

SUMMARY
A novel phage infecting Escherichia coli was isolated during a large-scale screen for phages that may be used for therapy of mastitis in cattle. The 77,554 bp genome of the phage, named phiEco32, was sequenced and annotated, and its virions were characterized by electron microscopy and proteomics. Two phiEco32-encoded proteins that interact with host RNA polymerase were identified. One of them is an ECF-family σ-factor that may be responsible for transcription of some viral genes. Another RNA polymerase-binding protein is a novel transcription inhibitor whose mechanism of action remains to be defined.
Keywords: bacteriophage, Podoviridae, E. coli, genome, MudPIT, RNA polymerase-binding proteins
INTRODUCTION
Bacteriophages (phages) are the most abundant and diverse form of life on Earth and exert a major influence over the microbial world.1,2 To date, more than 400 phage genomes have been completely sequenced (NCBI, National Center for Biotechnology Information – July 2007). Comparative analysis has provided important insights into diversity and evolution of phage genomes and the functions of various phage genes.3,4 The actual number of different phages on the planet has been estimated at ~1031.3 Nevertheless, for a given host, the number of different phages (defined here as phages that rely on different, non-homologous mechanisms of genome replication, host shut-off, and viral gene expression strategies) appears to be finite. For example, finding a truly new phage for Escherichia coli (E. coli) is quite rare and most new isolates are simple variations of known phages with identical sets of genes and more than 90% identity at the DNA level. Therefore, most phages infecting E. coli must have already been isolated during the many years of study of this model bacterium. In contrast, the genomic analysis of mycobacterial and Thermus phages continues to reveal novel genomes5,6, reflecting that our knowledge of phages infecting these less studied hosts is at its infancy. On the other hand, the comparative analysis of phages infecting different hosts indicates that i) there is a significant genetic exchange between phages and ii) similar mechanisms (for example, factor-dependent antitermination of transcription of long operons containing the structural proteins of many siphoviruses such as phage λ7) are often used to regulate coordinate expression of viral genes. However, phage regulators that perform apparently similar regulatory functions are often non-homologous.8 Identification and comparative mechanistic analysis of such regulators can be instructive as it reveals how common regulatory responses of bacterial RNA polymerase, an essential cellular molecular machine, are elicited by interactions with very different proteins.
We are engaged in a program of isolating phages infecting E. coli and determining the mechanisms of gene expression control that these phages use. To this end, a large collection of phages from the Eliava Institute, Tbilisi, Republic of Georgia, was surveyed for the presence of previously unreported phages. We hypothesized that the relative isolation of the Institute may help identify new phages. Here we report the identification of one such phage, E. coli phage phiEco32, and its genomic and proteomic analyses.
RESULTS
Isolation of phiEco32 and virion morphology
PhiEco32 was isolated in 2004 from the Kura river in Tbilisi, Georgia. It was later found to lyse 95% of E. coli strains isolated from cows suffering from acute mastitis and is a component of a polyvalent phage preparation currently being tested in experimental mastitis phage therapy trials (TG, KS, IJM, to be published). Its latent period at 37°C in rich media is 30 – 35 min. Virions of phiEco32 (Fig. 1) belong to the family Podoviridae and have a C3 morphotype9. The dimensions of phiEco32 are ~ 145 × 44 nm for the head and ~ 13 × 8 nm for the tail, with short, possibly kinked tail fibers folded on the tail.
Figure 1. Morphology of phiEco32 virions.
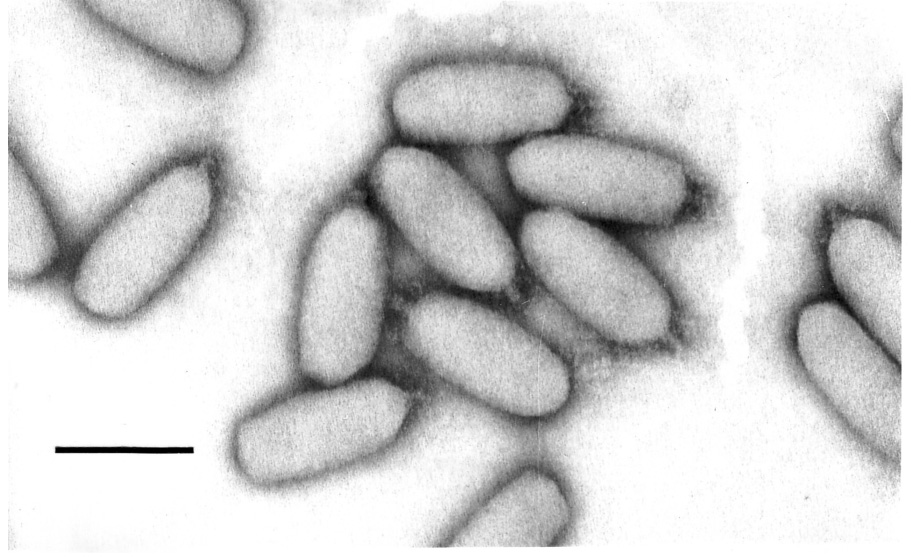
Phage phiEco32 virions stained with phosphotungstate and revealed by electron microscopy. Final magnification is ×297,000; the bar represents 100 nm. The virions appear oval due to flattening of the central part of the capsid.
Overview of the phiEco32 genome
The genome of phage phiEco32 consists of 77,554 bases pairs with a G+C content of 42.27%, which is significantly lower than the G+C content of the E. coli host (50–51%). The initial sequence of the phiEco32 genome assembled as a circle. However, restriction enzyme digests suggested both that the genome was linear and lacked cohesive ends, and also that there was ~200 bp of additional sequence not present in the assembled genome. The sequence of the right end of the genome (corresponding to bp 77,362–77,554) was determined by primer walking using phiEco32 genomic DNA as template. The exact left end could not be accurately determined by this procedure as no abrupt drop-off in the intensity of peaks on the electrophoregram was reproducibly observed. A SalI fragment that corresponded to the left genome end was therefore isolated, ligated to HincII-cut pUC19 and subjected to sequencing using a plasmid-specific primer. The first non-plasmid base determined by this procedure corresponds to bp 1 of the phiEco32 genome. A similar procedure at the right genome end confirmed that bp 77,554 is the terminal nucleotide. The results of this analysis reveal a 193-bp direct repeat that defines the genome ends. Neither repeat is predicted to be part of a coding sequence. The replication strategy of phiEco32 may thus be similar to that of the T7 phage group, with formation of either circular or linear concatemeric DNA during infection to allow duplication of genome ends. The observation that the right genome end sequence of the phiEco32 genome is constant while a minor fraction of the left end is heterogeneous suggests that the initial cleavage by phiEco32 terminase may release the mature right end. The mature left end must then be a product of duplication of the next terminal repeat in a concatemeric molecule or, as apparently occurs in a few packaged genomes, be created by a less-specific cleavage in the next genome of the concatemer. Preparations of deletion mutants, in particular, of T7 also show some heterogeneity at their left genome ends (IJM, unpublished observation). The close similarity of the large terminase subunit (ORF7, Table 1) to their P22 family counterparts is consistent with the blunt ends of the phiEco32 genome and perhaps with the heterogeneity seen at the left genome end (P22- like phages package by a headful mechanism. However, it is not consistent with the T7-like sequence-specific cleavage and direct repeats at the phiEco32 genome ends. Terminases that generate the direct repeats characteristic of the T7 phage family fall into a cluster distinct from the P22-like headful enzymes.10 However, both phiEco32 ORF7 and the P22 terminase family are also closely related in sequence to the PaP3 terminase, which has been described as generating 5′ protruding cohesive ends.11 Terminases that generate the latter type of genome ends also usually cluster in a group distinct from the P22 family10, perhaps suggesting that both the phiEco32 and PaP3 enzymes have only recently acquired sequence-specific cleavage activity.
Table 1.
Sequence similarity and predicted molecular function for gene products of phiEco32
| ORF/names | ORF strand/positiona | ORF length (aa) | GenBank ID and taxonomic origin of the best database match | GeneBank ID and taxonomic origin of the best database match among phages (if different) | Predicted molecular function | Comments |
|---|---|---|---|---|---|---|
| 1 | 296‥496 | 66 | Weak SD element | |||
| 2 | 489‥629 | 46 | 77118167 Escherichia coli phage K1F | |||
| 3 | 565‥744 | 59 | Weak SD element | |||
| 4 | 698‥952 | 84 | ||||
| 5 | 945‥1640 | 231 | ||||
| 6 | 1697‥2170 | 157 | ||||
| 7 | 2240‥3781 | 513 | 27414483 Pseudomonas aeruginosa phage PaP3 | Terminase large subunit | ||
| 8 | 3843‥6086 | 747 | 27476052 Pseudomonas aeruginosa phage PaP3 | Portal protein | ||
| 9 | 6096‥6332 | 78 | Weak SD element | |||
| 10 | 6332‥7417 | 361 | 27414480 Pseudomonasaeruginosa phage PaP3 | Scaffolding protein | ||
| 11 | 7459‥8517 | 352 | 78696363 Bradyrhizobium sp. | 27414479 Pseudomonas aeruginosa phage PaP3 | Major head protein | |
| 12 | 8529‥9041 | 170 | 90587664 Flavobacterium johnsoniae | Bacterial Ig-like domain | ||
| 13 | 9143‥9895 | 250 | 113876452 Sinorhizobium medicae | 27476047 Pseudomonas aeruginosa phage PaP3 | Conserved hypothetical protein | |
| 14 | 9905‥12547 | 880 | 45686334 Escherichia coli phage T1 | Putative tail fiber | ||
| 15 | 12588‥14756 | 722 | 33770533 Yersinia enterocolitica phage PY54 | Tail fiber | ||
| 16 | 14849‥15067 | 72 | Holin | |||
| 17 | 15097‥15588 | 163 | 33340418 Salmonella phage Felix01 | Lysis protein (muraminidase) | ||
| 18 | 15601‥16404 | 267 | 52139914 Escherichia coli phage JS98 | Putative structural protein | ||
| 19 | 16414‥19431 | 1005 | 27414473 Pseudomonas aeruginosa phage PaP3 | Bacterial surface proteins containing Ig-like region | ||
| 20 | 19475‥20443 | 322 | 77864687 Burkholderia cepacia phage Bcep176 | Putative tail tip fiber protein and a host specificity determinant | ||
| 21 | 20445‥21476 | 343 | 22126074 Yersinia pestis | 33770532 Yersinia enterocolitica phage PY54 | Conserved hypothetical protein | |
| 22 | 21489‥22271 | 260 | 29243594 Pseudomonas putida phage gh-1 | Virion protein with transglycosylase SLT domain | ||
| 23 | 22291‥23343 | 350 | ||||
| 24 | 23356‥24324 | 322 | 27476040 Pseudomonas aeruginosa phage PaP3 | Putative DNA injection protein | ||
| 25 | 24338‥25993 | 551 | A predicted coiled-coil protein | |||
| 26 | 26061‥30482 | 1473 | ||||
| 27 | − (30552‥30659) | 35 | ||||
| 28 | − (30672‥30833) | 53 | 32453596 Escherichia coli phage RB69 | Conserved hypothetical protein | ||
| 29 | − (30842‥31069) | 75 | ||||
| 30 | − (31062‥31460) | 132 | 149408178 Pseudomonas aeruginosa phage PA11 | Conserved hypothetical protein | ||
| 31 | − (31450‥31902) | 150 | 33340391 Salmonella phage Felix01 | HNH endonuclease | Weak SD element | |
| 32 | − (31889‥32068) | 59 | ||||
| 33 | − (32082‥32900) | 272 | 27414457 Pseudomonas aeruginosa phage PaP3 | 5’-3’ exonuclease similar to the N-terminal exonuclease domain of DNA polymerase I | ||
| 34 | − (32887‥33780) | 297 | 45686300 Escherichia coli phage T1 | ATP-binding protein with Walker A motif; an ATPase or dNMP kinase | ||
| 35 | − (33777‥34169) | 130 | 88603084 Methanospirillum hungatei | 3172310 Mycobacteria phage D29 | Predicted GTP-binding protein | |
| 36 | − (34166‥34810) | 214 | 68229118 Frankia sp. | RNA polymerase ECF sigma factor | ||
| 37 | − (34833‥35273) | 146 | 94498600 Sphingomonas sp | 29135040 Pseudomonas aeruginosa phage phiKZ | Appr-1-p processing enzyme family, phosphatase | Weak SD element |
| 38 | − (35273‥35446) | 57 | 56412276 Salmonella enterica | |||
| 39 | − (35418‥35768) | 116 | 33340399 Salmonella phage Felix01 | Conserved hypothetical protein | ||
| 40 | − (35826‥36572) | 248 | ||||
| 41 | − (36883‥36995) | 35 | ||||
| 42 | − (36989‥37168) | 59 | No SD element | |||
| 43 | − (37149‥37364) | 71 | ||||
| 44 | − (37380‥37523) | 47 | 71834082 Escherichia coli phage JK06 | |||
| 45 | − (37520‥37687) | 55 | ||||
| 46 | − (37684‥37860) | 58 | No SD element | |||
| 47 | − (37857‥38249) | 130 | Match in an environmental sample, 44001214 | |||
| 48 | − (38431‥38607) | 58 | ||||
| 49 | − (38617‥38784) | 55 | ||||
| 50 | − (38784‥38912) | 42 | 81343940 Escherichia coli phage RTP | Conserved hypothetical protein | ||
| 51 | − (38968‥39159) | 63 | 5354344 Escherichia coli phage T4 | Conserved hypothetical protein | ||
| 52 | − (39159‥39524) | 121 | ||||
| 53 | − (39533‥41377) | 614 | 27414453 Pseudomonas aeruginosa phage PaP3 | DNA polymerase domain, lacking both 5’-3’ and 3’-5’ exonuclease domains. | ||
| 54 | − (41394‥41807) | 137 | ||||
| 55 | − (41809‥41967) | 52 | ||||
| 56 | − (41971‥42255) | 94 | ||||
| 57 | − (42257‥42514) | 85 | ||||
| 58 | − (42583‥42900) | 105 | 40549402 Pseudomonas aeruginosa phage PaP3 | |||
| 59 | − (42955‥43287) | 110 | 58336433 Lactobacillus acidophilus | 38640011 Aeromonas hydrophila phage Aeh1 | Conserved hypothetical protein | |
| 60 | − (43318‥44061) | 247 | 29376909 Enterococcus faecalis | 19343479 Roseobacter phage SIO1 | Phosphate starvation-inducible phoH-like, ATPase | |
| 61 | − (44058‥44243 | 61 | Weak SD element | |||
| 62 | − (44230‥44559) | 109 | 22298400 Thermosynechococcus elongatus | 82657960 Pseudomonas aeruginosa phage phiEL | NAD-dependent DNA ligase, minimal nucleotidyltransferas e domain as in some other phages and viruses | |
| 63 | − (44556‥44741) | 61 | Weak SD element | |||
| 64 | − (44741‥45397) | 218 | 149408182 Pseudomonas aeruginosa phage PA11 | Thymidylate synthase thyX/thy1 (flavin-dependent) | ||
| 65 | − (45413‥45688) | 91 | 114705060 Fulvimarina pelagi | 46401774 Escherichia coli phage T5 | Thiol-disulfide isomerase and thioredoxin | |
| 66 | − (45697‥45876) | 59 | ||||
| 67 | − (45937‥46500) | 187 | 88912807 Acidothermus cellulolyticus | 56693168 Lactobacillus plantarum phage LP65 | DNA-binding protein, DPS family of ferritin-like diiron-carboxylate proteins | |
| 68 | − (46493‥46822) | 109 | 34333203 Vibrio phage KVP40 | Conserved hypothetical protein | ||
| 69 | − (46895‥47104) | 69 | Weak SD element | |||
| 70 | − (47104‥47313) | 69 | Weak SD element | |||
| 71 | − (47313‥47510) | 65 | ||||
| 72 | − (47511‥48044) | 177 | 56750822 Synechococcus elongatus | 18996679 Pseudomonas aeruginosa phage phiKZ | dCTP deaminase | |
| 73 | − (48054‥49367) | 437 | Weak SD element | |||
| 74 | − (49369‥49926) | 185 | 27414446 Pseudomonas aeruginosa phage PaP3 | 3’-5’ exonuclease domain of type I DNA polymerase | ||
| 75 | − (49920‥51710) | 596 | 27414445 Pseudomonas aeruginosa phage PaP3 | Primase/helicase | ||
| 76 | − (51737‥51940) | 67 | Weak SD element | |||
| 77 | − (51930‥52349) | 139 | 15669708 Methanocaldococcus jannaschii | 27414443 Pseudomonas aeruginosa phage PaP3 | Conserved YtfP/UPF0131 family, predicted FAD-binding oxidoreductase | |
| 78 | − (52419‥52604) | 61 | ||||
| 79 | − (52601‥52849) | 82 | ||||
| 80 | − (52856‥54049) | 397 | 149408167 Pseudomonas aeruginosa phage PA11 | ATP-grasp enzyme, predicted amino acid ligase/glutaminyl transferase | ||
| 81 | − (54052‥54276) | 74 | ||||
| 82 | − (54257‥54490) | 77 | ||||
| 83 | − (54493-56523) | 676 | 149408163 Pseudomonas aeruginosa phage PA11 | Glutamine amidotransferase domain AUU start codon | ||
| 84 | − (56603‥57706) | 367 | 27414439 Pseudomonas aeruginosa phage PaP3 | Weak SD element | ||
| 85 | − (57703‥58320) | 205 | ||||
| 86 | − (58330‥59136) | 268 | 27414437 Pseudomonas aeruginosa phage PaP3 | Conserved hypothetical protein, marginal match to carboxylate-amine ligases, conserved catalytic histidine | ||
| 87 | − (59129‥60100) | 323 | ||||
| 88 | − (60111‥61310) | 399 | ||||
| 89 | − (61332‥61684) | 120 | ||||
| 90 | − (61687‥61839) | 50 | ||||
| 91 | − (62075‥62290) | 71 | 111116430 Escherichia coli APEC 01 | Weak SD element | ||
| 92 | − (62290‥62754) | 154 | 32453588 Escherichia coli phage RB69 | Conserved hypothetical protein | Weak SD element | |
| 93 | − (62754‥62987) | 77 | ||||
| 94 | − (63094‥63210) | 38 | ||||
| 95 | − (63219‥63467) | 82 | ||||
| 96 | − (63473‥63718) | 81 | Weak SD element | |||
| 97 | − (63718‥63966) | 82 | Weak SD element | |||
| 98 | − (63968‥64198) | 76 | ||||
| 99 | − (64207‥64530) | 107 | Weak SD element | |||
| 100 | − (64520‥64741) | 73 | ||||
| 101 | − (65031‥65162) | 43 | Weak SD element | |||
| 102 | − (65159‥65404) | 81 | ||||
| 103 | − (65388‥65561) | 57 | 149408169 Pseudomonas aeruginosa phage PA11 | |||
| 104 | − (65571‥65792) | 73 | ||||
| 105 | − (65800‥66048) | 82 | ||||
| 106 | − (66050‥66331) | 93 | 81343973 Escherichia coli phage RTP | Putative phage lipoprotein | ||
| 107 | − (66436‥66615) | 59 | No SD element | |||
| 108 | − (66608‥66859) | 83 | 148734558 Escherichia coli phage TLS | Conserved hypothetical protein | ||
| 109 | − (66856‥67080) | 74 | ||||
| 110 | − (67077‥67253) | 58 | ||||
| 111 | − (67332‥67541) | 69 | 33340411 Salmonella phage Felix 01 | Conserved hypothetical protein | Weak SD element | |
| 112 | − (67531‥67671) | 46 | Weak SD element | |||
| 113 | − (68030‥68248) | 72 | ||||
| 114 | − (68249‥68506) | 85 | Weak SD element | |||
| 115 | − (68496‥68732) | 78 | ||||
| 116 | − (68722‥69018) | 98 | ||||
| 117 | − (69218‥69607) | 129 | Weak SD element | |||
| 118 | − (69582‥69713) | 43 | Weak SD element | |||
| 119 | − (69718‥70515) | 265 | 16565696 Escherichia coli | Agglutinating adhesin | No SD element | |
| 120 | − (70617‥71060) | 147 | ||||
| 121 | − (71130‥71339) | 69 | ||||
| 122 | − (71336‥71560) | 74 | 26989746 Pseudomonas putida | 17313260 Pseudomonas aeruginosa phage phiCTX | Conserved hypothetical protein | |
| 123 | − (71557‥71775) | 72 | ||||
| 124 | − (71887‥72786) | 299 | ||||
| 125 | − (74731‥75096) | 121 | ||||
| 126 | − (75189‥75470) | 93 | Weak SD element | |||
| 127 | − (75656‥75919) | 87 | ||||
| 128 | − (75944‥76816) | 290 |
A total of 128 ORFs could be predicted in the phiEco32 genome (Table 1, Fig. 2). Intergenic regions longer than 100 bases were scanned for additional genes by searching for similar sequences in Genbank and the database of unfinished microbial genomes at NCBI, but no additional ORFs were found. There are 51 cases of overlaps (from 1 to 65 bases long) between neighboring ORFs. The longest non-coding region (1,944 bases) lies between ORFs 124 and 125. The predicted ORFs encode putative proteins from 35 amino acids (ORF27 and ORF41) to 1,473 amino acids (ORF26), and more than 40% have sequence homologs in the databases (see below). Only ~70% of ORFs are preceded by recognizable Shine-Dalgarno sequences. About 15% of ORFs, in particular those that are short, have neither homologs in the databases nor well-defined Shine-Dalgarno sequences, and it remains to be seen whether they represent functional genes.
Figure 2. The phiEco32 genome.
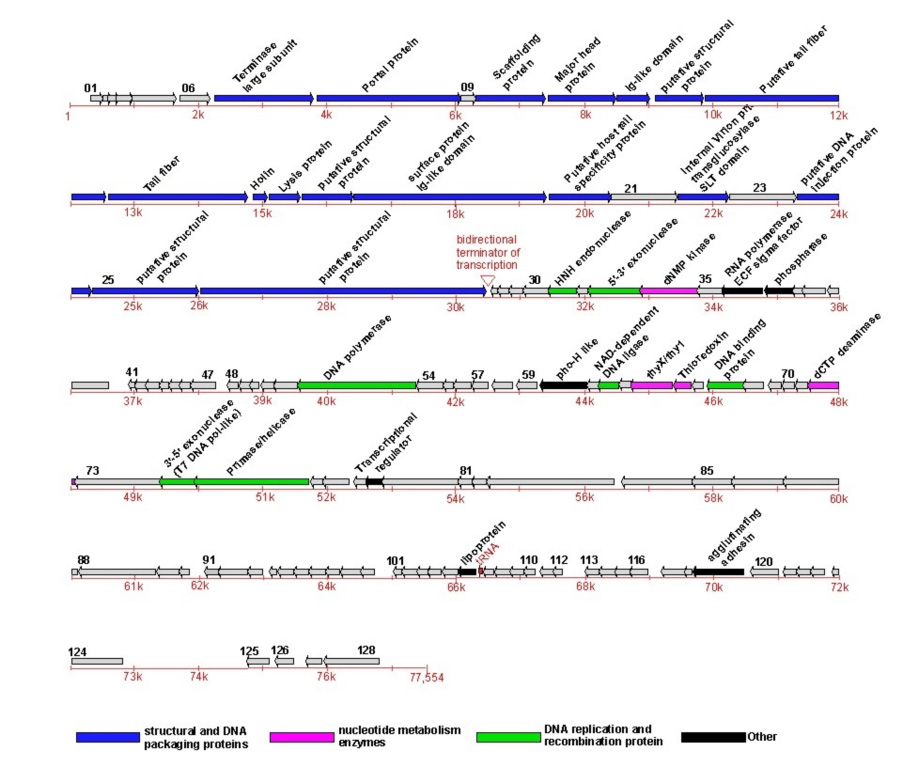
The phiEco32 genome is schematically presented with predicted ORFs indicated by arrows and numbered in black. The direction of an arrow indicates the direction of transcription. ORFs for whose products clear functional predictions can be made are highlighted by color (see Table 1 for more details); ORF79, which codes for a novel RNAP inhibitor identified in this work, is also indicated. The genes encoding phiEco32 virion proteins as identified by a combination of LC-ESI MS/MS and MudPIT are highlighted. The putative bidirectional terminator of transcription is indicated by a red wedge and the tRNA gene is also indicated by a red arrow and labeled in red. A scale of nucleotide numbers (red) runs below the ORFs.
Most phiEco32 ORFs start with the AUG codon, 13 ORFs use GUG, two ORFs start with UUG, and ORF83 is predicted to use AUU. Only two E. coli genes: infC12 and pcnB13 are known to use this codon for initiation; its predicted use in phiEco32 ORF83 is based on sequence alignments from the initiating Met through residue ~200 with phage PA11 ORF4, phage PaP3 ORF45, and various amidotransferases, including their conserved catalytic residues. UAA is the most common phiEco32 stop codon (82 ORFs), with 32 and 14 ORFs ending with, respectively, UGA and UAG. About one-fifth of phiEco32 genes (26 ORFs) are transcribed in one direction (designated as rightward in the genome map, Fig. 2) and the rest are transcribed leftward. Genes transcribed in the same direction are clustered, forming two regions of genes with similar orientations. There is no difference in average G+C content between the rightward- and leftward-transcribed regions.
Using Genskew (http://mips.gsf.de/services/analysis/genskew) a cumulative GC-skew plot follows the direction of transcription precisely. The maximum skew value is at bp 30,493, 11 bp after the end of the last rightward ORF, ORF26 and 166 bp downstream of the last leftward ORF, ORF27. An imperfect hairpin with a GAAA tetraloop is predicted for bps 30,490–30,517 that plausibly could serve as a bidirectional terminator of transcription. The minimum value of the cumulative GC-skew is at bp 77,232, in a noncoding region between the terminal repeat and ORF128. The corresponding maxima and minima for an AT-skew analysis are 30,570 and 74,460. Minima of both skews are often associated with origins and/or termini of bidirectional DNA replication (for a review, see Ref. 14), but this remains to be established for phiEco32. However, an origin near the left genome end would give the favorable result that transcription and replication on the phiEco32 genome are co-directional.
Using the tRNA scan-SE program, we identified one tRNA gene in the phiEco32 genome. The gene lies in the intergenic region between ORF106 and ORF107 (Fig. 2). The phiEco32 tRNA gene has a TCT anticodon and should recognize the arginine codon AGA. This codon is one of the rarest in the E. coli genome but is overrepresented in phiEco32 compared to the host, with a frequency (defined as in the EMBOSS package, i.e., the observed or extrapolated number of a corresponding codon per 1000 codons) of 10 in the phage but only 2 in E. coli. Thus, as is the case with other phages6,15, phiEco32-encoded tRNA may allow efficient translation of phage mRNA in the absence of sufficient amounts of corresponding cellular tRNA. However, the phage-coded tRNA may play a more subtle role as neither the major capsid nor scaffolding proteins genes, which are among those expected to be expressed at the highest levels during infection, contain the AGA codon.
Sequence analysis of predicted phiEco32 proteins
All predicted phiEco32 proteins were compared to proteins in sequence databases using PSI-BLAST, and to databases of unfinished genomes and environmental samples using TBLASTN. The results show that about 43% of phiEco32 proteins display sequence similarity to known proteins, and most of them match proteins with known molecular functions (Table 1).
PhiEco32 proteins involved in DNA replication and recombination
PhiEco32 codes for an assortment of replication and recombination factors, including a 5’-3’ exonuclease (gp33), DNA polymerase (gp53), 3’-5’ exonuclease (gp74), primase/helicase (gp75), and NAD-dependent DNA ligase (gp62). Gp53 is a member of DNA polymerase Family A (pfam00476) which shares significant similarity with the polymerase domain of E. coli DNA polymerase I. In most known Family A DNA polymerases, this domain is fused to one or two exonuclease domains, whereas the phiEco32 arrangement, where both exonucleases are represented by separate noncontiguous genes, was heretofore observed only once, in Pseudomonas phage PaP3. Finally, gp67, which is a member of a Dps family of DNA-binding proteins that is frequently encountered in bacterial genomes but rarely in phages, may also be involved in phage DNA replication. Gp67 is the only phiEco32 protein that belongs to a recognizable family of DNA-binding proteins.
Among phiEco32 replication proteins, gp33, gp53, gp74, and gp75 are most closely related to homologs from P. aeruginosa phage PaP3 (a close evolutionary relationship between these two phages of gammaproteobacteria is supported by analysis of several other phiEco32 genes; see Discussion). The remaining phiEco32 replication genes products show higher sequence similarity to various bacterial proteins, although the closest relative of putative phiEco32 thioredoxin (gp65) comes from phage T5.
PhiEco32 proteins involved in nucleotide metabolism
Phage genomes commonly encode enzymes of nucleotide salvage and modification.4 PhiEco32 codes for at least 4–5 proteins of this class--putative deoxynucleoside monophosphate kinase gp34, ADP-ribosylphosphate-processing phosphatase gp37, flavin-dependent thymidylate synthase gp64, thioredoxin-like protein gp65, and deoxycytidine triphosphate deaminase gp72. The phylogenetic affinities of these enzymes are different: closest gp34, gp37, and gp64 homologs come from three distinct families of tailed phages (siphovirus T1, myovirus phiKZ, and podovirus SiO1, respectively), gp72 is equally close to homologs from phiKZ and from cyanobacterium Synechococcus elongates, and gp65 contains a thioredoxin-like domain most closely related to proteins from Fulvimarina pelagi and other uncultured marine bacteria.
Structural proteins of phiEco32
We used mass-spectrometric analysis of purified virions to identify structural proteins of phiEco32. In one approach, virion components were separated by denaturing SDS-PAGE (Fig. 3), gel slices containing visible protein bands were treated with trypsin and digests were analyzed by ESI-MS/MS. Ten virion components, gp8, gp11, gp13-15, gp18-19, gp25-26, and gp58, were identified in this way (Fig. 3 and Table 1). Only six of these proteins have sequence similarities indicative of their structural roles, underscoring the importance of experimental analysis for comprehensive determination of the structural set of phage proteins.
Figure 3. Mass spectrometric analysis of virion proteins.
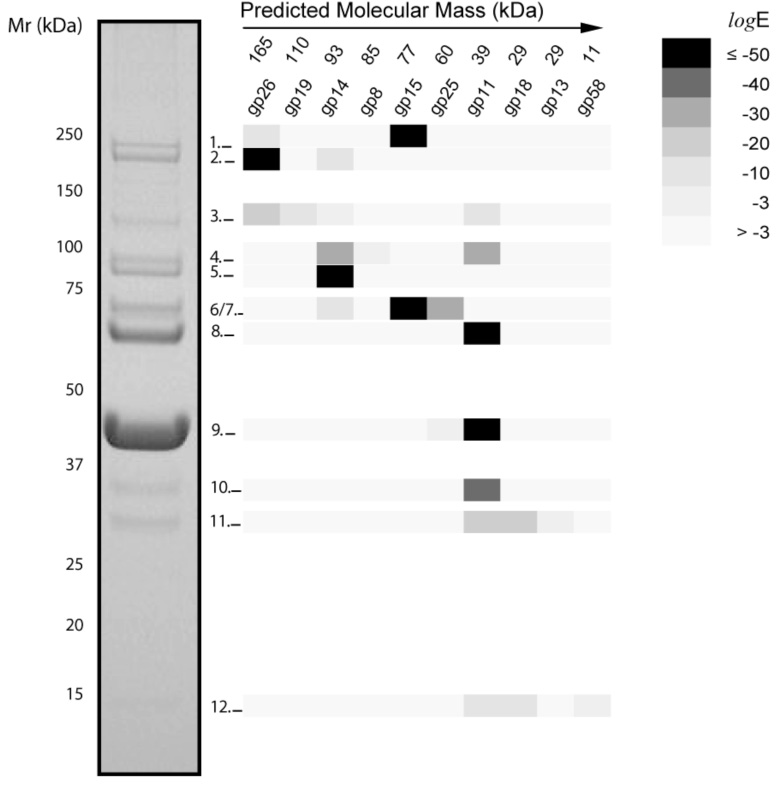
A highly purified preparation of phiEco32 virions was loaded onto a 4–12 % SDS-gel and after electrophoresis proteins were stained with Coomassie. Eleven bands were in-gel digested with trypsin and the resulting peptides were analyzed by LC/MS/MS. The proteins (identified by their “gp” number) detected in each band were plotted in a matrix coded by their expectation values as indicated. Numbers above the “gp” numbers are predicted molecular masses for each protein.
Most gel bands contained more than one protein (Fig. 3). The predicted molecular masses of the proteins and their apparent molecular masses in the gel were in approximate agreement, with a notable exception of tail fiber protein gp15, which was a predominant component of two distinct bands: band 6/7 has an apparent molecular weight of 70 kDa, which is close to the predicted molecular weight of gp15 (77 kDa); band 1, however, has an apparent MW above 200 kDa. This might indicate that gp15 is modified post-translationally and/or forms very stable trimers that were not completely dissociated before SDS-PAGE. The thermal stability of the trimeric tailspike protein of P22-like phages has been well-documented.16
As expected for the most abundant virion component, the major capsid protein is also found in more than one band. The majority is found in band 9 (Fig. 3) but the 352 amino acid capsid protein is also a significant component of band 8, which has an apparent size about 20 kDa larger. A protein of this size could be synthesized via a −1 frameshift during translation of ORF11 making a gp11-12 fusion protein of 528 amino acids. Programmed frameshifting is a common feature in long-tailed double-stranded DNA phages17 and also in the T7 family.18 Near the end of the ORF11 reading frame a potential slippery sequence (GGGAAAG) is present in the mRNA, this is the same sequence found in phage λ that allows synthesis of the essential gpGT fusion protein at a level about 3.5% of the non-frameshifted gpG product.17 In phiEco32 ORF12 has a very weak Shine-Dalgarno element (Table 1) and is unlikely to be synthesized as an independent protein. However, a gp11-12 fusion protein would place the bacterial Ig-like domain of gp12 on the phage capsid, a location that similar domains are found at in many other phages19, including T7. In the latter phage family the coding sequence for the frameshifted, minor capsid protein gp10B, is immediately followed by a typical class II transcription terminator. A similar stem-loop and oligoU stretch immediately follows the coding sequence mRNA for the phiEco32 gp11-12 fusion protein.
Highly-purified phiEco32 particles were also examined by MudPIT, a shotgun proteomics approach that allows analysis of complex protein mixtures without electrophoretic separation. Peptides matching twenty of the phiEco32-encoded proteins could be identified. Nine of these proteins are shared with the set of structural proteins identified by gel band analysis and correspond to the most abundant proteins in phiEco32 virions. The other eleven proteins detected by MudPIT are of lesser abundance and of lower molecular weight. Most of these proteins would run between bands 11 and 12, a portion of the gel that has no stained material (Fig. 3). Seven of these lower-abundance proteins (gp20-22, gp24, and gp67-68) are conserved in other phages and bacteria, and the predicted functions of gp20, gp22, and gp24 are consistent with their presence in the virions (Table 1). Other low-abundance proteins, such as the putative DNA-binding protein gp67, may be novel virion components, or their presence may be due to adventitious binding to virions.
Transcription regulation of phiEco32
The phiEco32 genome encodes a putative RNA polymerase (RNAP) σ factor, the product of gene 36. Gp36 is related to σ70–like factors of the ECF subfamily. Proteins belonging to this very diverse subfamily typically recognize promoters of genes whose products participate in specialized cellular adaptations and responses to various external stresses (hence the name ECF, for extracytoplasmic factors, reviewed in Ref. 20). To determine if gp36 indeed functions as a σ factor, E. coli cells with chromosomally-encoded protein A (PrA)-tagged RNAP β’ subunit were infected with phiEco32, RNAP was affinity-purified and its composition was compared to that of RNAP from uninfected cells. As can be seen from a gel presented in Fig. 4A, RNAP from infected cells contained two bands with apparent molecular masses of ~26 and ~10 kDa that were absent from the control lane. Mass spectrometric analysis revealed that the 26 kDa band was gp36 (E = 4.9×10−17), suggesting that this protein interacts with host RNAP, as expected of a σ factor. The 10 kDa band was identified as gp79 (E = 4.2×10−4), a small non-conserved protein of unknown function.
Figure 4. Polypeptide composition of RNAP affinity purified from phiEco32-infected cells.
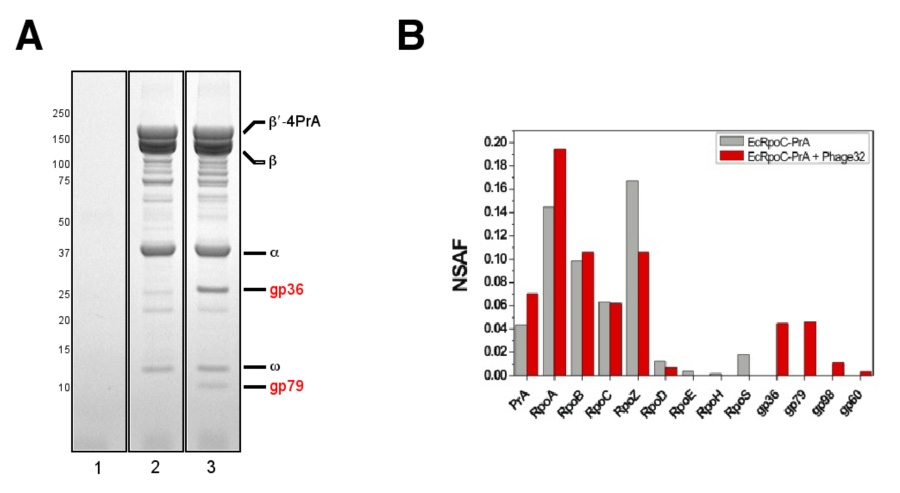
A. The β′-4PrA fusion protein, and co-isolating proteins, was affinity purified from E. coli 55 infected with phiEco32 (lane 1; control), E. coli 55rpoC::4PrA (lane 2), and E. coli 55rpoC::4PrA + phiEco32 (lane 3). The samples were analyzed by SDS-PAGE and bands due to proteins present in lane 3, but absent from lanes 1 and 2, were identified by MALDI MS.
B. NSAF values for proteins identified by MudPIT in RNAP samples affinity purified from both phiEco32-infected and uninfected cells are shown.
MudPIT analysis of PrA-tagged RNAP purified from uninfected and phiEco32-infected E. coli cells was also performed. In the uninfected sample, four different σ factors (σ70/σ24/σ32/σ38) were detected in substochiometric amounts (Fig. 4B). In contrast, only low levels of the primary host sigma factor, σ70, were identified in RNAP from infected cells. Two phage-specific proteins detected in gels, gp36 and gp79, were also found by MudPIT in this preparation, suggesting that one or both of these phage proteins may compete with endogenous σ factors for binding the RNAP core. In addition, the phiEco32 proteins gp98 and gp60 were detected in RNAP from infected cells. However, these two proteins were represented by single peptides and were present at lower levels; they may therefore be contaminants.
To validate the affinity purification results, the DNA encoding the two major phiEco32 proteins present in the RNAP preparation purified from infected cells, phiEco32 gp36 and gp79 were cloned in E. coli expression vectors, and recombinant proteins were purified and tested for their ability to bind to E. coli RNAP core. As can be seen from results of a native gel protein-protein interaction assay presented in Fig. 5, both gp36 and gp79 alone run off the gel (lanes 1 and 2), while RNAP core formed a characteristic slow-moving band20 (lane 3). Addition of gp36 to the core resulted in formation of a new sharp band with higher mobility (lane 4), a situation observed upon the holoenzyme formation with other σ factors, see for example, Ref. 20; in fact the mobility of the new band matched that of the σ70 holoenzyme, data not shown. Likewise addition of gp79 resulted in a formation of a band that moved just slightly slower than the fast-moving band formed upon the addition of gp36 (lane 5). We conclude that both gp36 amd gp79 can bind E. coli RNAP core in vitro, in agreement with affinity purification results, above.
Figure 5. PhiEco32 gp36 and gp79 bind E. coli RNAP core in vitro.
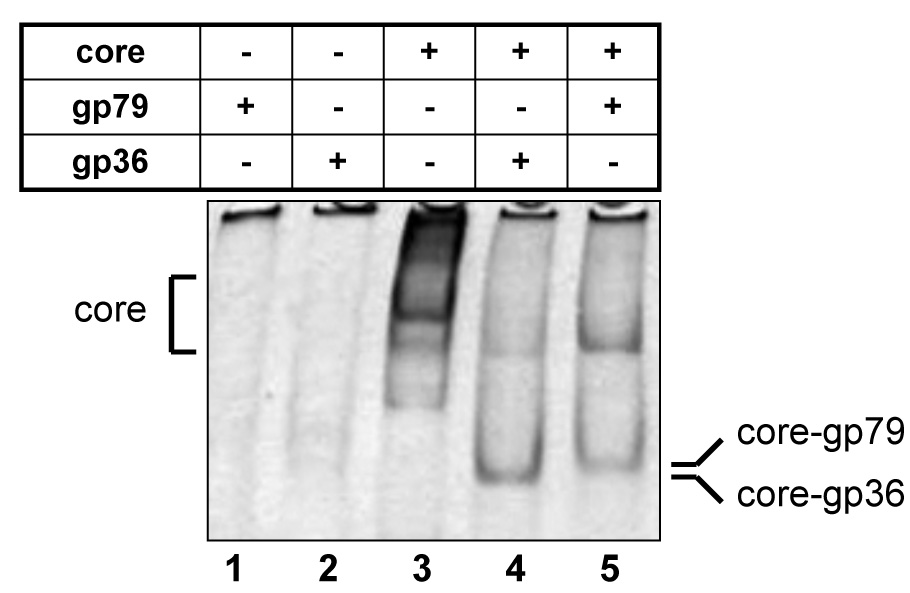
The indicated proteins were combined and loaded on a native polyacrylamide gel. After electrophoresis complexes were revealed by Coomassie-staining.
A complex containing gp36 and RNAP core did not transcribe from a strong σ70-dependent promoter T7 A1 (data not shown). The promoter specificity of the gp36-associated holoenzyme remains to be determined. Most likely, gp36 allows RNAP core to recognize those phiEco32 promoters that are distinct from early promoters that must be recognized by the σ70 holoenzyme. Recombinant gp79 efficiently inhibited promoter-dependent transcription by the σ70 holoenzyme (Fig. 6). Thus, gp79 may be a host transcription shut-off factor. The molecular mechanism of transcription inhibition by gp79 and the physiological role of this protein will be the subject of future investigations.
Figure 6. PhiEco32 gp79 inhibits abortive initiation by E. coli σ70 RNAP holoenzyme.
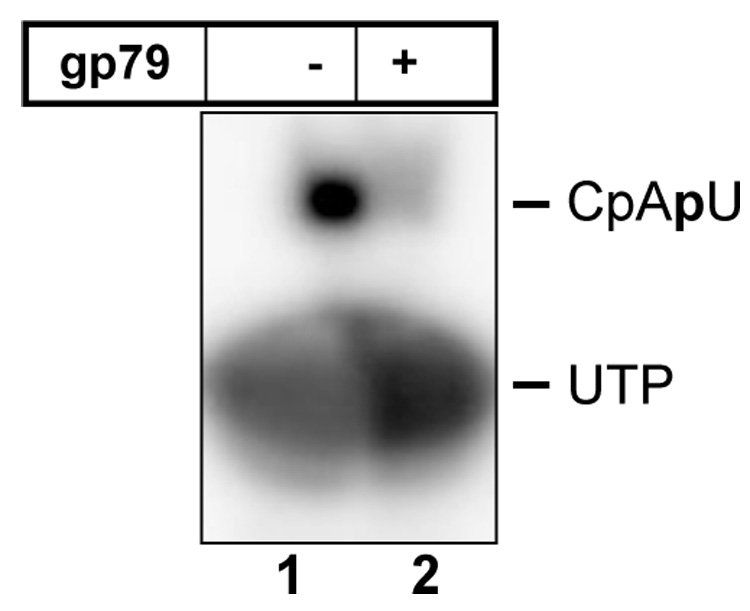
The results of abortive synthesis of the CpApU product from CpA and radioactively labeled UTP from the T7 A1 promoter-containing fragment in the presence or in the absence of gp79 are presented.
DISCUSSION
We report the isolation and initial characterization of phiEco32, a novel podophage infecting E. coli. The C3 type morphology of phiEco32 virions is quite rare, occurring in <1% of phage virions22; curiously, one of the first phages ever observed by electron microscopy23,24 also had C3 morphology. The closest known morphological relatives of phiEco32 are Salmonella enterica serotype Newport phage 7–1125, coliphage Esc-7-1126, Lactococcus lactis phage KSY127, and Vibrio vulnificus phage 71A-6.28 Phage Esc-7-11 has a head of 134 × 40–44 nm and its genome has been estimated to be 70.5 kbp.29 No sequence information is available for salmonellaphage 7–11. The genome sizes of salmonellaphage 7–11 and vibriophage 71A-6 have been estimated to be 93 and 143 kbp, respectively.30,31 No evidence for variation in phiEco32 head size, which is quite common in salmonellaphage 7–11, was found, suggesting that the two phages are distinct. The genome sizes of the Lactococcus phage KSY1 (79,232 bp) and phiEco32 (77,554 bp) are similar, but KSY1 differs from phiEco32 by genome sequence and structure, larger head (233 × 45 nm), elaborate fixation structures, and host range.
The 54 conserved ORFs in the phiEco32 genome encode proteins whose best matches in public databases come from a diverse set of bacteriophages and bacteria. Thus, the phiEco32 genome is a complex mosaic, a situation encountered with many other phages. Fourteen of the 54 conserved phiEco32 ORFs have proteins encoded by P. aeruginosa bacteriophage PaP3 as closest database relatives (Table 1, Fig. 7); four additional phiEco32 proteins have significant sequence similarity to PaP3 ORFs, two among these being the second-closest matches, with scores insignificantly lower than those of the best matches. Similar to phiEco32, the coding regions of PaP3 are divided into two oppositely transcribed groups. Within each PaP3 group, clusters of genes whose products show significant similarities to phiEco32 proteins are present (Fig. 2). The first cluster encodes nine phiEco32 proteins (gp7, gp8, gp10, gp11, gp13, gp17, gp18, gp19, and gp24), mostly involved in head assembly and DNA packaging. The second cluster encodes seven proteins, two of which, gp74 and gp75, are enzymes involved in DNA replication (two additional components of the DNA replication assembly, gp33 and gp53, also have closely related homologs in PaP3 but are located outside of this cluster). Five more proteins in the second cluster, gp77, gp80, gp83, gp84, and gp86, contain several conserved domains indicative of various enzymatic functions (Table 1). The preservation of this module between phiEco32 and PaP3 is in stark contrast with numerous gene insertions and deletions observed in other parts of the two genomes. A comparison of all putative proteins with predicted functional or structural roles in the two phages indicate that PaP3 genes form an almost perfect subset of phiEco32 genes. Only PaP3 proteins p14 and p25 do not appear to have homologs in the phiEco32 genome. Structural proteins in phiEco32 for which no homologs are seen in PaP3 include a bacterial Ig-like domain (gp12 or the gp11-12 fusion), and tail fiber proteins (gp14-15). PaP3 is a temperate phage of P. aeruginosa isolated from hospital sewage11. It is a member of Podoviridae owing to the presence of an icosahedral head, a short tail, and a linear dsDNA genome. However, the head of PaP3 is isometric and thus is clearly different from that of phiEco32. PaP3 is also a temperate phage, that has 5′-protruding cohesive ends and a genome (45,503 bp) that is much smaller that the phiEco32 genome. As phiEco32 also differs in that it has blunt ends with direct repeat, packaging and likely the mechanism of replication are therefore also different.
Figure 7. Comparison of phiEco32 and PaP3 genomes.
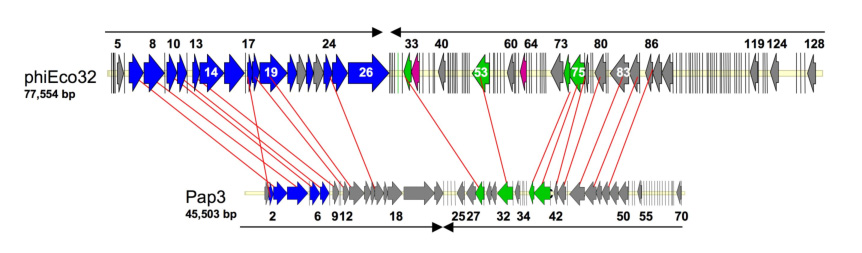
The phiEco32 and PaP3 genomes are drawn to scale with larger ORFs indicated as arrows, the direction of an arrow indicating the direction of transcription. Smaller ORFs have been reduced to bars to avoid cluttering the figure. The ORFs in phiEco32 are colored according to the functional predictions made (Table 1, Fig. 2). The ORFs in PaP3 are colored with a similar scheme using the annotations from the Genbank entry NC_004466. The homologous genes in the two genomes are connected by red lines. The light yellow bar in both genomes indicates the whole length of the genome. The black arrows (on top in PhiEco32 and below in PaP3) indicate the direction of transcription of the ORFs in that region.
The phiEco32 genome encodes more proteins involved in replication and control of gene expression than the PaP3 genome. The phiEco32 genome encodes an RNAP σ factor, gp36. Several phages, including the well-studied coliphage T4, are known to encode proteins that recruit the host RNAP core to promoters of late viral genes. However, these proteins are only very distantly related to σ factors of the σ70 family. To date, only one phage, B. anthracis Fah, has been shown to encode a bona fide σ factor with all four conserved regions characteristic of proteins of this family.32 PhiEco32 gp36 is also an unmistakable homolog of σ factors of the ECF subfamily. Gp36 binds host RNAP core and can displace the σ70 subunit from the holoenzyme in vitro (data not shown). Given the conservation of main domains characteristic of σ factors, the role of gp36 is unlikely to be limited to σ70 displacement; it is more likely that gp36-holoenzyme recognizes promoters of middle or late genes of the virus. Identification of these promoters will have to await the results of currently ongoing analysis of phiEco32 gene expression at various stages of transcription. Another phiEco32 protein that associates with host RNAP core is gp79, a novel transcription factor that inhibits σ70-dependent transcription in vitro. This protein may inhibit early phage (and host) transcription and/or help gp36 to effectively compete with σ70 for a common core-binding site. Identification of the RNAP site that gp79 binds to and determination of the molecular mechanisms of transcription inhibition may clarify the physiological role of this novel transcription inhibitor.
MATERIALS AND METHODS
Isolation and propagation of phiEco32
PhiEco32 was isolated in 2004 from the river Kura, in Tbilisi, Georgia by enrichment on an E. coli 55 strain recovered from cows suffering from mastitis. PhiEco32 was propagated in laboratory in rich medium using its natural host E. coli 55. Clarified lysates were concentrated by PEG and then purified by equilibrium CsCl density gradients.
Electron microscopy
Density-gradient purified phages were deposited on a grid with a carbon-coated Formvar film, negatively stained with 2 % potassium phosphotungstate (pH 7.0), and examined in a Philips EM300 electron microscope operated at 60 kV. Magnification was controlled with T4 phage tails (length 113 nm).
Sequence analysis
ORFs of the phiEco32 genome were predicted using the GeneMark server (http://exon.gatech.edu/GeneMark/heuristic_hmm2.cgi31) and Glimmer2.34 The PSI-BLAST program was used to detect the homologs of phiEco32 genes in the DNA and protein databases, with profile inclusion cutoff e-value in PSI-BLAST (-h parameter) set to 0.02. Both options of low-complexity filtering (-F parameter) and composition-based statistics (-t parameter) were sometimes adjusted during sequence similarity searches. tRNA genes were searched using the tRNAscan-SE server (http://selab.janelia.org/tRNAscan-SE/35).
The codon frequencies for phiEco32 and E. coli (NC_000913) genomes were calculated using the EMBOSS package.36
Strain construction and purification of PrA-tagged RNAP
A strain of E. coli 55 encoding four Fc-binding repeats of the protein A (4PrA affinity tag37) appended to the 3′ end of the rpoC (RNAP β′) gene was constructed using the method of gene gorging.38 The presence of the PrA tag was validated by both PCR analysis of genomic DNA and Western blot analysis of cell lysates. The E. coli 55 wild-type and E. coli 55 rpoC∷4PrA strains exhibited almost identical growth curves in rich medium and were productively infected by phiEco32 (data not shown).
To prepare phiEco32-infected biomass, wild-type E. coli 55 or E. coli 55 rpoC∷4PrA strains were grown at 37 °C in 4 l of LB until an A600 nm of 0.5 and were infected with phiEco32 at a multiplicity of infection of 10. Infection was halted 23 minutes post-infection by rapidly cooling the samples in icy water baths. Cells were harvested by centrifugation and washed once with ice-cold 10 % (v/v) glycerol. Finally, 1 ml of 20 mM Hepes (pH 7.4 at 4 °C) supplemented with 0.2 mg/ml PMSF, 4 µg/ml pepstatin was added per 10 g of cell paste, and cells were frozen in liquid nitrogen. Cells were lysed by cryogenic grinding using the Retsch MM 301 Mixer Mill (Retsch). Eight 2-min bursts of grinding (30 Hz) with 20 mm tungsten carbide grinding balls were performed in 25 ml jars. The jars were cooled in liquid nitrogen between each step.
Lysed cells (1 g) were suspended in 5 ml of extraction buffer (20 mM Hepes (pH 7.4), 2 mM MgCl2, 150 mM NaCl, 0.1 % (v/v) Tween 20, 0.2 mg/ml PMSF, 4 µg/ml pepstatin) supplemented with one protease inhibitor cocktail tablet (Roche Diagnostics). The lysate was treated with 300 Kunitz units of DNase I (Sigma-Aldrich) and 300 Kunitz units of RNase A (Sigma-Aldrich) at room temperature for 15 minutes with gentle agitation. Two successive 10-min centrifugation steps at 27,200 g were used to isolate the soluble fraction. The soluble fraction was incubated with 20 mg of Dynabeads (Invitrogen) cross-linked to rabbit IgG (MP Biomedicals) with gentle agitation for 5 minutes. Dynabeads were collected with a magnet and washed five times with wash buffer (20 mM Hepes (pH 7.4), 150 mM NaCl, 0.1 % (v/v) Tween 20). The β′-4PrA fusion protein and co-purifying proteins were eluted from the IgG-Dynabeads with 0.5 M NH4OH, 0.5 mM EDTA. The eluted proteins were frozen in liquid nitrogen and evaporated to dryness in a SpeedVac (Thermo Savant). Dried protein samples were dissolved in SDS-PAGE loading buffer and heated to 95 °C for 5 minutes. Iodoacetamide (25 mM) was added and the mixture incubated at room temperature to modify reduced cysteines. Samples were resolved by SDS-PAGE in 4–12 % (w/v) Bis-Tris polyacrylamide gels (Invitrogen). Proteins in the gel were visualized by Colloidal Coomassie staining with Gel Code Blue (Pierce).
Liquid chromatography and electrospray ionization mass spectrometry analysis of phiEco32 virion proteins
PhiEco32 virions were heated to 95°C for 2.5 minutes, treated with DNase I (0.3 Kunitz units; Sigma-Aldrich) for 15 minutes at room temperature, mixed with SDS-PAGE sample buffer, and heated to 95 °C for a further 2.5 minutes. The sample was treated with iodoacetamide (25 mM) at room temperature, resolved on a denaturing 4–12 % (w/v) Bis-Tris polyacrylamide gel (Invitrogen), and stained with Gel Code Blue (Pierce). Stained bands were excised, destained, and digested as described above. The resulting peptides were loaded onto a microcolumn packed with Poros 20R2 reverse phase resin (PerSeptive Biosystems), washed, and eluted in two steps using (i) 5 pmol MFL tripeptide in 25% (v/v) acetonitrile, 0.1% trifluoroacetic acid and (ii) 5 pmol MFL tripeptide in 90 % (v/v) acetonitrile, 0.1 % (v/v) trifluoroacetic acid. The eluates were pooled and evaporated to dryness in a SpeedVac (Thermo Savant). The resulting tryptic peptide samples were resuspended in water/methanol/acetic acid at 44:5:1 (v/v/v) and loaded onto a Magic MS C18 column (Michrom Bioresources). Peptides were separated using a methanol gradient from 10–100% in 9 minutes on a Smart System HPLC (GE Healthcare). The chromatographic eluate was directly infused at a rate of 2.1 µl/minute into an LCQ-Deca electrospray-ion trap mass spectrometer (Thermo Electron) through a 25 µm tapered-tip fused silica capillary. Ions were formed by an applied potential of +3.6kV and desolvation was assisted by maintaining the heated capillary at 135 °C and the use of a declustering potential of 40V across the tube lens. MS peaks above a signal threshold of 105 in MS mode were subsequently fragmented using the following parameters: three micro scans, automatic gain control set to 500ms or a maximal number of counts of 5×109, isolation window of 4 m/z units and relative collision energy of 35% (Thermo Electron nomenclature). MS/MS information was collected into separate files which were converted to DTA format using extract_msn.exe from Thermo Electron prior to database search.
Preparation of samples and MALDI MS mass spectrometry
Gel bands due to gp36 and gp79 were excised, destained, and digested with 25 ng/µl sequencing grade modified trypsin (Promega). Peptides were extracted on reverse phase resin (Poros 20 R2; PerSeptive Biosystems); eluted with 50 % (v/v) methanol, 20 % (v/v) acetonitrile, 0.1 % (v/v) trifluoroacetic acid and subjected to MALDI-MS analysis using an in-house constructed MALDI interface coupled to a Qq-TOF mass analyzer (Centaur, Sciex) and MALDI-MS/MS analysis using an in-house modified ion trap (LCQ DECA XP Plus, Thermo Electron).39,40 Proteins were identified using the algorithms “Profound”41 and “SonarMSMS”.42
Mass spectrometry analysis of phiEco32 virion proteins
The TCA-precipitated protein pellet from phage virion preparation was denatured, reduced, alkylated, and digested with endoproteinase LysC and trypsin (both from Roche Applied Science, Indianapolis, IN) as described.43 The peptide mixture was desalted offline on SPEC-PLUS PTC18 cartridges (Varian, Palo Alto, CA) and pressure-loaded onto a 100 µm fused silica column packed with 9 cm AQUA C18 reverse phase (Phenomenex, Torrance, CA) and 3 cm of strong cation exchange material (Whatman, Brentford, UK).44 The loaded and washed microcapillary column was installed in-line with a Quaternary 1100 series HPLC pump (Agilent Technologies, Santa Clara, CA), coupled to Deca-XP ion trap tandem mass spectrometer (ThermoElectron, San Jose, CA) and analyzed via a ten-step chromatography as described.43
The MS/MS datasets were searched using SEQUEST45 against a database combining 133 phiEco32 proteins, 29823 sequences from six E. coli genomes (CFT073, K12, O157H7_EDL933, O157H7, UTI89, W3110) downloaded from NCBI in July 2006, 177 sequences for usual protein contaminants, as well as randomized sequences for each of the non-redundant entries (i.e. the final database size was 60266 amino acid sequences). DTASelect/CONTRAST46 was used to select spectra/peptide matches with normalized difference in cross-correlation score (DeltCn) of at least 0.08, a minimum cross-correlation score (XCorr) of 1.8 for singly charged, 2.5 for doubly charged, and 3.5 for triply charged spectra, a maximum Sp rank of 10. In addition, peptides had to be fully tryptic and at least seven amino acids long. No peptide matching shuffled protein sequences passed this criteria set. Spectral counts were normalized against protein length47 and NSAF values were used to estimate relative protein levels in the samples.
Cloning, overexpression, and purification of phiEco32 gp36 and gp79
The DNA encoding phiEco32 gp36 was cloned in two steps. Firstly, the DNA encoding phiEco32 gp36 was PCR amplified using primers that appended NdeI and HindIII sites at the 5′ and 3′ ends of gene 36, respectively. The resultant PCR product was cleaved with NdeI and HindIII and a DNA fragment encoding gene 36 nucleotides 241–648 cloned between the NdeI and HindIII sites of a pET28a-based plasmid, creating pSKB2phiEco32gp36#1. pSKB2phiEco32gp36#1 was next cleaved with NdeI and a PCR product encoding gene 36 nucleotides 1–240, cleaved with NdeI, was cloned between the NdeI site of pSKB2phiEco32gp36#1, creating pSKB2phiEco32gp36. The DNA encoding phiEco32 gp79 was amplified using primers that appended NdeI and BamHI sites at the 5′ and 3′ ends of gene 79, respectively. The PCR product was cleaved with NdeI and BamHI and cloned between the NdeI and BamHI sites of a pET28a-based plasmid, creating pSKB2phiEco32gp79. All DNA manipulations were confirmed to be correct by sequencing.
To express gp36, E. coli BL21(DE3) cells ([F− ompT hsdSB(rB−mB−) gal dcm (DE3)]) carrying pSKB2phiEco32gp36 and pG-KJE8 (dnaK-dnaJ-grgE, groES-groEL (Takara BIO Inc., Japan)) plasmids were grown at 30°C in 300 ml LB with 50 µg/ml of kanamycin and 40 µg/ml of chloramphenicol. To induce chaperone synthesis L-arabinose (4 mg/ml) and tetracycline (10 ng/ml) were added to the medium. When the culture reached mid-log phase, expression of recombinant protein was induced by the addition of 0.1 mM IPTG and growth was continued for additional 2 h. Cells were harvested by centrifugation and resuspended in 9 ml of buffer A (20 mM phosphate buffer, pH 7.4, 50 mM NaCl) with 1 mg/ml lysozyme. After a 1-hour incubation on ice, cells were disrupted by sonication. Cleared cell lysate was applied on a 1 ml heparin-agarose column connected to a 1 ml chelating HiTrap column (both from GE Healthcare) charged with Ni2+. Material bound to the HiTrap column was washed with buffer B (20 mM phosphate buffer, pH 7.4, 0.5 M NaCl, 50 mM imidazole) and bound proteins were eluted with buffer A containing 250 mM imidazole. Fractions containing gp36 were concentrated on Centricon® Centrifugal Filter Units (Millipore), glycerol was added to a final concentration 50% and protein was stored at −20 °C.
Gp79 expression was carried out in E. coli BL21(DE3) cells carrying pSKB2phiEco32gp79 and pG-KJE8 plasmids. Cell growth conditions and protein purification protocol were the same as for gp36 but without preliminary purification on a heparin-agarose column.
In vitro transcription
His-tagged E. coli RNAP core and untagged recombinant σ70 subunit were prepared as described.48
Abortive initiation transcription reactions were performed in 10 µl of transcription buffer (40 mM Tris-HCl, 40 mM KCl, 10 mM MgCl2, 5 mM DTT, 100 µg/ml BSA) and contained 150 nM E. coli RNAP core enzyme, 300 nM recombinant σ70 and 600 nM of recombinant phiEco32 gp36 or gp79. Reactions were incubated for 15 min at 37 °C, followed by the addition of 20 nM of a PCR fragment containing the T7 A1 promoter, 100 µM initiating dinucleotide CpA, 10 µM of α-[32P]UTP (400 Ci/mmol). Reactions proceeded for 10 min at 37 °C and were terminated by the addition of an equal volume of denaturing loading buffer. The reaction products were resolved on a denaturing 6 M urea 20% (w/v) polyacrylamide gels and visualized using a PhosphorImager.
Protein complex analysis
12 pmol of E. coli RNAP core was incubated with 100 pmol of gp36 or 60 pmol of gp79 in 40 µl of transcription buffer for 10 min at 37 °C. Reaction products were separated by electrophoresis in a 5% native polyacrylamide gel (29:1) at 100 V for 1 h. The gel was stained with Coomassie G-250. The peptide composition of native-gel bands was established by SDS PAGE
Acknowledgements
This work was supported by NIH R01 grant GM59295, a new groups grant from the Presidium of Russian Academy of Sciences and a Russian Foundation for Basic Research grant (to KS), NIH R01 GM32095 grant (to IJM) and NIH grants RR00862 and RR022220 (to BTC). LFW was supported by an NIH grant GM61898 to Seth Darst.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Hendrix RW, Smith MC, Burns RN, Ford ME, Hatfull GF. Evolutionary relationships among diverse bacteriophages and prophages: all the world's a phage. Proc. Natl. Acad. Sci. USA. 1999;96:2192–2197. doi: 10.1073/pnas.96.5.2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wommack KE, Colwell RR. Virioplankton: viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 2000;64:69–114. doi: 10.1128/mmbr.64.1.69-114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hendrix RW. Bacteriophage genomics. Curr. Opin. Microbiol. 2003;6:506–511. doi: 10.1016/j.mib.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 4.Liu J, Glazko G, Mushegian A. Protein repertoire of double-stranded DNA bacteriophages. Virus Res. 2006;117:68–80. doi: 10.1016/j.virusres.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 5.Pedulla ML, Ford ME, Houtz JM, Karthikeyan T, Wadsworth C, Lewis JA, Jacobs-Sera D, Falbo J, Gross J, Pannunzio NR, Brucker W, Kumar V, Kandasamy J, Keenan L, Bardarov S, Kriakov J, Lawrence JG, Jacobs WR, Jr, Hendrix RW, Hatfull GF. Origins of highly mosaic mycobacteriophage genomes. Cell. 2003;113:171–182. doi: 10.1016/s0092-8674(03)00233-2. [DOI] [PubMed] [Google Scholar]
- 6.Naryshkina T, Liu J, Florens L, Swanson SK, Pavlov AR, Pavlova NV, Inman R, Minakhin L, Kozyavkin SA, Washburn M, Mushegian A, Severinov K. Thermus thermophilus bacteriophage phiYS40 genome and proteomic characterization of virions. J. Mol. Biol. 2006;364:667–677. doi: 10.1016/j.jmb.2006.08.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Semenova E, Djordjevic M, Shraiman B, Severinov K. The tale of two RNA polymerases: transcription profiling and gene expression strategy of bacteriophage Xp10. Mol. Microbiol. 2005;55:764–777. doi: 10.1111/j.1365-2958.2004.04442.x. [DOI] [PubMed] [Google Scholar]
- 8.Nechaev S, Severinov K. Bacteriophage-induced modifications of host RNA polymerase. Annu. Rev. Microbiol. 2003;57:301–322. doi: 10.1146/annurev.micro.57.030502.090942. [DOI] [PubMed] [Google Scholar]
- 9.Ackermann H-W, Eisenstark A. The present state of phage taxonomy. Intervirology. 1974;3:201–219. doi: 10.1159/000149758. [DOI] [PubMed] [Google Scholar]
- 10.Casjens SR, Gilcrease EB, Winn-Stapley DA, Schicklmaier P, Schmieger H, Pedulla ML, Ford ME, Houtz JM, Hatfull GF, Hendrix RW. The generalized transducing Salmonella bacteriophage ES18: complete genome sequence and DNA packaging strategy. J. Bacteriol. 2005;187:1091–1104. doi: 10.1128/JB.187.3.1091-1104.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tan Y, Zhang K, Rao X, Jin X, Huang J, Zhu J, Chen Z, Hu X, Shen X, Wang L, Hu F. Whole genome sequencing of a novel temperate bacteriophage of P. aeruginosa: evidence of tRNA gene mediating integration of the phage genome into the host bacterial chromosome. Cell. Microbiol. 2006;9:479–491. doi: 10.1111/j.1462-5822.2006.00804.x. [DOI] [PubMed] [Google Scholar]
- 12.Sacerdot C, Fayat G, Dessen P, Springer M, Plumbridge JA, Grunberg-Manago M, Blanquet S. Sequence of a 1.26-kb DNA fragment containing the structural gene for Escherichia coli initiation factor-IF3 – presence of an AUU initiator codon. EMBO J. 1982;1:311–331. doi: 10.1002/j.1460-2075.1982.tb01166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Binns N, Masters M. Expression of the Escherichia coli pcnB gene is translationally limited using an inefficient start codon: a second chromosomal example of translation initiated at AUU. Mol. Microbiol. 2002;44:1287–1298. doi: 10.1046/j.1365-2958.2002.02945.x. [DOI] [PubMed] [Google Scholar]
- 14.Nikolaou C, Almirantis Y. A study on the correlation of nucleotide skews and the positioning of the origin of replication: different modes of replication in bacterial species. Nucleic Acids Res. 2005;33:6816–6822. doi: 10.1093/nar/gki988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller ES, Kutter E, Mosig G, Arisaka F, Kunisawa T, Rüger W. The bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 2003;67:86–156. doi: 10.1128/MMBR.67.1.86-156.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen B, King J. Thermal unfolding pathway for the thermostable P22 tailspike endorhamnosidase. Biochemistry. 1991;30:6260–6269. doi: 10.1021/bi00239a026. [DOI] [PubMed] [Google Scholar]
- 17.Xu J, Hendrix RW, Duda RL. Conserved translational frameshift in dsDNA bacteriophage tail assembly genes. Mol. Cell. 2004;16:11–21. doi: 10.1016/j.molcel.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 18.Molineux IJ. The T7 group. In: Calendar R, editor. The Bacteriophages. Oxford University Press; 2005. pp. 277–301. [Google Scholar]
- 19.Fraser JS, Yu Z, Maxwell KL, Davidson AR. Ig-like domains on bacteriophages: a tale of promiscuity and deceit. J. Mol. Biol. 2006;359:496–507. doi: 10.1016/j.jmb.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 20.Helmann JD. The extracytoplasmic function (ECF) sigma factors. Adv. Microb. Physiol. 2002;46:47–110. doi: 10.1016/s0065-2911(02)46002-x. [DOI] [PubMed] [Google Scholar]
- 21.Severinova E, Severinov K, Fenyö D, Chait BT, Brody ET, Darst SA. Domain organization of the σ70 subunit of Escherichia coli RNA polymerase. J. Mol. Biol. 1996;263:637–647. doi: 10.1006/jmbi.1996.0604. [DOI] [PubMed] [Google Scholar]
- 22.Ackermann H-W. Frequency of morphological phage descriptions in the year 2000. Arch. Virol. 2001;146:843–857. doi: 10.1007/s007050170120. [DOI] [PubMed] [Google Scholar]
- 23.Ruska H. Morphologische Befunde bei der bakteriophagen Lvse. Arch. Gesamte Viruforsch. 1942;2:345–387. [Google Scholar]
- 24.Kottmann R. Morphologische Befunde aus taches vierges von Colikulturen. Arch. Gesamte Virusforsch. 1942;2:388–396. [Google Scholar]
- 25.Moazamie N, Ackermann H-W, Murthy MRV. Characterization of two Salmonella newport bacteriophages. Can. J. Microbiol. 1979;25:1063–1072. doi: 10.1139/m79-163. [DOI] [PubMed] [Google Scholar]
- 26.Ackermann H-W, Nguyen T-M, Delâge R. Un nouveau phage d'entérobactéries à tête allongée et queue courte. Ann Virol (Inst. Pasteur) 1981;132E:229–234. [Google Scholar]
- 27.Chopin A, Deveau H, Ehrlich SD, Moineau S, Chopin M-C. KSY1, a lactococcal phage with a T7-like transcription. Virology. 2007;365:1–9. doi: 10.1016/j.virol.2007.03.044. [DOI] [PubMed] [Google Scholar]
- 28.DePaola A, Motes ML, Chan AM, Suttle CA. Phages infecting Vibrio vulnificus are abundant and diverse in oysters (Crassostrea virginica) collected from the Gulf of Mexico. Appl. Environ. Microbiol. 1998;64:346–351. doi: 10.1128/aem.64.1.346-351.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grimont F, Grimont PAD. DNA relatedness among bacteriophages of the morphological group C3. Curr. Microbiol. 1981;6:65–69. [Google Scholar]
- 30.Grimont F, Grimont PAD. Characteristics of five bacteriophages of yellow-pigmented enterobacteria. Curr. Microbiol. 1981;5:61–66. [Google Scholar]
- 31.Khan AS, Khan AA, Nawaz MS, DePaola A, Andrews A, Cerniglia CE. DNA packaging and developmental intermediates of a broad host range Vibrio vulnificus bacteriophage 71A-6. Mol. Cell. Probes. 2001;15:61–69. doi: 10.1006/mcpr.2000.0344. [DOI] [PubMed] [Google Scholar]
- 32.Minakhin L, Semenova E, Liu J, Vasilov A, Severinova E, Gabisonia T, Inman R, Mushegian A, Severinov K. Genome and gene expression of Bacillus anthracis bacteriophage Fah. J. Mol. Biol. 2005;354:1–15. doi: 10.1016/j.jmb.2005.09.052. [DOI] [PubMed] [Google Scholar]
- 33.Besemer J, Borodovsky M. Heuristic approach to deriving models for gene finding. Nucleic Acid. Res. 1999;27:3911–3920. doi: 10.1093/nar/27.19.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27:4636–4641. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rice P, Longden I, Bleasby A. EMBOSS: The European Molecular Biology Open Software Suite. Trend. Genetics. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 37.Tackett AJ, Dilworth DJ, Davey MJ, O'Donnell M, Aitchison JD, Rout MP, Chait BT. Proteomic and genomic characterization of chromatin complexes at a boundary. J. Cell Biol. 2005;169:35–47. doi: 10.1083/jcb.200502104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herring CD, Glasner JD, Blattner FR. Gene replacement without selection: regulated suppression of amber mutations in Escherichia coli. Gene. 2003;311:153–163. doi: 10.1016/s0378-1119(03)00585-7. [DOI] [PubMed] [Google Scholar]
- 39.Krutchinsky AN, Zhang W, Chait BT. Rapidly switchable matrix-assisted laser desorption/ionization and electrospray quadrupole-time-of-flight mass spectrometry for protein identification. J. Am. Soc. Mass Spectrom. 2000;11:493–504. doi: 10.1016/S1044-0305(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 40.Krutchinsky AN, Kalkum M, Chait BT. Automatic identification of proteins with a MALDI-quadrupole ion trap mass spectrometer. Anal. Chem. 2001;73:5066–5077. doi: 10.1021/ac010682o. [DOI] [PubMed] [Google Scholar]
- 41.Zhang W, Chait BT. ProFound: an expert system for protein identification using mass spectrometric peptide mapping information. Anal. Chem. 2000;72:2482–2489. doi: 10.1021/ac991363o. [DOI] [PubMed] [Google Scholar]
- 42.Field HI, Fenyö D, Beavis RC. RADARS, a bioinformatics solution that automates proteome mass spectral analysis, optimises protein identification, and archives data in a relational database. Proteomics. 2002;2:36–47. [PubMed] [Google Scholar]
- 43.Florens L, Washburn MP. Proteomic analysis by multidimensional protein identification technology. Methods Mol. Biol. 2006;328:159–175. doi: 10.1385/1-59745-026-X:159. [DOI] [PubMed] [Google Scholar]
- 44.Washburn MP, Wolters D, Yates JR., III Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 45.Eng J, McCormack AL, Yates JR., III An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Amer. Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 46.Tabb DL, McDonald WH, Yates JR., III DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 2002;1:21–26. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zybailov B, Mosley AL, Sardiu ME, Coleman MK, Florens L, Washburn MP. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J. Proteome Res. 2006;5:2339–2347. doi: 10.1021/pr060161n. [DOI] [PubMed] [Google Scholar]
- 48.Kashlev M, Nudler E, Severinov K, Borukhov S, Komissarova N, Goldfarb A. Histidine-tagged RNA polymerase of Escherichia coli and transcription in solid phase. Methods Enzymol. 1996;274:326–334. doi: 10.1016/s0076-6879(96)74028-4. [DOI] [PubMed] [Google Scholar]