

Abstract
It is now apparent that naïve peripheral T cells are a dynamic population where active processes prevent inappropriate activation while supporting survival. The process of thymic education makes naïve peripheral T cells dependent on interactions with self-MHC for survival. However, as these signals can potentially result in inappropriate activation, various non-redundant, intrinsic negative regulatory molecules including Tob, Nfatc2, and Smad3 actively enforce T cell quiescence. Interactions among these pathways are only now coming to light and may include positive or negative crosstalk. In the case of positive crosstalk, self-MHC initiated signals and intrinsic negative regulatory factors may cooperate to dampen T cell activation and sustain peripheral tolerance in a binary fashion (on-off). In the case of negative crosstalk, self-MHC signals may promote survival through partial activation while intrinsic negative regulatory factors act as rheostats to restrain cell cycle entry and prevent T cells from crossing a threshold that would break tolerance.
Keywords: T cells, MHC, sensitization, desensitization, cell cycle, negative regulation, tolerance
The Influence of Self-MHC in T-Cell Quiescence and Survival
Self-MHC and naïve T cell survival
The role of self-peptides complexed to major histocompatibility complex proteins (self-MHC) to maintain homeostasis of naïve T cells has been the subject of extensive investigation over the past decade. This work has revealed apparent differences in the sensitivity of different T cell subsets to the absence of self-MHC. Perhaps the most informative experiments have attempted to characterize the expansion of peripheral lymphocytes in MHC-replete or MHC-deficient lymphopenic environments. The idea that “space” in the peripheral lymphoid organs drove lymphocyte proliferation arose from the observation that under conditions of lymphopenia such as those produced by non-myeloablative, lymphodepleting regimens in humans or mice, or under conditions encountered by adoptively transferred lymphocytes or bone marrow cells into lymphopenic hosts, T cells would expand without exogenous antigens. It is now apparent that this “lymphopenia-induced proliferation” or “homeostatic proliferation” (HP) results not only from “space”, but also from interactions of T cell receptors (TCR) with self-MHC, and from lessened competition for cytokines including interleukin-7 (IL-7), or in some cases, IL-2 and IL-15 (1–4).
Lessons from HP
The initial characterization of HP suggested that this process led T cells to acquire memory-like phenotypes (5). T cells generated by HP are not necessarily equivalent to memory T cells generated via responses to foreign antigen, as the frequency of effector cells appears to be lower with less robust molecular signaling profiles in T cells after HP than after response to foreign antigen (6–8). Nevertheless, HP and exposure to foreign antigen can generate functionally equivalent, protective CD8 T cell responses in the presence of CD4 T cell help (9), and many experiments evaluating the functional outcome of HP used unseparated T cell populations, so interpretations could be confounded by the fact that HP favors survival of memory T cells. Specifically, clonal competition promotes expansion of memory T cells at the expense of naïve T cells (10, 11); cytokine responsiveness similarly tilts the composition of the reconstituted population towards cells with memory phenotypes (12); and regulatory (CD4/CD25/FoxP3+) T cells (Treg) also can contribute to the balance of naïve and memory cells that repopulate a lymphopenic environment (13). More recent experiments with purified naïve T cells have allowed for the emergence of a model that defines the significance of self-MHC in naïve T cell homeostasis.
The environment that gives rise to HP is an essential determinant of the ultimate phenotype of surviving T cells. Specifically, the tempo of IL-7 mediated HP is slow (14), HP driven by constitutive levels of IL-15 is more rapid, generating memory phenotype cells (4), and HP driven by elevated levels of IL-2/IL-15 is rapid with differentiation into both effector and memory phenotype cells (2). The slow paced (IL-7-dependent) HP seems to be restrained by signals delivered through CD24 expressed in bone marrow derived dendritic cells (DC) (15), but differentiation from naïve T cells to memory phenotype T cells following rapid IL-2 and IL-15-dependent HP is more comparable to events driven by encounter with foreign antigen. In both cases, HP likely induces attenuation of intrinsic negative regulatory pathways that maintain T cells in a non-proliferative state.
Unlike HP in MHC-replete hosts, a majority of naïve T cells die or make at most a few divisions if the lymphopenic periphery is devoid of MHC (16–21), a result consistent with the premise that HP is itself partly driven by recognition of self-MHC complexes. Both CD4 and CD8 T cells undergo HP, although CD4 T cells expand less in response to MHC and CD8 T cells are more sensitive to its absence. In fact, while the requirement for self-MHC in naïve CD8 T cell survival is generally accepted (although it may not apply to all CD8 T cell subsets) (22), the delayed erosion of naïve CD4 T cells in lymphopenic hosts has led to some controversy regarding the significance of self-MHC signals in the survival of these cells.
Self-MHC and CD8 T cell homeostasis
The requirement for Class I MHC in CD8 T cell survival was first inferred by the inability of CD8 thymic emigrants to seed MHC-deficient peripheral organs (16), with the differential capacity of naïve and memory CD8 T cells to survive in an environment devoid of Class I MHC subsequently reported in a system using LCMV as a source of antigen to generate immunological memory (17). The requirement for MHC in survival of naïve CD8 T cells has since been confirmed in both transgenic and polyclonal T cells (see for example (23, 24)). An intriguing observation is that CD8 T cells do not die immediately after transfer to MHC-deficient environments. Rather, they survive and can respond to accessory cell signals for at least one week (23), and their eventual death is dependent on expression of the death factor protein Fas and its interaction with Fas ligand (FasL) (24). Rocha et al showed that survival of naïve CD8 cells was not only dependent on MHC expression, but also required the right MHC-restricting element (25). Moreover, while the concept that MHC expression is dispensable for survival of memory CD8 T cells is generally accepted, their experiments also showed that memory cells can only expand when antigen is presented in the context of the right MHC-restricting element, and at least HY-transgenic memory T cells underwent rapid erosion in an MHC-deficient environment (25). This suggests interactions with self-MHC by different memory T cell clones could result in different outcomes regarding HP and survival.
Self-MHC and CD4 T cell homeostasis
Although inevitably most CD4 T cells will die in an MHC-deficient environment, their half-life is estimated at three to four weeks (20, 26). One explanation for this invokes the possibility that the survival requirement for self-MHC is an acquired trait in naïve peripheral CD4 T cells. Specifically, Lantz and colleagues (26) proposed a model that separates naïve peripheral CD4 T cells into 3 stages based specifically on their requirement for self-MHC. Figure 1 illustrates this model, where we have added hypothetical fates for each stage of CD4 T cells after transfer to an MHC-deficient environment. Recent thymic emigrants represent a population of “Stage 1 CD4 T cells” that do not divide and have a half-life of about one week, regardless of the presence or absence of MHC. These recent thymic emigrants eventually mature into “stage 2 CD4 T cells” that can divide a few times by HP, suggesting maturation from stage 1 to stage 2 is preprogrammed and not driven by recognition of self antigen. “Stage 3 CD4 T cells” with a CD44hi phenotype appear about 2 weeks after HP in lymphopenic hosts and require MHC for extensive proliferation (50-fold more in MHC-positive than in MHC-deficient hosts). As a consequence of their proliferative capacity, stage 3, naïve CD4 T cells constitute the majority population (>80%) found after HP in lymphopenic, MHC-replete mice (26).
Figure 1. Maturation of CD4 thymic emigrants in the periphery.

CD4+ recent thymic emigrants can be subdivided into three stages of post-thymic maturation: Stage-1 cells have a half-life of about one week and do not divide. Maturation from stage-1 to stage-2 appears to be preprogrammed and independent from self MHC, leading stage-2 cells to undergo a few divisions by HP. Conversely, stage-3 cells require MHC for extensive proliferation. Under conditions of HP, CD44hi stage-3 cells appear within ~2 weeks and account for most of the cells seen in MHC-replete mice, but cells that can evade tuning or negative regulation survive in MHC-depleted hosts.
Self-MHC in desensitization vs. sensitization of naïve T cells
There is an intrinsic paradox to the model described above, since activation of potentially autoreactive T cells with sufficient affinity for self must be restrained while at the same time survival of T cells with lower affinity for self must be supported. Two explanations for the role of self-MHC in naïve peripheral T cell homeostasis have thus been advanced. Grossman and Paul (27) initially proposed that T cells with measurable reactivity for self-antigens were desensitized or ‘tuned’ by continued interactions in the periphery with MHC and self-peptides against which they were selected in the thymus. This may in part reflect a higher level of the self antigen in the periphery than in the thymus. Several reports indicate that there is a sufficiently high enough threshold for self-antigens in the periphery that autoreactivity can result. MHC dependent peripheral tuning of these T cells prevents this (7, 28, 29). In an alternative explanation proposed by Stefanova and Germain (30) self-MHC interactions in the periphery would keep T cells in a state of partial activation or ‘sensitization’ with survival as the outcome, allowing T cells to respond to foreign antigen. Stefanova and Germain base their hypothesis on data showing that interactions between TCR and self-MHC promote modest receptor clustering and CD3ζ chain phosphorylation (20, 30), and in the case of CD4 T cells, activation of Rap1 and Rac1 enhancing motility (31).
The observation that T cells undergoing HP in MHC-replete hosts can overcome tolerance could be used to argue in favor of sensitization as the dominant model. Specifically, lymphoid reconstitution by HP can lead to autoimmune phenotypes or T-cell mediated tumor rejection in several experimental models (32–36). However, release of autoreactive cells by HP may be the exception rather than the rule as autoimmunity after HP is generally restricted to strains with a propensity for immune mediated diseases such as the NOD mouse. In the case of tumor rejection, Bracci et al (36) showed that adoptive transfer of tumor-immune spleen cells to mice that were lymphodepleted by cyclophosphamide conditioning led to permanent tumor regression in 100% of mice, but naïve spleen cells were ineffective, suggesting the effect is dependent on T cells with a memory phenotype. We obtained comparable data in two independent models. First, we showed that adoptive transfer of potentially autoreactive T cells from pre-diabetic NOD donors to lymphopenic MHC-replete hosts (NOD-SCID) led to diabetes in 45 days, but adoptive transfer of wild type T cells from C57Bl/6 mice to lymphopenic MHC-replete hosts (B6-SCID) did not create a diabetic phenotype (Jubala, unpublished). Similarly, adoptive transfer of wild type T cells from C57Bl/6 mice to lymphopenic MHC-replete hosts (B6-SCID) delayed, but did not prevent growth of heterotopic syngeneic Lewis Lung carcinoma1.
Recent experiments from Reddy’s group used a conventional model of graft vs. host disease (GVHD) to test the hypothesis that HP released cells from tolerance. In their system, T cells were allowed to undergo HP for 2 weeks in lethally irradiated syngeneic hosts (B6) or in MHC-replete hosts (B6-SCID) prior to adoptive transfer into allogeneic Balb/c recipients. Intriguingly, allogeneic recipients that received HP T cells showed significantly better survival and less severe clinical GVHD than those that received allogeneic naïve T cells (37). The dampening of GVHD was due to the expanded HP cells and not to residual or newly generated Treg cells, arguing in favor of the desensitization or tuning model, although it is possible that this suppressive effect might be due to unique properties of alloreactivity. The conclusion that naïve, but not memory T cells were responsible for GVHD preceded these experiments (38–40).
The tuning and sensitization models are not mutually exclusive in MHC-replete hosts. Quantum signaling in response to varying avidity of TCR for self-peptides in polyclonal populations might direct tuning of naïve CD4 T cells under high affinity conditions and sensitization under low affinity conditions, respectively, restraining incipient, self-reactive cells and priming naïve cells to react against non-self antigens. Still, tuning and sensitization could be incompatible in the absence of MHC. If the dominant role of MHC is to tune naïve peripheral T cells, the predicted result of adoptive transfer experiments into MHC-deficient hosts would be self-reactivity (autoimmunity), while if the dominant role of MHC is to sensitize low affinity T cells, the predicted result of adoptive transfer experiments into MHC-deficient hosts would be T cell anergy or T cell death. Unfortunately, as mentioned above, both autoreactivity and T cell death have been reported in MHC depleted hosts such that distinguishing between these models may not be possible using this approach.
However, data from such MHC-deficient models provide more support for the tuning hypothesis. Specifically, Bhandoola and colleagues (7) reported rejection of MHC-positive skin grafts in approximately one third of MHC Class II-deficient animals that received naïve CD4 T cells with lymphocytic infiltrates detectable in the skin of all the animals, suggesting both that autoreactive cells could persist in the MHC-deficient environment and that they could then be activated upon encounter with self-MHC. We reached similar conclusions using mice that were doubly deficient for MHC Class I and MHC Class II. Our initial experiments were designed to exploit the predilection for survival of memory cells in the absence of self-MHC. As this model allows enrichment of antigen experienced memory T cells in vivo (17), we reasoned that ‘parking’ T cells from diabetic NOD mice in an MHC-deficient environment would render naïve (and presumably non-autoreactive) T cells unable to survive, leaving behind only the less frequent, antigen-experienced (and presumably autoreactive) T cells. This presented an unparalleled opportunity to study unique properties of diabetogenic T cells. While the experiments in diabetes-prone NOD mice bore out this prediction, we obtained similar results using diabetes-resistant B6 mice. This was somewhat unexpected, and it suggested that the absence of self-MHC led to the generation and/or survival of autoreactive T cells independent from the genetic background or the donor’s propensity for autoimmunity.
We expanded this observation to a tumor rejection model that allowed us to test survival and autoreactivity of T cells undergoing HP in an MHC-deficient environment. First, we verified that neither naïve T cells nor antigen experienced T cells (from mice challenged with Lewis Lung carcinoma) could mount effective anti-tumor responses after 2 weeks of HP in lymphopenic, MHC-replete hosts. In contrast, our experiments and those of Bracci (36) showed that adoptive transfer of tumor-immune T cells under these conditions led to tumor rejection. Next, we examined the behavior of these cells in MHC-deficient mice. Both naïve T cells and antigen experienced T cells showed delayed tumor growth or outright rejection of tumor challenge in MHC-deficient mice. When present, tumors were infiltrated by CD4 T cells, but persistence of both CD4 and CD8 T cell subsets in this environment required exposure to tumor in the donor. These observations could be explained by release of CD4 T cells from intrinsic negative regulation in the absence of MHC. In other words, the same mechanisms that restrain T cell proliferation might control “tuning”. In fact, Liu et al showed CD24 expressed in DCs controls proliferation and survival of CD4 and CD8 T cells transferred to lymphopenic hosts; in the absence of CD24, adoptively transferred T cells undergo unregulated and destructive HP leading to fulminant death (15).
Intrinsic Negative Regulators of T Cell Activation
Tuning might engage pathways that control the transition from G0 to G1 and G1 progression in T cells. It is now apparent that survival and quiescence are actively enforced in T cells (41–47). This paradigm has long been operative in other systems where survival requires signals from a substrate or matrix (avoidance of anoikis) (48, 49) and where quiescence is enforced by signals delivered when cells contact each other (50). Although the microenvironment that supports peripheral lymphocytes does not include a fixed matrix, these cells share many other characteristics with cells derived from the embryonic mesoderm (e.g., fibroblasts); hence, survival and quiescence must be enforced by alternative mechanisms.
The process of thymic education dictates that naïve peripheral T cells must interact with self-peptides presented by MHC (i.e., potential autoantigens) in order to survive, but the transition of positively selected (and thus potential autoreactive) thymocytes to the periphery presents an intriguing problem. Naïve, recent thymic emigrants express high levels of T cell receptors, so the avidity for self-MHC might trigger their activation and result in autoimmunity (Figure 1). To prevent such activation, quiescence is enforced by several interrelated negative regulatory pathways of lymphocyte, including molecules that dampen signaling such as Ian5 (51) and by negative transcriptional regulators such as the transducer of ErbB2-1 (Tob1) (52), nuclear factor of activated T cells-c2 (Nfatc2) (43), and Smad3, which is primarily responsible for integrating anti-proliferative and pro-survival signals delivered through the transforming growth factor-β (TGF-β) receptor (53–57).
Ian5 in negative regulation and survival
Ian5 (also known as GTPase of immunity-associated nucleotide binding protein 5 or Gimap5) is a member of a protein family consisting of GTP-binding proteins characterized by a common GTP-binding motif and coiled-coil motifs (58). One of the first lines of evidence that Ian5 was involved in supporting quiescence and survival came from the realization that the lyp mutation responsible for lymphopenia observed in the Biobreeding (BB) diabetes-prone (DP) rat resides in Ian5 (59). Several studies linked members of the IAN gene family with regulation of apoptosis; specifically, mouse Ian5 associates with anti-apoptotic proteins Bcl-2 and Bcl-xL (60) and T cells from Ian5-deficient rats show reduced mitochondrial integrity (61). Thymic development in the BB-DP rat is largely normal, but peripheral CD4+ T cells are reduced in number and peripheral CD8+ T cells are virtually absent. Moreover, the phenotype of surviving peripheral T cells indicates they are recent thymic emigrants (RTE), so death must occur rapidly after thymocytes enter the periphery (51, 62, 63). On the other hand, the Ian5 knockout mouse shows compromised survival of both peripheral T cells and double positive thymocytes (60).
An important observation linking negative regulation to survival of peripheral T cells was that T cells in the BB-DP rat showed upregulation of the activation proteins CD25 and OX40, increased spontaneous incorporation of BrdU, and reduced expression of the cell cycle inhibitor p27/Kip-1 (63, 64). T cells from Ian5-deficient rats spontaneously enter this state of partial or incomplete activation, and this is not simply a response to the lymphopenic environment, but rather it is due to elevated activity of MEK that in turn leads to constitutive activation of NFκB. Moreover, the phenotype is intrinsic to the T cells, as it occurs in bone marrow chimeras where Ian5 mutant T cells develop in a wild type environment (51). Thus, partial T cell activation in Ian5-deficient animals is not a consequence of lymphopenia but is rather part of the stimulus leading to cell death, providing one biochemical link between survival and quiescence pathways (42, 51).
Tob1 in negative regulation and survival
Tob1 is a member of the Btg/Tob family of transcriptional repressors (65). Regulation of Tob1 is multifaceted and includes shuttling between the nuclear and cytoplasmic compartments, phosphorylation, and ubiquitin-dependent degradation. These biochemical events are regulated respectively by constitutive nuclear import (mediated by a nuclear localization signal) and nuclear export in response to stimulatory signals (66), phosphorylation by Erk and Jnk mitogen activated protein kinases (67, 68), and ubiquitination by SCFSkp2 ubiquitin ligase (69).
In peripheral T cells, Tob1 overexpression enforces quiescence directly by supporting expression of the CDK inhibitor p27/Kip-1 and by silencing the IL-2 promoter, and indirectly by associating with and modulating the activity of SMAD transcription factors (52). Endogenous Tob1 is stabilized by partial antigen stimulation with altered peptide ligands, resulting in anergy (70). Although Tob1 deficiency does not produce gross immune defects, our analyses of T cell receptor signaling in T cells from Tob1-deficient mice show there is a demonstrably more facile progression into activation in Tob1-deficient T cells (Figure 2). Tob1-deficiency also lowers the thresholds for inflammation and tumor recognition in vivo. Specifically, when we examined the capacity of Tob1-deficient mice to reject or delay transplantable tumors as a measure of latent autoreactivity, these mice showed increased inflammatory tumor infiltrates (with consequent necrosis), reduced capillary density, and delayed growth of B16 melanoma as compared to wild type C57Bl/6 littermates (Figure 3).
Figure 2. T cells from Tob1-deficient mice are hyper-responsive.

(a) Competence induction in cells from WT, Tob1-deficient, or NFATc2-deficient mice was established by measuring the ability of cells to respond to limiting quantities of IL-2. Cells were treated without stimulation (U/S), with a limiting dose of anti-CD3 antibody (10 ng/ml, COMP), or with the same dose of antibody in the presence of a limiting dose of IL-2 (0.5 U/ml, STIM). Proliferation was measured using the CyQuant Cell proliferation assay kit. The Y-axis reflects fluorescence absorbance at 485 nm. Inset: steady state CDK4 levels were assessed by immunoblotting in unstimulated cells or in cells treated with anti-CD3 for 6 hr. β-actin was used as a loading control. (b) T cells from B6 (WT) or B6 Tob1 knockout mice (KO) were stimulated with increasing doses of anti-CD3. Cells were labeled at induction with CFSE and evaluated after 20 or 44 hours to track cell division. The Y-axis reflects the percent of cells with CFSE dilution measured using flow cytometry.
Figure 3. Tumor growth is delayed in Tob1−/− mice.
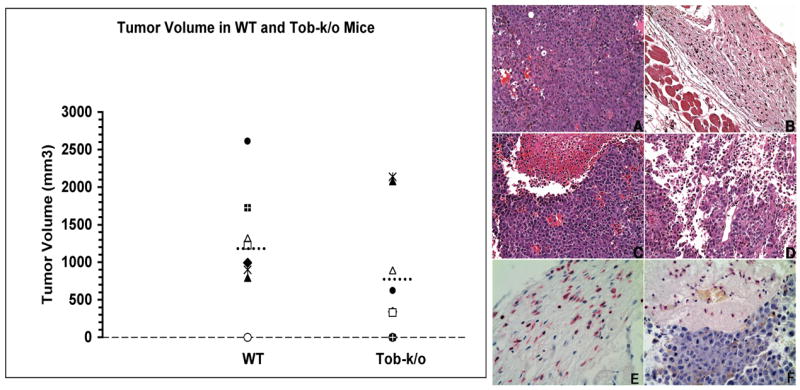
Left: Littermates (8 WT and 8 Tob1-k/o) from Tob1+/− heterozygous breedings were challenged sq with 2.5 × 104 B16 melanoma cells. Graph shows 3-D tumor volume for each pair 17 days after challenge. Dashed lines represent mean tumor volume. A trend was present for smaller tumors in Tob1-k/o. Right: Representative sq sections from wild type (A) or Tob1-deficient mice (B–F) 21 days after injection with B16 melanoma cells. There is a high mitotic rate, no inflammation, and visible vascularization of tumors in WT mice (A), in contrast to dermal and subcutaneous inflammatory infiltrates (B, C, D), hemorrhagic necrosis (C, D), severe edema and reduced vascularization (D) in Tob−/− mice. (H&E, 200X magnification). (E) and (F) show immunohistochemical staining for leukocytes using CD18 (red-staining cells). (E) is the same tumor as (B). (F) is the same tumor as (C). Note abundant leukocytes in the subcutis, extravasating neutrophils and inflammatory cells infiltrating the tumors (400X magnification). Gross measurable tumor volumes in the mice shown were (A) 1,320 mm3, (B) 0 mm3, (C) 891 mm3, and (D) 340 mm3.
Nfatc2 in negative regulation and survival
Nfatc2 is one of a family of proteins whose transcriptional activity is regulated by a cycle of dephosphorylation (by the calcium-dependent phosphatase calcineurin, or CN) and rephosphorylation (possibly by glycogen synthase kinase-3β, or GSK-3) (71). NFAT dephosphorylation leads to nuclear translocation and DNA binding, and rephosphorylation leads to rapid nuclear export. The role of Nfatc2 as a bona fide negative regulator was initially inferred by the observation that Nfatc2-deficient mice had marked splenomegaly and T cell hyper-responsiveness (72).
We identified a possible mechanism to explain the negative regulatory function of Nfatc2 (Figure 2), which functions as a strong repressor of CDK4 expression in isolated peripheral blood T cells and lymphocyte cell lines (43). The functional significance of this finding was confirmed in the Nfatc2-deficient mice that had enhanced CDK4 expression and activity restricted to the peripheral T cell pool, which shows dysregulated expansion in these animals (43, 44). An important finding from this work was that the association of Nfatc2 with AP-1 transcription factors was a critical determinant of its activity as a transcriptional activator or a transcriptional repressor.
This concept was subsequently confirmed in a series of elegant experiments by Rao’s group showing that low affinity TCR signals (perhaps akin to those seen by autoreactive cells) induce NFATc2 but fail to activate AP-1 or NFkB. This NFAT activation absent AP-1 is tolerogenic, and tolerance is mediated at least in part through enhanced expression of the Itch and Nedd components of the ubiquitination pathway (45, 46). Upregulation of these proteins leads to selective degradation of components that are essential for TCR signaling, such as phospholipase C-gamma (PLCγ) and PKCθ with concomitant induction of factors that suppress activation such as Cbl-b. While splenomegaly in the Nfatc2 knockout may result predominantly from failure to pull cells out of cell cycle during the transition from naïve to memory (43), Nfatc2 also might contribute to quiescence of naïve peripheral T cells and may skew their differentiation towards an inducible regulatory phenotype by supporting expression of Foxp3 (reviewed in (73)).
Smad3 in negative regulation and survival
SMAD proteins integrate signals from TGF-β into the system of intrinsic negative regulation (74). CDK inhibitors including Ink4 proteins and p27 are upregulated by TGF-β (75–77), and at least p27 also is strongly induced by Tob1 in lymphocytes (52). TGF-β signaling on naïve T cells leads to cell cycle arrest, apoptosis or prevents development of effector function in a context dependent manner (78–80). Recent findings have shown that it also is essential for the development of the T-helper-17 lineage (81). TGF-β has also been shown to be required for maintaining peripheral CD4/Foxp3 Treg cells, key mediators of peripheral tolerance (82).
The importance of Smad3 as a mediator of TGF-β-dependent T cell growth arrest cannot be overstated. T cells from Smad3-deficient mice have constitutively active phenotypes with consequent immune dysfunction (57) and they are resistant to downregulation of CDK4 upon exposure to TGF-β (83). CDK4 inhibition has been linked to the inability of T cells to respond to cytokine stimulus and enter cell cycle (84) and both Smad3 and retinoblastoma family proteins are directly inhibited by CDK4 phosphorylation (85). Thus, Smad3 inactivation might facilitate transition from the G0 to the G1 phase of the cell cycle, as evidenced by the observation that a mutant Smad3 that was resistant to CDK-phosphorylation rendered T cells resistant to antigenic priming and attenuation of Smad3 by RNA inhibition prevented tolerance induction (54).
TGF-β itself has a profound effect on naïve T cell homeostasis. Inhibition is mediated both upstream and downstream from CDK inhibitors, and while it modulates the activation and survival of both CD4 and CD8 T cell subsets, there are subtle to readily apparent differences in how it affects each of these populations (54, 79, 83). TGF-β1 deficient mice develop lethal autoimmunity early in life (86, 87). Breeding the TGF-β-deficiency into lymphocyte deficient backgrounds showed T cells mediate the lethality phenotype, as the lymphopenic mice did not develop autoimmune disease or inflammation (88, 89). Efforts to study the effects of TGF-β signaling on T cells have hence generated mouse models with a spectrum of phenotypes. Generally, TGF-β-mediated signals are dispensable for thymic development (90–93), whereas conditional deletion of TGF-β RII on T cells results in wasting disease and death by 3–5 weeks of age (91, 93).
Two recently developed models have made it possible to carefully study the effects of TGF-β signaling on T cells over a longer time span (92, 94). These models use a dominant negative form of the TGF-β RII (RII DN) under the control of CD2 or CD4 promoters. CD4 RII DN mice are healthy until 3–4 months of age, when they develop symptoms of autoimmune dysfunction including inflammatory bowel disease. Conversely, CD2 RII DN mice develop polyclonal CD8 T cell hyperproliferation between 2–9 months of age, but do not manifest inflammatory disease. CD8 T cells in the periphery are primarily CD44hi and have a greater percentage of cells in cycle as compared to wild type littermate controls (92). It should be noted, however, that TGF-β signaling via a TGF-β RII dominant negative transgene does not completely abrogate TGF-β signaling, which may explain why these animals do not have identical phenotypes to those seen in the TGF-β1 knockouts or in the conditional TGF-β deletion mutants.
TGF-β signals may act as counterbalance to IL-15 signals in the development of CD8 T cell memory (95). Specifically, CD8 T cells that do not receive TGF-β signals proliferate and develop a memory phenotype as measured by CD44 expression (92); TGF-β attenuates expression of the IL-2/IL15Rβ receptor (95); and it reduces IL-15 dependent expression of c-myc (96), which is necessary to support homeostasis of CD8 memory phenotype cells (97). Conversely, IL-15 can overcome the anti-proliferative effects of TGF-β on human peripheral blood mononuclear cells, even though these effects are resistant to IL-2 (98). Together, these results illustrate the existence of complex and finely orchestrated interactions between TGF-β-and IL-15-regulated pathways that control T cell survival, proliferation, and progression from naïve to memory phenotypes.
Despite its important cell-intrinsic effects, TGF-β also is important in extrinsic control of autoimmunity as a secreted cytokine and as a membrane-bound form on Treg cells. The extrinsic effects of TGF-β are diminished in the presence of CD28 co-stimulation (99, 100), perhaps allowing pathogen specific T cells to respond to contextually presented antigens, while preventing autoreactive cells from becoming activated.
This has presented a heretofore-insurmountable challenge in the development of strategies that employ cancer immunotherapy, where effector T cells are generally self-reactive and co-stimulation may be in short supply (101–103). Creative approaches to ablate these effects such as those used by Koehler et al. to show that CD28 co-stimulation can overcome TGF-β induced suppression of engineered, tumor specific CD8 and CD4 T cells (99) may be necessary to develop successful immunologic approaches to treat cancer as monotherapy, but these also will require strategies to ameliorate adverse autoimmunity that can be exacerbated in the absence of TGF-β signals (104).
Do Common Pathways Control Tuning by Self-MHC and Intrinsic Negative Regulation?
A connection between intrinsic negative regulation and survival is evident from the results described above regarding how Ian5-deficient T cells undergo apoptosis after they enter the peripheral circulation due to their progression to an incomplete state of activation. However, this phenotype is unique to the BB-DP rat, as deficiency in other intrinsic negative regulatory molecules alternatively leads to fulminant autoimmunity (TGF-β RII DN), expansion of the T cell compartment (Nfatc2 knockout) or no appreciable immune phenotype (Tob1 knockout). Tuning might engage molecules that control the transition from G0 to G1 and G1 progression, but there is a paucity of experiments to address the possible connection between these processes. We put this hypothesis to test using adoptive transfer of naive Tob1-knockout cells to MHC-deficient mice that were challenged 2 weeks later with Lewis Lung carcinoma cells. Unlike the results obtained using wild type naïve T cells, Tob1-knockout T cells did not prevent or delay tumor growth in the recipients, and the periphery showed a reverse phenotype with rapid erosion of CD4 T cells and protracted survival of CD8 T cells in peripheral lymphoid organs1. This suggests that, much like absence of CD24 in DCs, the loss of intrinsic negative regulation created by knocking out the Tob1 gene shifted the balance of proliferation and death, allowing CD8 T cells to persist but leading to rapid erosion of CD4 T cells.
The dramatically different phenotypes observed upon adoptive transfer of wild type T cells and Tob1-knockout T cells into MHC-deficient mice indicates that MHC-dependent tuning and intrinsic negative regulation of T cell proliferation use non-redundant and cooperative signaling pathways, and that the consequences of losing one or both pathways are handled differently by naïve peripheral CD4 and CD8 T cells. A mechanistic explanation for these distinct phenotypes has not been elucidated, but co-stimulation is one attractive possibility (73). In this scenario (Figure 4), the survival of CD4 or CD8 T cells in an MHC-barren environment would be consistent with different thresholds for co-stimulation by CD28. Specifically, CD28 signaling is more robust in CD4 cells than in CD8 cells (105–107) and can still be engaged by MHC-deficient dendritic cells (DC) that retain expression of CD80 and/or CD86 (108).
Figure 4. An integrated model of T cell fates in an MHC-less environment.
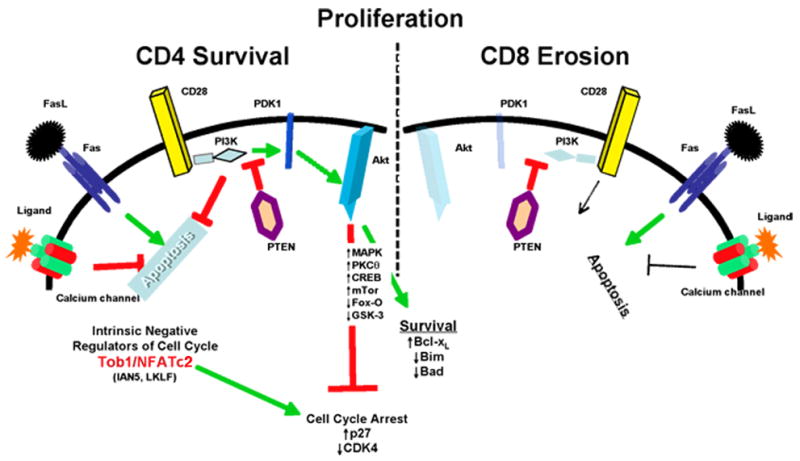
Lymphopenia in MHC-less mice induces proliferation of naïve CD4 and CD8 T cells, leading to upregulation of Fas receptors. We hypothesize that naïve CD4 cells survive because signals initiated by binding CD28, and transmitted mostly through the PI3K-dependent activation of Akt, antagonize the apoptotic impetus from Fas as well as the intrinsic signals from Tob1 and NFAtc2 that enforce cell cycle arrest. At the same time, CD28/PI3K signals tilt the balance of mitochondrial homeostasis towards survival by upregulating Bcl-xL and by downregulating Bim and Bad. PI3K also activates or inhibits additional pathways that similarly favor proliferation and survival. Each of these PI3K-mdeiated events is antagonized by PTEN through dephosphorylation of inositol phospholipids and by dephosphorylation of tyrosine residues in activating kinases. Finally, signals transmitted through calcium channels also increase resistance to Fas-mediated cell death. Naïve CD8 cells, in contrast, do not signal effectively through CD28, and thus show impaired survival when FasL engages Fas signaling. In this context, calcium signals offer only partial or no protection from cell death.
The importance of CD28 for optimal signal transduction has been recognized for some time (109, 110). CD28 is not necessary for T cell development in the thymus, and although CD28-deficient mice have reduced T cell help, they still mount effective cytolytic responses (e.g., against virus) (111). During productive interactions between T cells and APCs, the binding of CD28 to CD80 (B7) or CD86 (B7.1) has various signaling consequences. First, this interaction (and those of other adhesion molecules) may stabilize the immunological synapse. Second, CD28 appears to link TCR signaling with the phosphoinositol-3 kinase (PI3K) pathways, either independently or cooperatively (Figure 4) (112). Numerous roles have been characterized for PI3K signaling in T cells; phospholipid-inositol trisphosphate (PIP3) with phosphorylated inositol at position-3 promotes activation of phospholipid-dependent kinase 1 (PDK1) and protein kinase-B/Akt, which in turn activate mitogen activated protein kinase (MAPK) pathways, protein kinase C-theta (PCKθ), cyclic AMP response element binding protein (CREB), and mammalian target of rapamycin (mTOR), while inhibiting Forkhead Box protein O (FoxO) and glycogen synthase kinase-3 (GSK-3) (112–115). In addition to these signaling consequences, PI3K activation also promotes cell survival by modulating various Bcl-2 family members such as Bad (phosphorylation), Bcl-XL (upregulation), and Bim (inhibition).
PI3K signaling also seems to impair Fas-dependent death signals. Specifically, T cells that express activated Akt fail to assemble caspase-8 to the death-inducing signaling complex (DISC) (116). The lipid phosphorylation by PI3K is antagonized by the dual-specificity phosphatase PTEN (117), a potent prototypical tumor suppressor protein. Among other defects, developmental PTEN-deficiency in mice results in lymphoma, and haploinsufficiency (loss of a single allele) leads to death by fulminant autoimmunity (118). These PTEN-haploinsufficient mice show defective Fas-mediated apoptosis, and the phenotype of this defect resembles that of mice that carry an activated Akt allele (defective recruitment of caspase-8 to the DISC) (116), establishing a link between PI3K activation and repression of both intrinsic (mitochondrial-dependent) and extrinsic (death factor-dependent) apoptosis. Nevertheless, various alternative mechanisms might explain the differential sensitivity of T cell subsets to self-MHC and the importance of intrinsic negative regulatory factors in sensitization and tuning. We predict that this will be a fertile area of investigation for some time to come.
Acknowledgments
The authors thank Dr. Stephen Jameson for his careful review of the manuscript and helpful suggestions. We regret if meritorious references may have been omitted in the interest of space or brevity. The work was supported by grants R21DK63410, P30CA46934, and R01DK58722 from the National Institutes of Health.
Footnotes
CM Jubala et al, MHC-dependent desensitization of intrinsic anti-self reactivity. Submitted
References
- 1.Boyman O, Purton JF, Surh CD, Sprent J. Cytokines and T-cell homeostasis. Curr Opin Immunol. 2007;19:320–326. doi: 10.1016/j.coi.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 2.Cho JH, Boyman O, Kim HO, Hahm B, Rubinstein MP, Ramsey C, et al. An intense form of homeostatic proliferation of naive CD8+ cells driven by IL-2. J Exp Med. 2007;204:1787–1801. doi: 10.1084/jem.20070740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kamimura D, Bevan MJ. Naive CD8+ T cells differentiate into protective memory-like cells after IL-2 anti IL-2 complex treatment in vivo. J Exp Med. 2007;204:1803–1812. doi: 10.1084/jem.20070543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sandau MM, Winstead CJ, Jameson SC. IL-15 is required for sustained lymphopenia-driven proliferation and accumulation of CD8 T cells. J Immunol. 2007;179:120–125. doi: 10.4049/jimmunol.179.1.120. [DOI] [PubMed] [Google Scholar]
- 5.Goldrath AW, Bogatzki LY, Bevan MJ. Naive T cells transiently acquire a memory-like phenotype during homeostasis-driven proliferation. J Exp Med. 2000;192:557–564. doi: 10.1084/jem.192.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kieper WC, Jameson SC. Homeostatic expansion and phenotypic conversion of naive T cells in response to self peptide/MHC ligands. Proc Natl Acad Sci U S A. 1999;96:13306–13311. doi: 10.1073/pnas.96.23.13306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhandoola A, Tai X, Eckhaus M, Auchincloss H, Mason K, Rubin SA, et al. Peripheral expression of self-MHC-II influences the reactivity and self-tolerance of mature CD4(+) T cells: evidence from a lymphopenic T cell model. Immunity. 2002;17:425–436. doi: 10.1016/s1074-7613(02)00417-x. [DOI] [PubMed] [Google Scholar]
- 8.Goldrath AW, Luckey CJ, Park R, Benoist C, Mathis D. The molecular program induced in T cells undergoing homeostatic proliferation. Proc Natl Acad Sci U S A. 2004;101:16885–16890. doi: 10.1073/pnas.0407417101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamilton SE, Wolkers MC, Schoenberger SP, Jameson SC. The generation of protective memory-like CD8+ T cells during homeostatic proliferation requires CD4+ T cells. Nat Immunol. 2006;7:475–481. doi: 10.1038/ni1326. [DOI] [PubMed] [Google Scholar]
- 10.Troy AE, Shen H. Cutting edge: homeostatic proliferation of peripheral T lymphocytes is regulated by clonal competition. J Immunol. 2003;170:672–676. doi: 10.4049/jimmunol.170.2.672. [DOI] [PubMed] [Google Scholar]
- 11.Min B, Foucras G, Meier-Schellersheim M, Paul WE. Spontaneous proliferation, a response of naive CD4 T cells determined by the diversity of the memory cell repertoire. Proc Natl Acad Sci U S A. 2004;101:3874–3879. doi: 10.1073/pnas.0400606101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tan JT, Ernst B, Kieper WC, LeRoy E, Sprent J, Surh CD. Interleukin (IL)-15 and IL-7 jointly regulate homeostatic proliferation of memory phenotype CD8+ cells but are not required for memory phenotype CD4+ cells. J Exp Med. 2002;195:1523–1532. doi: 10.1084/jem.20020066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen S, Ding Y, Tadokoro CE, Olivares-Villagomez D, Camps-Ramirez M, Curotto de Lafaille MA, et al. Control of homeostatic proliferation by regulatory T cells. J Clin Invest. 2005;115:3517–3526. doi: 10.1172/JCI25463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Surh CD, Boyman O, Purton JF, Sprent J. Homeostasis of memory T cells. Immunol Rev. 2006;211:154–163. doi: 10.1111/j.0105-2896.2006.00401.x. [DOI] [PubMed] [Google Scholar]
- 15.Li O, Chang X, Zhang H, Kocak E, Ding C, Zheng P, et al. Massive and destructive T cell response to homeostatic cue in CD24-deficient lymphopenic hosts. J Exp Med. 2006;203:1713–1720. doi: 10.1084/jem.20052293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nesic D, Vukmanovic S. MHC class I is required for peripheral accumulation of CD8+ thymic emigrants. J Immunol. 1998;160:3705–3712. [PubMed] [Google Scholar]
- 17.Murali-Krishna K, Lau LL, Sambhara S, Lemonnier F, Altman J, Ahmed R. Persistence of memory CD8 T cells in MHC class I-deficient mice. Science. 1999;286:1377–1381. doi: 10.1126/science.286.5443.1377. [DOI] [PubMed] [Google Scholar]
- 18.Boursalian TE, Bottomly K. Survival of naive CD4 T cells: roles of restricting versus selecting MHC class II and cytokine milieu. J Immunol. 1999;162:3795–3801. [PubMed] [Google Scholar]
- 19.Viret C, Janeway CA., Jr MHC and T cell development. Rev Immunogenet. 1999;1:91–104. [PubMed] [Google Scholar]
- 20.Witherden D, van Oers N, Waltzinger C, Weiss A, Benoist C, Mathis D. Tetracycline-controllable selection of CD4(+) T cells: half-life and survival signals in the absence of major histocompatibility complex class II molecules. J Exp Med. 2000;191:355–364. doi: 10.1084/jem.191.2.355. [DOI] [PubMed] [Google Scholar]
- 21.Martin B, Bourgeois C, Dautigny N, Lucas B. On the role of MHC class II molecules in the survival and lymphopenia-induced proliferation of peripheral CD4+ T cells. Proc Natl Acad Sci U S A. 2003;100:6021–6026. doi: 10.1073/pnas.1037754100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dorfman JR, Germain RN. MHC-dependent survival of naive T cells? A complicated answer to a simple question. Microbes Infect. 2002;4:547–554. doi: 10.1016/s1286-4579(02)01571-x. [DOI] [PubMed] [Google Scholar]
- 23.Jabbari A, Harty JT. Cutting edge: differential self-peptide/MHC requirement for maintaining CD8 T cell function versus homeostatic proliferation. J Immunol. 2005;175:4829–4833. doi: 10.4049/jimmunol.175.8.4829. [DOI] [PubMed] [Google Scholar]
- 24.Markiewicz MA, Brown I, Gajewski TF. Death of peripheral CD8+ T cells in the absence of MHC class I is Fas-dependent and not blocked by Bcl-xL. Eur J Immunol. 2003;33:2917–2926. doi: 10.1002/eji.200324273. [DOI] [PubMed] [Google Scholar]
- 25.Tanchot C, Lemonnier FA, Perarnau B, Freitas AA, Rocha B. Differential requirements for survival and proliferation of CD8 naive or memory T cells. Science. 1997;276:2057–2062. doi: 10.1126/science.276.5321.2057. [DOI] [PubMed] [Google Scholar]
- 26.Grandjean I, Duban L, Bonney EA, Corcuff E, Di Santo JP, Matzinger P, et al. Are major histocompatibility complex molecules involved in the survival of naive CD4+ T cells? J Exp Med. 2003;198:1089–1102. doi: 10.1084/jem.20030963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grossman Z, Paul WE. Adaptive cellular interactions in the immune system: the tunable activation threshold and the significance of subthreshold responses. Proc Natl Acad Sci U S A. 1992;89:10365–10369. doi: 10.1073/pnas.89.21.10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kassiotis G, Zamoyska R, Stockinger B. Involvement of avidity for major histocompatibility complex in homeostasis of naive and memory T cells. J Exp Med. 2003;197:1007–1016. doi: 10.1084/jem.20021812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kieper WC, Burghardt JT, Surh CD. A role for TCR affinity in regulating naive T cell homeostasis. J Immunol. 2004;172:40–44. doi: 10.4049/jimmunol.172.1.40. [DOI] [PubMed] [Google Scholar]
- 30.Stefanova I, Dorfman JR, Germain RN. Self-recognition promotes the foreign antigen sensitivity of naive T lymphocytes. Nature. 2002;420:429–434. doi: 10.1038/nature01146. [DOI] [PubMed] [Google Scholar]
- 31.Fischer UB, Jacovetty EL, Medeiros RB, Goudy BD, Zell T, Swanson JB, et al. MHC class II deprivation impairs CD4 T cell motility and responsiveness to antigen-bearing dendritic cells in vivo. Proc Natl Acad Sci U S A. 2007;104:7181–7186. doi: 10.1073/pnas.0608299104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dummer W, Niethammer AG, Baccala R, Lawson BR, Wagner N, Reisfeld RA, et al. T cell homeostatic proliferation elicits effective antitumor autoimmunity. J Clin Invest. 2002;110:185–192. doi: 10.1172/JCI15175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu HM, Poehlein CH, Urba WJ, Fox BA. Development of antitumor immune responses in reconstituted lymphopenic hosts. Cancer Res. 2002;62:3914–3919. [PubMed] [Google Scholar]
- 34.Marleau AM, Sarvetnick N. T cell homeostasis in tolerance and immunity. J Leukoc Biol. 2005;78:575–584. doi: 10.1189/jlb.0105050. [DOI] [PubMed] [Google Scholar]
- 35.Brown IE, Blank C, Kline J, Kacha AK, Gajewski TF. Homeostatic proliferation as an isolated variable reverses CD8+ T cell anergy and promotes tumor rejection. J Immunol. 2006;177:4521–4529. doi: 10.4049/jimmunol.177.7.4521. [DOI] [PubMed] [Google Scholar]
- 36.Bracci L, Moschella F, Sestili P, La Sorsa V, Valentini M, Canini I, et al. Cyclophosphamide enhances the antitumor efficacy of adoptively transferred immune cells through the induction of cytokine expression, B-cell and T-cell homeostatic proliferation, and specific tumor infiltration. Clin Cancer Res. 2007;13:644–653. doi: 10.1158/1078-0432.CCR-06-1209. [DOI] [PubMed] [Google Scholar]
- 37.Maeda Y, Tawara I, Teshima T, Liu C, Hashimoto D, Matsuoka K, et al. Lymphopenia-induced proliferation of donor T cells reduces their capacity for causing acute graft-versus-host disease. Exp Hematol. 2007;35:274–286. doi: 10.1016/j.exphem.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 38.Anderson BE, McNiff J, Yan J, Doyle H, Mamula M, Shlomchik MJ, et al. Memory CD4+ T cells do not induce graft-versus-host disease. J Clin Invest. 2003;112:101–108. doi: 10.1172/JCI17601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen BJ, Cui X, Sempowski GD, Liu C, Chao NJ. Transfer of allogeneic CD62L-memory T cells without graft-versus-host disease. Blood. 2004;103:1534–1541. doi: 10.1182/blood-2003-08-2987. [DOI] [PubMed] [Google Scholar]
- 40.Chen BJ, Deoliveira D, Cui X, Le NT, Son J, Whitesides JF, et al. Inability of memory T cells to induce graft-versus-host disease is a result of an abortive alloresponse. Blood. 2007;109:3115–3123. doi: 10.1182/blood-2006-04-016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boise LH, Thompson CB. Hierarchical control of lymphocyte survival. Science. 1996;274:67–68. doi: 10.1126/science.274.5284.67. [DOI] [PubMed] [Google Scholar]
- 42.Lang JA, Kominski D, Bellgrau D, Scheinman RI. Partial activation precedes apoptotic death in T cells harboring an IAN gene mutation. Eur J Immunol. 2004;34:2396–2406. doi: 10.1002/eji.200324751. [DOI] [PubMed] [Google Scholar]
- 43.Baksh S, Widlund HR, Frazer-Abel AA, Du J, Fosmire S, Fisher DE, et al. NFATc2-mediated repression of cyclin-dependent kinase 4 expression. Mol Cell. 2002;10:1071–1081. doi: 10.1016/s1097-2765(02)00701-3. [DOI] [PubMed] [Google Scholar]
- 44.Frazer-Abel AA, Baksh S, Fosmire SP, Willis D, Pierce AM, Meylemans H, et al. Nicotine activates NFATc2 and prevents cell cycle entry in T cells. J Pharmacol Exp Ther. 2004;311:758–769. doi: 10.1124/jpet.104.070060. [DOI] [PubMed] [Google Scholar]
- 45.Heissmeyer V, Macian F, Im SH, Varma R, Feske S, Venuprasad K, et al. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat Immunol. 2004;5:255–265. doi: 10.1038/ni1047. [DOI] [PubMed] [Google Scholar]
- 46.Macian F, Garcia-Cozar F, Im SH, Horton HF, Byrne MC, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002;109:719–731. doi: 10.1016/s0092-8674(02)00767-5. [DOI] [PubMed] [Google Scholar]
- 47.Yusuf I, Fruman DA. Regulation of quiescence in lymphocytes. Trends Immunol. 2003;24:380–386. doi: 10.1016/s1471-4906(03)00141-8. [DOI] [PubMed] [Google Scholar]
- 48.Frisch SM. Evidence for a function of death-receptor-related, death-domain- containing proteins in anoikis. Curr Biol. 1999;9:1047–1049. doi: 10.1016/s0960-9822(99)80455-2. [DOI] [PubMed] [Google Scholar]
- 49.Frisch SM, Screaton RA. Anoikis mechanisms. Curr Opin Cell Biol. 2001;13:555–562. doi: 10.1016/s0955-0674(00)00251-9. [DOI] [PubMed] [Google Scholar]
- 50.Modiano JF, Ritt MG, Wojcieszyn J, Smith R., 3rd Growth arrest of melanoma cells is differentially regulated by contact inhibition and serum deprivation. DNA & Cell Biology. 1999;18:357–367. doi: 10.1089/104454999315259. [DOI] [PubMed] [Google Scholar]
- 51.Kupfer R, Lang J, Williams-Skipp C, Nelson M, Bellgrau D, Scheinman RI. Loss of a gimap/ian gene leads to activation of NF-kappaB through a MAPK-dependent pathway. Mol Immunol. 2007;44:479–487. doi: 10.1016/j.molimm.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 52.Tzachanis D, Freeman GJ, Hirano N, van Puijenbroek AA, Delfs MW, Berezovskaya A, et al. Tob is a negative regulator of activation that is expressed in anergic and quiescent T cells. Nat Immunol. 2001;2:1174–1182. doi: 10.1038/ni730. [DOI] [PubMed] [Google Scholar]
- 53.Letterio JJ. TGF-beta signaling in T cells: roles in lymphoid and epithelial neoplasia. Oncogene. 2005;24:5701–5712. doi: 10.1038/sj.onc.1208922. [DOI] [PubMed] [Google Scholar]
- 54.Li L, Iwamoto Y, Berezovskaya A, Boussiotis VA. A pathway regulated by cell cycle inhibitor p27Kip1 and checkpoint inhibitor Smad3 is involved in the induction of T cell tolerance. Nat Immunol. 2006;7:1157–1165. doi: 10.1038/ni1398. [DOI] [PubMed] [Google Scholar]
- 55.Classen S, Zander T, Eggle D, Chemnitz JM, Brors B, Buchmann I, et al. Human resting CD4+ T cells are constitutively inhibited by TGF beta under steady-state conditions. J Immunol. 2007;178:6931–6940. doi: 10.4049/jimmunol.178.11.6931. [DOI] [PubMed] [Google Scholar]
- 56.McKarns SC, Schwartz RH, Kaminski NE. Smad3 is essential for TGF-beta 1 to suppress IL-2 production and TCR-induced proliferation, but not IL-2-induced proliferation. J Immunol. 2004;172:4275–4284. doi: 10.4049/jimmunol.172.7.4275. [DOI] [PubMed] [Google Scholar]
- 57.Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, et al. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. Embo J. 1999;18:1280–1291. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nitta T, Takahama Y. The lymphocyte guard-IANs: regulation of lymphocyte survival by IAN/GIMAP family proteins. Trends Immunol. 2007;28:58–65. doi: 10.1016/j.it.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 59.MacMurray AJ, Moralejo DH, Kwitek AE, Rutledge EA, Van Yserloo B, Gohlke P, et al. Lymphopenia in the BB rat model of type 1 diabetes is due to a mutation in a novel immune-associated nucleotide (Ian)-related gene. Genome Res. 2002;12:1029–1039. doi: 10.1101/gr.412702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nitta T, Nasreen M, Seike T, Goji A, Ohigashi I, Miyazaki T, et al. IAN family critically regulates survival and development of T lymphocytes. PLoS Biol. 2006;4:e103. doi: 10.1371/journal.pbio.0040103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Keita M, Leblanc C, Andrews D, Ramanathan S. GIMAP5 regulates mitochondrial integrity from a distinct subcellular compartment. Biochem Biophys Res Commun. 2007;361:481–486. doi: 10.1016/j.bbrc.2007.07.048. [DOI] [PubMed] [Google Scholar]
- 62.Zadeh HH, Greiner DL, Wu DY, Tausche F, Goldschneider I. Abnormalities in the export and fate of recent thymic emigrants in diabetes-prone BB/W rats. Autoimmunity. 1996;24:35–46. doi: 10.3109/08916939608995355. [DOI] [PubMed] [Google Scholar]
- 63.Ramanathan S, Norwich K, Poussier P. Antigen activation rescues recent thymic emigrants from programmed cell death in the BB rat. J Immunol. 1998;160:5757–5764. [PubMed] [Google Scholar]
- 64.Moore JK, Scheinman RI, Bellgrau D. The identification of a novel T cell activation state controlled by a diabetogenic gene. J Immunol. 2001;166:241–248. doi: 10.4049/jimmunol.166.1.241. [DOI] [PubMed] [Google Scholar]
- 65.Jia S, Meng A. Tob genes in development and homeostasis. Dev Dyn. 2007;236:913–921. doi: 10.1002/dvdy.21092. [DOI] [PubMed] [Google Scholar]
- 66.Kawamura-Tsuzuku J, Suzuki T, Yoshida Y, Yamamoto T. Nuclear localization of Tob is important for regulation of its antiproliferative activity. Oncogene. 2004;23:6630–6638. doi: 10.1038/sj.onc.1207890. [DOI] [PubMed] [Google Scholar]
- 67.Maekawa M, Nishida E, Tanoue T. Identification of the Anti-proliferative protein Tob as a MAPK substrate. J Biol Chem. 2002;277:37783–37787. doi: 10.1074/jbc.M204506200. [DOI] [PubMed] [Google Scholar]
- 68.Suzuki T, J KT, Ajima R, Nakamura T, Yoshida Y, Yamamoto T. Phosphorylation of three regulatory serines of Tob by Erk1 and Erk2 is required for Ras-mediated cell proliferation and transformation. Genes Dev. 2002;16:1356–1370. doi: 10.1101/gad.962802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hiramatsu Y, Kitagawa K, Suzuki T, Uchida C, Hattori T, Kikuchi H, et al. Degradation of Tob1 mediated by SCFSkp2-dependent ubiquitination. Cancer Res. 2006;66:8477–8483. doi: 10.1158/0008-5472.CAN-06-1603. [DOI] [PubMed] [Google Scholar]
- 70.Yamashiro H, Odani Y, Hozumi N, Nakano N. Hierarchical signaling thresholds determine the fates of naive T cells: partial priming leads nai;ve T cells to unresponsiveness. Biochem Biophys Res Commun. 2002;299:148–154. doi: 10.1016/s0006-291x(02)02586-x. [DOI] [PubMed] [Google Scholar]
- 71.Kiani A, Rao A, Aramburu J. Manipulating immune responses with immunosuppressive agents that target NFAT. Immunity. 2000;12:359–372. doi: 10.1016/s1074-7613(00)80188-0. [DOI] [PubMed] [Google Scholar]
- 72.Xanthoudakis S, Viola JP, Shaw KT, Luo C, Wallace JD, Bozza PT, et al. An enhanced immune response in mice lacking the transcription factor NFAT1. Science. 1996;272:892–895. doi: 10.1126/science.272.5263.892. [DOI] [PubMed] [Google Scholar]
- 73.Sundrud MS, Rao A. New twists of T cell fate: control of T cell activation and tolerance by TGF-beta and NFAT. Curr Opin Immunol. 2007;19:287–293. doi: 10.1016/j.coi.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 74.Derynck R, Zhang Y, Feng X-H. Transcriptional Activators of TGF-[beta] Responses: Smads. Cell. 1998;95:737–740. doi: 10.1016/s0092-8674(00)81696-7. [DOI] [PubMed] [Google Scholar]
- 75.Massague J, Weinberg RA. Negative regulators of growth. Curr Opin Genet Dev. 1992;2:28–32. doi: 10.1016/s0959-437x(05)80317-x. [DOI] [PubMed] [Google Scholar]
- 76.Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, et al. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994;8:9–22. doi: 10.1101/gad.8.1.9. [DOI] [PubMed] [Google Scholar]
- 77.Quelle DE, Ashmun RA, Hannon GJ, Rehberger PA, Trono D, Richter KH, et al. Cloning and characterization of murine p16INK4a and p15INK4b genes. Oncogene. 1995;11:635–645. [PubMed] [Google Scholar]
- 78.Kehrl JH, Wakefield LM, Roberts AB, Jakowlew S, Alvarez-Mon M, Derynck R, et al. Production of transforming growth factor beta by human T lymphocytes and its potential role in the regulation of T cell growth. J Exp Med. 1986;163:1037–1050. doi: 10.1084/jem.163.5.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McKarns SC, Schwartz RH. Distinct effects of TGF-beta 1 on CD4+ and CD8+ T cell survival, division, and IL-2 production: a role for T cell intrinsic Smad3. J Immunol. 2005;174:2071–2083. doi: 10.4049/jimmunol.174.4.2071. [DOI] [PubMed] [Google Scholar]
- 80.Ranges GE, Figari IS, Espevik T, Palladino MA., Jr Inhibition of cytotoxic T cell development by transforming growth factor beta and reversal by recombinant tumor necrosis factor alpha. J Exp Med. 1987;166:991–998. doi: 10.1084/jem.166.4.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-[beta] induces development of the TH17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 82.Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. 2005;201:1061–1067. doi: 10.1084/jem.20042276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wolfraim LA, Walz TM, James Z, Fernandez T, Letterio JJ. p21Cip1 and p27Kip1 Act in Synergy to Alter the Sensitivity of Naive T Cells to TGF-{beta}-Mediated G1 Arrest through Modulation of IL-2 Responsiveness. J Immunol. 2004;173:3093–3102. doi: 10.4049/jimmunol.173.5.3093. [DOI] [PubMed] [Google Scholar]
- 84.Modiano JF, Mayor J, Ball C, Fuentes MK, Linthicum DS. Cdk4 expression and activity are required for cytokine responsiveness in T cells. J Immunol. 2000;165:6693–6702. doi: 10.4049/jimmunol.165.12.6693. [DOI] [PubMed] [Google Scholar]
- 85.Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature. 2004;430:226–231. doi: 10.1038/nature02650. [DOI] [PubMed] [Google Scholar]
- 86.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Diebold RJ, Eis MJ, Yin M, Ormsby I, Boivin GP, Darrow BJ, et al. Early-onset multifocal inflammation in the transforming growth factor beta 1-null mouse is lymphocyte mediated. Proc Natl Acad Sci U S A. 1995;92:12215–12219. doi: 10.1073/pnas.92.26.12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kobayashi S, Yoshida K, Ward JM, Letterio JJ, Longenecker G, Yaswen L, et al. {beta}2-Microglobulin-Deficient Background Ameliorates Lethal Phenotype of the TGF-{beta}1 Null Mouse. J Immunol. 1999;163:4013–4019. [PubMed] [Google Scholar]
- 90.Leveen P, Carlsen M, Makowska A, Oddsson S, Larsson J, Goumans MJ, et al. TGF-beta type II receptor-deficient thymocytes develop normally but demonstrate increased CD8+ proliferation in vivo. Blood. 2005;106:4234–4240. doi: 10.1182/blood-2005-05-1871. [DOI] [PubMed] [Google Scholar]
- 91.Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25:455–471. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 92.Lucas PJ, Kim SJ, Melby SJ, Gress RE. Disruption of T cell homeostasis in mice expressing a T cell-specific dominant negative transforming growth factor beta II receptor. J Exp Med. 2000;191:1187–1196. doi: 10.1084/jem.191.7.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Marie JC, Liggitt D, Rudensky AY. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity. 2006;25:441–454. doi: 10.1016/j.immuni.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 94.Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–181. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- 95.Lucas PJ, Kim SJ, Mackall CL, Telford WG, Chu YW, Hakim FT, et al. Dysregulation of IL-15-mediated T-cell homeostasis in TGF-beta dominant-negative receptor transgenic mice. Blood. 2006;108:2789–2795. doi: 10.1182/blood-2006-05-025676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Warner BJ, Blain SW, Seoane J, Massague J. Myc Downregulation by Transforming Growth Factor beta Required for Activation of the p15Ink4b G1 Arrest Pathway. Mol Cell Biol. 1999;19:5913–5922. doi: 10.1128/mcb.19.9.5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bianchi T, Gasser S, Trumpp A, MacDonald HR. c-Myc acts downstream of IL-15 in the regulation of memory CD8 T-cell homeostasis. Blood. 2006;107:3992–3999. doi: 10.1182/blood-2005-09-3851. [DOI] [PubMed] [Google Scholar]
- 98.Campbell JD, Cook G, Robertson SE, Fraser A, Boyd KS, Gracie JA, et al. Suppression of IL-2-induced T cell proliferation and phosphorylation of STAT3 and STAT5 by tumor-derived TGF beta is reversed by IL-15. J Immunol. 2001;167:553–561. doi: 10.4049/jimmunol.167.1.553. [DOI] [PubMed] [Google Scholar]
- 99.Koehler H, Kofler D, Hombach A, Abken H. CD28 Costimulation Overcomes Transforming Growth Factor-{beta}-Mediated Repression of Proliferation of Redirected Human CD4+ and CD8+ T Cells in an Antitumor Cell Attack. Cancer Res. 2007;67:2265–2273. doi: 10.1158/0008-5472.CAN-06-2098. [DOI] [PubMed] [Google Scholar]
- 100.Sung JL, Lin JT, Gorham JD. CD28 co-stimulation regulates the effect of transforming growth factor-[beta]1 on the proliferation of naive CD4+ T cells. International Immunopharmacology. 2003;3:233–245. doi: 10.1016/S1567-5769(02)00276-X. [DOI] [PubMed] [Google Scholar]
- 101.Ahmadzadeh M, Rosenberg SA. TGF-beta 1 attenuates the acquisition and expression of effector function by tumor antigen-specific human memory CD8 T cells. J Immunol. 2005;174:5215–5223. doi: 10.4049/jimmunol.174.9.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat Med. 2001;7:1118–1122. doi: 10.1038/nm1001-1118. [DOI] [PubMed] [Google Scholar]
- 103.Zhang Q, Yang XJ, Kundu SD, Pins M, Javonovic B, Meyer R, et al. Blockade of transforming growth factor-{beta} signaling in tumor-reactive CD8+ T cells activates the antitumor immune response cycle. Mol Cancer Ther. 2006;5:1733–1743. doi: 10.1158/1535-7163.MCT-06-0109. [DOI] [PubMed] [Google Scholar]
- 104.Fahlen L, Read S, Gorelik L, Hurst SD, Coffman RL, Flavell RA, et al. T cells that cannot respond to TGF-beta escape control by CD4(+)CD25(+) regulatory T cells. J Exp Med. 2005;201:737–746. doi: 10.1084/jem.20040685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kerstan A, Hunig T. Cutting edge: distinct TCR- and CD28-derived signals regulate CD95L, Bcl-xL, and the survival of primary T cells. J Immunol. 2004;172:1341–1345. doi: 10.4049/jimmunol.172.3.1341. [DOI] [PubMed] [Google Scholar]
- 106.Rowell EA, Walsh MC, Wells AD. Opposing roles for the cyclin-dependent kinase inhibitor p27kip1 in the control of CD4+ T cell proliferation and effector function. J Immunol. 2005;174:3359–3368. doi: 10.4049/jimmunol.174.6.3359. [DOI] [PubMed] [Google Scholar]
- 107.Wolfraim LA, Letterio JJ. Cutting edge: p27Kip1 deficiency reduces the requirement for CD28-mediated costimulation in naive CD8+ but not CD4+ T lymphocytes. J Immunol. 2005;174:2481–2484. doi: 10.4049/jimmunol.174.5.2481. [DOI] [PubMed] [Google Scholar]
- 108.Bettini M, Xi H, Kersh GJ. T cell stimulation in the absence of exogenous antigen: a T cell signal is induced by both MHC-dependent and -independent mechanisms. Eur J Immunol. 2003;33:3109–3116. doi: 10.1002/eji.200324067. [DOI] [PubMed] [Google Scholar]
- 109.Berridge MJ. Lymphocyte activation in health and disease. Crit Rev Immunol. 1997;17:155–178. doi: 10.1615/critrevimmunol.v17.i2.30. [DOI] [PubMed] [Google Scholar]
- 110.Janeway CA, Jr, Bottomly K. Signals and signs for lymphocyte responses. Cell. 1994;76:275–285. doi: 10.1016/0092-8674(94)90335-2. [DOI] [PubMed] [Google Scholar]
- 111.Shahinian A, Pfeffer K, Lee KP, Kundig TM, Kishihara K, Wakeham A, et al. Differential T cell costimulatory requirements in CD28-deficient mice. Science. 1993;261:609–612. doi: 10.1126/science.7688139. [DOI] [PubMed] [Google Scholar]
- 112.Kane LP, Weiss A. The PI-3 kinase/Akt pathway and T cell activation: pleiotropic pathways downstream of PIP3. Immunol Rev. 2003;192:7–20. doi: 10.1034/j.1600-065x.2003.00008.x. [DOI] [PubMed] [Google Scholar]
- 113.Okkenhaug K, Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nat Rev Immunol. 2003;3:317–330. doi: 10.1038/nri1056. [DOI] [PubMed] [Google Scholar]
- 114.Okkenhaug K, Vanhaesebroeck B. PI3K-signalling in B- and T-cells: insights from gene-targeted mice. Biochem Soc Trans. 2003;31:270–274. doi: 10.1042/bst0310270. [DOI] [PubMed] [Google Scholar]
- 115.Altman A, Villalba M. Protein kinase C-theta (PKCtheta): it’s all about location, location, location. Immunol Rev. 2003;192:53–63. doi: 10.1034/j.1600-065x.2003.00027.x. [DOI] [PubMed] [Google Scholar]
- 116.Jones RG, Elford AR, Parsons MJ, Wu L, Krawczyk CM, Yeh WC, et al. CD28-dependent activation of protein kinase B/Akt blocks Fas-mediated apoptosis by preventing death-inducing signaling complex assembly. J Exp Med. 2002;196:335–348. doi: 10.1084/jem.20020307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sulis ML, Parsons R. PTEN: from pathology to biology. Trends Cell Biol. 2003;13:478–483. doi: 10.1016/s0962-8924(03)00175-2. [DOI] [PubMed] [Google Scholar]
- 118.Di Cristofano A, Kotsi P, Peng YF, Cordon-Cardo C, Elkon KB, Pandolfi PP. Impaired Fas response and autoimmunity in Pten+/− mice. Science. 1999;285:2122–2125. doi: 10.1126/science.285.5436.2122. [DOI] [PubMed] [Google Scholar]