

Abstract
Chlorofusin is a recently isolated, naturally occurring inhibitor of p53−MDM2 complex formation whose structure is composed of a densely functionalized azaphilone-derived chromophore linked through the terminal amine of ornithine to a nine residue cyclic peptide. Herein we report the full details of the total synthesis of chlorofusin, resulting in the assignment of the absolute stereochemistry and reassignment of the relative stereochemistry of the complex chromophore. Condensation of each enantiomer of an azaphilone chromophore precursor with the Nδ-amine of a protected ornithinethreonine dipeptide, followed by a one-step oxidation/spirocyclization of the most reactive olefin provided all eight diastereomers of the fully elaborated chromophore−dipeptide conjugate. Comparison of the spectroscopic properties for these eight compounds and those of simpler models with that reported for the natural product allowed the full assignment of the (4R,8S,9R)-stereochemistry of the chlorofusin chromophore. The natural, but stereochemically reassigned, diastereomer of the dipeptide conjugate was incorporated in a convergent total synthesis of chlorofusin confirming the stereochemical reassignment and establishing its absolute stereochemistry. Similarly and enlisting the late stage convergent point in the total synthesis, the remaining seven diastereomers of the chromophore−dipeptide conjugates were individually incorporated into the 9-residue cyclic peptide of chlorofusin (4 steps each) providing all seven remaining possible chromophore diastereomers of the natural product.
Introduction
The tumor suppressor p53 is an important part of an innate cancer defense mechanism and acts as a transcription factor that initiates cell cycle arrest and apoptosis in response to stress such as DNA damage.1-4 The activity of p53 is modulated by MDM2 (HDM2), which tightly binds p53 preventing it from acting as a regulator of cell division5-7 and targeting it for nuclear export and degradation.8,9 Overexpression of MDM2 has been implicated in many cancers,10-16 defining the p53—MDM2 interaction as an attractive target for therapeutic intervention.4,17 An X-ray crystal structure of the N-terminal domain of MDM2 bound to a 15-residue transactivation domain of p53 revealed the structural details of their complex that is mediated by the interaction of three hydrophobic residues of a p53 α-helix with a hydrophobic cleft of MDM2.18 Molecules that bind the hydrophobic cleft of MDM2 disrupt this protein—protein interaction with p53, restoring its regulatory function and inhibiting tumor growth.4,19-22
Chlorofusin (1, Figure 1) was isolated from the fungal strain Microdochium caespitosum23 and found to disrupt the MDM2—p53 interaction by directly binding to the N-terminal domain of MDM2 (IC50 = 4.6 μM, KD = 4.7 μM).23,24 Thus, chlorofusin represents an exciting lead for antineoplastic intervention that acts by a rare disruption of a protein—protein interaction although the structural details of this inhibitory interaction derived from MDM2 binding have yet to be established.20,21,25 On the basis of extensive spectroscopic and degradation studies, the chlorofusin structure was proposed to consist of a densely functionalized, azaphilone-derived chromophore linked through the terminal amine of ornithine to a 27-membered cyclic peptide composed of nine amino acid residues.23 Two of the cyclic peptide amino acids possess a nonstandard or modified side chain, and four possess the d-configuration. Although the spectroscopic and degradation studies of chlorofusin permitted the identification of the cyclic peptide structure and connectivity, the two asparagine residues Asn3 and Asn4 were only established to have opposite stereochemistries (l and d) and their respective assignments were not possible. In earlier studies, we reported the synthesis of the two cyclic peptide diastereomers bearing either the l-Asn3/d-Asn4 or d-Asn3/l-Asn4 stereochemistry and correlation of the former with the spectroscopic properties (1H and 13C NMR) of the natural product.26 In that work, four key subunits were assembled, sequentially coupled, and cyclized to provide the nine residue cyclic peptide. The coupling and macrocyclization sites were chosen to minimize the use of protecting groups and maximize the convergency of the synthesis, and a deliberate late-stage incorporation of the subunit bearing the two asparagine residues allowed convenient access to both diastereomers required to assign the absolute stereochemistry. Concurrent with this disclosure, Searcey reported the synthesis of the l-Asn3/d-Asn4 diastereomers incorporating either a d-ADA8 or l-ADA8 residue,27 and recently Nakata28 has also reported a synthesis of this cyclic peptide.
Figure 1.
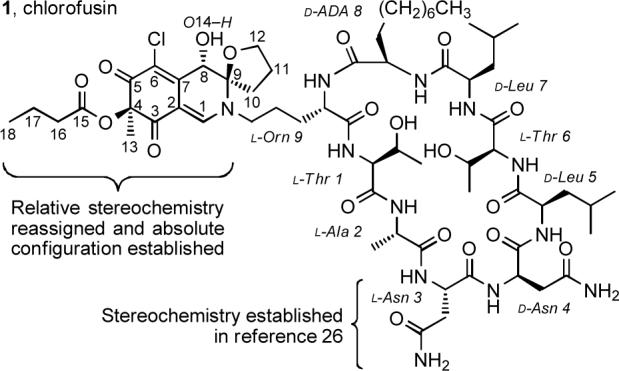
Chlorofusin.
Similarly, the chlorofusin spectroscopic studies conducted by Williams provided the structure and an assigned relative stereochemistry for the unusual azaphilone-derived chromophore, but did not permit an assignment of its absolute stereochemistry (Figure 2). The relative stereochemistry was assigned using gradient 1D NOE studies in which irradiation of the proton attached to C8 with a mixing time of 50 ms resulted in NOEs to the protons attached to C10 as well as C12, whereas a shorter mixing time of 25 ms resulted in NOEs to only the protons attached to C10. In addition, irradiation of the C4-Me protons provided a long range NOE to the proton attached to C8, but only if conducted with very extended mixing times (500 ms). These results suggested that the protons attached to C8, C10 and C4-Me all lie on the same face of the chromophore and led to the relative stereochemical assignment depicted in Figure 2.
Figure 2.
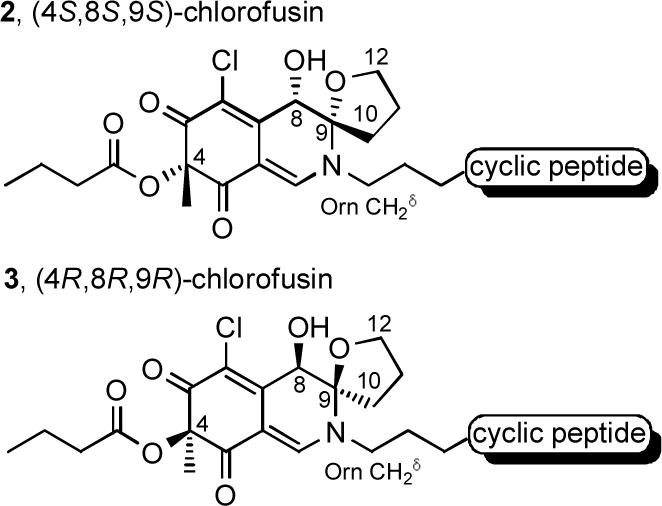
Original chromophore relative stereochemical assignment.23 No assignment of absolute stereochemistry was made.
Herein, we report the full details of studies leading to a reassignment of this relative stereochemistry for the chlorofusin chromophore that is still, but less obviously, consistent with these experimentally observed NOEs, as well as the assignment of the chromophore absolute configuration (4R,8S,9R). A total synthesis of this revised chlorofusin structure provided material displaying spectroscopic properties indistinguishable from that reported for the natural product confirming the new chromophore structural assignment and establishing the accuracy of our earlier l-Asn3/d-Asn4 cyclic peptide stereochemical assignment. A preliminary account of the first stage of these studies was disclosed29 shortly following the report of Yao30 which claimed to have completed a total synthesis of the natural product. Although this group similarly reassigned the chromophore relative stereochemistry, they misassigned the absolute stereochemistry overlooking key diagnostic spectroscopic differences that distinguish their unnatural (4S,8R,9S)-diastereomer 4 (Figure 3) from the natural (4R,8S,9R)-diastereomer. In extending our previously disclosed studies which provided all four 4R-diastereomers of chlorofusin, we have now additionally incorporated the four 4S-diastereomers of the chlorofusin chromophore (enantiomeric series) into fully elaborated chlorofusin diastereomers. Thus, in addition to natural chlorofusin, the remaining seven chlorofusin chromophore diastereomers [(4R,8R,9R), (4R,8S,9S), (4R,8R,9S), (4S,8S,9S), (4S,8R,9R), (4S,8S,9R) and (4S,8R,9S)] including the (4R,8R,9R)/(4S,8S,9S)-diastereomers proposed by Williams as well as the Yao misassigned (4S,8R,9S)-diastereomer were synthesized as key analogues of the natural product and the comparison of their spectral properties with that reported for chlorofusin provide unambiguous support for the structural reassignment.
Figure 3.
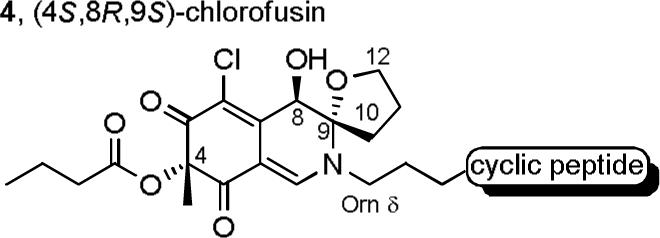
Stereochemical assignment made by Yao and co-workers.30
The chlorofusin chromophore is derived from a class of compounds referred to as azaphilones, so named for their ability to readily condense with ammonia or primary amines.31 In light of this, at least two potential routes to chlorofusin could be envisioned: (1) condensation of an appropriate azaphilone with the full cyclic peptide followed by chromophore oxidation and spirocycle formation, or (2) construction of the fully functionalized chromophore appended to a smaller peptide fragment followed by peptide coupling and macrocyclization to afford chlorofusin (Scheme 1). The former requires the conduct of an azaphilone chromophore oxidative spirocyclization on a complex, late stage synthetic intermediate with control of the resulting relative and absolute stereochemistry or effective separation and characterization of the resulting diastereomers, whereas the latter requires that the chromophore N,O-spiroketal in the smaller peptide fragment not unravel during the late stages of the ensuing cyclic peptide synthesis. Both routes require the same precursor azaphilone allowing us to potentially explore either route as the work progressed.
Scheme 1.

Since the absolute configuration of the chromophore was unknown and the relative stereochemical assignment came under question as the work progressed, the routes pursued in our first generation synthesis permitted access to both enantiomeric series and to all possible diastereomers, albeit optimized to access the original diastereomer assigned by Williams. As discussed herein, confidence in a requisite reassignment of the relative stereochemistry emerged with the observation of diagnostic spectroscopic distinctions (1H and 13C NMR) made possible by access to all diastereomers. Due to concerns of an unambiguous stereochemical assignment as the studies progressed, the strategy of early-stage versus late-stage chromophore oxidative spirocyclization with clear stereochemical assignments for all diastereomers became the focus of our efforts and was effectively conducted on the azaphilone adduct with an ornithine—threonine dipeptide. Beautifully, even an assignment of the chromophore absolute configuration emerged from diagnostic distinctions observed in the spectroscopic properties of the series of all eight diastereomers derived from the chromophore adduct (four diastereomers of the two enantiomeric series) that was confirmed by the total synthesis of the natural product. In retrospect, a late-stage chromophore elaboration, like that of Yao,30 may have failed to produce sufficient quantities of the natural product for detection and may not have allowed an unambiguous stereochemical assignment given that the natural product was found to possess a contrathermodynamic N,O-spiroketal stereochemistry.
Results and Discussion
The Chromophore Azaphilone Precursor: Route 1
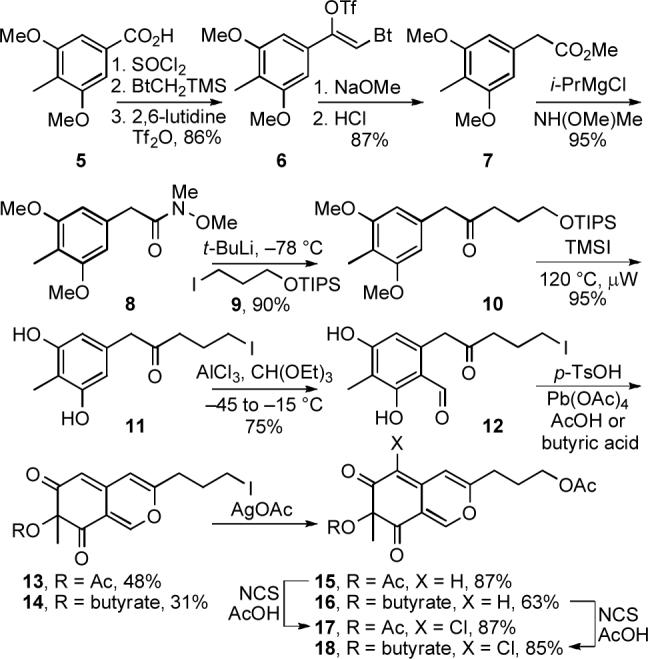
The initial approach to the key intermediates 17 and 18 used to explore the C8—C9 oxidative spirocyclization followed established routes to access azaphilones bearing a range of C9 substituents,32-34 and began with the one carbon homologation of commercially available 3,5-dimethoxy-4-methylbenzoic acid (5) (Scheme 2). The crude acid chloride derived from 5 was alkylated with TMSCH2Bt to afford the ketone adduct (SOCl2, reflux; TMSCH2Bt, THF, reflux, 6 h)35 and subsequent treatment with 2,6-lutidine and Tf2O provided 6 (2,6-lutidine, Tf2O, CH2Cl2, 0 to 23 °C, 2 h, 86%, three steps). The enol triflate 6 was converted to the methyl ester 7 by treatment with NaOMe followed by acidic hydrolysis of the resulting ynamine (NaOMe, MeCN, reflux, 12 h; 12 N HCl, MeOH, reflux, 24 h, 87%, two steps). After conversion to Weinreb amide 8 (NHMe(OMe), i-PrMgCl, THF, −20 °C, 30 min, 95%),36 the lithium reagent derived from 9 was added to 8 to provide the ketone 10 (9, t-BuLi, −78 °C, 2 h, Et2O—THF, 90%). Selective deprotection of the two methyl ethers of 10 in the presence of the silyl ether using BBr3, BF3·Et2O, AlCl3, LiI, TMSI, or PhSH was not successful. In all cases, the silyl ether was found to preferentially or non-productively undergo cleavage. However, the two methyl ethers were cleaved and the TIPS-protected primary hydroxyl group was displaced cleanly with iodide to provide 11 in remarkably high yields when 10 was treated with TMSI under microwave irradiation (TMSI, MeCN, 120 °C, 50 min, normal absorption, 95%). Formylation of 11 provided 12 (AlCl3, CH(OEt)3, toluene, −45 to −15 °C, 1 h, 75%)34 and onepot oxidative cyclization of 12 in acetic acid gave the azaphilone 13 while conducting the same reaction in butyric acid provided 14 (p-TsOH, AcOH or butyric acid, 95 °C, 2 h; Pb(OAc)4, 15 °C, 40 min, 48% for 13, 31% for 14).34 Displacement of the primary iodide of 13 and 14 with acetate (AgOAc, AcOH, 55 °C, 2 h, 87% for 15, 63% for 16)37 gave 15 and 16 whose regioselective C6-chlorination (NCS, AcOH, 23 °C, 24 h, 87% for 17, 85% for 18)38,39 afforded the chloroazaphilones 17 and 18.
Scheme 2.
The Chromophore Azaphilone Precursor: Route 2
The analogous intermediate 28 was also prepared by a more concise route based on the work of Porco.38 Although a one-step conversion40,41 of commercially available 2,4-dimethoxy-3-methylbenzaldehyde to 6-bromo-2,4-dimethoxy-3-methylbenzaldehyde suffered from poor conversions, a more controlled two-step procedure42,43 was adopted beginning with the condensation of 2,4-dimethoxy-3-methylbenzaldehyde (19) with N,N'-dimethylethylenediamine (toluene, 130 °C, 3 h, 95%) (Scheme 3). The resulting dimethylimidazolidine 20 was regioselectively lithiated (t-BuLi, Et2O, −55 to 0 °C, 1 h) and subjected to electrophilic bromination followed by aminal hydrolysis (CF2Br2, −78 to 23 °C, 18 h; 2 N HCl (aq), 30 min, 72%) to provide 6-bromo-2,4-dimethoxy-3-methylbenzaldehyde (21) in good yield. This two-step procedure offers more expeditious access to 21 than a previously reported four-step synthesis.38 Removal of the aryl methyl ethers proceeded in high yield to give 22 (BBr3, CH2Cl2, −78 to 23 °C, 88%). Sonogashira coupling of aryl bromide 22 and alkyne 23 provided the azaphilone precursor 24 (Pd(Cl)2(PPh3)2, CuI, Et3N, DMF, 70 °C, 16 h, 73%). Oxidative cyclization of 24 using Porco's methodology38 generated the azaphilone 25 in good yield (Au(OAc)3, CF3COOH, dichloroethane, 4 min; IBX, Bu4NI, 4 h, 72%). Acylation of the tertiary alcohol of 25 to give 26 (butyric anhydride, DMAP, CH2Cl2, 24 h, 91%), regioselective C6-chlorination yielding 27 (NCS, AcOH, 36 h, 89%) and removal of the TBDPS protecting group (HF—pyridine, THF, 2 h, 72%) proceeded smoothly to provide azaphilone 28. The racemic azaphilones 25, 26, 27 and 28 could be chromatographically separated into their constituent enantiomers, and the most effective chiral phase resolution was achieved with 27 (α = 1.33). The absolute stereochemistry of 25−28 was assigned based on the sign of their optical rotation (αD) and the sign of the longest wavelength Cotton effect (350−370 nm) in their CD spectra (Figure 4).44-46 This assignment was further confirmed by application of Porco's asymmetric oxidative cyclization47 of the OTIPS variant of 24 conducted using the catalyst generated from Cu(I) and (−)-sparteine to provide the corresponding azaphilone (94% ee), matching the assignment made based on CD. As fate would dictate and since (+)-sparteine is not yet readily accessible, this asymmetric route currently only provides access to the chromophore unnatural enantiomer series.
Scheme 3.
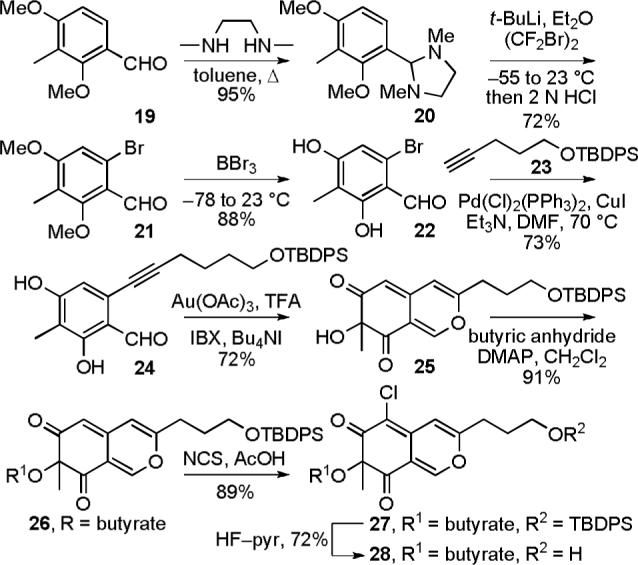
Figure 4.

CD spectra (0.2 mM in MeOH) and αD23 for azaphilones 25−28 (R = butyrate).
Oxidative Spiroketalization
With azaphilones 15 and 17 in hand, we briefly examined their direct oxidative spiroketalization. Thus, selective epoxidation or dihydroxylation of the C8—C9 double bond followed by oxonium ion formation upon epoxide opening or water elimination was anticipated to install the C8 alcohol and lead to subsequent C9-spiroketalization with the pendant side chain alcohol. Confident that the Williams proposed C8—C9 stereochemistry with the two oxygen substituents syn would be the thermodynamically most stable, conducting the reaction under conditions of reversible ketal formation would favor generation of the assigned C8—C9 syn versus anti diastereomers. Thus, a variety of oxidizing agents were explored to effect this transformation. Whereas subjecting azaphilone 15 to the Sharpless asymmetric dihydroxylation protocol48 or that of oxidative spiroketalization with Re2O749 did not lead to identifiable products, treatment of 15 with DMDO50 led to epoxidation of the desired C8—C9 double bond in ca. 40% yield (Equation 1). However, the labile epoxide product51 quickly decomposed and proved challenging to work with or even purify. In hopes of trapping the labile epoxide, the analogous azaphilone 29 possessing the unprotected primary alcohol was treated with DMDO alone or followed with addition of a catalytic amount of acid (HCl, HOAc, PPTs, CSA).52 Although the formation of the intermediate epoxide was confirmed by 1H NMR,51 the subsequent spiroketal formation was not observed and the intermediate epoxide was non-productively consumed. Consequently, we focused our efforts on conducting the oxidative spirocyclization following amine introduction with substrates such as 34 avoiding the imminent issue of C9-isomerization during amine conjugation.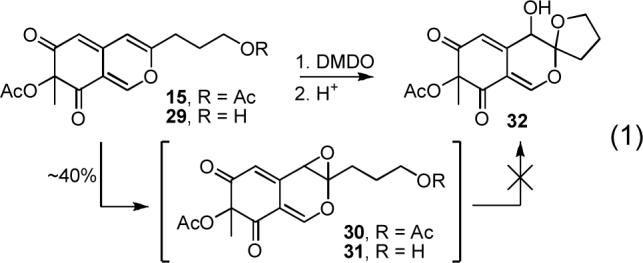
Chromophore Model Studies
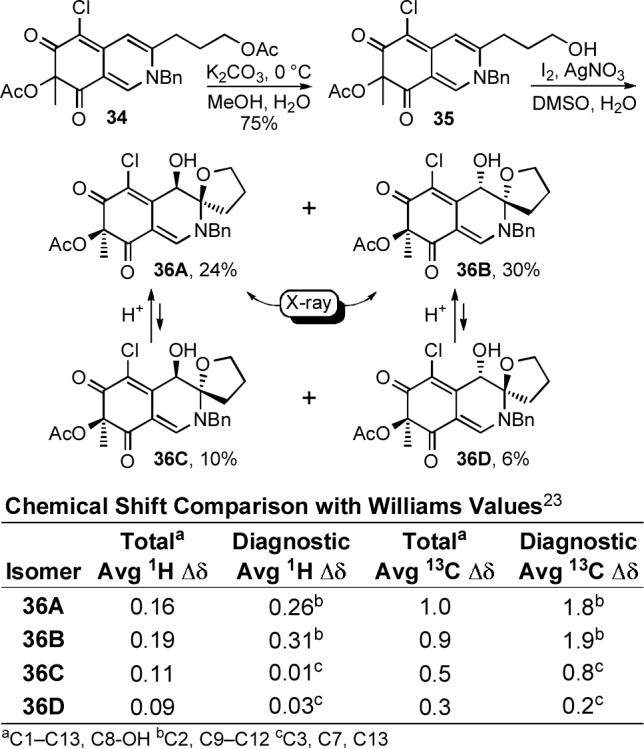
We explored a variety of conditions for condensation of the azaphilones 15 and 17 with benzylamine to probe the compatability of the condensation with solvents that may be required for effective dissolution of the cyclic peptide or a smaller peptide fragment. Although condensation of azaphilone 15 with benzylamine did provide 33 in modest yield (CH2Cl2, 1−16 h, 54%), condensation of chloroazaphilone 17 provided 34 in excellent yield (CH2Cl2, 1 h, 99%) establishing the productive role of the electron-withdrawing C6-chlorine substituent (Equation 2). Selective hydrolysis of the primary acetate (K2CO3, H2O, MeOH, 0 °C, 30 min, 75%) followed by oxidative spirocyclization of 35 with I2 assisted by AgNO3 in the presence of H2O (I2, AgNO3, H2O, DMSO, 2 d) allowed access to all four diastereomers (36A—36D) of the simplified chlorofusin chromophore (Scheme 4). 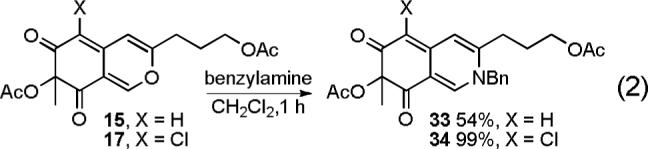
Scheme 4.
The development of these conditions followed unsuccessful efforts to effect a direct oxidative spirocyclization enlisting DMDO, m-CPBA, peroxyimidate, peroxycarbamate, peroxyisourea, singlet oxygen, TMSOOTMS, as well as Shi, Sharpless, and Davis asymmetric epoxidation methods or osmium (OsO4), rhenium (Re2O7), and ruthenium (RuO4) based dihydroxylation reactions. Hypervalent iodine oxidizing reagents including Koser's reagent provided mixtures of 38,53 the oxidized form of the desired product, and 39,53 representing the allylic oxidation product (Equation 3) although the yields of 38 were consistently low (10−15%) and the use of alternative reagents have not yet significantly improved on this. Nonetheless, the modest success with such reagents initiated studies with related reagents that first activate the C8—C9 double bond by reversible bromonium or iodonium ion formation.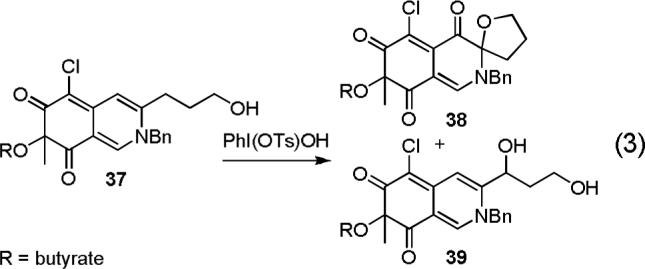
Thus, early efforts exploring a bromonium ion initiated spirocyclization, induced upon treatment of 35 with NBS under anhydrous conditions, provided the α-bromo N,O-spiroketal as a 1:1 mixture of C4-diastereomers resulting from clean trans addition across the C8—C9 double bond. Treatment of 35 with NIS under such conditions failed to provide analogous products upon workup despite the slow disappearance of starting material under the reaction conditions. However, inclusion of water in the reaction mixture led directly to the desired syn α-hydroxy N,O-spiroketal presumably resulting from water displacement of the iodide while premature workup of the reaction allowed isolation of two isomers of the unstable intermediate α-iodo N,O-spiroketal. Switching from NIS to I2 served to increase the yield of the syn products and significantly, also led to the production of trans α-hydroxy N,O-spiroketal products. Although a range of solvents support the reaction (e.g., CH3CN—H2O, DMF—H2O), DMSO—H2O provided the syn cycloadducts in the best yield along with fewer undesired side products. Finally, the use of Ag(I) salts (AgOTf, AgNO3, AgClO4, CF3CO2Ag) in the mixture accelerated the reaction, improved the conversions and minimized a competitive oxidation reaction. Thus, the reaction appears to be initiated by fast reversible iodonium ion formation and subsequent slow iodoetherification with N,O-spiroketal formation followed by slow Ag(I)-assisted iodide displacement by H2O providing 36A—36D directly. Notably, the lone pair of the endocyclic amine is tied up in the π-system of the chromophore as a vinylogous amide, and accounts for the relative stability of the iodonium intermediate, the predominance of C8/C9 syn products arising from SN2-like trans intramolecular opening of the iodonium ion, and the high degree of planarity at the nitrogen center observed in the X-ray crystal structures (Figure 5). The major products (54%), in which the C8 and C9 oxygen substituents are syn possessing the C8/C9 stereochemistry found in the Williams assignment, represent those that formally arise from a trans iodoetherification reaction followed by water SN2 displacement of the iodide. The structures of 36A and 36B, the major products of the oxidative spirocyclization reaction, were established by X-ray crystallography54 and the C8 and C9 oxygen substituents of both compounds were found to be oriented syn with respect to one another with the C8-OH occupying an axial orientation. N,O-Ketal equilibration studies unambiguously established the structure of the remaining two minor anti diastereomers. The 1H NMR signals of the tetrahydrofuran ring were found to be diagnostic of the relative orientation of the C8 and C9 substituents. Accordingly, chemical shifts of the C10-H signals for the two anti isomers 36C (m, 2.25 ppm) and 36D (m, 2.26 ppm) are similar to each other and to the C10-H signal reported for chlorofusin (br m, 2.38 ppm), but are distinct from the C10-H signals for 36A (m, 1.89) and 36B (m, 1.81). Similarly the C11-H signals for 36C (m, 2.01 ppm) and 36D (m, 2.00 ppm) possess chemical shifts that are closer to that of chlorofusin (m, 2.0−2.2 ppm) than the two syn isomers 36A (m, 1.79 ppm; m, 1.96 ppm) and 36B (m, 1.76 ppm; m, 1.82 ppm). The C12-H signals for 36C (m, 3.80 ppm; m, 3.87 ppm) and 36D (m, 3.77 ppm; m, 3.83 ppm) were also similar to one another and chlorofusin (q, 3.78 ppm; m, 4.02 ppm), and different from those of the syn isomers 36A (m, 3.97 ppm; m, 4.18 ppm) and 36B (m, 3.97 ppm; m, 4.18 ppm), but the correlation with the anti isomers was more tentative. The 13C NMR data exhibited similar trends, the most striking difference appearing with C10 signals for 36C (30.6 ppm) and 36D (30.5 ppm) being very similar to each other and to chlorofusin (30.3 ppm), but 4 ppm farther upfield than the C10 signals for the two syn isomers 36A (34.5 ppm) and 36B (35.0 ppm). A complete tabular comparison of the spectroscopic properties of 36A—36D (Tables S1 and S2) and bar graph representations of the chemical shift differences (Δδ, Figures S4 and S5) relative to chlorofusin are provided in the Supporting Information and they, including the diagnostic distinctions highlighted above, were the first indication that the C8 and C9 oxygen substituents of chlorofusin may be oriented anti with respect to one another, and not syn as originally reported. These may also be summarized more simply as observed chemical shift differences averaged over the entire chromophore or restricted to the more diagnostic comparisons as presented in Scheme 4, both of which indicate that the anti diastereomer 36D provides the best match with the chlorofusin chromophore. However, we were concerned about potential perturbation of the chromophore NMR signals by the appended benzyl group and elected to examine the additional, more comparable system 42 incorporating a N-butyl substituent as well as a C4 butyrate versus acetate. Significantly, the observations with 36A—36D initiated a switch in our strategy from targeting the total synthesis of chlorofusin to one that could also unambiguously establish the chromophore stereochemistry. As such, the efforts were refocused on preparing and spectroscopically characterizing all possible chromophore diastereomers rather than exclusively targeting the Williams syn isomer.
Figure 5.

X-Ray structures of 42A and 42D.
Thus, butylamine was condensed with chloroazaphilone 18 to give 40 in excellent yield (CH2Cl2, 1 h, 99%). Hydrolysis of the primary acetate (K2CO3, H2O, MeOH, 0 °C, 30 min, 91%) followed by the single step oxidative spirocyclization of 41 (I2, AgNO3, H2O, DMSO, 2 d) provided 42A—42D possessing the n-butyl substitution (Scheme 5). As with 36A—36D, the NMR data collected from the anti isomers 42C and 42D provided a better match with chlorofusin than the syn isomers 42A or 42B (Supporting Information Tables S3 and S4 and Figures S8 and S9). An X-ray crystal structure of 42A, one of the major products of the spirocyclization reaction representing the Williams assigned diastereomer and the closest match by NMR to 36A, confirmed that the C4, C8 and C9 oxygen substituents were all syn while an X-ray of 42D, the closest match with chlorofusin by NMR, confirmed that the relative orientation of the C8 and C9 oxygen substituents is anti and that the C4 methyl group is cis to the C8-OH and trans to the C9 oxygen of the spirotetrahydrofuran (Figure 5).55 N,O-Ketal equilibration studies established the related syn/anti pairs and completed the unambiguous stereochemical assignments for the entire series. As the similarity of the model to the chlorofusin chromophore increased, the trends in the NMR data distinguishing the C8/C9 syn diastereomers from the anti diastereomers became more predominant. For C10-H, 42A (m, 1.85 ppm; m, 1.94 ppm) and 42B (m, 1.82 ppm; m, 2.00 ppm) each display two signals, neither of which is within 0.3 ppm of chlorofusin (br m, 2.38 ppm), whereas the signals for the two anti isomers 42C (m, 2.37 ppm) and 42D (m, 2.39 ppm) differ from chlorofusin by 0.01 ppm. Similarly, the C12-H signals for the syn isomers 42A (dd, 4.03 ppm; m, 4.22 ppm) and 42B (dd, 4.04 ppm; dd, 4.22 ppm) are much farther downfield than the analogous signals for 42C (dd, 3.82 ppm; m, 3.95 ppm), 42D (dd, 3.80 ppm; dd, 3.95 ppm) and chlorofusin (q, 3.78 ppm; m, 4.02 ppm). An additional and important trend emerged linking 42B with 42D, which share the same relative stereochemistry at C8. The signals for C8-H observed in the spectra of 42A (d, 4.48 ppm) and 42C (d, 4.49 ppm) are similar to one another, whereas the signals for 42B (d, 4.54 ppm) and 42D (d, 4.53 ppm) are not only similar to one another, but also to that of chlorofusin (d, 4.53 ppm). This, along with other more subtle differences, distinguishes the two anti diastereomers with 42D, not 42C, being representative of the stereochemistry found in chlorofusin. Finally, the C8-OH signal in this series appears to distinguish 42A (d, 6.18 ppm), 42B (d, 6.11 ppm), 42C (d, 6.41 ppm), and 42D (d, 6.25 ppm) as well as link the relative stereochemistry of 42D with that of chlorofusin (d, 6.26 ppm).
Scheme 5.
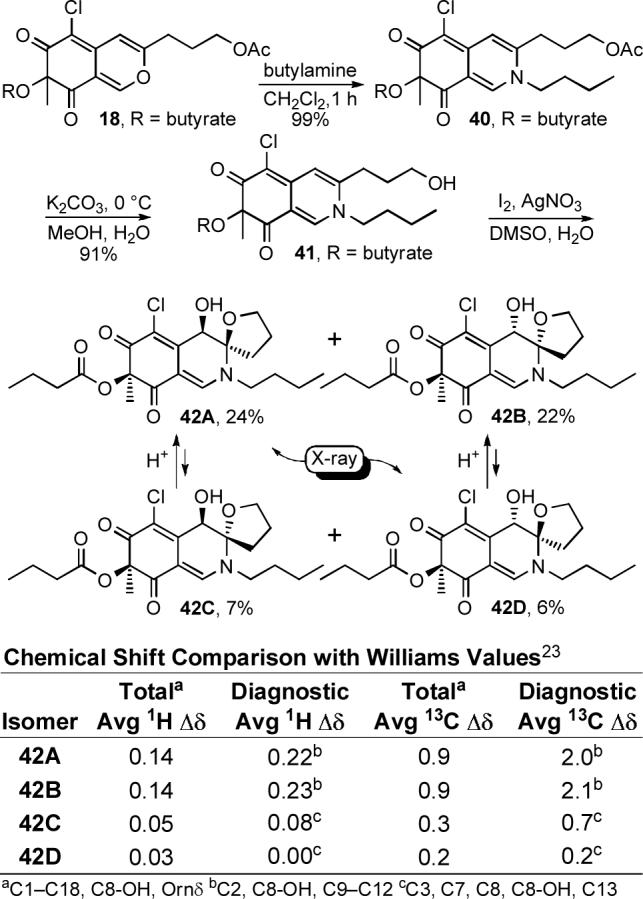
The 13C NMR show similar trends to the observations made with 36A—36D. However, the differences between the syn and anti compounds become even greater in this series for C10, C11, C12 and C9 in the absence of a proximal phenyl group. With X-ray structures of representative C8/C9 syn and anti diastereomers in hand, the diagnostic differences seen in their C10 13C NMR chemical shifts can be attributed to its axial (syn) or equatorial (anti) orientation. For C10, the chemical shifts of 42C (30.2 ppm) and 42D (30.2 ppm) became even closer to that of chlorofusin (30.3 ppm), but remained over 4 ppm farther upfield than the C10 signals for the syn isomers 42A (34.6 ppm) and 42B (35.0 ppm). The butylamine replacement for the Orn side chain also allowed a comparison of the chemical shift of the methylene adjacent to the chromophore ring system, with the anti isomers 42C (50.5 ppm) and 42D (50.6 ppm) matching that of chlorofusin (50.5 ppm) as opposed to 42A (49.7 ppm) and 42B (49.8 ppm).
Although intuitively surprising but which now may be expected from the X-ray crystal structure of 42D (Figure 5) that shows an unobstructed path between C8-H and C4-Me (4.761 Å, C8-H—C13), the ROESY NMR for 42D exhibited a weak cross-peak between C4-Me and C8-H as observed by Williams as a long range NOE in chlorofusin. Also observed in its X-ray structure is the diaxial orientation of the C8 and C9 oxygen substituents. In this conformation, the equatorial C8-H can exhibit NOEs to both C10-H and C12-H as observed by Williams for chlorofusin. Although these NOEs were not quantitated, it is notable that C8-H is closer to C10-H (2.484 Å) than C12-H (2.592 Å) in this X-ray consistent with such implications in the Williams work. In fact, the C8-OH is axial and the C8-H is equatorial in the X-ray structures for 36A, 36B, 42A and 42D indicating that the C8-H NOEs observed by Williams would be possible with all four diastereomers. This dominance of the C8-OH axial orientation arises presumably from the A(1,3)-strain an equatorial C8-alcohol would experience with the C6-chloride which is further destabilized by the electronegative nature of the two interacting substituents (Cl/OH). With this understanding of the conformational properties of the chromophore diastereomers and with the characterization data from both the 36 and 42 model systems in hand, the stereochemistry of the chlorofusin chromophore was confidently reassigned as either the (4S,8R,9S) or (4R,8S,9R) anti diastereomer (42D enantiomers).
l-Orn-l-Thr Azaphilone Conjugation and Elaboration to All 8 Chromophore Diastereomers
At this stage, we were convinced that a final stage chromophore coupling to the intact cyclic peptide followed by oxidative spirocyclization would be difficult to implement. Not only would this require the separation, characterization, and unambiguous stereochemical assignments of the four diastereomers generated in each enantiomeric series, but the reassigned anti isomer represented a minor, contrathermodynamic product of the cyclization reaction in our model systems. As such, we elected to take the less ambitious, more manageable step of linking and elaborating the chromophore on a smaller dipeptide constituting l-Orn9-l-Thr1 of chlorofusin examining the two enantiomeric series and their four possible diastereomers. Not only would this provide intermediates with more manageable physical and spectroscopic properties and a third model on which to carefully reassess the chlorofusin stereochemical assignments, but it provided an opportunity to probe the chromophore absolute stereochemical assignment.
To this end, dipeptide 44 was prepared from the commercially available amino acid derivatives Fmoc-l-Orn(Mtt)-OH and l-Thr-OBn using a standard coupling protocol (EDCI, HOAt, DMF, 24 h, 82%) followed by deprotection of the Nδ-amine of ornithine (CF3CO2H, CH2Cl2, 1 h, 97%), Equation 4. Condensation of 44 with either (R)-28 or (S)-28 (NaHCO3, DMF, CH2Cl2, 24 h; 1 N HCl, 4 h, 84% for 45, 71% for 46) provided 45 and 46, respectively (Scheme 6). While incorporation of simple amines into the azaphilone core proceeded in high yield with simply CH2Cl2 as the reaction solvent, condensation with 44 required more specialized conditions. Solubility considerations led to the use of a DMF as a co-solvent and additive studies revealed that the early stages of the incorporation were accelerated by addition of NaHCO3 and subsequent treatment with 1 N HCl (aq) drove the reaction to completion. Oxidative spirocyclization of 45 (I2, AgNO3, H2O, DMSO, 3 d) provided 47A—47D and similar treatment of 46 provided 48A—48D. The 1H and 13C NMR spectra of all eight diastereomers were fully assigned (Tables S5—S8, Figures S11—S14) using COSY, HMQC, HMBC and ROESY NMR data and N,O-ketal equilibration studies defined the related syn/anti pairs in each enantiomeric series. Notably, the equilibration studies requiring 5% TFA—HOAc (25 °C) revealed that the interconversions, like those in the 36 and 42 series, are slow even under these strongly acidic conditions and that the N,O-spiroketals are remarkably robust. Additionally, resubjecting 47B or 47C to the oxidative spirocyclization reaction conditions did not provide isomerized products suggesting that the syn products are both thermodynamically and kinetically favored and that the anti products generated are stable to the reaction conditions.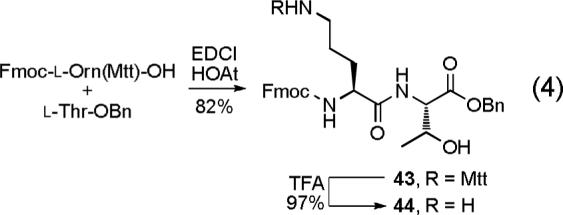
Scheme 6.

Stereochemical Assignment of Chlorofusin
By using the diagnostic spectroscopic distinctions observed in the 47 and 48 series, having assigned the C4 absolute stereochemistry by CD (see Figure 4),44-46 and enlisting N,O-ketal equilibrations to define the syn/anti pairs in each enantiomeric series, the complete relative and absolute stereochemical assignments for all eight diastereomers were unambiguously established. Moreover, only diastereomer 47D (4R,8S,9R) matched all the spectroscopic properties reported for the chlorofusin chromophore. In short, the two syn and two anti diastereomers in each enantiomeric series are readily distinguishable by the diagnostic 1H NMR (C10-H, C11-H, C12-H, and C8-OH) and 13C NMR (C2 and C9—C12) chemical shifts and supported by kinetic and thermodynamic predominance of the syn isomers. The two anti diastereomers in each enantiomeric series are most readily distinguished by the diagnostic 1H NMR (C8-H, C13-H, and C8-OH) and 13C NMR (C3, C7, C8, and C13) chemical shifts although there are more subtle spectroscopic distinctions as well. These are discussed in detail in the Supporting Information where Figures S11—S14 graphically quantitate these distinctions. In both series, it was the anti diastereomers 47D and 48D (chromophore enantiomers) that most closely matched chlorofusin. Additionally, the (4R,8S,9R)-diastereomer 47D provided a near perfect match with the spectroscopic properties reported for the chlorofusin chromophore, whereas the (4S,8R,9S)-diastereomer 48D (chromophore enantiomer) proved readily distinguishable by both the 1H NMR chemical shift and multiplicity of the ornithine CH2δ adjacent to the chromophore (δ 3.45, m, 2H for 47D vs δ 3.41 and 3.52, two m, 1H each for 48D; chlorofusin = δ 3.42, t, 2H). This final multiplicity distinction between 47D and 48D allowed the absolute stereochemical assignment for the chlorofusin chromophore (Figure 6). This assignment of the absolute configuration for chlorofusin is especially significant given that our examination of the extensive series of known azaphilones revealed that both C4 configurations are found naturally with roughly equivalent representations.
Figure 6.
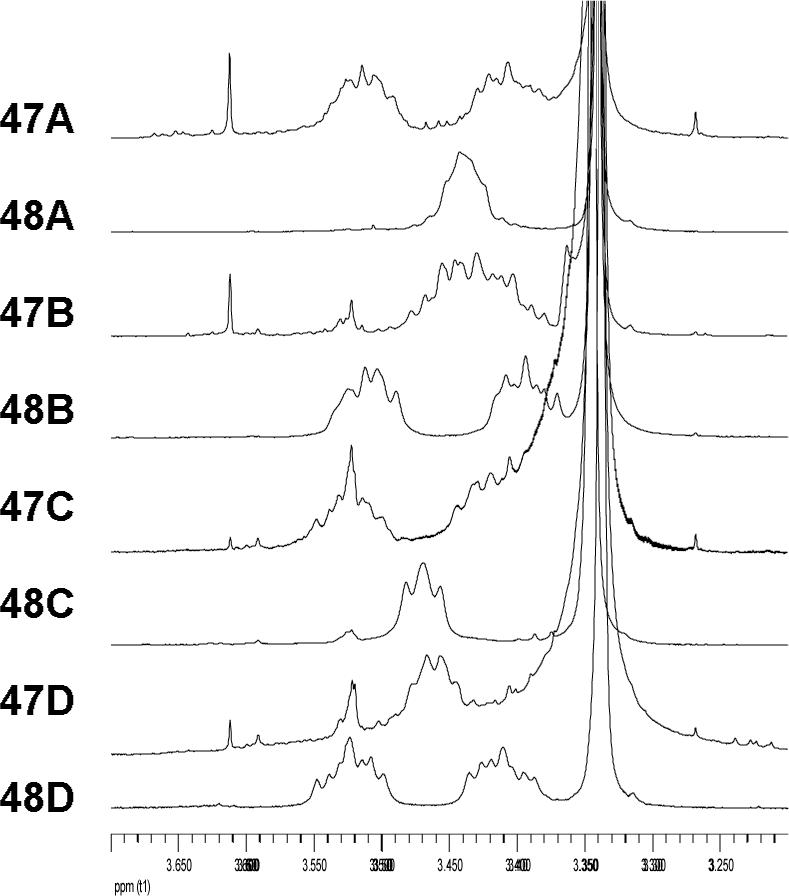
Ornithine CH2δ 1H NMR signals for 47A—47D and 48A—48D.
As previously discussed, the anti diastereomers adopt a conformation with both the C8 and C9 oxygen substituents axial (see Figure 5) and their equatorial C8-H can exhibit NOEs to both C10-H and C12-H consistent with data for chlorofusin. The syn diastereomers adopt a conformation in which the C8-OH is axial, the C9 oxygen substituent is equatorial, and the C8-H is also equatorial. These isomers similarly exhibit C8-H to C10-H and C12-H NOEs. However, an important ramification of the conformational features of the syn and anti diastereomers where each possess an equatorial C8-H is the counterintuitive observation that all four diastereomers possess nearly equivalent C4-Me to C8-H distances (5.42—5.55 Å) and that it is actually closest (5.42 Å) for the anti isomer we assigned to chlorofusin. The ROESY NMR data (300 ms mixing time) for these eight diastereomers reveals an NOE only between C8-H and C4-Me (and not C4-Me/C8-OH) when those groups are cis with respect to one another (47A, 47C, 48A and 48C) and a correlation between C4-Me and both C8-H and C8-OH when C4-Me is trans with respect to C8-H (47B, 47D, 48B and 48D). Thus, regardless of the relative stereochemistry of the chromophore, a NOE is seen between C4-Me and C8-H making the reassignment of the chromophore relative stereochemistry consistent with Williams' spectroscopic characterization of the natural product. Importantly, the NOE data collected and reported by Williams is impeccable including that of the long range C4-Me/C8-H NOE, but it did not prove diagnostic of a C4-Me/C8-H cis stereochemistry.
Comparison of the CD spectra of all eight diastereomers shows that the region between 250−500 nm is dependent solely on the stereochemistry of the chromophore with nearly equal and opposite spectra observed for 47A and 48A, for 47B and 48B, for 47C and 48C and for 47D and 48D (Figures 7 and 8). Of particular note is the sign of the longest wavelength Cotton effect (395−410 nm).56 As with azaphilones 25—28, a positive longest wavelength Cotton effect is diagnostic of the C4-R stereochemistry and a negative Cotton effect is diagnostic of the C4-S stereochemistry.44-46
Figure 7.
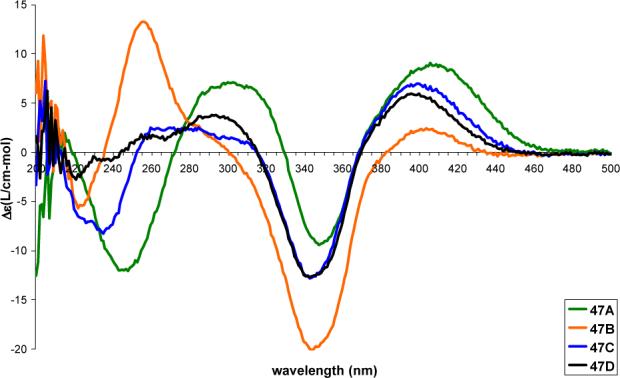
CD spectra (0.2 mM in MeOH) of 47A—47D.
Figure 8.
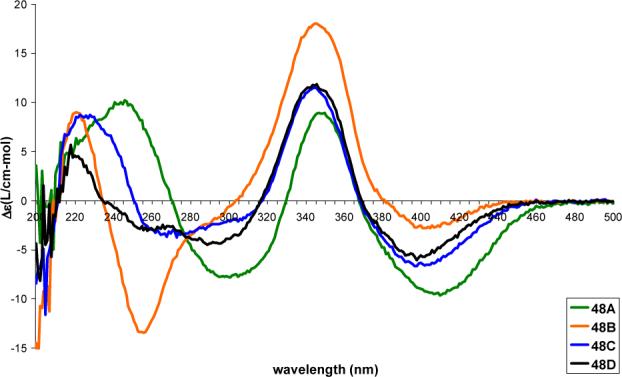
CD spectra (0.2 mM in MeOH) of 48A—48D.
Total Synthesis of Chlorofusin and its Three 4R-Diastereomers
With the chromophore stereochemistry confidently assigned, the (4R,8S,9R)-diastereomer 47D was incorporated into a total synthesis of chlorofusin. As a prelude to our initial assemblage of chlorofusin, one of the two anti diastereomers (47C) in the natural 4R-enantiomeric series was exposed to many of the reaction conditions and recovered unchanged. Its treatment with EDCI—HOAt and NaHCO3 in DMF for 16 h followed by exposure to workup conditions (dilute with EtOAc, wash with 1 N HCl (2×), aq. NaHCO3 (2×), saturated aq. NaCl, and drying over Na2SO4) provided a quantitative recovery of 47C with no detectable isomerization to its more stable syn isomer (47A). This would be representative of the peptide coupling and macrocyclization reaction conditions and their workup. Extended treatment (16 h) with H2/Pd—C does slowly hydrogenate the material, but it does not isomerize it. We minimized this potentially competitive reaction during the benzyl ester and Cbz deprotections using short reaction times (4 h). The Fmoc deprotection with piperidine (40 min) proceeded uneventfully providing the free amines of 47A—47D, each of which was isolated by chromatography, without detection of isomerization or other conceivable problems. As such, the stability of the N,O-ketal is greater than one might intuitively suspect and this may be attributed to the fact that the nitrogen is tied up as a vinylogous amide. Two empirical observations additionally highlight this. First, the isolation of chlorofusin was conducted with TFA (0.01%) in the reverse phase HPLC solvent system.23 Second, the conditions required for N,O-ketal isomerization are strongly acidic (e.g., 5% TFA—HOAc or 1:1 TFA—CH2Cl2, 5 h) and they do not isomerize under the conditions of a mild aqueous acid workup. Thus, a projected concern over the chromophore stability did not materialize, thus simplifying the path forward.
Peptide 61, synthesized in a convergent sequence (Scheme 7), was protected in a manner that avoids treatment of late-stage chromophore-containing intermediates with strong acid that might promote C9 N,O-spiroketal isomerization. Fmoc deprotection of 47D (piperidine, CH2Cl2, DMF, 40 min) and coupling of the free amine 62 with heptapeptide 61 (EDCI, HOAt, NaHCO3, DMF, 0 to 23 °C, 24 h, 55%) provided 63 (Scheme 8). Concurrent benzyl ester deprotection and Cbz removal (H2, Pd/C, THF—DMF, 4 h) followed by macrocyclization of 64 (EDCI, HOAt, NaHCO3, DMF, 0 to 23 °C, 40 h, 60%) afforded synthetic chlorofusin (1), which displayed spectroscopic properties indistinguishable from that reported for natural chlorofusin. Among the most notable of these spectroscopic correlations was the single signal for the ornithine CH2δ (δ 3.42, t, 2H for natural and synthetic chlorofusin) that readily distinguishes the natural diastereomer from the (4S,8R,9S)-diastereomer incorporating the chromophore enantiomer (δ 3.42 and 3.50, two m, 1H each) that was prepared and miscorrelated by Yao.30
Scheme 7.
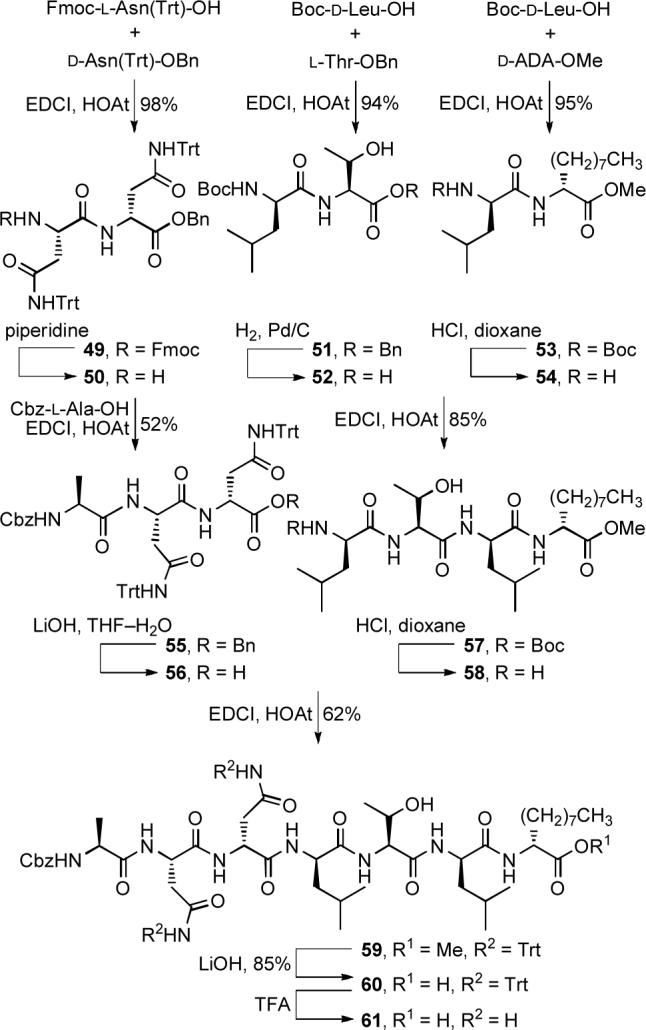
Scheme 8.
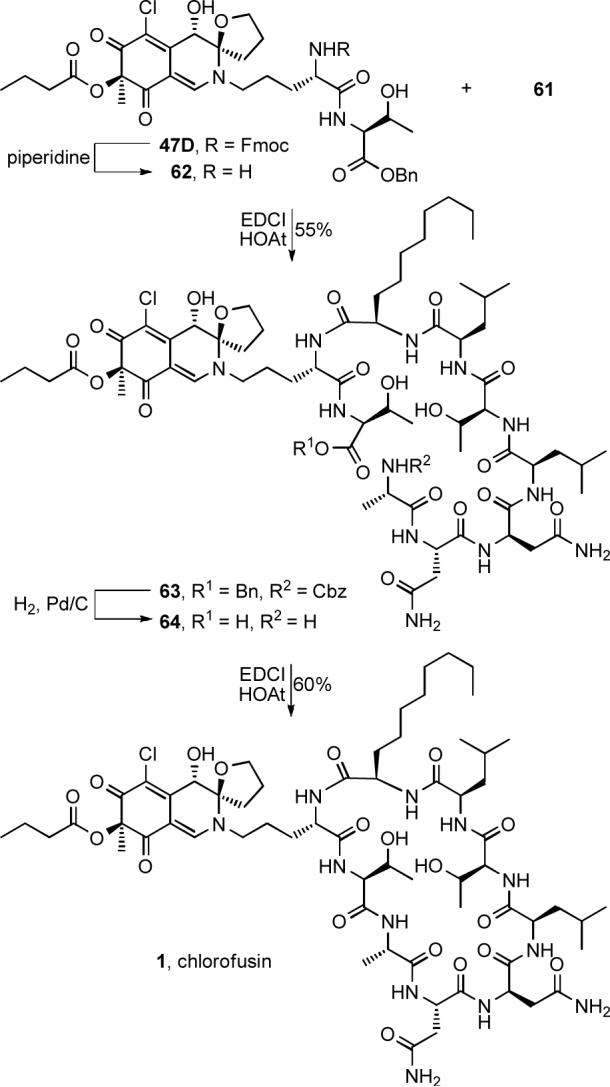
In addition to the natural (4R,8S,9R)-diastereomer constituting chlorofusin, the (4R,8R,9R)-diastereomer 3 proposed by Williams as well as the (4R,8S,9S)- and (4R,8R,9S)-diastereomers of chlorofusin (65 and 66) were also prepared by this route from 47A—47C. Notably, this entailed simply the three steps of Fmoc deprotection of the chromophore dipeptide precursor and EDCI-promoted coupling with the same heptapeptide 61, concurrent Cbz and benzyl ester deprotection, and a final macrolactamization to provide diastereomers 3, 65 and 66 with purification of a single intermediate in addition to the final products (Scheme 9). This late stage divergence in a convergent total synthesis facilitated the assemblage of the collective set of diastereomers and the anticipated non-correlation of their spectroscopic properties with that reported for the natural product provided further firm support for the new structural assignment. The spectroscopic distinctions in these isomers mirrored those observed with the dipeptide conjugates 47A—47D with the two syn and two anti diastereomers in each enantiomeric series readily distinguishable by the diagnostic 1H NMR (C10-H, C11-H, C12-H, and C8-OH) and 13C NMR (C2 and C9—C12) chemical shifts. The two anti diastereomers in each enantiomeric series are most readily distinguished by the diagnostic 1H NMR (C8-H, C13-H, and C8-OH) and 13C NMR (C3, C7, C8, and C13) chemical shifts along with more subtle spectroscopic distinctions.
Scheme 9.
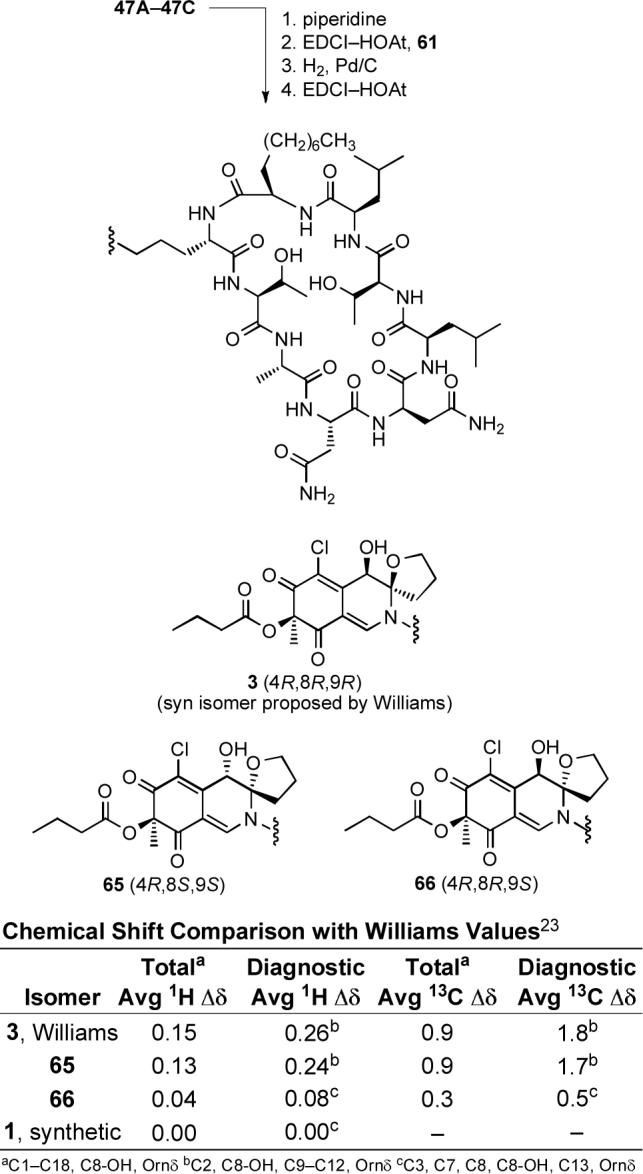
Following the completion of this work and our chlorofusin structural reassignment and upon the Yao report30 that claimed to have prepared chlorofusin (4S,8R,9S-diastereomer), we re-examined a sample of authentic chlorofusin provided by Dr. Stephen Wrigley in 2003 (aged and of unknown quality) that failed to provide a discernable 1H NMR spectrum in 2003. Now, with an intimate knowledge of the chromatographic and physical properties of such compounds in hand, the processing of the remaining material (<1 mg) provided a sample that exhibited a CD spectrum indistinguishable (sign and magnitude) from synthetic 1 confirming the absolute configuration assignment (Figure 10). Moreover, its CD spectrum was readily distinguishable from the two possible syn diastereomers (4R,8R,9R and 4R,8S,9S) and more subtly distinguishable from the alternative anti (4R,8R,9S)-diastereomer 66 (Figure 9). Like the simple azaphilone chromophore precursors 25−28, the dipeptide azaphilone conjugates 45 and 46, as well as the fully elaborated chromophore dipeptides 47 and 48, the longest wavelength (395−415 nm) CD Cotton effect is positive for all four 4R chlorofusin diastereomers as well as the natural product firmly establishing the absolute configuration assignment.44-46
Figure 10.
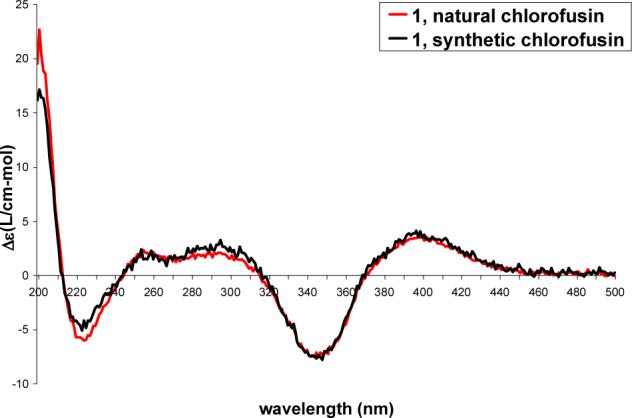
CD spectra of synthetic versus natural chlorofusin.
Figure 9.
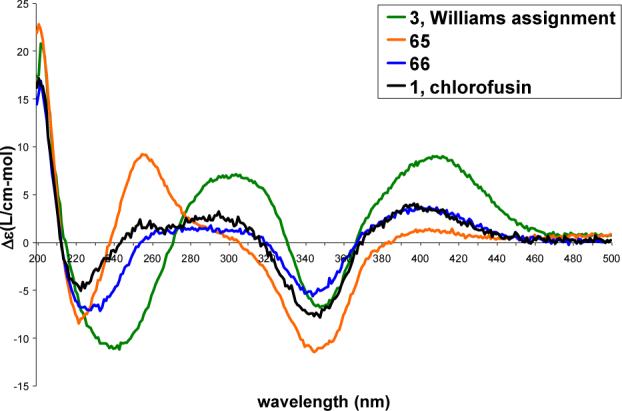
CD spectra (0.2 mM in MeOH) of 3, 65, 66 and 1.
Just as significantly, the sample provided an 1H NMR spectrum of a sufficient quality to confirm that it represented the authentic natural product (Supporting Information). Illustrated in Figure 11 is the spectral overlay of a diagnostic region of the 1H NMR spectra of this sample of natural chlorofusin with the four 4R diastereomers illustrating the clear correlation with only the synthetic (4R,8S,9R)-diastereomer 1 and the clear distinctions with any of the alternative 4R diastereomers in this enantiomeric series. Significantly, the syn diastereomer 3 proposed by Williams is readily discernable from natural chlorofusin not only by CD (Figure 9), but also by 1H NMR (Figure 11). Additionally, the comparisons in this region of the 1H NMR spectra clearly confirm the stereochemical purity and integrity of each isomer indicating that each, including the less stable anti diastereomers, is unaffected by the synthetic sequence required for their incorporation into the final products.
Figure 11.

1H NMR overlay of natural chlorofusin with all four C4-R chlorofusin chromophore diastereomers.
Synthesis of All Four 4S-Diastereomers of Chlorofusin
With the Yao disclosure of the synthesis and miscorrelation of 4 with chlorofusin,30 we elected to extend our studies to include the preparation and characterization of all four 4S-chromophore diastereomers of chlorofusin including 4. The preparation of this enantiomeric series simply required the late-stage individual incorporation of 48A—48D into the full chlorofusin structure and again benefited from the convergent nature of the total synthesis providing diastereomers 2, 67, 68 and 4 in four steps of which 4 corresponds to the Yao structure (Scheme 10). Though operationally short, the final materials are technically challenging to handle (SiO2 and C18-reverse phase chromatography) making their synthesis more challenging than our expression of access in four steps might inadvertently convey.
Scheme 10.
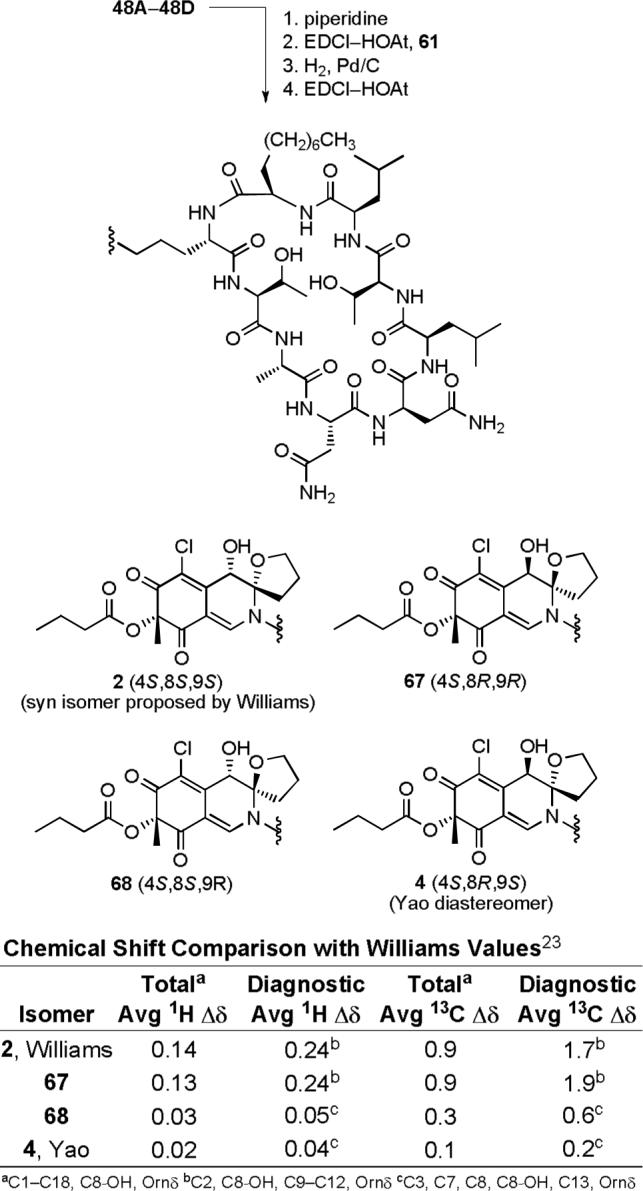
With 2, 67, 68 and 4 in hand, their spectroscopic properties proved readily distinguishable from the natural product. Each displayed an anticipated long wavelength negative Cotton effect opposite that observed with the natural product (Figure 12). Moreover, each 4S-diastereomer provided an essentially identical, but of opposite sign, CD spectrum (250−500 nm) to the corresponding 4R-diastereomer indicating that it is the chromophore, and not the cyclic peptide, that establishes the sign and magnitude of the CD spectrum in this region. Notably, that of 4 which bears the chlorofusin chromophore enantiomer is nearly identical to that of chlorofusin, but is clearly of the opposite sign.
Figure 12.
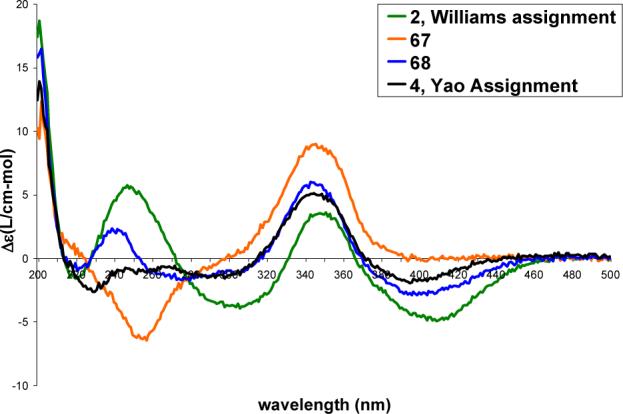
CD spectra (0.2 mM in MeOH) of 2, 67, 68 and 4.
Similarly, the 1H NMR and 13C NMR spectroscopic properties of each diastereomer 2, 67, 68 and 4 were distinguishable from those of chlorofusin (see Supporting Information Table S11 and S12). This is most apparent in the region represented with the spectral overlay with the natural product in Figure 13. The Yao diastereomer 4, bearing the enantiomer of the chromophore found in natural chlorofusin, is most readily distinguishable by its ornithine CH2δ signal which appears as two multiplets of 1H each (δ 3.41 and 3.52), but also exhibits more subtle differences including C8-OH (δ 6.22 vs 6.26), Orn9 α-CH (δ 4.56 vs 4.59), Leu5 α-CH (δ 4.45 vs 4.48), Thr1 α-CH (δ 3.68 vs 3.66), Asn3 β-CH2 (δ 2.57, dd vs 2.62, app t), C10-H (δ 2.40, m vs 2.38, br m) and C11-H (δ 2.04, m vs 2.0−2.2, br m). Similarly, the alternative syn diastereomer 2 proposed by Williams and in this enantiomeric series also exhibited spectroscopic properties readily distinguishable from the natural product. Interestingly, while the ornithine CH2δ signals for 1, 65, 66 and 3 follow the trend observed with 47A—47D appearing as a single multiplet of 2H in the 1H NMR when the stereochemistry at C8 is S and as two multiplets of 1H each when the stereochemistry at C8 is R, it was observed that the ornithine CH2δ signal for 2, 67, 68 and 4 appeared as distinguishable multiplets of 1H each independent of the C8 stereochemistry.
Figure 13.

1H NMR overlay of natural chlorofusin with all four C4-S chlorofusin chromophore diastereomers.
Conclusion
Full details of the first total synthesis of chlorofusin are reported resulting in the reassignment of its chromophore relative stereochemistry and the establishment of its chromophore absolute stereochemistry. Careful spectroscopic characterization of each of the four chlorofusin model chromophore diastereomers in two distinct series suggested the initial Williams assignment might not prove accurate and revealed that the key spectroscopic feature leading to the original assignment does not distinguish the four possible diastereomers. Subsequent preparation and characterization of all four diastereomers of a dipeptide conjugate with both enantiomers of the chlorofusin chromophore confirmed the required relative stereochemistry reassignment and permitted an assignment of its absolute configuration. This was unambiguously established with the total synthesis of not only chlorofusin, which displayed spectroscopic and physical properties indistinguishable from the natural product, but also all seven alternative chlorofusin chromophore diastereomers, each of which displayed spectroscopic properties readily distinguishable from the natural product. This included the diastereomer advanced by Yao30 as being identical with spectroscopic properties reported for natural chlorofusin as well as both enantiomeric chromophores of the original Williams assignment.23 This study with the comprehensive characterization of all possible isomers was accomplished enlisting a convergent synthetic strategy with a key late-stage divergent introduction of the eight chromophore-dipeptide conjugates requiring only four steps for completion of the synthesis and facilitating the total synthesis of the full series of eight chromophore diastereomers. Currently, efforts are underway to synthesize the chromophore stereoselectively as well as to probe the mechanism by which chlorofusin disrupts the interaction of p53 and MDM2.
Acknowledgments
Dedicated to Professor E. J. Corey on the occasion of his 80th birthday. We gratefully acknowledge the financial support of the National Institutes of Health (CA41101) and the Skaggs Institute for Chemical Biology. We are especially grateful to Dr. S. S. Pfeiffer for initial studies on the chromophore synthesis (Scheme 2) and both Dr. S. S. Pfeiffer and Dr. P. Desai for initial studies on the chlorofusin cyclic peptide.26
Footnotes
Supporting Information. Full experimental details are provided. This material is available free of charge via the internet at http://pubs.acs.org.
Supplementary Material
References
- 1.Levine AJ, Momand J, Finlay CA. Nature. 1991;351:453. doi: 10.1038/351453a0. [DOI] [PubMed] [Google Scholar]
- 2.Vousden KH. Cell. 2000;103:691. doi: 10.1016/s0092-8674(00)00171-9. [DOI] [PubMed] [Google Scholar]
- 3.Shen Y, White E. Adv. Cancer Res. 2001;82:55. doi: 10.1016/s0065-230x(01)82002-9. [DOI] [PubMed] [Google Scholar]
- 4.Chéne P. Nat. Rev. Cancer. 2003;3:102. doi: 10.1038/nrc991. [DOI] [PubMed] [Google Scholar]
- 5.Momand J, Zambetti GP, Olson DC, George D, Levine AJ. Cell. 1992;69:1237. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, Marechal V, Levine AJ. Mol. Cell. Biol. 1993;13:4107. doi: 10.1128/mcb.13.7.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Nature. 1993;362:857. doi: 10.1038/362857a0. [DOI] [PubMed] [Google Scholar]
- 8.Haupt Y, Maya R, Kazaz A, Oren M. Nature. 1997;387:296. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 9.Kubbutat MHG, Jones SN, Vousden KH. Nature. 1997;387:299. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 10.Fakharzadeh SS, Trusko SP, George DL. EMBO J. 1991;10:1565. doi: 10.1002/j.1460-2075.1991.tb07676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown DR, Thomas CA, Deb SP. EMBO J. 1998;17:2513. doi: 10.1093/emboj/17.9.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Capoulade C, Paillerets B. B.-d., Lefrere I, Ronsin M, Feunteun J, Tursz T, Wiels J. Oncogene. 1998;16:1603. doi: 10.1038/sj.onc.1201702. [DOI] [PubMed] [Google Scholar]
- 13.Juven-Gershon T, Oren M. Mol. Med. 1999;5:71. [PMC free article] [PubMed] [Google Scholar]
- 14.Leite KR, Franco MF, Srougi M, Nesrallah LJ, Nesrallah A, Bevilacqua RG, Darini E, Carvalho CM, Meirelles MI, Santana I, Camara-Lopes LH. Mod. Pathol. 2001;14:428. doi: 10.1038/modpathol.3880330. [DOI] [PubMed] [Google Scholar]
- 15.Polsky D, Bastian BC, Hazan C, Melzer K, Pack J, Houghton A, Busam K, Cordon-Cardo C, Osman I. Cancer Res. 2001;61:7642. [PubMed] [Google Scholar]
- 16.Eymin B;, Gazzeri S, Brambilla C, Brambilla E. Oncogene. 2002;21:2750. doi: 10.1038/sj.onc.1205359. [DOI] [PubMed] [Google Scholar]
- 17.Zhang R, Wang H. Curr. Pharm. Des. 2000;6:393. doi: 10.2174/1381612003400911. [DOI] [PubMed] [Google Scholar]
- 18.Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Science. 1996;274:948. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 19.Bullock AN, Fersht AR. Nat. Rev. Cancer. 2001;1:68. doi: 10.1038/35094077. [DOI] [PubMed] [Google Scholar]
- 20.Fischer PM, Lane DP. Trends Pharmacol. Sci. 2004;25:343. doi: 10.1016/j.tips.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 21.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. Science. 2004;303:844. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 22.Zhang R, Mayhood T, Lipari P, Wang Y, Durkin J, Syto R, Gesell J, McNemar C, Windsor W. Anal. Biochem. 2004;331:138. doi: 10.1016/j.ab.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 23.Duncan SJ, Grueschow S, Williams DH, McNicholas C, Purewal R, Hajek M, Gerlitz M, Martin S, Wrigley SK, Moore M. J. Am. Chem. Soc. 2001;123:554. doi: 10.1021/ja002940p. [DOI] [PubMed] [Google Scholar]; J. Am. Chem. Soc. 2002;124:14503. Correction. [Google Scholar]
- 24.Duncan SJ, Cooper MA, Williams DH. Chem. Commun. 2003:316. doi: 10.1039/b211889k. [DOI] [PubMed] [Google Scholar]
- 25.Boger DL, Desharnais J, Capps K. Angew. Chem. Int. Ed. 2003;42:4138. doi: 10.1002/anie.200300574. [DOI] [PubMed] [Google Scholar]
- 26.Desai P, Pfeiffer SS, Boger DL. Org. Lett. 2003;5:5047. doi: 10.1021/ol036083g. [DOI] [PubMed] [Google Scholar]
- 27.Malkinson JP, Zloh M, Kadom M, Errington R, Smith PJ, Searcey M. Org. Lett. 2003;5:5051. doi: 10.1021/ol0360849. For additional studies, see: Woon ECY, Arcieri M, Wilderspin AF, Malkinson JP, Searcey M. J. Org. Chem. 2007;72:5146. doi: 10.1021/jo070450a.
- 28.Mori T, Miyagi M, Suzuki K, Shibasaki M, Saikawa Y, Nakata M. Heterocycles. 2007;72:275. [Google Scholar]
- 29.Lee SY, Clark RC, Boger DL. J. Am. Chem. Soc. 2007;129:9860. doi: 10.1021/ja073264v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qian W-J, Wei W-G, Zhang Y-X, Yao Z-J. J. Am. Chem. Soc. 2007;129:6400. doi: 10.1021/ja072225g. For preceding studies see: Wei W-G, Zhang Y-X, Yao Z-J. Tetrahedron. 2005;61:11882. Wei W-G, Yao Z-J. J. Org. Chem. 2005;70:4585. doi: 10.1021/jo050414g. Wei W-G, Qian W-J, Zhang Y-X, Yao Z-J. Tetrahedron Lett. 2006;47:4171.
- 31.Powell ADG, Robertson A, Whalley WB. Chem. Soc. Spec. Publ. 1956;5:27. [Google Scholar]
- 32.Chong R, King RR, Whalley WB. J. Chem. Soc. C. 1971:3566. doi: 10.1039/j39710003566. [DOI] [PubMed] [Google Scholar]
- 33.Chong R, King RR, Whalley WB. J. Chem. Soc. D. 1969:1512. [Google Scholar]
- 34.Suzuki T, Okada C, Arai K, Awaji A, Shimizu T, Tanemura K, Horaguchi T. J. Heterocycl. Chem. 2001;38:1409. [Google Scholar]
- 35.Katritzky AR, Lam JN. Heteroatom Chem. 1990;1:21. [Google Scholar]
- 36.Williams JM, Jobson RB, Yasuda N, Marchesini G, Dolling U-H, Grabowski EJ. J. Tetrahedron Lett. 1995;36:5461. [Google Scholar]
- 37.Takahata H, Takamatsu T, Yamazaki T. J. Org. Chem. 1989;54:4812. [Google Scholar]
- 38.Zhu J, Germain AR, Porco JA., Jr. Angew. Chem. Int. Ed. 2004;43:1239. doi: 10.1002/anie.200353037. [DOI] [PubMed] [Google Scholar]
- 39.Fuchs JR, Funk RL. J. Am. Chem. Soc. 2004;126:5068. doi: 10.1021/ja049569g. [DOI] [PubMed] [Google Scholar]
- 40.Comins DL, Brown JD. J. Org. Chem. 1989;54:3730. [Google Scholar]
- 41.Comins DL, Brown JD. J. Org. Chem. 1984;49:1078. [Google Scholar]
- 42.Snieckus V. Chem. Rev. 1990;90:879. [Google Scholar]
- 43.Harris TD, Roth GP. J. Org. Chem. 1979;44:2004. [Google Scholar]
- 44.Whalley WB, Ferguson G, Marsh WC, Restivo RJ. J. Chem. Soc., Perkin Trans. 1. 1976:1366. [Google Scholar]
- 45.Steyn PS, Vleggaar R. J. Chem. Soc., Perkin Trans. 1. 1976:204. [PubMed] [Google Scholar]
- 46.Chen FC, Manchand PS, Whalley WB. J. Chem. Soc. C. 1971:3577. doi: 10.1039/j39710003577. [DOI] [PubMed] [Google Scholar]
- 47.Zhu J, Grigoriadis NP, Lee JP, Porco JA., Jr. J. Am. Chem. Soc. 2005;127:9342. doi: 10.1021/ja052049g. [DOI] [PubMed] [Google Scholar]
- 48.Hashiyama T, Morikawa K, Sharpless KB. J. Org. Chem. 1992;57:5067. [Google Scholar]
- 49.D'Souza LJ, Sinha SC, Lu SF, Keinan E, Sinha SC. Tetrahedron. 2001;57:5255. [Google Scholar]
- 50.Adam W, Curci R, Edwards JO. Acc. Chem. Res. 1989;22:205. [Google Scholar]
- 51.For 30: 1H NMR (500 MHz, CDCl3) [.delta] 7.64 (s, 1H), 6.31 (s, 1H), 5.72 (s, 1H), 4.14 (t, J = 6.3 Hz, 2H), 2.46 (t, J = 7.5 Hz, 2H), 2.16 (s, 3H), 2.07 (s, 3H), 1.96 (m, 2H), 1.54 (s, 3H); HR ESI-TOF m/z 351.1072 (M + H+, C17H19O8 requires 351.1074). (Representative Diastereomer) Treatment of 29 with DMDO followed by concentration provided a crude 1H NMR indicating the consumption of starting material and appearance of signals corresponding to epoxide 31 by analogy to 30.
- 52.Corey EJ, Kang M, Desai MC, Ghosh AK, Houpis JN. J. Am. Chem. Soc. 1988;110:649. doi: 10.1021/ja00210a083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.For 38: 1H NMR (600 MHz, CD3OD) δ 7.94 (s, 1H), 7.38 (m, 5H), 4.83 (d, J = 15.1 Hz, 1H), 4.75 (d, J = 15.3 Hz, 1H), 4.21 (dd, J = 14.7, 6.9 Hz, 1H), 4.07 (dd, J = 14.9, 7.0 Hz, 1H), 2.53 (td, J = 14.1, 7.1 Hz, 1H), 2.38 (t, J = 7.2 Hz, 2H), 2.21 (m, 1H), 2.01 (m, 2H), 1.63 (m 2H), 1.53 (s, 3H), 0.97 (t, J = 7.4 Hz, 3H); 13C NMR (CD3OD, 150 MHz) δ 190.6, 190.0, 189.4, 172.9, 148.6, 134.9, 134.3, 129.5 (2C), 129.0, 128.2 (2C), 120.4, 102.1, 97.2, 84.5, 71.2, 54.4, 35.2, 33.0, 25.1, 23.1, 18.4, 13.7; IR (film) νmax 2984, 1737, 1374, 1242, 1046, 917, 734 cm−1; HR ESI-TOF m/z 458.1383 (M + H+, C24H24ClNO6 requires 458.1365). (Representative Diastereomer) For 39: 1H NMR (500 MHz, CDCl3) δ 7.80 (s, 1H), 7.37−7.45 (br m, 3H), 7.19 (d, J = 7.0 Hz, 2H), 7.10 (s, 1H), 5.26 (d, J = 16.3 Hz, 1H), 5.10 (d, J = 16.4 Hz, 1H), 5.00 (dd, J = 9.1, 4.7 Hz, 1H), 3.82 (m, 1H), 3.75 (m, 1H), 2.44 (dt, J = 7.3, 3.3 Hz, 2H), 2.28 (m, 1H), 2.20 (m, 1H), 1.67 (m, 2H), 1.56 (s, 3H), 0.97 (t, J = 7.4 Hz, 3H), the two OH signals were not observed; 13C NMR (CDCl3, 125 MHz) δ 193.5, 185.6, 173.3, 148.8, 143.4, 142.4, 134.1, 129.9 (2C), 129.5, 126.6 (2C), 115.1, 114.1, 105.0, 85.0, 77.2, 58.4, 57.1, 39.2, 35.4, 23.1, 18.4, 13.8; MALDI-TOF m/z 460.15 (M + H+, C24H26ClNO6 requires 460.15). (Representative Diastereomer)
- 54.Atomic coordinates for the two C8/C9 syn spirocycles 36A (CCDC646443) and 36B (CCDC646442) have been deposited with the Cambridge Crystallographic Data Center.
- 55.Atomic coordinates for the C8/C9 syn spirocycle 42A (CCDC678265) and the C8/C9 anti spirocycle 42D (CCDC646441) have been deposited with the Cambridge Crystallographic Data Center.
- 56.There is an interesting shift in the longest wavelength CD Cotton effect from 360 nm for the azaphilones 25−28 (Figure 4), to 375−380 nm for the (dipeptide) amine conjugates 45 and 46, to 395−410 nm for 47A—47D and 48A—48D.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.