

Abstract
Extracellular signal-regulated kinase (Erk)1/2 activity signals myeloid cell differentiation induced by 12-O-tetradecanoyl-phorbol-13-acetate (TPA). Previously, we reported that Erk1/2 activation (phosphorylation) induced by TPA required reactive oxygen species (ROS) as a second messenger. Here, we hypothesized that ROS generated in response to TPA inhibit Erk1/2-directed phosphatase activity, which leads to an increase phosphorylation of Erk1/2 to signal p21WAF1/Cip1-mediated growth arrest in ML-1 cells. Incubation of ML-1 cells with TPA resulted in a marked accumulation of phosphorylated Erk1/2, and is subsequent to H2O2 generation. Interestingly, post-TPA-treatment with N-acetylcysteine (NAC) stimulated a marked and a rapid dephosphorylation of Erk1/2, suggesting a regeneration of Erk1/2-directed phospahatase activity by NAC. ROS generation in ML-1 cells induced by TPA was suggested to occur in the mitochondrial electron transport chain (METC) based on the following observations: (i) undifferentiated ML-1 cells not only lack p67-phox and but also express a low level of p47-phox key components required for NADPH oxidase enzymatic activity, (ii) pretreatment with DPI, an inhibitor of NADH- and NADPH-dependent enzymes, or rhein, an inhibitor of complex I, blocked the ROS generation, and (iii) examination of the microarray analysis data and Western blot analysis data revealed an induction of MnSOD expression at both mRNA and protein levels in response to TPA. MnSOD is a key member of the mitochondrial defense system against mitochondrial-derived superoxide. Together, this study suggested that TPA stimulated ROS generation as a second messenger to activate Erk1/2 via a redox-mediated inhibition of Erk1/2-directed phosphatase in ML-1 cells.
Reactive oxygen species (ROS) are generated from partial reduction of molecular oxygen. Superoxide (O2-·), the first member of ROS, is generated when a single electron interacts and partially reduces molecular oxygen (Reid and Durham, 2002). Dismutation of O2-· forms hydrogen peroxide (H2O2) which is then reduced to water by the enzymatic actions of catalase, gluthathione peroxidase, or other peroxidases. Following the discovery of ROS, the primary focus was on the oxidative damage elicited by ROS within cells (Agarwal and Sohal, 1994a, 1994b). However, in the last several years, physiological roles have been attributed to ROS generation in cells (Chen et al., 1995; Sundaresan et al., 1995; Bae et al., 1997; Bogoyevitch et al., 2000; Rinna et al., 2006). In a recent study, we demonstrated that TPA-induced ROS generation signaled expression of p21WAF1/Cip1 in human acute myeloid leukemia cells THP-1 (Traore et al., 2005). p21WAF1/Cip1, a 21-kDa protein and inhibitor of cyclin-dependent kinases is a critical downstream effector in the p53-specific pathway of cell cycle control. However, p21 can also be induced to express by p53-independent pathways during initiation of terminal differentiation, and has been implicated in mediating cell cycle arrest in response to extracellular factors including phorbol esters, irradiation, H2O2, tumor necrosis factor-α, and transforming growth factor-β (TGF-β) (Michieli et al., 1994; Akashi et al., 1995; Datto et al., 1995; Biggs et al., 1996; Droge, 2002; Traore et al., 2005).
Potential sources of ROS in phagocytic cells include NADPH oxidase and the mitochondrial electron transport chain (METC) (Forman and Torres, 2002; Ichikawa et al., 2004). Mitochondria-generated cellular energy, ATP, is synthesized through oxidative phosphorylation, a process during which the METC accepts electrons from NADH and FADH2 (Weiss et al., 1991; Devin and Rigoulet, 2006). As electrons are passed down the METC, molecular oxygen is eventually converted to water (Ichikawa et al., 2004). O2-· is generated when a single electron prematurely leaves the METC and partially reduces molecular oxygen. ROS generated in the METC during the oxidative phosphorylation activity has been shown to modulate multiple molecular processes, signaling pathways, and transcription factor activities (Kim et al., 1999; Ichikawa et al., 2004; Kim et al., 2005).
The concept that ROS can stimulate cellular signal transduction cascades has been continuously advancing in recent years (Chen et al., 1995; Sundaresan et al., 1995; Yoshizumi et al., 2000; Traore et al., 2005; Traore et al., 2007). Numerous studies have indicated that ROS play a role as messengers or effector molecules to activate members of a large family of enzymes known as the mitogen-activated protein kinases (MAPKs) including JNK and p38 in response to various extracellular stimuli including TNF-alpha and lysophosphatidic acid (Chen et al., 1995; Yoshizumi et al., 2000; Kamata et al., 2005). In our previous studies, we have shown that ROS was required for TPA-induced extracellular signal-regulated kinase (Erk)1/2 activation in ML-1 and THP-1 cells (Traore et al., 2005; Traore et al., 2007). Furthermore, studies have shown that JNK, P38, and Erk1/2 are phosphorylated by treatment with exogenous H2O2 and this event can be inhibited by pretreatment with anti-oxidants (Abe et al., 1996; Lo et al., 1996; Guyton et al., 1996b). Despite these well-documented findings, the mechanism by which ROS activates MAPKs is not well understood. However, one well-accepted model is that ROS transiently inactivates regulatory phosphatases via interaction with catalytically active, redox sensitive cysteines in the active site (Denu and Tanner, 1998; Kamata et al., 2005).
While our previous studies identified the importance of ROS-mediated signaling in ERK-dependent THP-1 cell growth inhibition and ICAM-1 expression in ML-1 cells, we did not identify the mechanism of Erk1/2 activation in response to TPA (Traore et al., 2005; Traore et al., 2007). In this study, we investigated the mechanism of ROS-mediated Erk1/2 activation induced by TPA in human myeloid leukemia cells ML-1. We found that TPA-induced PKC-stimulated ROS generation inactivates Erk1/2-directed phosphatases and promotes a transient and reversible activation of Erk1/2 cells to signal growth inhibition in ML-1 cells.
Materials and Methods
Reagents and antibodies
The pharmacologic inhibitors for MEK (PD98059 and U0126) were purchased from Sigma (St. Louis, MO). The pharmacologic inhibitor for PKC (GF109203X) was obtained from ALEXIS Biochemicals (San Diego, CA). The pharmacologic inhibitor for mitochondria complex I and NADPH oxidase diphenyleneiodium (DPI), the competitive inhibitor of mitochondria complex I (rhein) (Kean et al., 1971; Floridi et al., 1989), the inhibitor for mitochondria complex I (rotenone), and the ROS scavenger N-acetylcysteine (NAC) were purchased from Sigma. Anti-human phospho-Erk1/2 antibodies were purchased from Cell Signaling Technology, Inc. (Beverly, MA). Anti-human p21WAF1/Cip1 antibodies were purchased from BD Pharmingen (San Diego, CA). Anti-human p47-phox antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and anti-human p67-phox antibodies were from BD-Transduction Laboratories (Lexington, KY).
Cell culture
Human myeloid leukemia ML-1 cells were obtained from Dr. Ruth Craig, Dartmouth School of Medicine. Human myeloid leukemia THP-1 cells were purchased from American Type Cell Collection (ATCC). All cell culture media and supplements were obtained from Invitrogen (Carlsband, CA). Cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, antibiotics (50 U/ml penicillin and 50 μg/ml streptomycin) in 150 cm2 tissue culture flasks, and maintained at 37°C in a humidified 5% CO2 atmosphere. Experiments were routinely carried out using cells in the log phase of growth. 12-O-tetradecanoyl-phorbol-13-acetate (TPA) (Sigma Chemical Co., St. Louis, MO) was dissolved in DMSO to obtain a 100 μM stock solution and further diluted before use. For all experiments, cells were cultured at an initial density of 5 × 105 cells/ml and treated with TPA for various concentrations and times as indicated. For negative controls, the cells were incubated in the absence of TPA, in medium containing an equivalent 1% (v/v) DMSO. Viability was determined by hemocytometer counts of trypan blue-impermeable cells.
Microarray analysis
ML-1 cells were grown at density of 105 cells/ml in 150 ml dish and treated with vehicle (DMSO) or 5 nM TPA (n = 3) 6 h, 3 and 6 days. Total RNA was extracted using Trizol reagent and purified with the Qiagen Rneasy minikit. The purified RNA was subjected to Affymetrix oligonucleotide microarray analysis using Human Genome U133 2.0 Plus Array Chip. Six replicates, including three controls and three TPA treated ML-1 cells samples were examined.
RT-PCR
RNA samples representing ML-1 cells incubated in the presence or absence of TPA for 6 h. Total RNA was extracted using Trizol (Invitrogen) according to the manufacturer’s instructions. Total RNA (2 μg) was used for cDNA synthesis. RT-PCR analyses were performed using assay probe sets from Applied Biosystems (Foster City, CA). Assays were performed by using the ABI 7000 Taqman system (Applied Biosystems). GAPDH was used for normalization. The cycle threshold (CT) value indicates the number of PCR cycles that are necessary for the detection of a fluorescence signal exceeding a fixed threshold. The fold change (FC) was calculated by using the following formulas: ΔCT = CT (GAPDH) – CT (target gene) and FC = 2-(ΔCT2-ΔCT1), in which ΔCT1 represents the highest CT value among all the samples and ΔCT2 represents the value of a particular sample.
Western-blot analysis
Cells were lysed in buffer containing 50 mM Tris-HCl (pH 7.4), 100 mM NaCl, 50 mM sodium fluoride, 5 mM EDTA, 40 mM β-glycerophosphate, 1 mM sodium orthovanadate, 10-4 M phenylmethylsulfonyl fluoride, 10-6 M leupeptin, 10-6 M pepstatin A, 1% (v/v) Triton X-100. Lysates were clarified by centrifugation at 13,000g for 10 min. Samples, each containing 40 μg of cell lysates in loading buffer, were subjected to SDS/PAGE (12% gel) electrophoresis and proteins were transferred to a PVDF membrane (Immobilon; Millipore, Bedford, MA). Immunoblotting was performed using anti-human p47-phox, p67-phox, MnSOD and p21WAF1/CIP1 diluted 1:1,000. After washing the blots, horseradish-conjugated anti-IgG, diluted 1:2,000, was added to the blots. Immunoreactivity was detected by chemiluminescence (CL) (Perkin-Elmer, Life Sciences, MA).
Determination of phospho-Erk1/2 y FACS analysis
The levels of phospho-Erk1/2 were determined using FACS-Scan analysis. Cells (1 × 106) were washed twice in PBS, fixed in 2% formaldehyde in PBS and permeabilized with ice cold 90% methanol. Cell were resuspended in PBS containing 0.1% sodium azide and 5% FBS for 30 min, incubated with (Alexa-fluor 488)-conjugated anti-phospho-Erk1/2 antibodies for 1 h, washed twice in PBS and analyzed by FACS-Scan analysis. Negative controls were stained with isotype-matched (Alexa-Fluor 488)-conjugated IgG and compensation was adjusted using the single-stained cell samples. The fluorescence intensities were determined using Cellquest software (Becton Dickinson, Bedford, MA).
Measurement of ROS generation by
Luminol-derived chemiluminescence (CL)
The generation of cellular ROS, in particular H2O2, was assessed using luminol-derived CL as previously described (Traore et al., 2005; Traore et al., 2007). One million cells were preincubated with luminol (10 μM) and horseradish peroxidase (10 μg/ml) in complete PBS (PBS with 0.5 mM MgCl2, 0.7 mM CaCl2, 0.1% glucose) for 10 min followed by TPA treatment. The CL was measured continuously in a Berthold LB9505 luminometer (Pforzheim, Germany) at 37°C for 50 min.
FACS analysis using flurophore hydroethidine
To detect intracellular ROS, hydroethidine was used as probe. ML-1 cells were first incubated for 5 min with a 0.5 μM hydroethidine Sigma at 37°C followed by 30 min incubation with TPA or DMSO as control. The reaction was stopped by incubating cells for 10 min on ice followed by two washes with PBS. ROS was determined within 1 h on a FACSCaliber flow cytometry (BD Biosciences, San Jose, CA) using fluorescent intensity of excitation at 490 nm and emission at 520 nm.
Protein phosphatase activity
The cellular phosphatase activity was assessed using the 3, 6-fluorescein diphosphate (FDP) dependent fluoregenic phosphatase substrate assay (AnaSpec, San Jose, CA). ML-1 cells were incubated in media in the presence or absence of TPA for 1 h. Cells were harvested, washed twice with PBS and lysed in buffer containing 1% Triton X-100, 10 μg/ml aprotinin, 10 10 μg/ml leupeptin, 100 μg/ml PMSF, and 10 μg/ml pepstatin. Phosphatase activity assay was conducted according to manufacturer protocol using phosphatase assay buffer without DTT (AnaSpec).
Results
TPA treatment inhibits ML-1 cell proliferation by upregulating p21WAF1/cip1 expression
One of the required cellular processes necessary for differentiation to occur is the inhibition of cell growth. In our previous report, we demonstrated that incubation of human myeloid THP-1 cells with TPA resulted in growth arrest at G1 phase of the cell cycle (Traore et al., 2005). In this study, in order to better understand the mechanism of TPA-induced growth arrest, we examined the effect of TPA on another human myeloid cell line, ML-1 cells. The cell proliferation assay data shown in Figure 1 indicated that TPA inhibits the proliferation of both ML-1 and THP-1 cells for up to 72 h. Examination of microarray analysis data demonstrated that TPA induces the expression of negative regulators and inhibits the expression of positive regulators of the cell cycle (Table 1). In Figure 2, data revealed an increase in p21WAF1/Cip1 expression at the mRNA and protein levels in ML-1 cells in response to TPA.
Fig. 1.
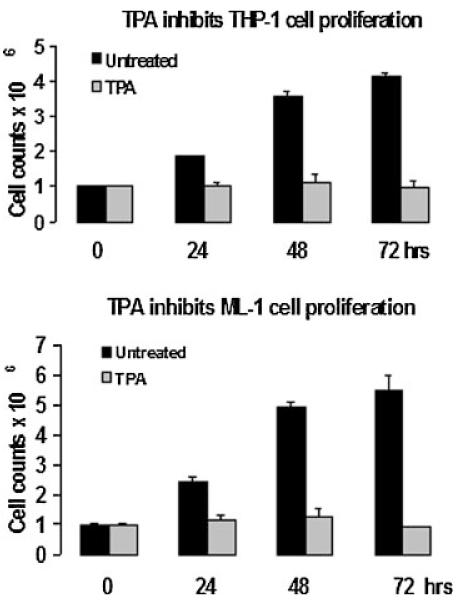
Effects of TPA on human myeloid cell proliferation. One million ML-1 or THP-1 cells were incubated in the presence or absence of 5 and 20 nM TPA respectively for the indicated times. Cells were harvested each day for 3 days and counted using a hemocytometer to determine the cell number in each sample.
TABLE 1.
mRNA Levels of Genes Involved in Cell Cycle Control
| Negative regulators | Fold change | Positive regulators | Fold change |
|---|---|---|---|
| CDKN1A (p21WAF1) | 3.78 | ANAPC5 | -2.62 |
| CYLD | 3.76 | ANAPC7 | -2.16 |
| BRCA2 | -2.20 | ||
| E2F2 | -2.17 | ||
| MCM2 | -3.89 | ||
| MCM5 | -2.30 | ||
| MCM6 | -3.14 | ||
| MYC | -6.36 | ||
| WEE1 | -2.10 | ||
| WT1 | -5.54 |
ML-1 cells were incubated in the growth medium in the presence or absence of TPA for 6 h. Total RNA was extracted purified, normalized, and analyzed by microarray analysis using a human genome U133 2.0 Plus Array Chip.
Fig. 2.
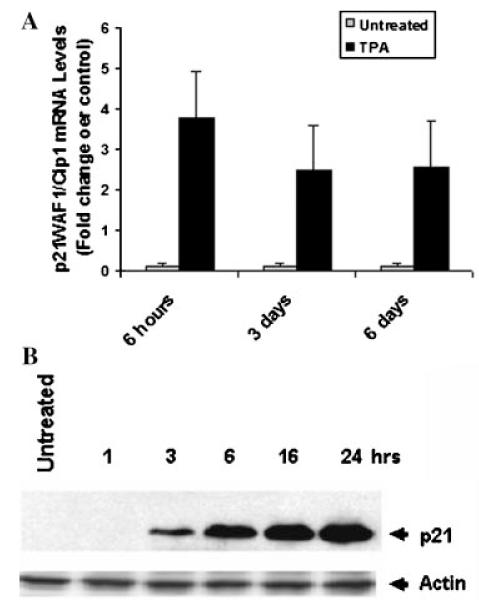
TPA induces p21WAF1/Cip1 expression in ML-cells. ML-1 cells were incubated in the presence or absence of 5 nM TPA for indicated times. A: p21WAF1/Cip1 mRNA levels were determined by examination of microarray analysis data. B: Total proteins were extracted and 40 μg of total protein were analyzed by Western blot analysis using mouse anti-human p21WAF1/Cip1 antibodies. The figure depicts representative results from one of three replicate experiments.
TPA induces p21WAF1/Cip1 expression via a PKC, ROS, and MAPK dependent mechanism
ROS-mediated Erk1/2 activation has been implicated in signaling p21WAF1/cip1 expression induced by TPA in human myeloid cells THP-1 (Traore et al., 2005). Using GF109203X (a PKC inhibitor), N-acetylcysteine, NAC (an ROS scavenger), and DPI (an inhibitor of NADH and NADPH dependent enzymes) we assessed the role of PKC and ROS in p21WAF1/Cip1 expression induced by TPA in ML-1 cells. Pretreatment with GF109203X, NAC, and DPI effectively blocked p21WAF1/Cip1 expression (Fig. 3). Moreover, p21WAF1/cip1 expression was also suppressed by U0126 (a MEK inhibitor) (Fig. 3). These findings indicated the involvement of PKC, ROS, and the MAPK cascade in p21WAF1/cip1 expression induced by TPA in ML-1 cells and support our previous report on the mechanism of TPA-induced growth arrest in THP-1 cells (Traore et al., 2005).
Fig. 3.
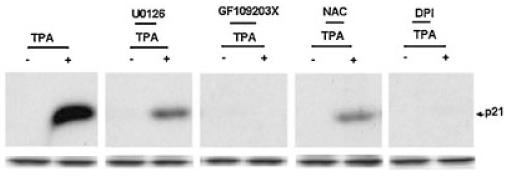
Effects of GF109203X, U0126, NAC, and DPI on p21WAF1/Cip1 protein expression. ML-1 cells were pretreated with 5 μM GF109203X (GF), 10 μM U0126, 10 μM DPI, or 25 mM NAC for 10 min followed by incubation with 5 nM TPA for 6 h. Total proteins were extracted and analyzed by Western blot analysis as indicated above.
TPA induces ROS generation in absence of p67-phox in ML-1 cells
Previous studies have indicated that TPA induces ROS generation in undifferentiated U937 and THP-1 cells (Datta et al., 2000; Traore et al., 2005). Data shown in Figure 4A illustrates the detection of extracellular H2O2 by luminol-dependent CL within minutes following addition of TPA to ML-1 cells and its inhibition by GF109203X and NAC assuring the involvement of PKC and the effectiveness of NAC as scavenger of ROS generated in response to TPA in ML-1 cells. Similarly, increased intracellular ROS was also detected using dihydroethidine-dependent fluorescence in ML-1 cells as a result of the addition of TPA (Fig. 4B).
Fig. 4.
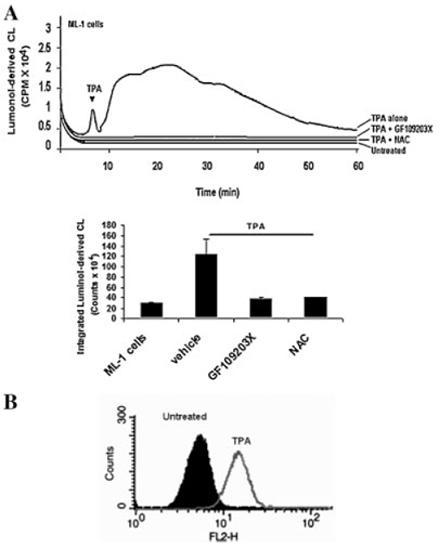
Role of PKC in TPA-induced ROS generation in ML-1 cells. The generation of ROS in ML-1 cells in response to TPA was assessed by: (A) luminol-derived CL or (B) flow cytometry analysis using flurophore hydroethidine. Cells were preincubated with GF109203X or NAC for 10 min followed by 5 nM TPA treatment for 50 min. Luminol-derived luminescence of 1 million cells was measured in Berthold luminometer for 1 h. The CL in (A) are CPM × 104 over the indicate time period. The data in bar graph is the CL from three cell samples integrated over 50 min (means ± SD, n = 3). Analysis of the data by one way ANOVA indicates that the effect of GF109203X and NAC was significant (P < 0.01). The luminol-dependent CL of untreated cells is at the level of the instrument background. B. Cells were preincubated in presence of hydroethidine for 5 min followed by incubated in the presence or in the absence of 5 nM TPA for 30 min. Cells were harvested, washed with PBS and analyzed by flow cytometry as indicated in materials and methods.
Major sources of ROS in differentiated myeloid cells include NADPH oxidase (Forman and Torres, 2001) and the METC (Rembish and Trush, 1994). Following the TPA-induced differentiation of ML-1 cells to macrophages, the mitochondria are also a cellular source of ROS (Li and Trush, 1998; Li et al., 1998; Li et al., 1999b). To determine the involvement of NADPH oxidase in the ROS generation induced by TPA, we examined the levels of p47-phox and p67-phox protein (two key components of NADPH oxidase) in undifferentiated (untreated) and 3 days TPA-treated THP-1 and ML-1 cells. Western blot analysis data shown in Figure 5 indicated a low level of p47-phox and a complete absence p67-phox expression in undifferentiated (untreated) ML-1 cells. However, both components p47-phox and p67-phox levels were detectable in undifferentiated THP-1 cells, and were further increased following differentiation in both THP1 and ML-1 cells. The expression of p47-phox and p67-phox observed following addition of TPA first occurs after 24 h of incubation with TPA in ML-1 cells (data not shown). In addition, to determine the involvement of mitochondria as the source of ROS we assessed the effect of DPI, rotenone and rhein (a competitive inhibitor of mitochondria complex I) (Kean et al., 1971; Floridi et al., 1989). As shown in Figure 6, DPI and rhein inhibited H2O2-dependent luminol-derived CL in both the THP1 and ML-1 cells following the addition of an equivalent concentration of TPA. Interestingly, rotenone a mitochondria complex I inhibitor increased luminol-derived CL induced by TPA in ML-1 cells, an event that is also blocked by DPI (Fig. 6). While DPI can inhibit both NADPH oxidase and complex I of the METC (Li and Trush, 1998), ML-1 cells lack the p67-phox component necessary for NADPH oxidase enzymatic activity (Fig. 5).
Fig. 5.
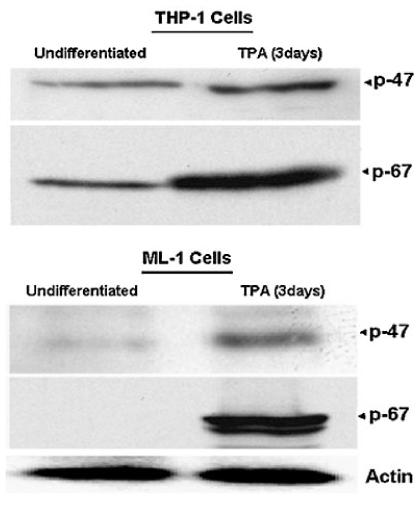
Determination of p47-phox and p67-phox expression in undifferentiated and differentiated ML-1 and THP-1 cells. ML-1 and THP-1 cells were incubated in growth medium in the presence or absence of 5 or 20 nM TPA respectively for 3 days. Total proteins were extracted and 40 μg of proteins were analyzed by Western blot analysis using anti-human p47-phox and p67-phox antibodies. The figure depicts representative results from one of three replicate experiments.
Fig. 6.
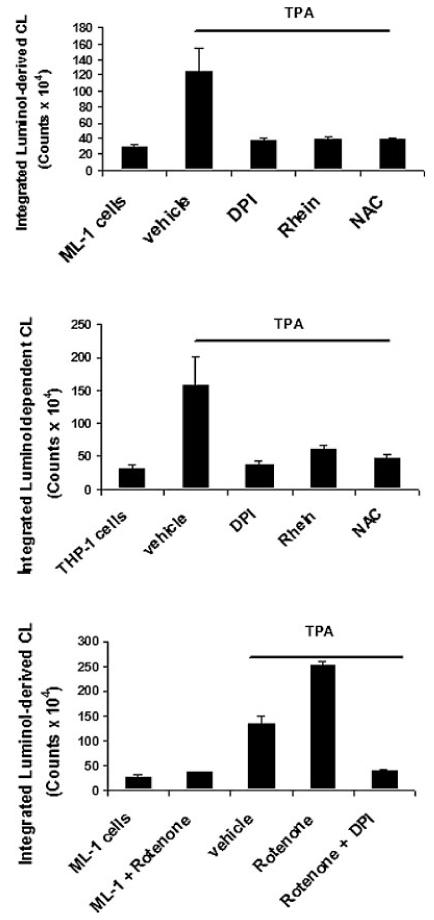
Effects of DPI, rotenone, rhein, and NAC on luminol-derived CL in ML-1 and THP-1 cells. Cells were preincubated with 10 μM DPI, 10 μM rotenone, 40 μM rhein, or 25 mM NAC for 10 min follows by incubation with 5 or 20 nM TPA respectively for 50 min. Luminoldependent luminescence of 1 million cells was measured in Berthold luminometer for 1 h. The CL are in CPM × 104 over the indicate time period. The data is the CL from three cell samples integrated over 50 min (means ± SD, n = 3). The luminol-dependent CL of untreated cells is at the level of the instrument background.
MnSOD, a key member of the mitochondrial defense system against METC-derived O2-· (Kim et al., 2005), expression was induced at both the mRNA and protein levels (Fig. 7). MnSOD expression was further linked to PKC, MAPK, and ROS as pretreatment with GF109203X, PD9059, U0126, and DPI also blocked MnSOD expression (Fig. 8). Taken together, the above findings indicated that TPA-induced ROS generation in ML-1 cells occurred in absence of p67-phox a key component of NADPH oxidase. The data also suggested mitochondria ETC at least in part as the source of the observed low level of ROS generated in ML-1 cells in response to TPA.
Fig. 7.
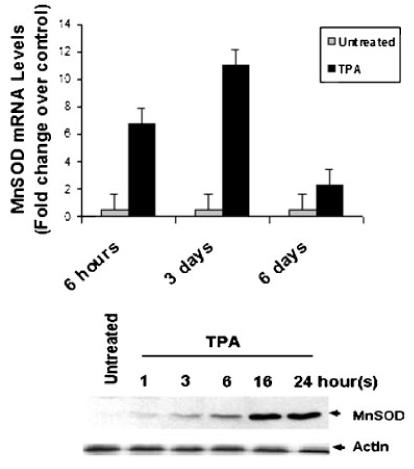
TPA induces MnSOD expression at mRNA and protein levels in ML-1 cells. Three million cells were incubated in the presence or in the absence of 5 nM TPA for various times. MnSOD mRNA levels were determined by examination of microarray analysis data. MnSOD protein levels were determined by Western blot analysis using mouse anti-human MnSOD antibodies. The figure depicts representative results from one of three replicate experiments.
Fig. 8.
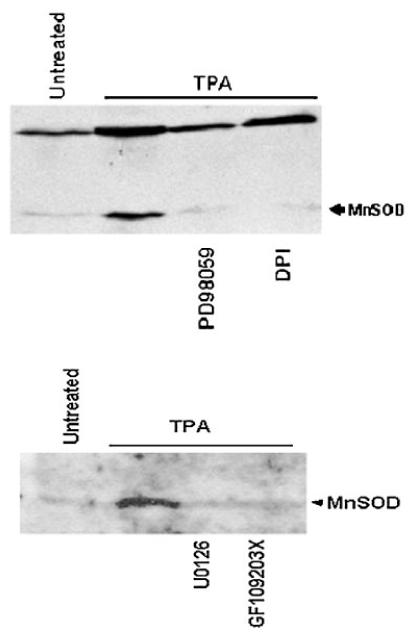
The effects of GF10920X, PD9059, U0126, and DPI on TPA induced MnSOD expression in ML-1 cells. Three million cells were pretreated with 5 μM GF109203X, 25 μM PD98059, 10 μM U0126, or 10 μM DPI for 20 min follow by incubation with 5 nM TPA for 24 h. MnSOD protein levels were determined by Western blot analysis as indicated above.
Kinetics of Erk1/2 phosphorylation induced by TPA correlates with ROS generation
As we have previously shown, Erk1/2 is important in signaling both the early events (growth inhibition) as well as the overall differentiation of ML-1 cells (He et al., 1999). H2O2 has been shown to play a role in the activation of MAPKs (p38, Erk1/2, and JNK) induced by extracellular stimuli including TNF-alpha, TPA, and lysophosphatidic acid (Lo et al., 1996; Bouaouina et al., 2004; Kamata et al., 2005; Traore et al., 2005; Traore et al., 2007). To understand the mechanism of Erk1/2 activation induced by TPA in ML-1 cells, we first examined the kinetics of Erk1/2 phosphorylation induced by TPA. Several independent experiments were conducted using Western blot and flow cytometry analysis. Treatment with TPA resulted in a marked increase in phosphorylation (activation) of Erk1/2 within 15 min of incubation with TPA (data not shown). The highest level of phospho-Erk1/2 induced by TPA was observed 1 h after incubation with TPA (Fig. 9A and B) subsequent to H2O2 generation induced by TPA (Fig. 4), which could be blocked by pretreatment with NAC (Fig. 10A). The level of Erk1/2 phosphorylation 6 h after TPA treatment was much less than at 3 h and correlated with increase in DUSP6 protein levels (Fig. 9A and B). In addition, incubation with exogenous H2O2 also induced Erk1/2 phosphorylation (Fig. 9B), assuring the effectiveness of H2O2 in mediating of Erk1/2 phosphorylation in ML-1 cells. These observations indicated that Erk1/2 activation (phosphorylation) in response to TPA is rapid and required ROS generation in ML-1 cells. However, the Erk1/2 deactivation (dephosphorylation) is a slow process and occurs 3–6 h after TPA treatment.
Fig. 9.
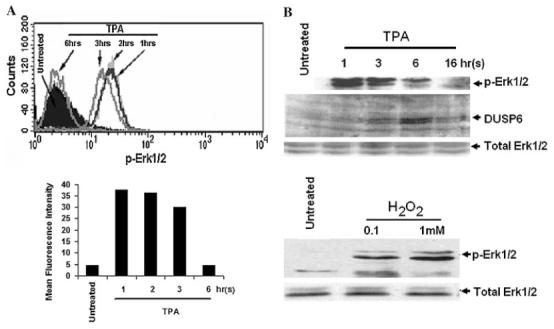
Erk1/2 activation induces by TPA and H2O2 treatment. Three millions cells were incubated in the presence of TPA for various times, or in the presence of 0.1 or 1 mM H2O2 for 1 h. Cells were harvested, and the levels of phospho-Erk1/2 were determined (A) by flow cytometry analysis or (B) by Western blot analysis as described in the Section “Materials and Methods.” The figure depicts representative results from one of three replicate experiments.
Fig. 10.
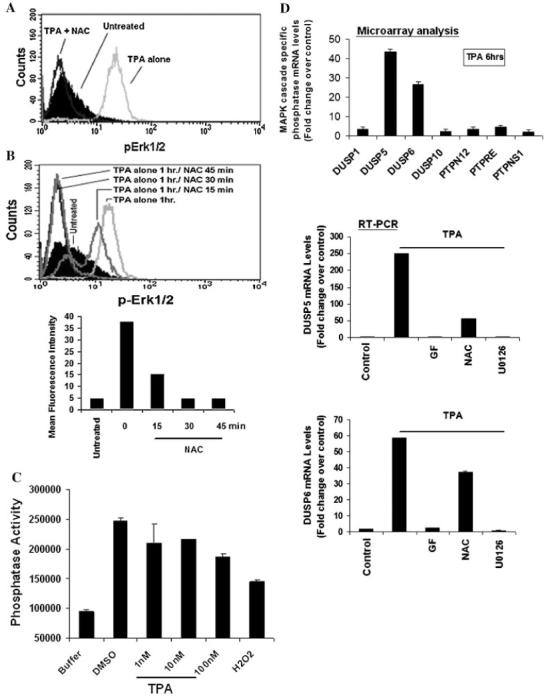
ROS-dependent inactivation of Erk 1/2-directed phosp hatases mediates Erk 1/2 activation induced by TPA.Using flow cytometry analysis, a cell based assay was used to assess the levels of intracellular phospho-Erk1/2 in various experimental conditions. A: Cells were preincubated in the presence of NAC for 10 min followed by incubation with TPA for 1 h. B: Cells were incubated in media containing TPA for 1 h followed by incubation with 25 mM NAC for 15, 30, or 45 min. Cells were then harvested, washed, fixed, permeabilized, and stained using (Alexa-Fluor 488)-labeled anti-phospho-Erk1/2 and assessed by flow cytometry. Data were analyzed by ModFILT statistical software. The figure depicts representative results from one of three replicate experiments. C: Cells were incubated in media containing TPA or 100 nM H2O2 for 1 h. After harvest, cells were lyzed in Triton X-100 containing buffer and analyzed for tyrosine phosphatase activity using FDP-derived fluorescence phosphatase assay. The fluorescent intensity were measure at Ex/Em = (485 ± 20)/(528 ± 20). D: ML-1 cells were incubated with TPA or DMSO (vehicle) in the presence or absence of 5 mM GF109203X, 10 μM U0126, or 25 mM NAC for 6 h. Total RNA were extract and 2 μg of total RNA were analyzed by RT-PCR as described in materials and methods sections. Results are expressed as means ± SD (n = 3).
TPA and H2O2 inactivates Erk1/2-directed phosphatase activity
Erk1/2 activity is controlled by a critical balance between the activities of an activating kinase MEK and inhibitory phosphatases activity (Gronda et al., 2001; Nika et al., 2004). Given the reported sensitivity of protein phosphatases to the cellular redox state (Lo et al., 1996; Lee et al., 1998; Kamata et al., 2005), we examined whether the modulation of Erk1/2-directed phosphatases activity by ROS played a role in TPA-induced Erk1/2 increased phosphorylation (activation) in ML-1 cells. A cell-based assay was utilized to assess Erk1/2 directed phosphate activity by monitoring the kinetics of intracellular Erk1/2 dephosphorylation and in the presence or absence of the anti-oxidant (reducing agent) NAC. ML-1 cells were incubated in media containing TPA for 1 h, to reach maximal levels of intracellular Erk1/2 phosphorylation (Fig. 10). One hour post-TPA-treated cells were then incubated in the presence of 25 mM NAC for various times and the levels of phospho-Erk1/2 were determined by flow cytometry (Fig. 10B). The kinetics of Erk1/2 dephosphorylation in the absence (Fig. 9A) or in the presence (Fig. 10B) of NAC would then be indicative of Erk1/2-directed phosphatase activity. As shown in Figure 10B, the post-TPA-treatment with NAC stimulated a marked and rapid dephosphorylation of Erk1/2 within minutes of treatment with NAC, presumably in response to a rapid regeneration of the phosphatase activity by NAC. In addition, a direct analysis of phosphotyrosine phosphatase activity revealed a dose dependent decrease of cellular phosphotyrosine phosphatase activity in response to TPA and H2O2 in ML-1 cells (Fig. 10C). Together, our data suggested that Erk1/2 activation induced by TPA occurred at least in part in response to a ROS-mediated inactivation of Erk1/2-directed phosphatase activity in ML-1 cells.
DUSP5 and DUSP6 expression induced by TPA occurs in response Erk1/2 activation
To further understand the mechanism of Erk1/2 regulation in response TPA, we examined whether Erk1/2 inactivation which occurred 6 h after its activation by TPA was associated with de novo synthesis of delayed Erk1/2-driected phosphatase in ML-1. Examination of microarray analysis data and RT-PCR data revealed a significant induction of DUSP5 and DUSP6 (dual specific phosphatases) expression at mRNA levels 6 h after TPA treatment (Fig. 10D). This event could be blocked by pretreatment with PKC and MEK inhibitors, and by NAC, ROS scavenger (Fig. 10D), suggesting the role of PKC and Erk1/2 activation in this process. Furthermore, Western blot analysis data shown in Figure 9B indicates that the kinetic of Erk1/2 dephosphorylation (inactivation) which occurs 3–6 h after treatment with TPA correlates with an increase accumulation of DUSP6 protein in ML-1 cells. These observations suggested that the induction of DUSP6 protein expression, 3–6 h following Erk1/2 activation may have occurred to regulate Erk1/2 activity in ML-1 cells.
Discussion
A key role of low-level cellular ROS that has recently developed is in mediating signal transduction systems (Lo et al., 1996; Bouaouina et al., 2004; Kamata et al., 2005; Traore et al., 2005; Traore et al., 2007). For example, H2O2 generation in response to extracellular stimuli has been implicated as a second messenger that initiates activation of protein kinase cascades (Robinson et al., 1999). Intracellular ROS has been implicated in playing a role in the signal transduction of various biological processes (Fernandez et al., 1995; Goossens et al., 1999; Li et al., 1999a; Bogoyevitch et al., 2000; Majumder et al., 2000; Forman and Torres, 2001; Droge, 2002; Forman et al., 2002; Butow and Avadhani, 2004; Cadenas, 2004; Darley-Usmar, 2004; Ichikawa et al., 2004; Andreyev et al., 2005; Kim et al., 2005). Quagliaro et al. (Quagliaro et al., 2005) have shown that intermittent high glucose enhances mitochondria-derived ROS to signal ICAM-1, VCAM-1, and E-selectin expression in human umbilical vein endothelial cells, and support our previous finding that TPA-induced ROS signals ICAM-1 expression in ML-1 cells (Traore et al., 2007).
Growth arrest is a critical event in order for myeloid cell differentiation to occur (Tanigawa et al., 1983; Selvakumaran et al., 1996; Adachi et al., 1997; Kovanen et al., 2000; Gazitt et al., 2001). In this study, we demonstrate that TPA stimulates ROS generation to signal growth arrest in ML-1 cells. Our suggestion that mitochondria may be the source of the ROS is based on the observations that undifferentiated ML-1 cells lack a key component of NADPH oxidase, p67-phox; second, this ROS generation is inhibited by agents that effectively inhibit complex I activity, DPI and rhein (Kean et al., 1971; Floridi et al., 1989). In data shown in Figure 6, rotenone increased luminol-derived CL in ML-1 cells in response to TPA was blocked by DPI. This observation is consistent with previous reports that rotenone can enhances mitochondria-derived ROS generation in many cell types including dopaminergic primary culture (Li et al., 2003; Radad et al., 2006). Moreover, MnSOD, a key mitochondrial enzyme against METC-derived O2-· was induced in response to TPA. The induction of MnSOD expression has previously been reported in other cell lines when stimulated with TPA (Kim et al., 1999; Majumder et al., 2001; Porntadavity et al., 2001). MnSOD expression induced by TPA in ML-1 cells required not only PKC but also was dependent on MAPK activity.
The data presented in this and other previous studies suggest that TPA signaling is transduced via various isoforms of PKC (Darbon et al., 1986; Datta et al., 2000; Traore et al., 2005). Many different isoforms of PKC are known to interact with components of the mitochondria (Fernandez et al., 1995; Majumder et al., 2000; Majumder et al., 2001). We previously reported that PKC could interact with mitochondria in rat alveolar macrophages (Rembish et al., 1994). Our present as well as our previous studies have demonstrated that PKC inhibitors block both ROS generation as well as downstream signaling events in both THP-1 and ML-1 cells (Traore et al., 2005; Traore et al., 2007).
Much of our current understanding of ROS-dependent signaling is based on experiments in which exogenous H2O2 has been added to cells (Guyton et al., 1996a, 1996b, 1996c, 1996d). H2O2 is a diffusible form of ROS that can transverse from the outside in or the inside out of cells. The H2O2-dependent extracellular luminol-derived CL is consistent with PKC-stimulated mitochondrial-derived H2O2 being able to diffuse from the mitochondria, passing through the cytoplasm and crossing the plasma membrane. As this ROS is passing through the cell it can then interact with target molecules. One mechanism by which ROS can regulate cellular signaling is via interaction with critical sites of redox sensitive proteins (Kamata et al., 2005).
Many redox sensitive proteins and enzymes contain redox sensitive cysteine thiolate anions (Cys-S-) (Georgiou and Masip, 2003). H2O2 can oxidize redox-sensitive cysteine residues to cysteine sulfenic acid or a disulfide that can be readily reduced back to cysteine by various cellular reductants (Lee et al., 1998; Rhee et al., 2000). Protein tyrosine phosphatases (PTPs) contain this essential cysteine residue in their active sites. With a pKa value between 4.7 and 5.4, this make them susceptible to inactivation by low level of oxidants such as H2O2 (Lee et al., 1998; Robinson et al., 1999; Rhee et al., 2000; Bouaouina et al., 2004; Kamata et al., 2005; Rinna et al., 2006). Inactivation of MAPK specific phosphatases in response to H2O2 has been shown to stimulate the activation of MAPK family members (JNK, p38, and Erk1/2) induced by various extracellular stimuli (Lee et al., 1998; Robinson et al., 1999; Rhee et al., 2000; Bouaouina et al., 2004; Kamata et al., 2005). In this study, we have demonstrated that ROS (H2O2) generated in mitochondria response to TPA transiently inactivates Erk1/2-directed phosphatases activity which stimulates Erk1/2 phosphorylation in ML-1 cells. Similarly, in cardiomyocytes, mitochondrial ROS was suggested to initiate p38 phosphorylation in response to hypoxia (Kulisz et al., 2002) Moreover, in phagocytic cells, ROS was shown to mediate TPA-regulated tyrosine phosphorylation and phopholipase C activation by inhibiting phosphotyrosine phosphatase activity (Zor et al., 1993). Treatment with NAC (reductant) quickly restored the total phosphatases activity resulting to a rapid dephosphorylation of Erk1/2 in ML-1 cells. The restoration of the phosphatase activity by NAC may have occurred by the regeneration of sulfydryl groups on critical cysteines in these phosphatases previously oxidized by H2O2. This observation is further supported by previous report that oxidized and purified PTP1B enzyme could be effectively reactivated by treatment with thioredoxin (Trx) (Lee et al., 1998). Thus, Trx might be a physiological electron donor for PTP1B as well as for other PTPs (Lee et al., 1998). Furthermore, the expression of de novo Erk1/2-directed phosphatase protein DUSP6, 3–6 h following Erk1/2 activation could have occurred in response to lost phosphatase activity to regulate Erk1/2 activity in ML-1 cells. Thus, Erk1/2-directed phosphatases appear to function as critical cellular “molecular switches.”
In conclusion, the findings of this study demonstrate that following the addition of TPA to ML-1 cells that there is a PKC-dependent generation of cellular ROS. This ROS (H2O2) can then stimulate Erk1/2 activation via a redox mechanism associated with a reversible inactivation of Erk1/2-directed phosphatases (Fig. 11). This leads to the downstream activation of p21 expression resulting in cell growth inhibition and ICAM1 expression (Traore et al., 2005; Hu and Mivechi, 2006; Traore et al., 2007).
Fig. 11.
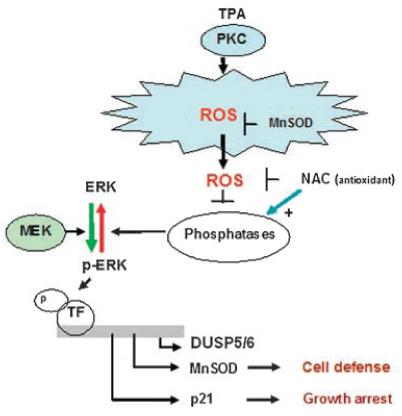
Proposed mechanism of TPA-induced Erk1/2 regulation during early events of ML-1 cell differentiation to macrophages.
Acknowledgments
We wish to acknowledge the financial support of the National Institute of Environmental Health Sciences: RO1 ES03760 and training grant T32 ES 07141 and a pilot grant from the Johns Hopkins Center in Urban Environmental Health (P30 ES 03819) and NIH grants HL081205 (S.B.) and GM079239 (S.B.).
Contract grant sponsor: National Institute of Environmental Health Sciences (NIEHS)
Contract grant numbers: T32 ES 07141, RO1 ES 03760, HL081205.
Contract grant sponsor: The Johns Hopkins Center in Urban Environmental Health
Contract grant number: P30 ES 03819.
Abbreviations
- TPA
12-O-tetradecanoyl-phorbol-13-acetate
- NAC
N-acetyl cysteine
- ROS
reactive oxygen species
- Erk
extracellular signal-regulated kinase
Literature Cited
- Abe J, Kusuhara M, Ulevitch RJ, Berk BC, Lee JD. Big mitogen-activated protein kinase 1 (BMK1) is a redox-sensitive kinase. J Biol Chem. 1996;271:16586–16590. doi: 10.1074/jbc.271.28.16586. [DOI] [PubMed] [Google Scholar]
- Adachi M, Roussel MF, Havenith K, Sherr CJ. Features of macrophage differentiation induced by p19INK4d, a specific inhibitor of cyclin D-dependent kinases. Blood. 1997;90:126–137. [PubMed] [Google Scholar]
- Agarwal S, Sohal RS. Aging and protein oxidative damage. Mech Ageing Dev. 1994a;75:11–19. doi: 10.1016/0047-6374(94)90024-8. [DOI] [PubMed] [Google Scholar]
- Agarwal S, Sohal RS. DNA oxidative damage and life expectancy in houseflies. Proc Natl Acad Sci USA. 1994b;91:12332–12335. doi: 10.1073/pnas.91.25.12332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi M, Hachiya M, Osawa Y, Spirin K, Suzuki G, Koeffler HP. Irradiation induces WAF1 expression through a p53-independent pathway in KG-1 cells. J Biol Chem. 1995;270:19181–19187. doi: 10.1074/jbc.270.32.19181. [DOI] [PubMed] [Google Scholar]
- Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 2005;70:200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, Rhee SG. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem. 1997;272:217–221. [PubMed] [Google Scholar]
- Biggs JR, Kudlow JE, Kraft AS. The role of the transcription factor Sp1 in regulating the expression of the WAF1/CIP1 gene in U937 leukemic cells. J Biol Chem. 1996;271:901–906. doi: 10.1074/jbc.271.2.901. [DOI] [PubMed] [Google Scholar]
- Bogoyevitch MA, Ng DC, Court NW, Draper KA, Dhillon A, Abas L. Intact mitochondrial electron transport function is essential for signalling by hydrogen peroxide in cardiac myocytes. J Mol Cell Cardiol. 2000;32:1469–1480. doi: 10.1006/jmcc.2000.1187. [DOI] [PubMed] [Google Scholar]
- Bouaouina M, Blouin E, Halbwachs-Mecarelli L, Lesavre P, Rieu P. TNF-induced beta2 integrin activation involves Src kinases and a redox-regulated activation of p38 MAPK. J Immunol. 2004;173:1313–1320. doi: 10.4049/jimmunol.173.2.1313. [DOI] [PubMed] [Google Scholar]
- Butow RA, Avadhani NG. Mitochondrial signaling: The retrograde response. Mol Cell. 2004;14:1–15. doi: 10.1016/s1097-2765(04)00179-0. [DOI] [PubMed] [Google Scholar]
- Cadenas E. Mitochondrial free radical production and cell signaling. Mol Aspects Med. 2004;25:17–26. doi: 10.1016/j.mam.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Chen Q, Olashaw N, Wu J. Participation of reactive oxygen species in the lysophosphatidic acid-stimulated mitogen-activated protein kinase kinase activation pathway. J Biol Chem. 1995;270:28499–28502. doi: 10.1074/jbc.270.48.28499. [DOI] [PubMed] [Google Scholar]
- Darbon JM, Issandou M, Delassus F, Bayard F. Phorbol esters induce both intracellular translocation and down-regulation of protein kinase C in MCF-7 cells. Biochem Biophys Res Commun. 1986;137:1159–1166. doi: 10.1016/0006-291x(86)90347-5. [DOI] [PubMed] [Google Scholar]
- Darley-Usmar V. The powerhouse takes control of the cell; the role of mitochondria in signal transduction. Free Radic Biol Med. 2004;37:753–754. doi: 10.1016/j.freeradbiomed.2004.05.026. [DOI] [PubMed] [Google Scholar]
- Datta R, Yoshinaga K, Kaneki M, Pandey P, Kufe D. Phorbol ester-induced generation of reactive oxygen species is protein kinase cbeta-dependent and required for SAPK activation. J Biol Chem. 2000;275:41000–41003. doi: 10.1074/jbc.M009322200. [DOI] [PubMed] [Google Scholar]
- Datto MB, Li Y, Panus JF, Howe DJ, Xiong Y, Wang XF. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc Natl Acad Sci USA. 1995;92:5545–5549. doi: 10.1073/pnas.92.12.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: Evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- Devin A, Rigoulet M. Mechanisms of mitochondrial response to variations in energy demand in eukaryotic cells. Am Physiol Cell Physiol. 2006;292:C52–C58. doi: 10.1152/ajpcell.00208.2006. [DOI] [PubMed] [Google Scholar]
- Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Fernandez E, Cuenca N, Garcia M, De Juan J. Two types of mitochondria are evidenced by protein kinase C immunoreactivity in the Muller cells of the carp retina. Neurosci Lett. 1995;183:202–205. doi: 10.1016/0304-3940(94)11151-8. [DOI] [PubMed] [Google Scholar]
- Floridi A, Castiglione S, Bianchi C. Sites of inhibition of mitochondrial electron transport by rhein. Biochem Pharmacol. 1989;38:743–751. doi: 10.1016/0006-2952(89)90226-8. [DOI] [PubMed] [Google Scholar]
- Forman HJ, Torres M. Redox signaling in macrophages. Mol Aspects Med. 2001;22:189–216. doi: 10.1016/s0098-2997(01)00010-3. [DOI] [PubMed] [Google Scholar]
- Forman HJ, Torres M. Reactive oxygen species and cell signaling: Respiratory burst in macrophage signaling. Am J Respir Crit Care Med. 2002;166:S4–S8. doi: 10.1164/rccm.2206007. [DOI] [PubMed] [Google Scholar]
- Forman HJ, Torres M, Fukuto J. Redox signaling. Mol Cell Biochem. 2002;234-235:49–62. [PubMed] [Google Scholar]
- Gazitt Y, Reddy SV, Alcantara O, Yang J, Boldt DH. A new molecular role for iron in regulation of cell cycling and differentiation of HL-60 human leukemia cells: Iron is required for transcription of p21(WAF1/CIP1) in cells induced by phorbol myristate acetate. J Cell Physiol. 2001;187:124–135. doi: 10.1002/1097-4652(2001)9999:9999<::AID-JCP1061>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Georgiou G, Masip L. Biochemistry. An overoxidation journey with a return ticket. Science. 2003;300:592–594. doi: 10.1126/science.1084976. [DOI] [PubMed] [Google Scholar]
- Goossens V, De Vos K, Vercammen D, Steemans M, Vancompernolle K, Fiers W, Vandenabeele P, Grooten J. Redox regulation of TNF signaling. Biofactors. 1999;10:145–156. doi: 10.1002/biof.5520100210. [DOI] [PubMed] [Google Scholar]
- Gronda M, Arab S, Iafrate B, Suzuki H, Zanke BW. Hematopoietic protein tyrosine phosphatase suppresses extracellular stimulus-regulated kinase activation. Mol Cell Biol. 2001;21:6851–6858. doi: 10.1128/MCB.21.20.6851-6858.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyton KZ, Gorospe M, Kensler TW, Holbrook NJ. Mitogen-activated protein kinase (MAPK) activation by butylated hydroxytoluene hydroperoxide: Implications for cellular survival and tumor promotion. Cancer Res. 1996a;56:3480–3485. [PubMed] [Google Scholar]
- Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J Biol Chem. 1996b;271:4138–4142. doi: 10.1074/jbc.271.8.4138. [DOI] [PubMed] [Google Scholar]
- Guyton KZ, Spitz DR, Holbrook NJ. Expression of stress response genes GADD153, c-jun, and heme oxygenase-1 in H2O2- and O2-resistant fibroblasts. Free Radic Biol Med. 1996c;20:735–741. doi: 10.1016/0891-5849(95)02151-5. [DOI] [PubMed] [Google Scholar]
- Guyton KZ, Xu Q, Holbrook NJ. Induction of the mammalian stress response gene GADD153 by oxidative stress: Role of AP-1 element. Biochem J. 1996d;314:547–554. doi: 10.1042/bj3140547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Wang X, Gorospe M, Holbrook NJ, Trush MA. Phorbol ester-induced mononuclear cell differentiation is blocked by the mitogen-activated protein kinase kinase (MEK) inhibitor PD98059. Cell Growth Differ. 1999;10:307–315. [PubMed] [Google Scholar]
- Hu Y, Mivechi NF. Association and regulation of heat shock transcription factor 4b with both extracellular signal-regulated kinase mitogen-activated protein kinase and dual-specificity tyrosine phosphatase DU SP26. Mol Cell Biol. 2006;26:3282–3294. doi: 10.1128/MCB.26.8.3282-3294.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa H, Kokura S, Aw TY. Role of endothelial mitochondria in oxidant production and modulation of neutrophil adherence. J Vasc Res. 2004;41:432–444. doi: 10.1159/000081466. [DOI] [PubMed] [Google Scholar]
- Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- Kean EA, Gutman M, Singer TP. Studies on the respiratory chain-linked nicotinamide adenine dinucleotide dehydrogenase. XXII. Rhein, a competitive inhibitor of the dehydrogenase. J Biol Chem. 1971;246:2346–2353. [PubMed] [Google Scholar]
- Kim A, Murphy MP, Oberley TD. Mitochondrial redox state regulates transcription of the nuclear-encoded mitochondrial protein manganese superoxide dismutase: A proposed adaptive response to mitochondrial redox imbalance. Free Radic Biol Med. 2005;38:644–654. doi: 10.1016/j.freeradbiomed.2004.10.030. [DOI] [PubMed] [Google Scholar]
- Kim HP, Roe JH, Chock PB, Yim MB. Transcriptional activation of the human manganese superoxide dismutase gene mediated by tetradecanoylphorbol acetate. J Biol Chem. 1999;274:37455–37460. doi: 10.1074/jbc.274.52.37455. [DOI] [PubMed] [Google Scholar]
- Kovanen PE, Junttila I, Takaluoma K, Saharinen P, Valmu L, Li W, Silvennoinen O. Regulation of Jak2 tyrosine kinase by protein kinase C during macrophage differentiation of IL-3-dependent myeloid progenitor cells. Blood. 2000;95:1626–1632. [PubMed] [Google Scholar]
- Kulisz A, Chen N, Chandel NS, Shao Z, Schumacker PT. Mitochondrial ROS initiate phosphorylation of p38 MAP kinase during hypoxia in cardiomyocytes. Am J Physiol Lung Cell Mol Physiol. 2002;282:L1324–L1329. doi: 10.1152/ajplung.00326.2001. [DOI] [PubMed] [Google Scholar]
- Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- Li L, Lorenzo PS, Bogi K, Blumberg PM, Yuspa SH. Protein kinase Cdelta targets mitochondria, alters mitochondrial membrane potential, and induces apoptosis in normal and neoplastic keratinocytes when overexpressed by an adenoviral vector. Mol Cell Biol. 1999a;19:8547–8558. doi: 10.1128/mcb.19.12.8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, Robinson JP. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem. 2003;278:8516–8525. doi: 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- Li Y, Trush MA. Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem Biophys Res Commun. 1998;253:295–299. doi: 10.1006/bbrc.1998.9729. [DOI] [PubMed] [Google Scholar]
- Li Y, Zhu H, Kuppusamy P, Roubaud V, Zweier JL, Trush MA. Validation of lucigenin (bis-N-methylacridinium) as a chemilumigenic probe for detecting superoxide anion radical production by enzymatic and cellular systems. J Biol Chem. 1998;273:2015–2023. doi: 10.1074/jbc.273.4.2015. [DOI] [PubMed] [Google Scholar]
- Li Y, Zhu H, Trush MA. Detection of mitochondria-derived reactive oxygen species production by the chemilumigenic probes lucigenin and luminol. Biochim Biophys Acta. 1999b;1428:1–12. doi: 10.1016/s0304-4165(99)00040-9. [DOI] [PubMed] [Google Scholar]
- Lo YY, Wong JM, Cruz TF. Reactive oxygen species mediate cytokine activation of c-Jun NH2-terminal kinases. J Biol Chem. 1996b;271:15703–15707. doi: 10.1074/jbc.271.26.15703. [DOI] [PubMed] [Google Scholar]
- Majumder PK, Mishra NC, Sun X, Bharti A, Kharbanda S, Saxena S, Kufe D. Targeting of protein kinase C delta to mitochondria in the oxidative stress response. Cell Growth Differ. 2001;12:465–470. [PubMed] [Google Scholar]
- Majumder PK, Pandey P, Sun X, Cheng K, Datta R, Saxena S, Kharbanda S, Kufe D. Mitochondrial translocation of protein kinase C delta in phorbol ester-induced Cytochrome c release and apoptosis. J Biol Chem. 2000;275:21793–21796. doi: 10.1074/jbc.C000048200. [DOI] [PubMed] [Google Scholar]
- Michieli P, Chedid M, Lin D, Pierce JH, Mercer WE, Givol D. Induction of WAF1/CIP1 by a p53-independent pathway. Cancer Res. 1994;54:3391–3395. [PubMed] [Google Scholar]
- Nika K, Hyunh H, Williams S, Paul S, Bottini N, Tasken K, Lombroso PJ, Mustelin T. Haematopoietic protein tyrosine phosphatase (HePTP) phosphorylation by cAMP-dependent protein kinase in T-cells: Dynamics and subcellular location. Biochem J. 2004;378:335–342. doi: 10.1042/BJ20031244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porntadavity S, Xu Y, Kiningham K, Rangnekar VM, Prachayasitikul V, Clair DK. TPA-activated transcription of the human MnSOD gene: Role of transcription factors Sp-1 and Egr-1. DNA Cell Biol. 2001;20:473–481. doi: 10.1089/104454901316976109. [DOI] [PubMed] [Google Scholar]
- Quagliaro L, Piconi L, Assaloni R, Da Ros R, Maier A, Zuodar G, Ceriello A. Intermittent high glucose enhances ICAM-1, VCAM-1 and E-selectin expression in human umbilical vein endothelial cells in culture: The distinct role of protein kinase C and mitochondrial superoxide production. Atherosclerosis. 2005;183:259–267. doi: 10.1016/j.atherosclerosis.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Radad K, Rausch WD, Gille G. Rotenone induces cell death in primary dopaminergic culture by increasing ROS production and inhibiting mitochondrial respiration. Neurochem Int. 2006;49:379–386. doi: 10.1016/j.neuint.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Reid MB, Durham WJ. Generation of reactive oxygen and nitrogen species in contracting skeletal muscle: Potential impact on aging. Ann NY Acad Sci. 2002;959:108–116. doi: 10.1111/j.1749-6632.2002.tb02087.x. [DOI] [PubMed] [Google Scholar]
- Rembish SJ, Trush MA. Further evidence that lucigenin-derived chemiluminescence monitors mitochondrial superoxide generation in rat alveolar macrophages. Free Radic Biol Med. 1994;17:117–126. doi: 10.1016/0891-5849(94)90109-0. [DOI] [PubMed] [Google Scholar]
- Rembish SJ, Yang Y, Trush MA. Inhibition of mitochondrial superoxide generation in rat alveolar macrophages by 12-O-tetradecanoylphorbol-13-acetate: Potential role of protein kinase C. Res Commun Mol Pathol Pharmacol. 1994;85:115–129. [PubMed] [Google Scholar]
- Rhee SG, Bae YS, Lee SR, Kwon J. Hydrogen peroxide: A key messenger that modulates protein phosphorylation through cysteine oxidation. Sci STKE 2000. 2000:PE1. doi: 10.1126/stke.2000.53.pe1. [DOI] [PubMed] [Google Scholar]
- Rinna A, Torres M, Forman HJ. Stimulation of the alveolar macrophage respiratory burst by ADP causes selective glutathionylation of protein tyrosine phosphatase 1B. Free Radic Biol Med. 2006;41:86–91. doi: 10.1016/j.freeradbiomed.2006.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson KA, Stewart CA, Pye QN, Nguyen X, Kenney L, Salzman S, Floyd RA, Hensley K. Redox-sensitive protein phosphatase activity regulates the phosphorylation state of p38 protein kinase in primary astrocyte culture. J Neurosci Res. 1999;55:724–732. doi: 10.1002/(SICI)1097-4547(19990315)55:6<724::AID-JNR7>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Selvakumaran M, Liebermann D, Hoffman B. The proto-oncogene c-myc blocks myeloid differentiation independently of its target gene ornithine decarboxylase. Blood. 1996;88:1248–1255. [PubMed] [Google Scholar]
- Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- Tanigawa T, Takayama H, Takagi A, Kimura G. Cell growth and differentiation in vitro in mouse macrophages transformed by a tsA mutant of simian virus 40. I. Cellular response in proliferative and phagocytic activities to the shift of temperature differs depending on the culture state in mouse bone marrow cells transformed by the tsA640 mutant of simian virus 40. J Cell Physiol. 1983;116:303–310. doi: 10.1002/jcp.1041160307. [DOI] [PubMed] [Google Scholar]
- Traore K, Sharma RB, Burek CL, Trush MA. Role of ROS and MAPK in TPA-induced ICAM-1 expression in the myeloid ML-1 cell line. J Cell Biochem. 2007;100:1010–1021. doi: 10.1002/jcb.21101. [DOI] [PubMed] [Google Scholar]
- Traore K, Trush MA, George M, Jr, Spannhake EW, Anderson W, Asseffa A. Signal transduction of phorbol 12-myristate 13-acetate (PMA)-induced growth inhibition of human monocytic leukemia THP-1 cells is reactive oxygen dependent. Leuk Res. 2005;29:863–879. doi: 10.1016/j.leukres.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Weiss H, Friedrich T, Hofhaus G, Preis D. The respiratory-chain NADH dehydrogenase (complex I) of mitochondria. Eur J Biochem. 1991;197:563–576. doi: 10.1111/j.1432-1033.1991.tb15945.x. [DOI] [PubMed] [Google Scholar]
- Yoshizumi M, Abe J, Haendeler J, Huang Q, Berk BC. Src and Cas mediate JNK activation but not ERK1/2 and p38 kinases by reactive oxygen species. J Biol Chem. 2000;275:11706–11712. doi: 10.1074/jbc.275.16.11706. [DOI] [PubMed] [Google Scholar]
- Zor U, Ferber E, Gergely P, Szücs K, Dombrádi V, Goldman R. Reactive oxygen species mediate phorbol ester-regulated tyrosine phosphorylation and phospholipase A2 activation: Potentiation by vanadate. Biochem J. 1993;295:879–888. doi: 10.1042/bj2950879. [DOI] [PMC free article] [PubMed] [Google Scholar]