

Abstract
Retinoids are indispensable for the health of mammals, which cannot synthesize retinoids de novo. Retinoids are derived from dietary provitamin A carotenoids, like β-carotene, through the actions β-carotene 15,15′ monooxygenase (BCMO1). As the substrates for retinoid metabolizing enzymes are water insoluble, they must be transported intracellularly bound to cellular retinol-binding proteins. Our studies suggest that cellular retinol binding protein, type I (RBP1) acts as an intracellular sensor of retinoid status that, when present as apo-RBP1, stimulates BCMO1 activity and the conversion of carotenoids to retinoids. Cellular retinol binding protein, type II (RBP2), which is 56% identical to RBP1 does not influence BCMO1 activity. Studies of mice lacking BCMO1 demonstrate that BCMO1 is responsible for metabolically limiting the amount of intact β-carotene that can be absorbed by mice from their diet. Our studies provide new insights into the regulation of BCMO1 activity and the physiological role of BCMO1 in living organisms.
Keywords: β-carotene, cleavage, β-carotene 15, 15′ monooxygenase, retinoid, metabolism, cellular retinol binding protein
Retinoids (vitamin A, its natural metabolites and synthetic analogs) are required for survival of mammals and are needed to support vision, normal cell differentiation and proliferation, embryonic development, normal immune function and reproduction [1]. Aside from in vision, all of the essential actions of retinoids within the body are thought to involve the transcriptional activity of retinoic acid [1]. Retinoic acid is a potent transcriptional regulator whose effects are mediated by two families of nuclear receptors, the retinoic acid receptors (RARs) and the retinoid X receptors (RXRs) [2–4]. When retinoic acid is not available, RAR/RXR heterodimers bind to retinoic acid response elements in promoter of genes and form complexes with transcriptional corepressors resulting in chromatin condensation and transcriptional silencing. In the presence of retinoic acid, corepressors are released from the RAR/RXR heterodimer which is now free to recruit and bind coactivators resulting in chromatin decondensation and transcriptional activation. In excess of 500 genes may be responsive to retinoic acid [5].
Since animals lack the capacity for de novo synthesis of retinoids [6], they have an obligatory need to obtain retinoid from the diet as either preformed vitamin A or provitamin A carotenoids. The uptake and metabolism of provitamin A carotenoids in the intestine are similar to those of other dietary lipids, requiring micelle formation for uptake into the enterocyte and chylomicron assembly for secretion into the lymphatic system [7, 8]. Both passive [8] and active [9, 10] mechanisms of β-carotene uptake into enterocytes have been suggested. Upon entering the enterocyte, provitamin A carotenoids like β-carotene can be cleaved by β-carotene 15,15′-monooxygenase (BCMO1) to retinal and the resulting retinal can bind one of cellular retinol-binding proteins, type II (RBP2, also known as CRBP II) [11]. Retinal bound to RBP2 (retinal-RBP2) is then acted upon by retinal reductase and reduced to retinol which also binds to RBP2.
Subsequently, retinol bound to RBP2 (retinol-RBP2, holo-RBP2) can be esterified to long chain fatty acyl groups by lecithin:retinol acyl transferase (LRAT). The product of LRAT action, retinyl ester, is incorporated into chylomicrons along with the other dietary lipids before secretion into the lymphatic system. Similar β-carotene conversion processes to vitamin A occur in other retinoid target tissues, except in those tissues, cellular binding protein, type I (RBP1, also known as CRBP I) will take place of type II, as latter is only expressed in intestine while former is expressed ubiquitously [11]. The enzymes that are involved in this pathway have been studied individually. Yet, there is little comprehensive understanding of how these proteins interact to facilitate and regulate provitamin A carotenoid conversion to retinoid.
As an initial step towards understanding fully the regulation of retinoid synthesis from carotenoids, we utilized two approaches: 1) examination of key enzymes that are involved in this pathway, BCMO1, retinal reductase 1 (RalR1), and LRAT and effects of cellular retinol binding proteins on these enzymes utilizing in vitro enzyme assays; and 2) examination of β-carotene uptake and metabolism in mouse models that lack either BCMO1 or RBP2. Here we present evidence that BCMO1 is indeed a metabolic gate keeper that regulates the majority of β-carotene conversion to retinoids in the intestine and provide new insights into how BCMO1 activity may be regulated by cellular retinol binding proteins within the body.
Materials and Methods
Retinoids and carotenoids
All-trans- and 13-cis-retinal, all-trans-retinol, and β-carotene were purchased from Sigma (St. Louis, MO). β-Carotene was further purified on a deactivated alumina column before use as a substrate in enzyme assays. The synthetic retinoid, all-trans-9-(4-methoxy-2,3,6-trimethylphenyl)-3,7-dimethyl-2,4,6,8-nonatetraen-1-ol (TMMP-ROH, Ro12-0586) [12] was obtained from Dr. Christian Eckhoff (Hoffmann-LaRoche, Inc). Our HPLC internal standard for carotenoid measurements, echinenone, was purchased from CaroteNature (Lupsingen, Switzerland).
Identification and sequence analysis of a cDNA encoding human carotene cleavage enzyme
A blast search of GenBank employing the mBCMO1 cDNA sequence revealed two clones (AI673642 and AI886145, IMAGE Consortium) that likely encoded sequences for hBCMO1. Both clones were obtained and sequenced. Each clone contained distinct but partially overlapping sequences for hBCMO1. Two separate pieces containing 738 nucleotide including start site and the rest of the coding region were amplified and cloned into pCR 4 Blunt-TOPO vector (Invitrogen, Carlsbad, CA) to provide a single cDNA (hBCMO1/Topo) encoding a full open reading frame for hBCMO1. This open reading frame was subcloned into the bacterial expression vector, pET28b (Novagen, Madison, WI) and sequenced by the Columbia University Core DNA Sequencing facility in both directions to verify orientation and correctness of reading frame.
Expression and purification of RBP1 and RBP2
A cDNA encoding rat RBP2 in the bacterial expression vector pMon was obtained from Dr. Ellen Li at Washington University (St. Louise, MO), and RBP2 protein was purified according to a published method [13]. An open reading frame encoding mouse RBP1 was cloned into a bacterial expression vector, pET11a and purified as described earlier [14]. Later, we subcloned open reading frames of both RBP1 and RBP2 into another bacterial expression vector pET28b (Novagen, Madison, WI) to generate 3′ his-tagged proteins as they are easier to purify. Purified his-tag proteins did not differ from native recombinant proteins with regards to their binding properties towards retinal and retinol. Induction, expression, and purification of recombinant proteins with 3′ his-tag were performed according to the manufacturer’s recommendations (Novagen). Briefly, E.coli expressing a recombinant protein was sonicated, centrifuged at 12,000 g for 30 min and applied to a column packed with His·Bind resin (Novagen). Following extensive washing steps, the proteins were eluted from the column in 1 M imidazole and analyzed for purity on 12% SDS-PAGE.
Generation of holo-RBP1 and holo-RBP2
Holo-RBP1 and holo-RBP2 were generated according to the method described earlier [15]. Molar excess all-trans-retinol or all-trans-retinal was incubated with 1 mg of purified RBP1 or RBP2 over night at 4 °C on a rotating rack. The next morning, the cellular retinol binding proteins and retinoid mixtures were treated with activated charcoal slurry (2.5 %, 0.2 % dextran sulfate) to remove unbound retinoid. Formation of holo-RBP1 and holo-RBP2 was assessed spectrally scanning between 250 and 400 nm [16, 17].
Cloning and expression of RalR1
The open reading frame of human RalR1 was directionally cloned into pcDNA3.1D/V5-His-Topo vector (Invitrogen) following RT-PCR using human prostate total RNA (Clontech, Palo Alto, CA). Chinese hamster ovary (CHO) cells were transfected with the resulting plasmid (RalR1/pcDNA3.1) and selected under G418 to create a cell line (RalR1/CHO) that stably expresses RalR1 for in vitro enzyme assays.
Cloning and expression of LRAT
The open reading frame of human LRAT was directionally cloned into pcDNA3.1D/V5-his-Topo vector (Invitrogen) from a cDNA clone obtained from IMAGE Consortium (clone ID: 286177). The resulting plasmid was used to transfect CHO cells (LRAT/CHO) to generate LRAT enzyme for use in our studies.
Enzyme assays
Carotene cleavage enzyme activity was measured as described earlier [18, 19] Our standard assay mixture for determining BCMO1 activity included 15 μM β-carotene, 0.1 mM α-tocopherol, 0.5 mM DTT, 4 mM sodium cholate, and 15 mM nicotinamide in 100 mM Tricine buffer pH8.0. BCMO1 activity was determined by extracting and measuring production of all-trans-retinal by high performance liquid chromatography (HPLC) following incubation of the assay mixture, routinely for 1 h at 37°C. For some experiments, homogeneously purified RBP1 or RBP2 was added to the reaction mixture to examine the effects of these proteins on BCMO1 activity.
For RalR1 activity, microsomal fractions of RalR1/CHO cells were obtained using a method described earlier [20]. Briefly, cells were grown to confluence, washed three times with ice cold PBS, and scraped into a buffer containing 25 mM Tris HCl pH7.5, 0.25 M sucrose, and 1 mM DTT. The cells were homogenized in a Dounce homogenizer and subjected to centrifugation at 12,000g for 15 min. The supernatant from the 12,000g centrifugation was removed and further centrifuged at 100,000g for 1 h at 4 °C. The resulting pellet (microsomes) was resuspended in the homogenization buffer lacking sucrose for use in enzyme assays. Our standard assay was conducted at 37 °C for 1 h in 10 mM Hepes pH 8.0, 150 mM KCl, 2 mM DTT containing 50–200 μg microsomal protein as an enzyme source and 2 mM NADPH as a cofactor. Either free retinal or retinal-RBP2 was used as a substrate in varying concentrations as described in the text. RalR1 activity was determined by measuring the production of retinol by HPLC using protocols described below.
LRAT activity was measured using the method reported by Shi et al. [21]. Briefly microsomes (50 – 100 μg protein) prepared from LRAT/CHO cells were incubated with 2 μM retinol solubilized in dipalmitoylphosphatidyl choline (DPPC)/BSA mixture in 100 mM Tris HCl pH 9.0 for 30 min at 37 °C. The final reaction volume was 100 μl and the final concentrations of BSA and DPPC were 0.5% and 200 μM respectively. For some experiments, retinol was provided as holo-RBP1 or as holo-RBP2 instead of bound to BSA. The reaction was stopped by adding an equal volume of ethanol to the assay mixture. The retinoids were extracted into 2.5 ml hexane and analyzed by HPLC as described below.
HPLC analyses
Retinal, retinol, and β-carotene were separated on a 4.6 × 150 mm Supelcosil LC-Si column preceded by a silica guard column (Supelco Inc). For the mobile phase hexane:ethyl acetate: butanol (96.9:3:0.1, v:v) was employed at a flow rate of 0.8 ml/min. Retinal, retinol, and β-carotene were detected at 365, 325, and 450 nm with Waters 996 photodiodearray (PDA) detector. Retention times were established by comparison of unknown peaks to those of authentic standards as well as from the spectral properties of the unknowns. Quantitation of retinal and retinol was carried out by comparing the area under the peak of an unknown to those of known concentrations of standards. Loss during the extraction was accounted for by adjusting the recovery of internal standard, TMMP-ROH.
For determination of tissue levels of retinol, retinyl esters and β-carotene, a reverse phase HPLC method was employed using a 4.6 × 250 mm Ultrasphere C18 column (Beckman, Fullerton, CA) and a running solvent (acetonitrile:methanol:methylene chloride 70:15:15 (v/v)) flowing at 1.8 ml/min. Retinol and retinyl esters were detected at 325 nm and β-carotene at 450 nm. Quantitation was based on the comparison of the area under the peak and spectra of unknown samples to those of known amounts of standards. The internal standards employed to correct for loss during extraction were retinyl acetate for retinoids and echinenone for β-carotene.
Animal Maintenance and dietary regimens
All mice employed in our studies were treated and maintained according to the NIH Guide for the Care and Use of Laboratory Animals, and all experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Washington. Detailed description of BCMO1-deficient mice (B6;129S6-Bcmo1tm1Dnp) generation is reported elsewhere [22]. All mice employed in our studies were in a mixed 129sv/C59BL/6 genetic background. For all of our studies, heterozygous mice (BCMO+/−) were mated to generate wild type (BCMO+/+), heterozygous (BCMO+/−) and homozygous (BCMO−/−) animals. During the breeding and lactation periods, all mice were maintained on breeder chow that contained 28 IU vitamin A/g diet (PicoLab Mouse Diet 20, PMI international). After weaning, the mice were placed on regular chow diet which contains 25 IU vitamin A/g diet for 3 weeks. At 6 weeks of age, all mice (16 mice/genotype, 8 male and 8 female/diet group) were put on a special diet containing β-carotene (Rovimix β–carotene 10%, DSM) as its only source of vitamin A (referred to in the text as β-carotene diet) for either 4 or 7 weeks. This diet was formulated based on the AIN-93G formulation but modified to contain a slightly higher fat content (9.1% fat instead of 7.1 %) and 100 μg β-carotene/g diet as a sole source of vitamin A (containing no preformed vitamin A). This level of β-carotene was chosen since we wanted to achieve 28 IU vitamin A/g diet from β-carotene, assuming a 12:1 conversion ratio [23]. At the start of the diet regimen and 4 and 7 weeks later, 8 mice (4 male, 4 female) from each genotype were sacrificed after a 4 h fast. Blood, liver, lung, kidney, intestine, testes or ovaries, and three fat depots (inguinal, retroperitoneal, perigonadal) were dissected and snap frozen in liquid N2. Tissues were stored in −70 °C until retinoid/carotenoid and triglyceride analyses.
In a separate experiment, we investigate the role of RBP2 in β-carotene uptake and metabolism in the intestine. For this purpose, RBP2-deficient mice [24] were obtained from Dr. Ellen Li (Washington University, St. Louis, MO). RBP2-deficient and genetically matched C57BL/6 wild type mice (8 male per genotype) were initiated onto the β-carotene containing diet at 6 weeks of age and allowed to consume this diet for 4 weeks prior to their sacrifice. Blood and tissues were collected and analyzed for retinoid and carotenoid concentrations as described above for BCMO1-deficient mice.
Results
Cloning and characterization of human BCMO1 (hBCMO1)
To obtain a cDNA for hBCMO1, we searched the human EST database using the cDNA sequence of murine BCMO1. Two clones were identified and obtained (AI886145, AI673642) from the IMAGE consortium. Upon sequencing, we determined that both clones contained only partial, but overlapping sequences. PCR was performed to join the two sequences to create a full-length cDNA encoding a 63 kDa protein [25]. The open reading frame of the cDNA was subcloned into bacterial expression vector, pET28b (hBCMO1/pET). E. coli transformed with hBCMO1/pET were cultured to induce hBCMO1, purified to homogeneity using His·Bind resin, and used for in vitro enzyme assays. Under our standard assay conditions, purified hBCMO1 demonstrated robust activity. The specific activity of purified hBCMO1 was 16-fold higher compared to that of 12,000 g supernatant of bacterial homogenate before purification (25.3 nmol retinal/mg protein/h vs. 1.6 nmol/mg protein/h). The catalytic properties of hBCMO1 were similar to those we reported for mouse BCMO1 [18] and to those Lindquist and Andersson reported for human BCMO1 [26] with respect to substrate- and protein-dependence and inhibition by chelators such as o-phenanthroline and α,α′-bipyridyl (data not shown).
Effects of cellular retinol binding proteins on human BCMO1 activity
Because the product of human BCMO1 catalytic activity, retinal, is known to bind to RBP2 in the intestine and RBP1 in other vitamin A metabolizing tissues, we wondered whether RBP1 and/or RBP2 can influence BCMO1 activity by sequestering products thus changing the equilibrium of the enzyme reaction. RBP1 and RBP2 were expressed in E.coli and purified to homogeneity as described in materials and methods. Retinal production from β-carotene was measured in the presence of a constant amount of purified hBCMO1 and varying levels of purified apo-RBP1/2 (Figure 1). Since retinal is not soluble in an aqueous environment, BSA was used in our control assays as BSA can non-specifically bind and solubilize retinoids. As seen in Figure 1, RBP1 but not RBP2 or BSA enhanced retinal production in a RBP1 protein-dependent manner.
Figure 1.
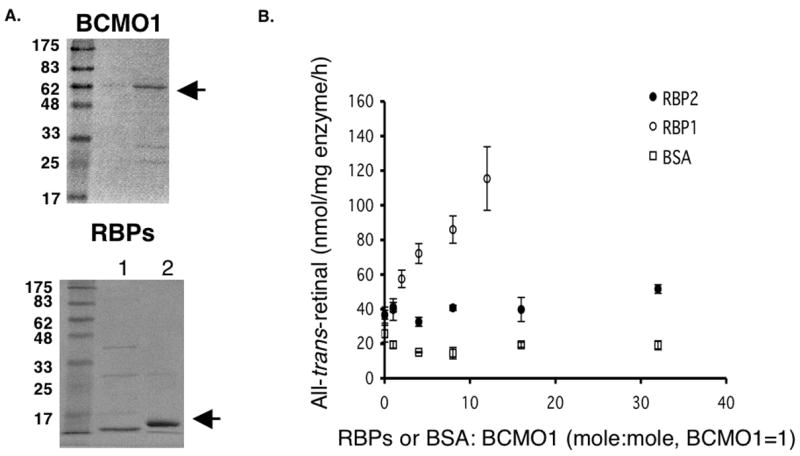
Effects of cellular retinol binding proteins on BCMO1 activity. (A) Purified human BCMO1, RBP1 and RBP2 were analyzed on SDS-PAGE and stained with Coomassie. (B) Purified human BCMO1 (2.6 μg) was incubated with 15 μM β-carotene in our standard assay conditions in the presence of varying concentrations of RBP1, RBP2 or BSA as indicated. The production of all-trans-retinal from β-carotene was measured by HPLC.
Cloning and characterization of RalR1
RalR1 is a microsomal enzyme that is expressed in various tissues, including intestine where human BCMO1 is also expressed [27, 28]. Moreover, RalR1 was active at pH 8 where BCMO1 catalyzes β-carotene cleavage most actively. Thus, we wondered whether RalR1 might be a retinal reductase acting immediately down stream to BCMO1. Using its reported sequence (accession number: AF167438), we cloned RalR1 from human prostate total RNA by RT-PCR and transfected CHO cells with the resulting plasmid (RalR1/pcDNA3). CHO cells were then selected under G418 to create a cell line that constitutively express RalR1 (RalR1/CHO). Microsomal fractions of RalR1/CHO cells were used for an enzyme source for in vitro assays. First, we examined the pH dependence of RalR1 activity (Figure 2). As reported earlier by others (23), unlike other dehydrogenase/reductases, RalR1 performed reduction of retinal at basic pHs much better than at an acidic pH.
Figure 2.
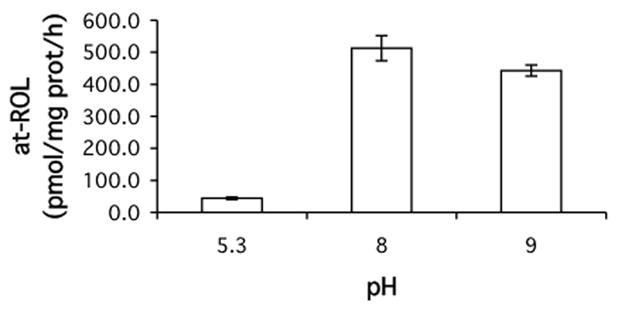
Effects of pH on hRalR1 activity. Human RalR1 activity was measured at three different pHs as indicated. Microsomes (90 μg protein) prepared from RalR1/CHO cells were incubated with 5 μM retinal-RBP2 as a substrate and 2 mM NADPH as a cofactor in buffers of three different pH for 1 h at 37 °C, and the production of retinol was measured by HPLC.
In intestine, retinal formed from β-carotene cleavage is likely bound to RBP2 and thus, we studied substrate specificity of RalR1 and effects of the presence of excess apo-RBP2 on RalR1 activity (Figure 3). RalR1 used both free- and RBP2 bound-retinal. Apparent Km and Vmax for free retinal were 3.7 μM and 588.2 pmol/mg protein/h and those for retinal-RBP2 were 0.13 μM and 181.8 pmol/mg protein/h. Moreover, the presence of apo-RBP2 did not affect RalR1 activity after a 1:1 molar ratio of retinal to RBP2 has been achieved. These data suggest that RalR1 recognizes retinal-RBP2 as its substrate and that the dissociation of retinal from RBP2 is not required for enzyme activity.
Figure 3.
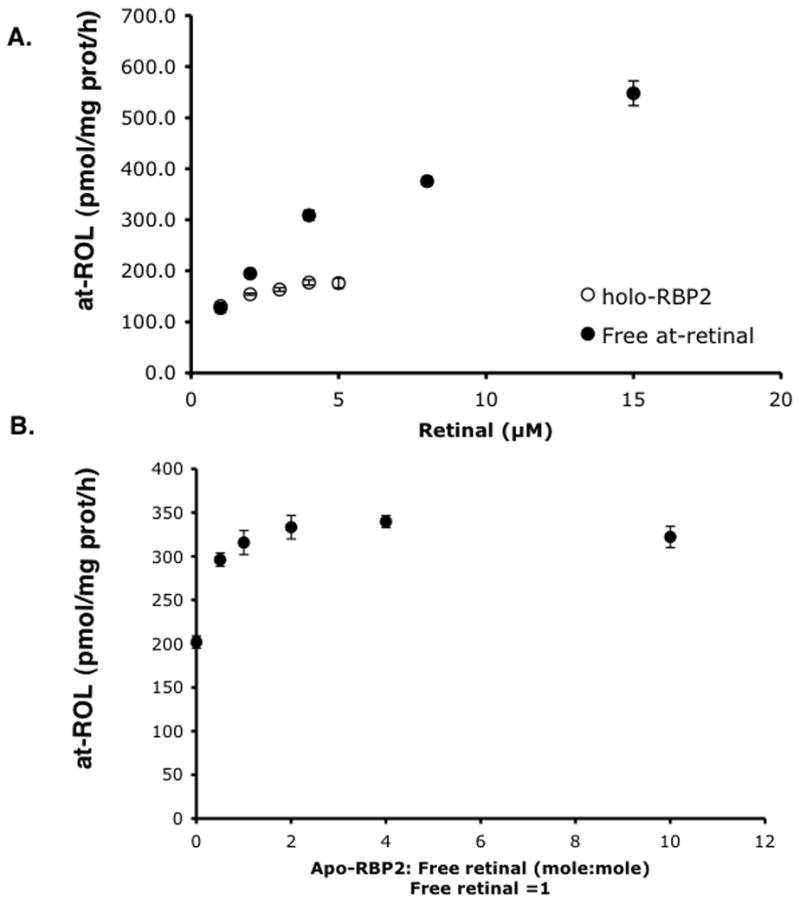
Effects of RBP2 on human RalR1. (A) Microsomes (108 μg protein) prepared from RalR1/CHO cells were incubated at 37 °C for 1 h with various concentrations of either free- or RBP2 bound-retinal (holo-RBP2) as indicated. Production of retinol was measured by HPLC. (B) In the above assay conditions, increasing concentrations of apo-RBP2 was added to a constant level of free retinal (2 μM), and the production of retinol was measured by HPLC.
Effects of CRBPs on LRAT activity
Before being incorporated into chylomicrons or storage within tissues, retinal generated from β-carotene cleavage must be reduced to retinol by retinal reductase and esterified to retinyl ester by LRAT or possibly other acyltransferases. During this process, RBP1 and RBP2 are proposed to channel retinal and retinol to appropriate enzymes involved in this metabolic process. Since we observed that the retinal reductase RalR1 not only recognizes but, based on apparent Km values, also prefers holo-RBP2 to free retinal as a substrate, we asked whether RBP1 or 2 can influence LRAT activity. To standard assay mixture, we added increasing amounts of apo-RBP1 or apo-RBP2 while keeping the holo-RBP1 or holo-RBP2 concentration constant at 2 μM (Figure 4. A and B, respectively). While increasing amounts of apo-RBP2 had little influence on LRAT activity, apo-RBP1 greatly inhibited LRAT activity in a dose-dependent manner when holo RBP1 is used as a substrate, resulting in a greater than 60% reduction of activity at 1:1 apo to holo cellular retinol binding protein ratio (Figure 4A). When holo-RBP2 was used as a substrate, increasing amounts of both apo-RBP1 and apo-RBP2 inhibited LRAT activity. However, inhibition by apo-RBP1 was greater than that of apo-RBP2, and no retinyl esters were produced when 6 times of apo-RBP1 to holo RBP2 was added to the reaction mixture.
Figure 4.
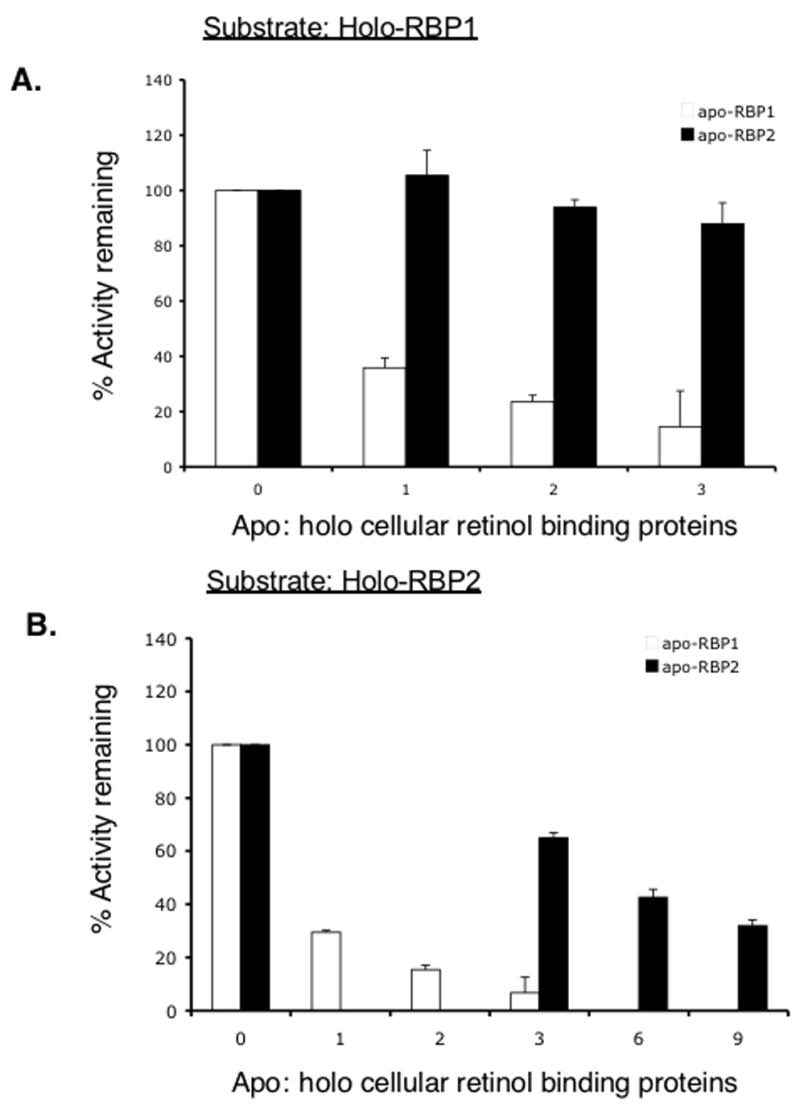
Effects of apo-RBP1 and apo-RBP2 on human LRAT activity. Microsomes (50–100 μg protein) prepared from LRAT/CHO cells were incubated with 2 μM holo-RBP1 (A) or holo-RBP2 (B) for 30 min in the absence or presence of apo-RBP 1 or 2 as indicated. X-axis represents the mole to mole ratio of apo− to holo-cellular retinol binding proteins at the start of the reaction (0 represents absence of apo-RBP 1 or 2). □, apo-RBP1; ■, apo-RBP2
β-carotene conversion is blocked in BCMO1-deficient mice
To explore the in vivo role of BCMO1 in β-carotene metabolism, we studied BCMO1-deficient mice generated on a mixed 129sv/C57BL/6 genetic background. All of our experiments were carried out using wild type, heterozygous or homozygous littermates for the BCMO1 mutation that were obtained from crosses of heterozygous males and females. Mice of each genotype (consisting of an equal number of male and female mice) were fed diet containing β-carotene (100 μg β-carotene/g diet) as a sole source of vitamin A for 4 or 7 weeks. In our facility, breeder chow contains 28 IU vitamin A/g diet (regular chow, 25 IU/g diet) and thus we matched this amounts with β-carotene, assuming a 12:1 conversion ratio [23]. All mice gained weight steadily through out the experimental period (Table 1) and no clinical signs of retinoid-deficiency were observed. Body weights, liver weights and total fat pad weights were all comparable among three genotypes at 0, 4 and 7 weeks on the diet (Table 1).
Table 1.
Body weight, liver weight and total fat pad weight of wild type, heterozygous and homozygous BCMO1-deficient mice fed β-carotene diet. Numbers represents mean ± SD for 4–5 animals/group. Baseline measurements were carried out at 6 weeks of age and β-carotene diet (100 μg β-carotene/g diet) was provided to mice for 4 and 7 weeks. Body, liver, and fat pad weights were not statistically different among all three genotypes at all time points.
| Male | Female | |||||
|---|---|---|---|---|---|---|
| Baseline | 4 weeks | 7 weeks | Baseline | 4 weeks | 7 weeks | |
| Weight (g)
| ||||||
| WT | 22.41 ± 0.81 | 26.29 ± 2.82 | 28.98 ± 1.66 | 17.88 ± 1.45 | 20.29 ± 1.16 | 20.05 ± 1.03 |
| Het | 22.67 ± 1.25 | 27.40 ± 2.79 | 28.15 ± 5.89 | 17.65 ± 0.66 | 18.88 ± 1.82 | 21.72 ± 0.94 |
| KO | 21.11± 1.49 | 25.93 ± 4.87 | 30.06 ± 3.67 | 18.18 ± 0.76 | 18.43 ± 1.38 | 20.17 ± 2.19 |
|
| ||||||
| Liver weight (g)
| ||||||
| WT | 1.36 ± 0.28 | 1.13 ± 0.13 | 1.07 ± 0.15 | 0.95 ± 0.11 | 0.95 ± 0.09 | 0.84 ± 0.08 |
| Het | 1.28 ± 0.19 | 1.21 ± 0.11 | 0.99 ± 0.25 | 0.87 ± 0.06 | 0.74 ± 0.14 | 0.86 ± 0.04 |
| KO | 1.14 ± 0.24 | 1.11 ± 0.22 | 1.15 ± 0.06 | 0.93 ± 0.23 | 0.89 ± 0.16 | 0.78 ± 0.14 |
|
| ||||||
| Fat pads weights (g)
| ||||||
| WT | 0.60 ± 0.15 | 1.37 ± 0.54 | 1.64 ± 0.28 | 0.36 ± 0.07 | 0.55 ± 0.15 | 0.75 ± 0.34 |
| Het | 0.71 ± 0.27 | 1.52 ± 0.59 | 1.78 ± 1.13 | 0.53 ± 0.10 | 0.57 ± 0.27 | 0.86 ± 0.21 |
| KO | 0.60 ± 0.19 | 1.29 ± 0.80 | 1.67 ± 0.73 | 0.66 ± 0.26 | 0.58 ± 0.29 | 0.75 ± 0.12 |
Plasma retinol levels were similar and stable regardless of genotype or diet duration and, as observed in other studies [29, 30], were uniformly lower in female mice in comparison to male mice (Figure 5, A and B). In most tissues, except for liver and lung, retinol levels were lower after administration of the β-carotene diet regardless of genotype. For liver and lung, tissue retinol levels were maintained through out the study period (Table 2). Liver retinyl ester stores did not change during the experimental period for all three genotypes (Figure 5, C and D) while lung retinyl esters decreased significantly on the β-carotene diet regardless of the genotype (Figure 6, A and B). Retinyl ester stores also decreased significantly in three fat depots analyzed in all three genotypes (Figures 7, 8). In contrast, β-carotene was detected in tissues of BCMO1-deficient mice examined and the level increased in time dependent manner after initiation onto the β-carotene diet. This was unlike the wild type and heterozygous mice where β-carotene concentrations were either always very low or undetectable in liver, lung, kidney, and intestine. In addition, β-carotene was only detected in plasma and fat depots for the BCMO1-deficient mice and not in these tissues from heterozygous or wild type mice (Table 3).
Figure 5.

Plasma retinol and liver retinyl esters for wild type, heterozygous and homozygous BCMO1-deficient mice. The bar graphs provide the mean ± SD from 4–5 animals/group. Baseline (0 weeks on diet) values are for 6 week-old mice. The β-carotene diet (100 μg β-carotene/g diet) was provided for 4 or 7 weeks starting at 6 weeks-of-age. One-way ANOVA followed by post-hoc analysis (Bonferoni) was performed to determine whether β-carotene diet changes retinoid levels significantly in different tissues for each genotype. *, p<0.05. Black bar, wild type mice (WT); gray bar, heterozygous mice; white bar, homozygous BCMO1-deficient mice (KO).
Table 2.
Tissue retinol concentrations of wild type, heterozygous and homozygous BCMO-deficient mice. Numbers represent mean ± SD from 4–5 animals/group. Baseline measurements were performed at 6 weeks of age and β-carotene diet was provided to mice for 4 and 7 weeks. Different superscript characters represent statistically significant changes after diet initiation within the same genotype (P<0.05). Differences among genotypes at each time point are depicted as different superscript numbers.
| Tissue retinol (μg/g tissue)
| ||||||
|---|---|---|---|---|---|---|
| Male | Female | |||||
| Baseline | 4 weeks | 7 weeks | Baseline | 4 weeks | 7 weeks | |
| Liver
| ||||||
| WT | 7.10 ± 1.7 | 18.04 ± 8.9 | 16.09 ± 10.8 | 9.53 ± 3.5 | 30.69 ± 14.6 | 28.18 ± 13.7 |
| Het | 10.42 ± 2.2 | 14.90 ± 5.2 | 16.68 ± 7.4 | 10.64 ± 0.7 | 15.95 ± 10.8 | 21.89 ± 11.0 |
| KO | 9.18 ± 5.4 | 20.87 ± 23.1 | 13.30 ± 5.5 | 10.46 ± 2.4 | 15.99 ± 9.3 | 17.52 ± 8.6 |
|
| ||||||
| Lung
| ||||||
| WT | 3.46 ± 2.8 | 4.55 ± 2.3 | 7.30 ± 6.3 | 3.25 ± 2.2a | 4.49 ± 1.2a,b | 15.23 ± 6.9c,1 |
| Het | 3.18 ± 3.0 | 4.69 ± 3.4 | 4.40 ± 0.9 | 5.43 ± 2.7 | 3.29 ± 1.2 | 8.16 ± 5.31,2 |
| KO | 4.90 ± 4.3 | 2.84 ± 0.3 | 6.28 ± 3.3 | 4.31 ± 1.6 | 4.59 ± 2.9 | 4.33 ± 1.32 |
|
| ||||||
| Intestine
| ||||||
| WT | 2.10 ± 1.4 | 0.43 ± 0.2 | 0.53 ± 0.11 | 1.87 ± 0.8 | 0.36 ± 0.3 | 0.83 ± 1.2 |
| Het | 5.04 ± 2.1a | 0.32 ± 0.1b | 0.47 ± 0.1a,b,1,2 | 2.67 ± 2.2 | 0.10 ± 0.0 | 0.21 ± 0.2 |
| KO | 2.95 ± 1.9 | 0.20 ± 0.1 | 0.17 ± 0.13 | 4.80 ± 2.9a | 0.12 ± 0.1b | 0.03 ± 0.0b |
|
| ||||||
| Testis | Ovaries | |||||
|
| ||||||
| WT | 0.06 ± 0.0a | 0.09 ± 0.0b | 0.74 ± 0.3 | 0.77 ± 0.2 | 0.97 ± 0.3 | |
| Het | 0.06 ± 0.0a | 0.07 ± 0.0b | 0.74 ± 0.1 | 0.64 ± 0.1 | 0.70 ± 0.2 | |
| KO | 0.08 ± 0.0 | 0.07 ± 0.0 | 0.74 ± 0.2a | 0.59 ± 0.1a,b | 0.48 ± 0.0 b | |
|
| ||||||
| Epididymal fat | Periuterine fat | |||||
|
| ||||||
| WT | 0.99 ± 0.2a | 0.47 ± 0.1b | 0.34 ± 0.1b,1 | 1.15 ± 0.1a | 0.54 ± 0.1b | 0.56 ± 0.4b |
| Het | 0.88 ± 0.3a | 0.40 ± 0.1b | 0.30 ± 0.0b,1,2 | 0.93 ± 0.1a | 0.48 ± 0.1b | 0.31 ± 0.1c |
| KO | 0.93 ± 0.3a | 0.39 ± 0.1b | 0.19 ± 0.0b,2 | 1.00 ± 0.1a | 0.41 ± 0.1b | 0.25 ± 0.1b |
|
| ||||||
| Retroperitoneal fat
| ||||||
| WT | 0.84 ± 0.1a | 0.45 ± 0.1b | 0.33 ± 0.1b,1 | 1.22 | 0.80 ± 0.1 | 0.95 ± 0.8 |
| Het | 0.89 ± 0.4a | 0.6 ± 0.1a,b | 0.27 ± 0.1b,2 | 0.80 ± 0.1a | 0.67 ± 0.1a | 0.23 ± 0.1b |
| KO | 0.79 ± 0.1a | 0.46 ± 0.0b | 0.15 ± 0.0c,3 | 1.02 ± 0.2a | 0.67 ± 0.0b | 0.18 ± 0.0c |
|
| ||||||
| Inguinal fat
| ||||||
| WT | 0.78 ± 0.1a | 0.55 ± 0.1b | 0.31 ± 0.1c | 0.81 ± 0.1a | 0.49 ± 0.0b | 0.45 ± 0.2b,1 |
| Het | 0.77 ± 0.3a | 0.59 ± 0.1a | 0.28 ± 0.1b | 0.77 ± 0.1a | 0.50 ± 0.0b | 0.29 ± 0.1c,1,2 |
| KO | 0.67 ± 0.1a | 0.45 ± 0.1b | 0.20 ± 0.0c | 0.93 ± 0.2a | 0.46 ± 0.1b | 0.13 ± 0.0c,2 |
Figure 6.
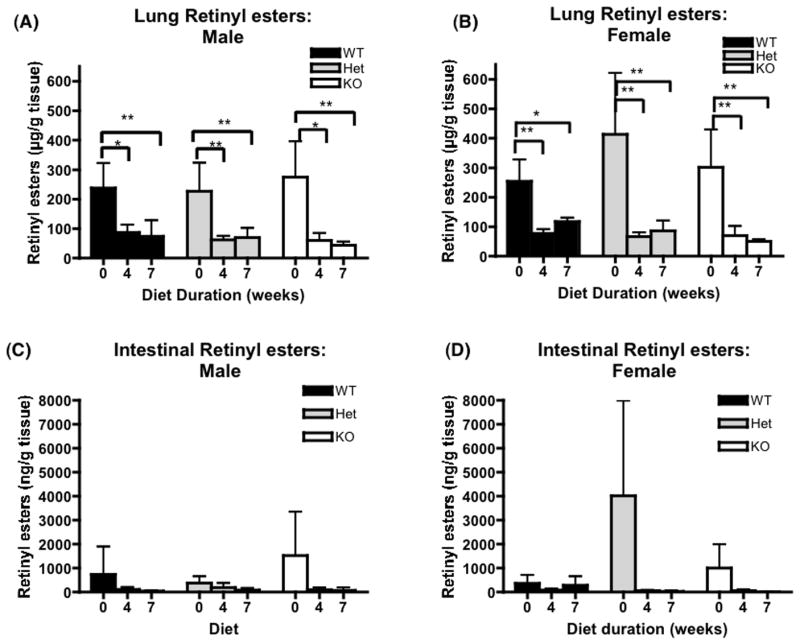
Lung and intestinal retinyl esters levels for wild type, heterozygous and homozygous BCMO1-deficient mice. The bar graphs provide the mean ± SD from 4–5 animals/group. Baseline (0 weeks on diet) values are from 6 week-old mice. The β-carotene diet (100 μg β-carotene/g diet) was provided for 4 or 7 weeks starting at 6 weeks-of-age. One-way ANOVA followed by post-hoc analysis (Bonferoni) was performed to determine whether β-carotene diet consumption changes significantly retinoid levels in different tissues for each genotype. *, P<0.05; **, P<0.01. Black bar, wild type mice (WT); gray bar, heterozygous mice; white bar, BCMO1-deficient mice (KO).
Figure 7.
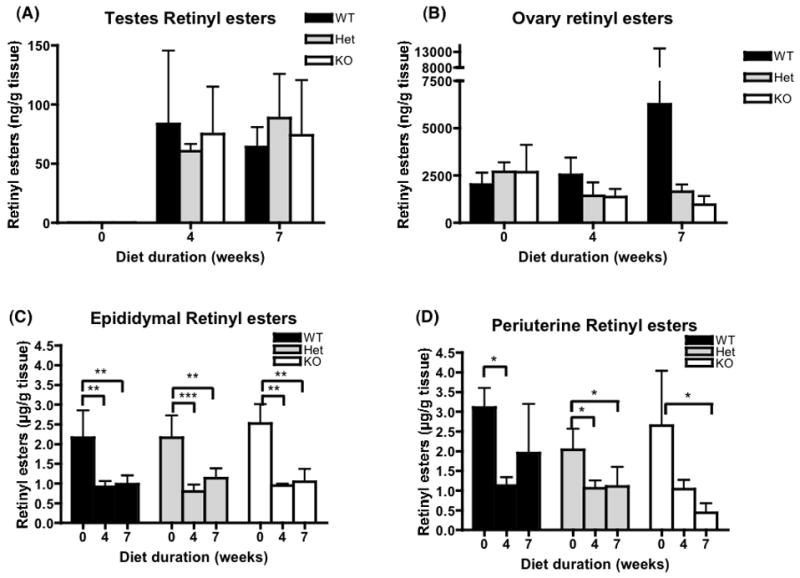
Retinyl ester concentrations in testes, ovaries, and perigonadal fat pads for wild type, heterozygous and homozygous BCMO1-deficient mice. The bar graphs provide the mean ± SD from 4 animals/group. Baseline (0 weeks on diet) values are from 6 week-old mice. β-carotene diet (100 μg β-carotene/g diet) was provided for 4 or 7 weeks starting at 6 weeks-of-age. One-way ANOVA followed by post-hoc analysis (Bonferoni) was performed to determine whether β-carotene diet changes significantly retinoid levels in different tissues for each genotype. **, P<0.01; ***, P<0.001. Black bar, wild type mice WT; gray bar, heterozygous mice; white bar, BCMO1-deficient mice (KO).
Figure 8.

Retinyl ester concentrations in retroperitoneal and inguinal fat pads for wild type, heterozygous and homozygous BCMO1-deficient mice. The bar graphs provide mean ± SD for 4 animals/group. Baseline (0 weeks on diet) values are from 6 weeks-old mice. β-carotene diet (100 μg β-carotene/g diet) was provided for 4 or 7 weeks starting at 6 weeks-of-age. *, P<0.05; **, P<0.01. Black bar, wild type mice (WT); gray bar, heterozygous mice; white bar, BCMO1-deficient mice (KO).
Table 3.
Plasma and tissue β-carotene concentrations of wild type, heterozygous and homozygous BCMO1-deficient mice. Numbers represent mean ± SD from 4–5 animals/group. β-carotene was not detected at the baseline (6 weeks of age) in any of the mice examined. ND, not detected.
| Male | Female | |||||
|---|---|---|---|---|---|---|
| Baseline | 4 weeks | 7 weeks | Baseline | 4 weeks | 7 weeks | |
| Plasma (μM)
| ||||||
| WT | ND | ND | ND | ND | ND | ND |
| Het | ND | ND | 0.02 ± 0.1 | ND | ND | ND |
| KO | ND | 0.21 ± 0.1 | 0.56 ± 0.8 | ND | 0.21 ± 0.1 | 0.22 ± 0.1 |
|
| ||||||
| Liver (ng/g tissue)
| ||||||
| WT | ND | 94.06 ± 188.1 | 90.41 ± 202.2 | ND | 181.04 ± 209.1 | ND |
| Het | ND | 211.57 ± 199.3 | 96.31 ± 215.3 | ND | 210.60 ± 249.3 | 102.6 ± 229.4 |
| KO | ND | 578.92 ± 154.8 | 770.32 ± 392.4 | ND | 967.94 ± 552.7 | 2152.9±1573.8 |
|
| ||||||
| Kidney (ng/g tissue)
| ||||||
| WT | ND | 3.38 ± 6.8 | ND | ND | 4.60 ± 9.2 | ND |
| Het | ND | 9.90 ± 9.0 | 7.67 ± 8.2 | ND | 21.99 ± 1.1 | 8.24 ± 11.4 |
| KO | ND | 83.68 ± 48.4 | 130.95 ± 60.2 | ND | 236.65 ± 200.0 | 156.13 ± 137.8 |
|
| ||||||
| Lung (ng/g tissue)
| ||||||
| WT | ND | ND | 54.37 ± 121.6 | ND | ND | ND |
| Het | ND | ND | 141.41 ± 193.7 | ND | ND | ND |
| KO | ND | ND | 326.70 ± 192.2 | ND | ND | 423.04 ± 113.2 |
|
| ||||||
| Intestine (ng/g tissue)
| ||||||
| WT | ND | 66.54 ± 47.0 | 36.74 ± 21.7 | ND | 47.02 ± 16.8 | 18.06 ± 31.3 |
| Het | ND | 88.63 ± 91.1 | 61.98 ± 41.2 | ND | 25.45 ± 17.1 | 21.88 ± 20.3 |
| KO | ND | 225.98 ± 202.9 | 107.49 ± 84.1 | ND | 103.01 ± 42.9 | 58.84 ± 4.0 |
|
| ||||||
| Testes (ng/g tissue) | Ovaries (ng/g tissue) | |||||
|
| ||||||
| WT | ND | ND | ND | ND | ND | ND |
| Het | ND | 5.43 ± 12.1 | 27.39 ± 18.5 | ND | ND | ND |
| KO | ND | 98.14 ± 49.3 | 493.05 ± 451.0 | ND | 573.63 ± 152.8 | 1122.2 ± 935.0 |
|
| ||||||
| Epididymal fat (ng/g tissue) | Periuterine fat (ng/g tissue) | |||||
|
| ||||||
| WT | ND | ND | ND | ND | ND | ND |
| Het | ND | ND | ND | ND | ND | ND |
| KO | ND | 128.65 ± 112.4 | 226.38 ± 45.1 | ND | 333.52 ± 143.9 | 638.84 ± 66.4 |
|
| ||||||
| Retroperitoneal fat (ng/g tissue)
| ||||||
| WT | ND | ND | ND | ND | ND | ND |
| Het | ND | ND | ND | ND | ND | ND |
| KO | ND | 217.43 ± 20.0 | 325.47 ± 211.1 | ND | 406.92 ± 224.3 | 659.66 ± 93.5 |
|
| ||||||
| Inguinal fat (ng/g tissue)
| ||||||
| WT | ND | ND | ND | ND | ND | ND |
| Het | ND | ND | ND | ND | ND | ND |
| KO | ND | 163.49 ± 157.6 | 340.46 ± 54.5 | ND | 389.55 ± 198.2 | 989.91 ± 462.8 |
CRBP II does not affect uptake of β-carotene in intestine
Since RBP2 binds the down stream metabolites of β-carotene, retinal and retinol, we investigated whether RBP2 is needed for optimal retinyl ester formation and absorption from dietary β-carotene. For this purpose, we placed both wild type (C57BL/6) and RBP2-deficient mice on the β-carotene diet for 4 weeks and analyzed plasma, intestine, liver, lung and 3 adipose depots for retinoid and carotenoid concentrations (Table 4). As expected, in both strains of mice β-carotene were not detected in adipose, liver and plasma. Retinoid values between the RBP2-deficient and wild type animals were similar for all tissues examined, while retinyl esters concentrations, except for liver, were lower in most tissues after 4 weeks on the β-carotene diet regardless of genotype.
Table 4.
Plasma and tissue retinoid concentrations of wild type and RBP2-deficient mice. Numbers represent mean ± SD from 6–8 male mice/group. Retinoid values are μg/g tissue except for plasma (μM). Baseline measurements were carried out at 6 weeks of age and β-carotene diet was provided for 4 weeks. Different superscript characters represent statistically significant changes after diet initiation (P<0.05). Differences among genotypes at each time point is depicted as superscript numbers (p<0.05).
| Retinol (μg/g tissue) | Retinyl esters (μg/g tissue) | |||
|---|---|---|---|---|
| Baseline | 4 weeks | Baseline | 4 weeks | |
| Plasma (μM)
| ||||
| WT | 1.28 ± 0.1 | 1.35 ± 0.1 | ||
| KO | 1.18 ± 0.2 | 1.23 ± 0.3 | ||
|
| ||||
| Liver
| ||||
| WT | 8.12 ± 2.5 | 16.76 ± 12.7 | 239.32 ± 238.1 | 303.05 ± 327.2 |
| KO | 12.45 ± 5.9 | 18.35 ± 12.8 | 201.88 ± 211.7 | 181.70 ± 41.0 |
|
| ||||
| Lung
| ||||
| WT | 4.25 ± 3.61 | 4.24 ± 2.1 | 61.31 ± 27.5 | 48.57 ± 7.8 |
| KO | 9.74 ± 3.72 | 7.19 ± 4.9 | 62.55 ± 12.9a | 41.47 ± 18.5b |
|
| ||||
| Intestine
| ||||
| WT | 1.03 ± 0.9a | 0.18 ± 0.2b | 1.41 ± 1.4 | 0.27 ± 0.3 |
| KO | 1.37 ± 1.0a | 0.18 ± 0.1b | 1.24 ± 1.8 | 0.27 ± 0.3 |
|
| ||||
| Epididymal fat
| ||||
| WT | 0.76 ± 0.2a | 0.39 ± 0.1b | 2.64 ± 0.7a | 0.63 ± 0.2b,1 |
| KO | 0.62 ± 0.1 | 0.46 ± 0.1 | 3.07 ± 0.9a | 1.11 ± 0.2b,2 |
|
| ||||
| Retroperitoneal fat
| ||||
| WT | 0.43 ± 0.1 | 0.36 ± 0.1 | 4.40 ± 2.9a | 1.04 ± 0.4b |
| KO | 0.43 ± 0.2 | 0.39 ± 0.0 | 1.44 ± 0.5 | 0.63 ± 0.1 |
|
| ||||
| Inguinal fat
| ||||
| WT | 0.51 ± 0.2 | 0.92 ± 0.4 | 0.92 ± 0.4a | 0.37 ± 0.1b |
| KO | 0.43 ± 0.1 | 1.15 ± 0.3 | 1.15 ± 0.3a | 0.39 ± 0.1b |
Discussion
Our data extend understanding of provitamin A carotenoid conversion to retinoid in several important ways. First, our in vitro studies establish that apo-RBP1 but not apo-RBP2 stimulates BCMO1 activity leading to increased rates of retinal formation from β-carotene. In addition, the retinal reductase, RalR1, which is expressed in enterocytes, is able to use retinal-RBP2 more effectively than free retinal. Collectively, our data suggest a metabolic role for intestinal RalR1 in the formation of retinoid from β-carotene. Second, our in vivo data establish that intestinal BCMO1 is a “gate keeper” that limits the amount of dietary β-carotene that is absorbed intact into the body from the intestine. Finally, other in vivo data indicate that RBP2 is not needed to facilitate optimal retinoid formation from dietary β-carotene in the intestine and/or retinoid absorption into the body.
Apo-RBP1 but not apo-RBP2 stimulates BCMO1 activity
Coordinated expression of BCMO1 activity and RBP2 mRNA was reported for chicken and rat intestine suggesting that the two proteins are regulated in a manner that facilitates metabolic and possibly physical interactions between the proteins [31, 32]. Thus, we investigated possible interactions between BCMO1 and cellular retinol binding proteins. Our data indicate that apo-RBP2 concentration does not influence BCMO1 activity directly, while increased apo-RBP1 concentration enhanced enzymatic activity. Holo-RBP1 or holo-RBP2 did not influence BCMO1 activity (data not shown). In accordance with this in vitro observation, RBP2-deficient mice accumulated similar amounts of retinoid in tissues upon administration of the β-carotene diet in comparison to wild type mice. At present, we do not understand the biochemical mechanism underlying the observed differential effects of the cellular retinol binding proteins on BCMO1 activity. Although the two binding proteins are structurally very similar, sharing 56% sequence identity, and bind both retinol and retinal with similar affinity, their tissue expression patterns and expression levels are very different. RBP2 expression is confined to intestine whereas RBP1 is expressed ubiquitously in retinoid metabolizing tissues other than intestine. In addition, the concentration of RBP2 is relatively high, reaching 1% of total intestinal protein [11], compared to a much lower concentration of RBP1 present in tissues where it is expressed. Thus, it is likely that even after the ingestion of retinoid/provitamin A carotenoid-rich foods, all retinol/retinal present in the enterocyte will be bound to RBP2. Moreover, the intracellular ratio of apo− to holo-RBP2 will change considerably between meals as ingestion of retinoid containing foods will give rise to a decreased ratio of apo− to holo-RBP2 while enterocyte secretion of chylomicron-containing retinoid into the lymphatic system will give rise to an increased ratio. In contrast, in a state of retinoid-sufficiency, tissue RBP1 concentrations are closely matched to the concentration of free retinol in the tissue [33]. It has been reported that the total RBP1 (apo + holo) concentration in tissues does not change considerably over wide ranges of retinoid status, resulting in changes in the apo− to holo-RBP1 ratio in response to changes in retinoid status in the tissue [33]. Thus, it would be reasonable that the changes in apo− to holo-RBP1 ratio can act as a sensor or barometer for retinoid status in tissues, while changes in ratio between apo− and holo-RBP2 cannot. We propose that these differences between the two binding proteins might explain our observation of differential effects of the two proteins on BCMO1 activity. From the perspective of retinoid physiology, the main function of intestine is to maintain optimal absorption of retinoid arriving from the diet at various concentrations depending on the specific foods consumed in the diet. This is unlike other retinoid metabolizing tissues which must be able to respond to the changes in retinoid status in that specific tissue, drawing from retinoid stores under conditions of dietary retinoid-deficiency and accumulating retinoid stores in times of dietary retinoid-sufficiency. Thus, it would be very beneficial to the organism for BCMO1 to be activated by increased levels of apo−RBP1 (retinoid-deficiency) allowing for the use of tissue provitamin A carotenoids for synthesis of retinoid. On the other hand, an increase in apo−RBP2 concentration (for instance between meal times) would not elicit this same response resulting in a relatively constant supply of retinoid being formed by the intestine from provitamin A carotenoids.
RalR1 may act as a physiologically significant retinal reductase in the intestine
Retinal produced in the intestine from carotenoids by BCMO1 is mainly reduced to retinol and esterified to retinyl esters before packaging into chylomicrons [34]. Enzymatic activities that carry out reduction of retinal have been reported for both cytosolic and microsomal fractions of intestine [11]. However, because of the high concentrations of RBP2 in the intestine, it was suggested that the microsomal enzyme that utilizes retinal-RBP2 is likely a more physiologically relevant enzyme. From the literature, we have identified a microsomal retinal reductase RalR1 that is expressed in intestine and uses retinal in the presence of high concentrations of RBP1 [28]. However, the effects of RBP2, the major intracellular retinol-binding protein present in the intestine, on this enzyme had not been studied. Thus, we wondered whether this enzyme could be an intestinal retinal reductase that acts downstream to BCMO1. Our data demonstrate that the enzyme is optimally active at the same pH range as BCMO1 and uses retinal-RBP2 even more efficiently than free-retinal, based on the apparent Km values and Vmax/Km ratios we determined. Moreover, the enzyme activity was not affected by the presence of excess amounts of apo-RBP2. These characteristics of RalR1 make it a good candidate as a retinal reductase that acts in the intestine directly downstream to BCMO1 to process retinoid formed from dietary β-carotene.
The differential effects of RBP1 and RBP2 on BCMO1 activity also appear to apply to another retinoid metabolizing enzyme, LRAT. Our study confirms and extends a previous report that activity of LRAT is regulated differentially by apo-RBP1 and apo-RBP2 [35]. Using homogenates obtained from cells stably transfected with a human LRAT cDNA, we demonstrate that apo-RBP1 is a strong inhibitor of LRAT activity while apo-RBP2 is not. An increased ratio of apo-RBP1 to holo RBP1 or to holo-RBP2 greatly suppresses LRAT activity, whereas an increased ratio of apo-RBP2 to holo-RBP1 or to holo-RBP2 does not illicit similar effects on LRAT. We postulate that this difference can be explained in the same manner as for the differential affects of RBP1 and RBP2 on BCMO1 activity. If the ratio of apo− to holo-RBP1 acts as a barometer of cellular retinoid status in tissues other than intestine, when retinoid intake is sufficient (indicated by a low apo− to holo-RBP1 ratio), most retinol will be esterified and stored in the tissue. However, when cellular retinoid levels decrease, this will result in increased levels of apo-RBP1 and a strong inhibition of LRAT activity; and hence, channel retinol to be oxidized to retinoic acid for use in maintaining retinoid-dependent actions. In contrast, in the intestine, dietary retinoid is absorbed through its incorporation into chylomicron as retinyl ester. The ratio of apo− to holo-RBP2 can change quickly in the intestine in response to dietary retinoid intake. Hence, in order to assure optimal dietary retinoid uptake into the body it is important that LRAT remain active to allow for esterification to take place over the wide range of retinoid concentrations that may be encountered in the diet. Moreover, our data from studies of RBP2-deficient mice maintained on β-carotene as the sole source of dietary retinoid suggest that absence of RBP2 does not affect retinyl ester formation in intestine or retinoid levels in the liver or other extrahepatic tissues.
BCMO1 acts as a metabolic “gate keeper” regulating intestinal absorption of intact β-carotene in the mouse
Ubiquitous expression of BCMO1 in tissues suggests that it is involved in providing an alternative source of retinoid in times of impaired dietary intake, in addition to retinol secreted from hepatic retinoid stores bound to retinol-binding protein (RBP4, also known as RBP). This possibility is in agreement with the observation that humans can accumulate substantial amounts of carotenoid in tissues, including testis, ovary, liver, and kidney, where mRNA for BCMO1 is detected [36, 37]. The BCMO1-deficient mice also accumulated β-carotene in a time-dependent manner in these same tissues when these mice are fed β-carotene in the diet; whereas wild type and heterozygous animals do not. This suggests that β-carotene can be used as an alternative source of retinoid in these tissues. Plasma and intestinal β-carotene was also higher in BCMO1-deficient mice in comparison to wild type and heterozygous mice. This is consistent with the notion that BCMO1 plays a major role in conversion of β-carotene to retinoid in the intestine. However, BCMO1 may not be the sole enzyme responsible for conversion of β-carotene to retinoid as BCMO1-deficient mice are able to maintain hepatic retinoid levels constant even when maintained on a β-carotene diet which has no source of preformed retinoid. Thus, our data suggest that one or more other enzymes, possibly including eccentric cleavage enzymes, may have roles in conversion of β-carotene to retinoid. This observation is somewhat different than that reported by Hessel et al. who observed that BCMO1−/− mice, which had been depleted of hepatic retinoid during gestation and weaning (i.e. the dams were fed a very low vitamin A diet, 0.15 IU/g) and subsequently fed a β-carotene supplemented diet containing the same very low levels of preformed retinoid, did display significant decrease in hepatic retinoid stores (19). This discrepancy between the two studies is likely due to differences in levels of hepatic retinoid stores present at the beginning of the studies and/or the shorter duration of β-carotene feeding in our study (7 weeks vs. 16 weeks).
From our data, we can conclude that β-carotene is not absorbed well by mice from the diet as extrahepatic retinoid levels decrease in all tissues when the mice were maintained on β-carotene as the sole source of retinoid, regardless of BCMO1 genotype. The same phenomenon was observed for RBP2-deficient mice as well as for C57BL/6 wild type mice. This occurred despite increasing the dietary fat content to 9.1% in the purified diets used in our investigations (w/w, 23% of total calories from fat). It is well established that β-carotene absorption by humans is more efficient with a higher fat content in the diet and this has been suggested to be one of the reasons why retinoid-deficiency occurs in developing countries even though foods rich in β-carotene are abundant in the diet [38, 39]. It is possible that an even higher fat content in our β-carotene diet would have increased absorption of β-carotene by the mice and thus provide higher retinoid accumulation in tissues. However, a high dietary fat content could induce obesity and relate nutritional diseases, and we felt that this might complicate the interpretation of our data. Consequently, we chose a dietary formulation for study that is based on the AIN-93G diet which was determined to be nutritionally adequate to promote growth of rodents [40]. For this diet, we observed that total cholesterol as well as triglyceride levels in plasma were statistically identical for all of our experimental mice, regardless of genotypes (data not shown). This was also the case for body weights and total fat pad weights (Table 1) which were not different for the different geneotypes studied. These data indicate that the diet used in our study did not induce obesity or related metabolic diseases. This finding contrasts with that of Hessel et al. who reported an accumulation of triglyceride in BCMO1−/− mice fed a much higher level of β-carotene in the diet than was employed in our studies (10-times the amount of β-carotene present in our diet) (19). It is reported that the water soluble form of β-carotene beadlet used by Hessel et al. is much more efficiently absorbed in intestine than other forms of β-carotene, resulting in 28 times higher blood response in preruminant calves [41]. Thus, the observation of Hessel et al. could arise because of the higher levels of β-carotene in the diet and/or the longer duration of the study (7 versus 16 weeks). Both of these differences in experimental design would lead to the accumulation of larger amounts of beta-carotene in the liver. Another possible explanation might be that when BCMO1−/− mice age, this phenotype shows more prominently since our study was carried out in relatively young mice (13 weeks-of-age versus 19 and 28 weeks-of-age). These differences between the two studies require further investigation.
Acknowledgments
This project was supported by grants DK061310, DK068437, DK079221 (to WSB), DK065719 (to JP) from the National Institutes of Health and 2003-35200-13789 from USDA Cooperative State Research Education and Extension Service (to JP).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sporn MB, Roberts AB, Goodman DS. The Retinoids : Biology, Chemistry, and Medicine. 2. Raven Press; New York: 1994. [Google Scholar]
- 2.Chambon P. A decade of molecular biology of retinoic acid receptors. Faseb J. 1996;10:940–954. [PubMed] [Google Scholar]
- 3.Evans RM. The nuclear receptor superfamily: a rosetta stone for physiology. Mol Endocrinol. 2005;19:1429–1438. doi: 10.1210/me.2005-0046. [DOI] [PubMed] [Google Scholar]
- 4.Pennimpede T, Cameron D, Petkovich M. Regulation of murine embryonic patterning and morphogenesis by retinoic acid signaling. Adv Dev Biol. 2006;16:65–104. [Google Scholar]
- 5.Balmer JE, Blomhoff R. Gene expression regulation by retinoic acid. J Lipid Res. 2002;43:1773–1808. doi: 10.1194/jlr.r100015-jlr200. [DOI] [PubMed] [Google Scholar]
- 6.Vieira A. Retinoid Endocrinology: From Metabolism to Cellular signaling. In: Quinn, Kagan, editors. Subcellular Biochemistry: Fat soluble Vitamins. Plenum Press; New York: 1998. pp. 29–51. [PubMed] [Google Scholar]
- 7.Rock CL. Carotenoids: biology and treatment. Pharmacol Ther. 1997;75:185–197. doi: 10.1016/s0163-7258(97)00054-5. [DOI] [PubMed] [Google Scholar]
- 8.Hollander D, Ruble PE., Jr beta-carotene intestinal absorption: bile, fatty acid, pH, and flow rate effects on transport. Am J Physiol. 1978;235:E686–691. doi: 10.1152/ajpendo.1978.235.6.E686. [DOI] [PubMed] [Google Scholar]
- 9.During A, Dawson HD, Harrison EH. Carotenoid transport is decreased and expression of the lipid transporters SR-BI, NPC1L1, and ABCA1 is downregulated in Caco-2 cells treated with ezetimibe. J Nutr. 2005;135:2305–2312. doi: 10.1093/jn/135.10.2305. [DOI] [PubMed] [Google Scholar]
- 10.During A, Hussain MM, Morel DW, Harrison EH. Carotenoid uptake and secretion by CaCo-2 cells: beta-carotene isomer selectivity and carotenoid interactions. J Lipid Res. 2002;43:1086–1095. doi: 10.1194/jlr.m200068-jlr200. [DOI] [PubMed] [Google Scholar]
- 11.Ong DE. Cellular retinoid-binding proteins. In: Sporn MB, Roberts AB, Goodman DS, editors. The Retinoids : biology, chemistry, and medicine. Raven Press; New York: 1994. [Google Scholar]
- 12.Napoli JL. Quantification of physiological levels of retinoic acid. Methods Enzymol. 1986;123:112–124. doi: 10.1016/s0076-6879(86)23015-3. [DOI] [PubMed] [Google Scholar]
- 13.Levin MS, Li E, Gordon JI. Structure-function analyses of mammalian cellular retinol-binding proteins by expression in Escherichia coli. Methods Enzymol. 1990;189:506–520. doi: 10.1016/0076-6879(90)89329-g. [DOI] [PubMed] [Google Scholar]
- 14.Vogel S, Mendelsohn CL, Mertz JR, Piantedosi R, Waldburger C, Gottesman ME, Blaner WS. Characterization of a new member of the fatty acid-binding protein family that binds all-trans-retinol. J Biol Chem. 2001;276:1353–1360. doi: 10.1074/jbc.M005118200. [DOI] [PubMed] [Google Scholar]
- 15.Grippo J, Sherman M. Retinoid-binding proteins in embryonal carcinoma cells. Methods Enzymol. 1990;190:148–155. doi: 10.1016/0076-6879(90)90019-w. [DOI] [PubMed] [Google Scholar]
- 16.Ong DE. Purification and partial characterization of cellular retinol-binding protein from human liver. Cancer Res. 1982;42:1033–1037. [PubMed] [Google Scholar]
- 17.MacDonald PN, Ong DE. Binding specificities of cellular retinol-binding protein and cellular retinol-binding protein, type II. J Biol Chem. 1987;262:10550–10556. [PubMed] [Google Scholar]
- 18.Paik J, During A, Harrison EH, Mendelsohn CL, Lai K, Blaner WS. Expression and Characterization of a Murine Enzyme Able to Cleave beta -Carotene. THE FORMATION OF RETINOIDS. J Biol Chem. 2001;276:32160–32168. doi: 10.1074/jbc.M010086200. [DOI] [PubMed] [Google Scholar]
- 19.During A, Nagao A, Hoshino C, Terao J. Assay of beta-carotene 15,15′-dioxygenase activity by reverse-phase high-pressure liquid chromatography. Anal Biochem. 1996;241:199–205. doi: 10.1006/abio.1996.0400. [DOI] [PubMed] [Google Scholar]
- 20.Paik J, Vogel S, Piantedosi R, Sykes A, Blaner WS, Swisshelm K. 9-cis-retinoids: biosynthesis of 9-cis-retinoic acid. Biochemistry. 2000;39:8073–8084. doi: 10.1021/bi992152g. [DOI] [PubMed] [Google Scholar]
- 21.Shi YQ, Hubacek I, Rando RR. Kinetic mechanism of lecithin retinol acyl transferase. Biochemistry. 1993;32:1257–1263. doi: 10.1021/bi00056a009. [DOI] [PubMed] [Google Scholar]
- 22.Hessel S, Eichinger A, Isken A, Amengual J, Hunzelmann S, Hoeller U, Elste V, Hunziker W, Goralczyk R, Oberhauser V, von Lintig J, Wyss A. CMO1-deficiency abolishes vitamin A production from β-carotene and alters lipid metabolism in mice. Journal of Biological Chemistry. 2007 doi: 10.1074/jbc.M706763200. [DOI] [PubMed] [Google Scholar]
- 23.Yeum KJ, Russell RM. Carotenoid bioavailability and bioconversion. Annu Rev Nutr. 2002;22:483–504. doi: 10.1146/annurev.nutr.22.010402.102834. [DOI] [PubMed] [Google Scholar]
- 24.Zhang XEL, Lu J, Tso P, Blaner WS, Levin MS, Li E. Increased neonatal mortality in mice lacking cellular retinol-binding protein II. J Biol Chem. 2002;277:36617–36623. doi: 10.1074/jbc.M205519200. [DOI] [PubMed] [Google Scholar]
- 25.Yan W, Jang GF, Haeseleer F, Esumi N, Chang J, Kerrigan M, Campochiaro M, Campochiaro P, Palczewski K, Zack DJ. Cloning and characterization of a human beta,beta-carotene-15,15′-dioxygenase that is highly expressed in the retinal pigment epithelium. Genomics. 2001;72:193–202. doi: 10.1006/geno.2000.6476. [DOI] [PubMed] [Google Scholar]
- 26.Lindqvist A, Andersson S. Biochemical Properties of Purified Recombinant Human beta -Carotene 15,15′-Monooxygenase. J Biol Chem. 2002;277:23942–23948. doi: 10.1074/jbc.M202756200. [DOI] [PubMed] [Google Scholar]
- 27.Nelson PS, Clegg N, Arnold H, Ferguson C, Bonham M, White J, Hood L, Lin B. The program of androgen-responsive genes in neoplastic prostate epithelium. Proc Natl Acad Sci U S A. 2002;99:11890–11895. doi: 10.1073/pnas.182376299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kedishvili NY, Chumakova OV, Chetyrkin SV, Belyaeva OV, Lapshina EA, Lin DW, Matsumura M, Nelson PS. Evidence that the human gene for prostate short-chain dehydrogenase/reductase (PSDR1) encodes a novel retinal reductase (RalR1) J Biol Chem. 2002;277:28909–28915. doi: 10.1074/jbc.M202588200. [DOI] [PubMed] [Google Scholar]
- 29.Quadro L, Blaner WS, Salchow DJ, Vogel S, Piantedosi R, Gouras P, Freeman S, Cosma MP, Colantuoni V, Gottesman ME. Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. Embo J. 1999;18:4633–4644. doi: 10.1093/emboj/18.17.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Bennekum AM, Wei S, Gamble MV, Vogel S, Piantedosi R, Gottesman M, Episkopou V, Blaner WS. Biochemical basis for depressed serum retinol levels in transthyretin-deficient mice. J Biol Chem. 2001;276:1107–1113. doi: 10.1074/jbc.M008091200. [DOI] [PubMed] [Google Scholar]
- 31.Tajima S, Goda T, Takase S. Coordinated distribution patterns of three enzyme activities involved in the absorption and metabolism of beta-carotene and vitamin A along the villus-crypt axis of chick duodenum. Life Sci. 1999;65:841–848. doi: 10.1016/s0024-3205(99)00311-2. [DOI] [PubMed] [Google Scholar]
- 32.During A, Nagao A, Terao J. beta-carotene 15,15′-dioxygenase activity and cellular retinol-binding protein type II level are enhanced by dietary unsaturated triacylglycerols in rat intestines. J Nutr. 1998;128:1614–1619. doi: 10.1093/jn/128.10.1614. [DOI] [PubMed] [Google Scholar]
- 33.Harrison E, Blaner W, Goodman D, Ross A. Subcellular localization of retinoids, retinoid-binding proteins, and acyl-CoA:retinol acyltransferase in rat liver. J Lipid Res. 1987;28:973–981. [PubMed] [Google Scholar]
- 34.Goodman DS, Blomstrand R, Werner B, Huang HS, Shiratori T. The intestinal absorption and metabolism of vitamin A and beta-carotene in man. J Clin Invest. 1966;45:1615–1623. doi: 10.1172/JCI105468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herr FM, Ong DE. Differential interaction of lecithin-retinol acyltransferase with cellular retinol binding proteins. Biochemistry. 1992;31:6748–6755. doi: 10.1021/bi00144a014. [DOI] [PubMed] [Google Scholar]
- 36.Parker RS. Carotenoids in human blood and tissues. J Nutr. 1989;119:101–104. doi: 10.1093/jn/119.1.101. [DOI] [PubMed] [Google Scholar]
- 37.Kaplan LA, Lau JM, Stein EA. Carotenoid composition, concentrations, and relationships in various human organs. Clin Physiol Biochem. 1990;8:1–10. [PubMed] [Google Scholar]
- 38.de Pee S, West CE, Muhilal, Karyadi D, Hautvast JG. Lack of improvement in vitamin A status with increased consumption of dark-green leafy vegetables. Lancet. 1995;346:75–81. doi: 10.1016/s0140-6736(95)92111-7. [DOI] [PubMed] [Google Scholar]
- 39.Takyi EE. Children’s consumption of dark green, leafy vegetables with added fat enhances serum retinol. J Nutr. 1999;129:1549–1554. doi: 10.1093/jn/129.8.1549. [DOI] [PubMed] [Google Scholar]
- 40.Reeves PG. Components of the AIN-93 diets as improvements in the AIN-76A diet. J Nutr. 1997;127:838S–841S. doi: 10.1093/jn/127.5.838S. [DOI] [PubMed] [Google Scholar]
- 41.Bierer TL, Merchen NR, Erdman JW., Jr Comparative absorption and transport of five common carotenoids in preruminant calves. J Nutr. 1995;125:1569–1577. doi: 10.1093/jn/125.6.1569. [DOI] [PubMed] [Google Scholar]