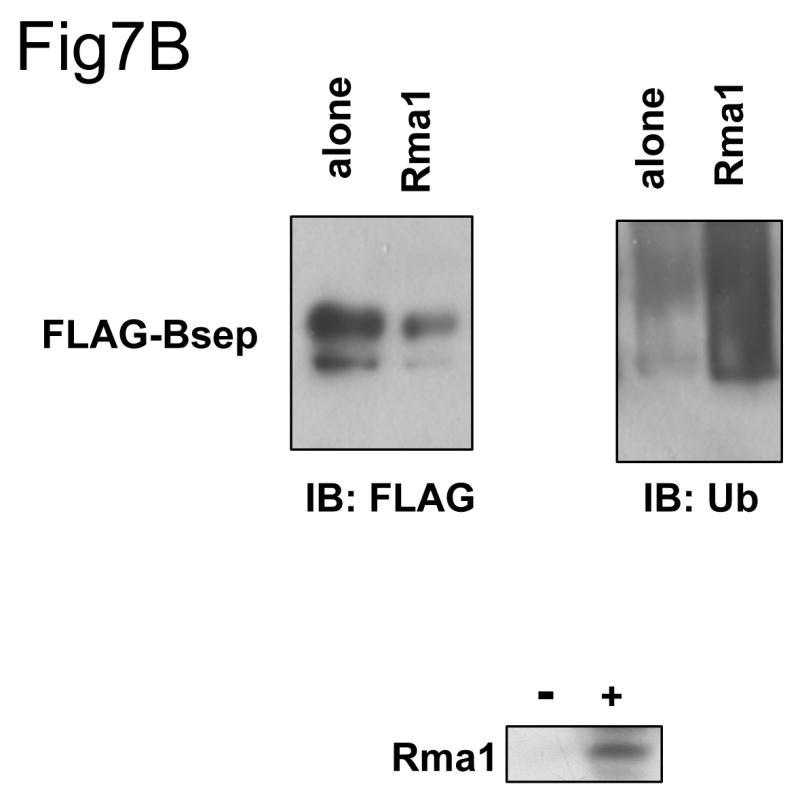
(A) The HEK 293 cells were transfected with wt GFP-Bsep, G238V and ΔGly alone or co-transfected with the cDNAs encoding the ERAD E3s HRD1, TEB4, Rma1 and myc-CHIP. The level of GFP-Bsep was analyzed by immunoblotting with an anti-GFP antibody. β-actin was also probed to indicate the equal loading of lysates. The expression of the ERAD E3s is analyzed by immunoblotting with specific antibodies to the E3s. (B) The HEK 293 cells were transfected with FLAG-Bsep alone or co-transfected with the Rma1 cDNA. FLAG-Bsep was immunoprecipitated and the immunoprecipitate was immunoblotted with anti-FLAG and anti-ubiquitin (Ub) antibody respectively. ECL was used to visualize the protein of interest. IB, immunoblot.
Since overexpression of exogenous ERAD E3s may have nonspecific effects, we used RNA interference to suppress expression of the endogenous ERAD E3s and asked if this would affect the level of expression of the wt and mutant GFP-Bseps. As can be seen in Fig. 8A, the siRNAs reduced levels of endogenous TEB4, Rma1 and CHIP, albeit to different extents. Reduction of TEB4 and Rma1 levels, but not those of CHIP, stabilized G238V. siRNA-mediated reduction in TEB4, Rma1 and CHIP had little or no effect on the levels of wt GFP-Bsep or ΔGly. These data suggest that TEB4 and Rma1 may function as the E3s targeting G238V for degradation but do not do so for the wt Bsep and ΔGly proteins.
Fig.8. The suppression of the ERAD E3 stabilizes different Bsep mutants.


(A) The HEK 293 cells were transfected with wt GFP-Bsep, G238V and ΔGly. The cells were co-transfected with the control siRNA or siRNA against TEB4, Rma1 and CHIP. (B) The HEK 293 cells were transfected with wt GFP-Bsep, R1286Q and ΔGly. The cells were co-transfected with a control shRNA or shRNA against HRD1. The level of GFP-Bsep was analyzed by immunoblotting with an anti-GFP antibody. β-actin was also probed to indicate the equal loading of lysates. The suppression of the ERAD E3s is analyzed by immunoblotting with specific antibodies. ECL was used to visualize the protein of interest.
In contrast, the shRNA-mediated decrease in HRD1 E3 levels caused a substantial increase in ΔGly levels but had no effect on either wt GFP-Bsep or the PFIC II mutant R1286Q (Fig.8B). Similar data was also seen for other PFIC mutants (data not shown). These results are consistent with the possibility that HRD1 might recognize a misfolded structure in Bsep that is distinct from the aberrations caused by the PFIC II mutants.
Finally, we attempted to detect possible physical interactions between Bsep mutants and the ERAD E3s or other components of the ER quality control system by co-immunoprecipitation. We transfected HEK cells with wt FLAG-Bsep, G982R and ΔGly, and isolated FLAG-Bsep by immunoprecipitation. We probed for co-precipitated ER quality control components by immunoblotting (Fig. 9). As can be seen, in all cases co-precipitation was weak or undetectable. Weak binding was seen between the sugar-binding ER chaperone calnexin and wt Bsep and G982R but not with the nonglycosylated ΔGly mutant, as expected. The ATPase p97, which extracts the retro-translocated proteins from the ER membrane [20], and a component of the retro-translocation complex HERP [21, 22] primarily associate with G982R as expected, which is a strong ERAD substrate. However, other detected interactions (cytosolic chaperone Hsc70, E3 CHIP and the putative retro-translocation channel derlin-1) were weak and sufficiently similar among the three substrates that we could not be sure of their specificity. We have tried a number of detergents (including digitonin, deoxyBigChap, NP-40 and Triton X-100) and solubilization conditions but could not detect any specific interaction between the endogenous HRD1, TEB4 and Rma1 E3s and Bsep mutant substrates such as G238V and ΔGly (data not shown). This may reflect the transient nature of the E3-substrate interaction. More experiments are needed to characterize the interacting proteins of Bsep mutants.
Fig.9. The ERAD substrate G982R interacts with multiple components of the ER quality control system.
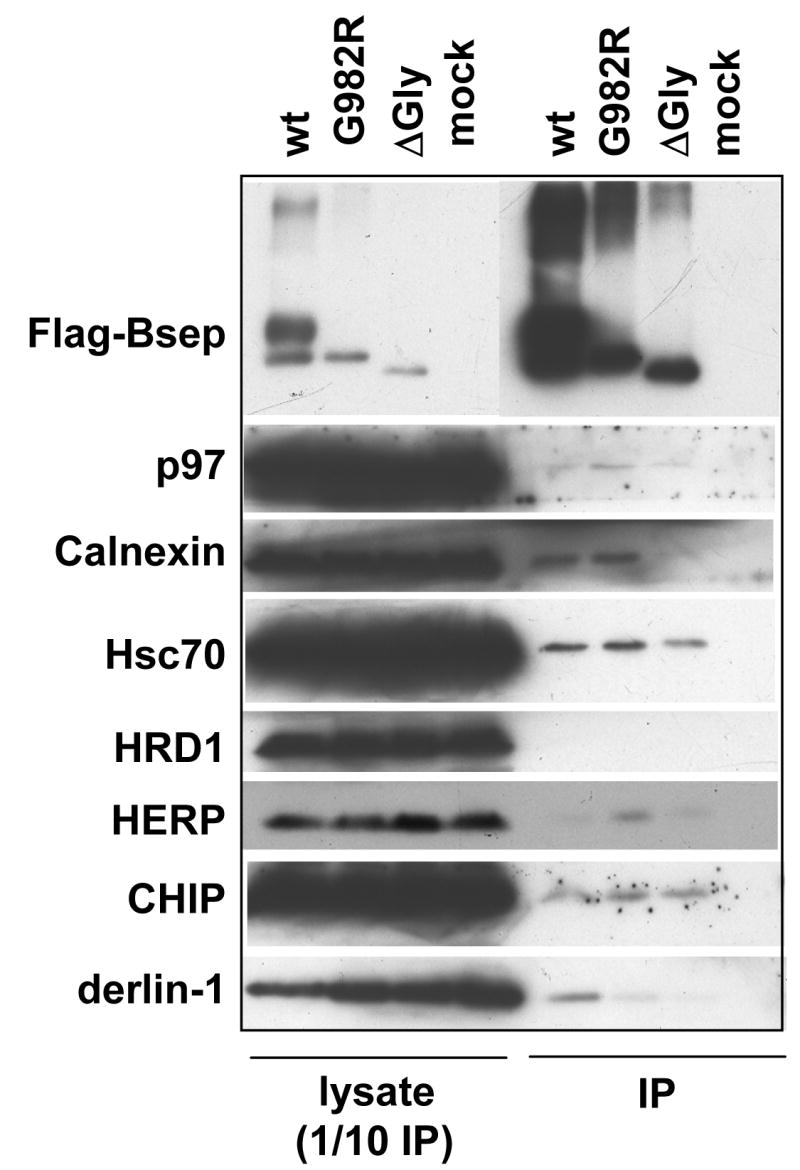
The HEK 293 cells were transfected with wt FLAG-Bsep, G982R and ΔGly. Untransfected HEK cells were used as a negative control (mock). The wt and mutant FLAG-Bsep were isolated by immunoprecipitation (IP) and eluted from Protein G agarose with a three-FLAG peptide. Any co-precipitated protein was probed by immunoblotting. The probed proteins include p97, calnexin, Hsc70, HRD1, HERP, CHIP and derlin-1. An aliquot of cell lysate was also analyzed, which corresponds to the one tenth of the material used for IP. ECL was used to visualize the protein of interest.
Discussion
In this study, we have characterized the effect of five missense mutations and one deletion mutation on the folding and stability of rat Bsep. Taken together, the data from this study show that G238V, D482G, G982R, R1153C, R1286Q and ΔGly mutations cause retention of Bsep in the ER to different extents. The core-glycosylation of the PFIC II mutants is consistent with our fluorescence data that show these mutants concentrating in the ER in HEK cells (Supplement Fig.1). Although weakly expressed, a significant portion of D482G is expressed as mature glycosylated protein, which is consistent with previous reports showing that this mutant can be expressed at the cell surface and also retains bile salt transport function [8-10]. These data together show that D482G is likely to be only a weakly misfolded mutant.
Except for R1153C, the PFIC II mutants are short-lived, displaying distinct half-lives as measured by both cycloheximide chase and pulse-chase analyses. Mutants like G982R and R1286Q are degraded with a half-life of about 1 hr. For mutants D482G and G238V, only a portion of immature glycosylated protein can be converted to mature glycosylated protein over the chase period, while a large pool of immature glycosylated protein is degraded. This suggests that D482G and G238V are partly misfolded. This notion is supported by the data showing that conditions favoring protein folding, such as low temperature or the addition of glycerol, stabilize D482G and G238V and increase the fraction of the mature glycosylated form and cell surface expression (Fig.4).
Proteasomes, rather than lysosomes, provide the major degradation pathway for the PFIC II mutants, while lysosome also moderately contributes to the degradation of D482G. This may reflect a situation that D482G can traffic through the secretory pathway and at different stages of the secretory pathway it can be sorted to proteasome or lysosome for degradation. Kagawa and co-workers have recently also analyzed the degradation of D482G. Comparable results were seen for the stabilization of D482G by proteasome inhibitor MG132 in MDCK cells [13]. However, bafilomycin A1, a lysosomal proton pump inhibitor, did not affect stability of Bsep protein. It is not clear whether this discrepancy was due to the different cell lines used or a lower sensitivity of assay in the MDCK cells. Some mutations of CFTR have been shown to facilitate the lysosome targeting and degradation of the CFTR protein [23].
The PFIC II mutant R1286Q appear to be more heavily ubiquitinated than the wt Bsep (Fig.6 and Supplement Fig. 2). R1286Q is also rapidly degraded (Fig. 3). These data suggest that the ubiquitin-proteasome pathway is responsible for the degradation of this mutant. In addition, G238V, D482G, G982R and ΔGly become relatively more ubiquitinated, compared with wt Bsep, when the function of the proteasome is inhibited, consistent with the notion that these mutant Bseps are misfolded and thus unstable. Taken together, our data support the conclusion that the PFIC II mutants and ΔGly are retained in the ER and degraded by the ubiquitin-proteasome pathway, i.e. these mutants are subject to the ERAD pathway.
R1153C is an interesting mutant, in that it is stable yet retained in the ER. The R1153C Bsep protein resembles two mutants of the yeast ABC transporter Ste6p, Ste6-13p and Ste6-90p, which are retained in the ER and are hyperstable [24]. In contrast, a large number of Ste6 mutants, such as Ste6-166p, are highly unstable and are degraded by the ERAD pathway. Two different ER quality control mechanisms have been suggested: 1) rapid degradation by the ubiquitin-proteasome system; 2) retention in the ER to give more time for proper folding and/or to sequester proteins from the secretory pathway. The R1153C mutant might be an example of this latter mechanism. R1153C most likely adopts a misfolded, bile salt transport-inactive conformation, since the mutant protein fails to transport taurocholate when expressed in Sf9 membrane vesicles [8]. The misfolded R1153C is retained in the ER for continued folding attempts or sequestration instead of being degraded by the ERAD pathway.
ERAD is a component of the ER quality-control system. In ERAD, a misfolded protein is ubiquitinated, retro-translocated out of the ER and finally degraded by the proteasome. HRD1, TEB4, gp78, Rma1 and CHIP are the known E3s in the ERAD pathway [25]. In eukaryotes, ERAD is evolutionally conserved. Budding yeast cells primarily use just two ERAD E3s, Hrd1 and Doa10, which are orthologues of the human E3s HRD1 and TEB4, respectively [26]. In yeast, Hrd1 primarily targets substrates with misfolded or unfolded domains located in the ER lumen, whereas Doa10 primarily targets substrates with misfolded or unfolded domains located in the cytosol [27]. In mammalian cells, the specificity of the ERAD E3s is less clear. CHIP and Rma1 have been previously reported to function in the degradation of CFTR ΔF508, which has its mutation located at the cytoplasmic side [28, 29].
In this paper, we found that expression of exogenous E3s leads to the degradation of both wt and mutant Bsep. This may reflect a situation in which the overexpressed E3 non-specifically ubiquitinates the Bsep proteins and targets them for degradation. Consistent with this notion, overexpression of the ERAD E3 Rma1 leads to increased ubiquitination of wt Bsep. In contrast, siRNA-mediated knockdown of endogenous Rma1 and TEB4 stabilizes G238V but not ΔGly, whereas the knockdown of HRD1 levels significantly stabilizes ΔGly but not R1286Q. These data suggest that the ERAD E3s have specificity towards differently misfolded Bseps. The PFIC II mutants studied in this paper all have their mutations located at cytoplasmic side, whereas ΔGly has its mutation located at lumenal side (Fig.1). Therefore, in mammalian cells HRD1 may also usually target proteins with misfolded or unfolded domains in the lumen of the ER, as occurs most often with yeast Hrd1, whereas Rma1 and TEB4 might target proteins with lesions facing the cytosol, as is true for the yeast Doa10. There are exceptions to these rules, such as the ability of HRD1 to target the soluble p53 protein [30], but our preliminary specificity data suggest that mammalian and yeast E3s might follow similar trends.
Finally we presented evidence that the ERAD substrates such as Bsep G982R weakly interact with components of the ER quality control system. The ER quality control is a complex system, which consists of folding enzymes (the ER and cytosolic chaperons), the ubiquitination machineries (the ERAD E2s and E3s) and degradation machineries (the ER retro-translocation channel such as derlin-1 and membrane extraction machineries such as p97). Although the detected interaction is weak, some of them may be more specific to Bsep mutants. For example, p97 and HERP primarily associate with G982R, which is a strong ERAD substrate (Fig.5A). Although calnexin interacts with both wt Bsep and G982R, the ratio of calnexin bound to Bsep is likely to be higher for G982R, since its expression level is lower than wt Bsep. The same may be true for Hsc70 and CHIP.
In summary, we have analyzed the modification and stability of a number of PFIC II mutants, most of which are short-lived and degraded by the ERAD pathway. Different ERAD E3s appear to recognize different mutants of Bsep. In the future, manipulation of the ER quality control system, such as inhibition of specific ERAD E3s, might be combined with chemical or pharmacological chaperones to stabilize the PFIC II mutants and restore cell surface expression of Bsep. This may be particularly pertinent to the rescue of those mutants that are partly misfolded but still functional, such as the D482G Bsep mutant.
Supplementary Material
After transient transfection with GFP-Bsep, HEK cells were processed for immunofluorescence using an antibody against calnexin and imaged to localize the GFP fusion protein and the ER of HEK cells. In each panel, the en face image is shown. The left, middle and right columns show the image of calnexin (red), GFP-Bsep (green) and merged image, respectively. Bar=5μm
The HEK 293 cells were transfected with wt and mutant FLAG-Bsep for 24 hrs. FLAG-Bsep was immunoprecipitated, and the immunoprecipitate was immunoblotted with an anti-FLAG and an anti-ubiquitin (Ub) antibody, respectively. FLAG-Bsep and polyubiquitinated Bsep (Ubn-Bsep) are indicated. IP, immunoprecipitate. IB, immunoblot.
Acknowledgments
This work was supported by a NIH Career Development Award DK 066121 to L.W. and a grant GM 046904 to M.H.. J.L.B and C.J.S. are supported by DK025636. Confocal images were obtained by the Morphology core of the Yale Liver Center P30 334989. We thank Drs. Emmanuel Wiertz, Yuval Reiss, Ze'ev Ronai, Cam Patterson, Rasmus Hartmann-Petersen, Koichi Kokame and Yihong Ye for generously providing us critical reagents. L.W. also acknowledges gratitude to Dr. Xingwang Deng for provision of lab space.
References
- 1.Alrefai WA, Gill RK. Bile acid transporters: structure, function, regulation and pathophysiological implications. Pharm Res. 2007;24(10):1803–23. doi: 10.1007/s11095-007-9289-1. [DOI] [PubMed] [Google Scholar]
- 2.Stieger B, Meier Y, Meier PJ. The bile salt export pump. Pflugers Arch. 2007;453(5):611–20. doi: 10.1007/s00424-006-0152-8. [DOI] [PubMed] [Google Scholar]
- 3.Gerloff T, et al. The sister P-glycoprotein represents the canalicular bile salt export pump of mammalian liver. J Biol Chem. 1998;273:10046–10050. doi: 10.1074/jbc.273.16.10046. [DOI] [PubMed] [Google Scholar]
- 4.Strautnieks SS, et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nature Genetics. 1998;20:233–238. doi: 10.1038/3034. [DOI] [PubMed] [Google Scholar]
- 5.Jansen PL, et al. Hepatocanalicular bile salt export pump deficiency in patients with progressive familial intrahepatic cholestasis. Gastroenterology. 1999;117:1370–1379. doi: 10.1016/s0016-5085(99)70287-8. [DOI] [PubMed] [Google Scholar]
- 6.Bull LN, et al. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet. 1998;18:219–224. doi: 10.1038/ng0398-219. [DOI] [PubMed] [Google Scholar]
- 7.deVree JML, et al. Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci U S A. 1998;95:282–287. doi: 10.1073/pnas.95.1.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang L, Soroka CJ, Boyer JL. The role of bile salt export pump mutations in progressive familial intrahepatic cholestasis type II. J Clin Invest. 2002;110(7):965–72. doi: 10.1172/JCI15968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Plass JR, et al. A progressive familial intrahepatic cholestasis type 2 mutation causes an unstable, temperature-sensitive bile salt export pump. J Hepatol. 2004;40(1):24–30. doi: 10.1016/s0168-8278(03)00483-5. [DOI] [PubMed] [Google Scholar]
- 10.Hayashi H, et al. Two common PFIC2 mutations are associated with the impaired membrane trafficking of BSEP/ABCB11. Hepatology. 2005;41(4):916–24. doi: 10.1002/hep.20627. [DOI] [PubMed] [Google Scholar]
- 11.Hayashi H, Sugiyama Y. 4-phenylbutyrate enhances the cell surface expression and the transport capacity of wild-type and mutated bile salt export pumps. Hepatology. 2007;45(6):1506–16. doi: 10.1002/hep.21630. [DOI] [PubMed] [Google Scholar]
- 12.Lam P, et al. Levels of plasma membrane expression in progressive and benign mutations of the bile salt export pump (Bsep/Abcb11) correlate with severity of cholestatic diseases. Am J Physiol Cell Physiol. 2007;293(5):C1709–16. doi: 10.1152/ajpcell.00327.2007. [DOI] [PubMed] [Google Scholar]
- 13.Kagawa T, et al. Phenotypic differences in PFIC2 and BRIC2 correlate with protein stability of mutant Bsep and impaired taurocholate secretion in MDCK II cells. Am J Physiol Gastrointest Liver Physiol. 2008;294(1):G58–67. doi: 10.1152/ajpgi.00367.2007. [DOI] [PubMed] [Google Scholar]
- 14.Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83(1):121–7. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- 15.Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–33. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 16.Tsai B, Ye Y, Rapoport TA. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat Rev Mol Cell Biol. 2002;3(4):246–55. doi: 10.1038/nrm780. [DOI] [PubMed] [Google Scholar]
- 17.Arteaga MF, et al. An amphipathic helix targets serum and glucocorticoid-induced kinase 1 to the endoplasmic reticulum-associated ubiquitin-conjugation machinery. Proc Natl Acad Sci U S A. 2006;103(30):11178–83. doi: 10.1073/pnas.0604816103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature J1 - Nat(Lond) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 19.Brown CR, Hong-Brown LQ, Welch WJ. Correcting temperature-sensitive protein folding defects. J Clin Invest. 1997;99(6):1432–44. doi: 10.1172/JCI119302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414(6864):652–6. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- 21.Schulze A, et al. The ubiquitin-domain protein HERP forms a complex with components of the endoplasmic reticulum associated degradation pathway. J Mol Biol. 2005;354(5):1021–7. doi: 10.1016/j.jmb.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 22.Ye Y, et al. Inaugural Article: Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc Natl Acad Sci U S A. 2005;102(40):14132–8. doi: 10.1073/pnas.0505006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma M, et al. Misfolding diverts CFTR from recycling to degradation: quality control at early endosomes. J Cell Biol. 2004;164(6):923–33. doi: 10.1083/jcb.200312018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loayza D, et al. Ste6p mutants defective in exit from the endoplasmic reticulum (ER) reveal aspects of an ER quality control pathway in Saccharomyces cerevisiae. Mol Biol Cell. 1998;9(10):2767–84. doi: 10.1091/mbc.9.10.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kostova Z, Tsai YC, Weissman AM. Ubiquitin ligases, critical mediators of endoplasmic reticulum-associated degradation. Semin Cell Dev Biol. 2007;18(6):770–9. doi: 10.1016/j.semcdb.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huyer G, et al. Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J Biol Chem. 2004;279(37):38369–78. doi: 10.1074/jbc.M402468200. [DOI] [PubMed] [Google Scholar]
- 27.Vashist S, Ng DT. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol. 2004;165(1):41–52. doi: 10.1083/jcb.200309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meacham GC, et al. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat Cell Biol. 2001;3(1):100–5. doi: 10.1038/35050509. [DOI] [PubMed] [Google Scholar]
- 29.Younger JM, et al. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell. 2006;126(3):571–82. doi: 10.1016/j.cell.2006.06.041. [DOI] [PubMed] [Google Scholar]
- 30.Yamasaki S, et al. Cytoplasmic destruction of p53 by the endoplasmic reticulum-resident ubiquitin ligase ‘Synoviolin’. Embo J. 2007;26(1):113–22. doi: 10.1038/sj.emboj.7601490. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
After transient transfection with GFP-Bsep, HEK cells were processed for immunofluorescence using an antibody against calnexin and imaged to localize the GFP fusion protein and the ER of HEK cells. In each panel, the en face image is shown. The left, middle and right columns show the image of calnexin (red), GFP-Bsep (green) and merged image, respectively. Bar=5μm
The HEK 293 cells were transfected with wt and mutant FLAG-Bsep for 24 hrs. FLAG-Bsep was immunoprecipitated, and the immunoprecipitate was immunoblotted with an anti-FLAG and an anti-ubiquitin (Ub) antibody, respectively. FLAG-Bsep and polyubiquitinated Bsep (Ubn-Bsep) are indicated. IP, immunoprecipitate. IB, immunoblot.