

SUMMARY
Stim1 responds to depletion of ER Ca2+ stores by rearranging from tubular structures throughout the ER into punctate structures near the plasma membrane, where it activates Orai store-operated Ca2+ entry (SOCE) channels. However, the mechanism and structural determinants of the localization and reversal of Stim1 puncta formation are poorly understood. Using HEK293 cells expressing enhanced yellow fluorescent protein-tagged Stim1 (EYFP-Stim1), we show that the basis for SOCE termination is reversal of punctate Stim1 localization which absolutely depends on SOCE-dependent store refilling. We also describe rapid, store-independent reversal of EYFP-Stim1 punctae by the myosin light chain kinase (MLCK) inhibitor ML-9. ML-9 similarly inhibited SOCE and Ca2+ release-activated Ca2+ current. Reversal by ML-9 resulted in full re-establishment of tubular EYFP-Stim1 localization. A constitutively active EF-hand mutant of EYFP-Stim1 was also reversed by ML-9, regardless of Ca2+ store content. Inhibition by ML-9 was not due to MLCK inhibition, since other MLCK-inhibitors had no effect. Lastly, we provide evidence that EYFP-Stim1 punctae form in specific predetermined cellular loci. We conclude that SOCE is tightly coupled to Stim1 puncta formation, and both SOCE and puncta formation involve a dynamic, reversible signaling complex that likely consists of components in addition to Stim1 and Orai channels.
Keywords: Stim1, ML-9, inhibitors, store-operated channels
INTRODUCTION
Store-operated Ca2+ entry (SOCE) is initiated when intracellular stores, located in the endoplasmic reticulum (ER) or a specialized component of it, release their stored Ca2+ and become depleted (Parekh and Putney, Jr. 2005; Smyth et al. 2006). ER Ca2+ stores can become depleted physiologically as a consequence of signaling mechanisms, such as those involving G-protein coupled or tyrosine kinase receptors, that result in activation of inositol 1,4,5-trisphosphate receptors (IP3R), intracellular Ca2+ release channels located in the ER membrane. Ca2+ that enters the cell by means of SOCE can enter the ER and replenish the intracellular stores via sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pumps located in the ER membrane. Thus, SOCE is important for reestablishing Ca2+ store content to maintain physiological ER function as well as to maintain a readily releasable pool of Ca2+, which acts as an important second messenger in a variety of cellular functions.
It is now known that SOCE involves an orchestration of signaling molecules in the ER and the plasma membrane (PM) (Smyth et al. 2006). Stromal interaction molecule 1 (Stim1) resides in the membrane of the ER and has an EF-hand domain that extends into the ER lumen (Dziadek and Johnstone 2007); this luminal EF-hand allows Stim1 to sense decreases in ER Ca2+ content (Stathopulos et al. 2006). Under conditions of full ER Ca2+ stores, Stim1 is localized throughout the ER network in structures organized by the microtubule network (Smyth et al. 2007), but when Ca2+ stores are depleted, Stim1 rearranges into punctate structures close to the PM while still remaining in the ER membrane (Liou et al. 2005; Zhang et al. 2005; Mercer et al. 2006). Stim1 then in some manner activates members of the Orai family (Orai1, 2, and 3) of SOCE channels, resulting in Ca2+ entry into the cell (Feske et al. 2006; Vig et al. 2006; Zhang et al. 2006; Mercer et al. 2006; Soboloff et al. 2006).
Fundamental to the physiological role of SOCE is the fact that the process must be reversible. Thus, as Ca2+ stores are refilled SOCE should shut down to prevent Ca2+ overload of the cell. In turn, near-PM localization of Stim1 that occurs as a result of store depletion should also be reversed by store refilling to terminate Orai-mediated Ca2+ entry. In the current study we investigate and compare the physiological process of Stim1 reversal that rigorously depends upon Ca2+ store content, and a novel pharmacological reversal process that is independent of Ca2+ store content. In addition, we demonstrate that the localization of Stim1 puncta is not a random process, but appears to be predetermined by unknown molecular and structural elements.
RESULTS
Reversal of EYFP-Stim1 Localization by Store Refilling Through SOC channels
We have investigated the reversibility of the store depletion-induced rearrangement of EYFP-Stim1 into near-plasma membrane punctae by simultaneous measurements of TIRFM and intracellular Ca2+ concentrations. For these experiments cells were co-transfected with plasmids encoding EYFP-Stim1 and the m5 muscarinic receptor. As shown in Figure 1A, treatment of these cells with 300 μM carbachol in the presence of nominal extracellular Ca2+ caused a rapid increase in intracellular Ca2+ concentration due to Ca2+ release, which was quickly followed by an increase in TIRFM fluorescence intensity due to rearrangement of EYFP-Stim1 into near-PM punctae. The addition of 50 μM atropine to terminate carbachol signaling caused a small drop in both the Ca2+ and TIRFM signals, likely due to the presence of residual Ca2+ in the nominally Ca2+-free extracellular solution (∼10 μM). Importantly, restoration of extracellular Ca2+ to 1.8 mM caused a large increase in the intracellular Ca2+ concentration due to SOCE, and this was accompanied simultaneously by a rapid decrease in the TIRFM signal. A similar dependence on extracellular Ca2+ for reversal of Stim1 localization was reported by Varnai et al. (Varnai et al. 2007). This decrease in the near-plasma membrane localization of EYFP-Stim1 can be attributed to Ca2+ store refilling by SOCE; when the identical experiment was performed in the presence of 5 μM Gd3+, which inhibits SOCE, restoration of extracellular Ca2+ did not initiate SOCE and a decrease in the TIRFM signal was not observed (Fig. 1B). Store refilling in the absence of Gd3+ was further verified by the fact that addition of thapsigargin, which depletes Ca2+ stores by inhibiting SERCA pumps, caused a small Ca2+ release and a subsequent increase in the TIRFM signal (Fig 1A); these responses were not observed in the Gd3+-treated cells (Fig 1B) because stores had not been refilled. It has been demonstrated that when stores are full, EYFP-Stim1 exhibits constitutive comet-like movements when overexpressed in DT40 cells and that these movements cease as stores are depleted and EYFP-Stim1 rearranges into near-PM punctae (Baba et al. 2006). We observed similar constitutive movements of EYFP-Stim1 in HEK293 cells, and further found that these movements were restored when punctate EYFP-Stim1 localization was reversed by store refilling (Supplemental Movie 1).
Figure 1. Ca2+ store refilling reverses the rearrangement of EYFP-Stim1.
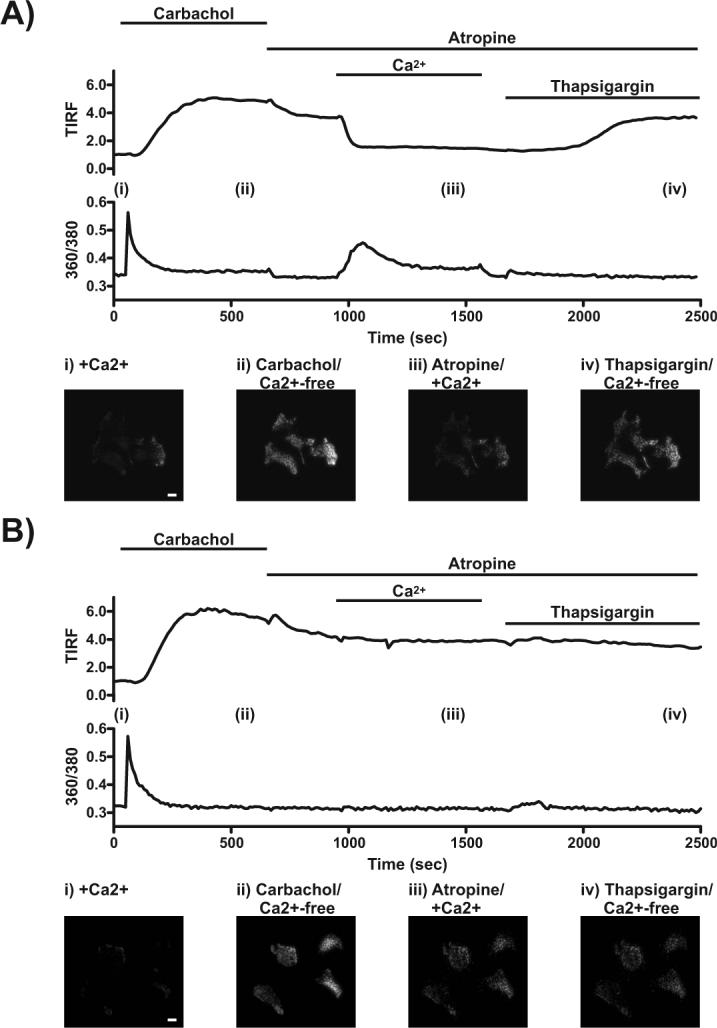
TIRFM fluorescence intensity and relative intracellular Ca2+ concentrations were measured simultaneously in the same HEK293 cells overexpressing EYFP-Stim1 and the m5 muscarinic receptor. As indicated, cells were treated with 300 μM carbachol in nominally Ca2+-free extracellular medium to deplete intracellular Ca2+ stores. Carbachol signaling was then terminated by the addition of 50 μM atropine, after which extracellular Ca2+ was restored to 1.8 mM. Fifteen minutes later the cells were treated with 2 μM thapsigargin to demonstrate that store refilling had occurred. This protocol was performed with cells treated in the absence (A) or presence (B) of 5 μM Gd3+ throughout. Note in (B) that SOCE did not occur upon restoration of extracellular Ca2+, and EYFP-Stim1 was not reversed. The upper traces show the TIRFM intensity profiles, and the bottom traces show the 360/380 fluorescence intensities representative of relative Ca2+ responses; each trace represents the average response of four cells measured in a single experiment. The bottom panels show TIRFM images taken at the times indicated (i-iv) in the intensity profiles. Scalebars = 10 μm.
It is apparent from the TIRFM imaging experiments of Fig 1 that store refilling causes a significant reversal of EYFP-Stim1 localization; however, it is difficult to determine from TIRFM images whether EYFP-Stim1 returns to the same structures characteristic of the store-replete condition. We therefore performed similar experiments by confocal microscopy. As shown in Figure 2A, EYFP-Stim1 is localized in tubular structures when Ca2+ stores are full, and it rearranges into punctate structures when Ca2+ stores are depleted by carbachol treatment. Store refilling with restoration of extracellular Ca2+ in the presence of atropine caused reversal of EYFP-Stim1 into tubular structures strikingly similar to those seen in the initial store-replete state. We have also observed, as shown by others (Luik et al. 2006; Xu et al. 2006; Varnai et al. 2007), that a CFP-tagged Orai1 (CFP-Orai1) is localized evenly throughout the plasma membrane when Ca2+ stores are replete, but rearranges into punctate structures that co-localize with those formed by EYFP-Stim1 when stores are emptied (Fig 2B). These CFP-Orai1 punctae were also reversed by Ca2+ store refilling, returning CFP-Orai1 to a localization similar to that seen in the initial store-replete state. As previously shown (Xu et al. 2006; Varnai et al. 2007) CFP-Orai1 did not rearrange to an appreciable degree upon store depletion when Stim1 was not also overexpressed (Supplemental Figure 1). Finally, Figure 2 illustrates a general finding that in some cells, Stim1 punctae appear small and discrete (Fig 2A), while in others, the punctae take on more of a patch-work appearance (Fig 2B, see also Fig 7C). The reason for these differences is not clear, but we would speculate that the larger structures arise with higher levels of EYFP-Stim1 expression. We do not generally observe these large patch-like aggregations of EYFP-Stim1 with TIRFM, which may indicate that significant areas of these structures lie beyond the Z-resolution of TIRFM.
Figure 2. Rearrangements of both Stim1 and Orai1 are reversed by Ca2+ store refilling.

A) Confocal images of HEK293 cells co-overexpressing EYFP-Stim1 and the m5 muscarinic receptor in the presence of 1.8 mM extracellular Ca2+ prior to store depletion (left panel), 10 minutes following treatment with 300 μM carbachol in nominally Ca2+-free extracellular solution (center panel), and 10 minutes following restoration of 1.8 mM extracellular Ca2+ in the presence of 50 μM atropine (right panel). B) The same protocol described in (A) was repeated with cells co-overexpressing EYFP-Stim1, CFP-Orai1, and the m5 muscarinic receptor. EYFP-Stim1 fluorescence is shown in the upper row, CFP-Orai1 is shown in the center row, and the bottom row shows merged images of EYFP-Stim1 (green) and CFP-Orai1 (red). Scalebars = 10 μm.
Figure 7. ML-9 reverses constitutive EYFP-D76N/D78N-Stim1 localization and SOCE activity.

A) Intracellular Ca2+ concentration was monitored in HEK293 cells overexpressing EYFP-D76N/D78N-Stim1 beginning in the presence of 1.8 mM extracellular Ca2+, followed at the time indicated by switch to nominally Ca2+-free extracellular solution. Extracellular Ca2+ was then restored, and 100 μM ML-9 was added. In the continuous presence of ML-9, extracellular Ca2+ was again removed and restored. Trace represents the average response of all the cells measured on a single coverslip; representative of 3 independent experiments. B) Time-lapse TIRFM was performed on a cell overexpressing EYFP-D76N/D78N-Stim1; 100 μM ML-9 was added at the time indicated in the fluorescence intensity profile (left panel). Right panel: representative TIRFM images taken at the times indicated (i and ii) in the intensity profile. Representative of 3 independent experiments. C) Confocal images of EYFP-D76N/D78N-Stim1-expressing cells in the absence (left panel) and presence (right panel) of 100 μM ML-9 in the presence of 1.8 mM extracellular Ca2+ throughout. Scalebars = 10 μm.
ML-9 Inhibits SOCE and Icrac
In an attempt to understand the underlying mechanism for Stim1 movement, we assessed the actions of a number of putative inhibitors of cytoskeletal function and molecular motors. One agent which gave particularly encouraging results was ML-9 ( 1-(5-Chloronaphthalene-1-sulfonyl)homopiperazine, HCl), a drug known to inhibit myosin light chain kinase (Saitoh et al. 1986; Saitoh et al. 1987). Previous reports demonstrated inhibition of SOCE by ML-9, and this was taken as evidence for a role of myosin light chain kinase in this signaling pathway (Watanabe et al. 1996; Norwood et al. 2000; Tran et al. 2001). We analyzed SOCE in HEK293 cells using a standard Ca2+ add-back assay, whereby intracellular Ca2+ stores were depleted by treating cells with thapsigargin in the presence of nominal extracellular Ca2+, followed by restoration of extracellular Ca2+ to 1.8 mM. When this assay was performed on cells that were treated with 100 μM ML-9 for 5 minutes prior to store depletion with thapsigargin, SOCE was nearly completely absent compared to untreated control cells (Fig. 3A). DMSO, as a vehicle control for ML-9, had no effect. SOCE recovered to the same magnitude as untreated controls when ML-9 was removed 5 minutes following addition of extracellular Ca2+, indicating that the inhibition of SOCE by ML-9 is rapidly reversible. The inhibition of SOCE by ML-9 was concentration dependent, with an IC50 of ∼10 μM (Fig. 3B; see also Fig. 6A). Note that there was a small increase in intracellular Ca2+ at the time of ML-9 addition; such an effect was previously reported and was attributed to release of Ca2+ from intracellular stores by ML-9 (Norwood et al. 2000). It is likely that this Ca2+ increase that we observed is also due to a partial intracellular Ca2+ release by ML-9, since the amount of Ca2+ mobilized by thapsigargin was reduced in ML-9-treated cells. We also determined whether ML-9 is effective when added after SOCE has been initiated. To test this, we performed the Ca2+ add-back assay as described in the absence of ML-9, and then added ML-9 five minutes following restoration of extracellular Ca2+. The addition of 100 μM ML-9 caused the intracellular Ca2+ concentration to rapidly return to its basal level (Fig. 3C). To evaluate the extent of inhibition statistically, the intracellular Ca2+ concentration following the addition of ML-9 was divided by the Ca2+ concentration just prior to ML-9 addition; thus, the data are represented as the proportion of SOCE that remains following ML-9 as a function of the amount of SOCE present just prior to ML-9. This evaluation over a range of concentrations revealed a concentration-dependent inhibition by ML-9 (IC50 = ∼16 μM; Fig. 3D; see also Fig. 6A) that was similar to that seen when ML-9 was added prior to activation of SOCE.
Figure 3. ML-9 dose-dependently inhibits SOCE and Icrac.

A) Relative intracellular Ca2+ concentrations were monitored in wildtype HEK293 cells treated with 100 μM ML-9 (red trace) or left untreated (black trace). Ca2+ stores were depleted with 2 μM thapsigargin in nominally Ca2+-free extracellular medium, and extracellular Ca2+ was restored to 1.8 mM 15 minutes later to reveal SOCE. ML-9 was removed at the end of the experiment to demonstrate reversibility of ML-9 inhibition. Each trace represents the averaged response of all cells measured on a single coverslip. B) The average peak SOCE responses above baseline from experiments performed as described in (A) were averaged for untreated control cells (n = 150; 5 coverslips), or cells treated with 1 μM (n = 78; 3 coverslips), 10 μM (n = 76; 3 coverslips) 50 μM (n = 84; 3 coverslips), or 100 μM (n = 64; 3 coverslips) ML-9. Data are reported as percent of untreated control ± SEM; * indicates significant difference compared to control (p < 0.001) based on one-way ANOVA. C) Experiments were performed as described in (A), but ML-9 was added 5 minutes following restoration of extracellular Ca2+ (red trace). D) For experiments performed as described in (C), the baseline-subtracted 340/380 ratio 5 minutes following ML-9 addition was divided by that just prior to addition. Data are reported as percent of untreated control ± SEM for untreated controls (n = 151; 5 coverslips), and for 1 μM (n = 90; 4 coverslips), 10 μM (n = 80, 3 coverslips), 50 μM (n = 61, 3 coverslips) and 100 μM (n = 74, 3 coverslips) ML-9; * indicates significant difference compared to control (p < 0.001) based on one-way ANOVA. E) Whole-cell patch clamp analysis was performed with a pipette solution containing 20 mM BAPTA and 25 μM IP3 to deplete intracellular Ca2+ stores. The cell was initially perfused with an extracellular solution containing 10 mM Ca2+. At the time indicated, perfusion was switched to a divalent-free solution (DVF), which resulted in the development of a Na+ current. Perfusion was then returned to 10 mM Ca2+, and 100 μM ML-9 was added at the time indicated. Note the decrease in the Ca2+ current upon ML-9 addition. In the continued presence of ML-9, perfusion was again switched to DVF. At the end of the experiment, 10 mM extracellular Ca2+ was restored and ML-9 was removed to demonstrate reversal of inhibition of the Ca2+ current. F) For experiments performed as described in (E), the peak Na+ currents at the initial switch to DVF in the absence of ML-9 (control) and that at the second switch to DVF in the presence of 100 μM ML-9 were averaged and are expressed as mean ± SEM (n = 6); * indicates significant difference compared to control (p < 0.005) based on t-test.
Figure 6. Inhibition of SOCE and Icrac by ML-9 is due to inhibition of Stim1 rearrangement.

A) SOCE experiments were performed in which ML-9 was added following restoration of extracellular Ca2+ as described in Figure 3C. As described in Figure 3D, SOCE following ML-9 addition as a percent of untreated control was calculated, and is plotted as a function of ML-9 concentration for HEK293 cells overexpressing unconjugated EYFP (black squares) and EYFP-Stim1 (blue triangles). B) TIRFM fluorescence intensity (black trace) and relative intracellular Ca2+ concentration (360/380 ratio; blue trace) were measured simultaneously in the same cell. Ca2+ stores were depleted with thapsigargin (Tg; 2 μM) in the presence of 1.8 mM extracellular Ca2+, and 100 μM ML-9 was added 15 minutes later. C) For the data shown in (B), the TIRFM (black trace) and 360/380 (blue trace) values beginning just prior to ML-9 addition were normalized to the same minimum and maximum values.
To confirm that the inhibition by ML-9 reflected a decrease in ion permeation of CRAC channels, rather than effects on membrane potential or calcium buffering, we carried out electrophysiological measurements of Icrac in HEK293 cells in the absence and presence of ML-9. It is difficult to reliably measure Icrac in these cells when Ca2+ is used as the charge carrier because the currents are very small, on the order of −0.5 pA/pF. However, a Na+ current can consistently be measured when the extracellular solution is switched to one that is free of all divalent cations. As shown in Figure 3E, when 25 μM IP3 and 20 mM BAPTA were included in the patch pipette to deplete intracellular Ca2+ stores, a Na+ current of approximately −3 to −4 pA/pF was measured when the extracellular solution was switched from 10 mM Ca2+ to divalent-free. The extracellular solution was then switched back to Ca2+, and 100 μM ML-9 was added. Notably, there was a decrease in the Ca2+ current when ML-9 was added, indicative of inhibition by ML-9. More importantly, the Na+ current was significantly inhibited when the extracellular solution was again switched to divalent-free in the continued presence of ML-9 (Fig. 3E and F). The residual current in the presence of ML-9 is not likely due to significant Icrac; upon switching to divalent-free solutions in the absence of store depletion, we generally observe a linear increase in membrane current of the order of 0.6 pA/pF (unpublished observations, and (DeHaven et al. 2007)). Thus, ML-9 similarly inhibits SOCE and Icrac in HEK293 cells.
ML-9 Reverses Stim1 Rearrangement
We hypothesized that ML-9 may inhibit SOCE and Icrac by blocking the rearrangement of Stim1 that occurs when Ca2+ stores are depleted. To test this, we monitored EYFP-Stim1 rearrangement by TIRFM in experiments carried out in a manner similar to the SOCE experiments described in Figure 3; i.e., Ca2+ stores were depleted with thapsigargin in nominally Ca2+-free extracellular solution, after which extracellular Ca2+ was restored to 1.8 mM. As seen in the control traces in Figure 4A, the fluorescence intensity measured by TIRFM significantly increased following store depletion with thapsigargin. Unexpectedly, we consistently observed a further, albeit relatively small, increase in TIRFM fluorescence when extracellular Ca2+ was restored; we do not currently know the cause of this Ca2+-induced increase in near-PM EYFP-Stim1 localization, and this is a topic of further investigation in our laboratory. On the other hand, cells treated with 100 μM ML-9 for 5 minutes prior to store depletion exhibited little to no increase in TIRFM fluorescence intensity following store depletion or upon Ca2+ add-back, and EYFP-Stim1 puncta were not formed (Fig. 4A). However, the TIRFM fluorescence intensity rapidly increased when ML-9 was removed, indicating that similar to the inhibition of SOCE, inhibition of EYFP-Stim1 rearrangement by ML-9 is reversible. Thus, the inhibitory properties of ML-9 in TIRFM experiments directly paralleled those observed in SOCE experiments. And since we found that ML-9 also blocked SOCE when added after its initiation, we examined the ability of ML-9 to reverse the near-PM localization of EYFP-Stim1 after rearrangement was induced by store depletion. In these TIRFM experiments, Ca2+ stores were depleted with thapsigargin in nominally Ca2+-free solution, resulting in a large TIRFM fluorescence intensity increase. Ca2+ was then restored to 1.8 mM (note again the small fluorescence intensity increase), and ML-9 (100 μM) was then added in the continued presence of 1.8 mM Ca2+ (Figure 4B). Addition of ML-9 at this time point caused the TIRFM fluorescence intensity to rapidly decrease to near basal levels, indicative of reversal of near-PM EYFP-Stim1 localization. Similar TIRFM responses and reversal by ML-9 were seen when stores were depleted with the receptor agonist, carbachol (data not shown) or with 400 nM ionomycin (Supplemental Figure 2). Thus, ML-9 effectively inhibited SOCE, Icrac, and EYFP-Stim1 rearrangement whether it was added prior to or after store depletion. Because the order of ML-9 addition in relation to store depletion did not appear to significantly influence the concentration dependence of SOCE inhibition, we determined the concentration dependence of the inhibition of EYFP-Stim1 rearrangement from TIRFM experiments in which ML-9 was added after store depletion as described in Figure 4B. This allowed us to normalize the fluorescence intensity after ML-9 addition to that just prior to ML-9 addition for each cell individually (Fig. 4C). However, we were surprised to find that the concentration dependence for inhibition of EYFP-Stim1 rearrangement (IC50 = ∼51 μM) was somewhat greater than that for SOCE. We will address this issue in a later section of this report.
Figure 4. ML-9 inhibits EYFP-Stim1 rearrangement.

A) Time-lapse TIRFM was performed on EYFP-Stim1 overexpressing HEK293 cells treated with 100 μM ML-9 or left untreated (control). The left panel shows TIRFM fluorescence intensity profiles for two control (black traces) and two ML-9-treated cells (red traces). Thapsigargin (Tg; 2 μM) was added in nominally Ca2+-free extracellular solution at the time indicated to deplete Ca2+ stores, and ML-9 was removed at the end of the experiment to demonstrate reversal of the ML-9 inhibition. The right panel shows representative TIRFM images taken at the times indicated (i-iii) in the intensity profile. B) TIRFM imaging was performed on three cells as described in (A), but ML-9 (100 μM) was added after store depletion with thapsigargin. C) The average baseline subtracted TIRFM fluorescence intensity 5 minutes following ML-9 addition was divided by that just prior to addition for experiments performed as described in (B) for untreated controls (n = 6; 2 coverslips), or cells treated with 1 μM (n = 11; 3 coverslips), 10 μM (n = 11, 3 coverslips), 50 μM (n = 14, 3 coverslips) and 100 μM (n = 10, 3 coverslips) ML-9. Data are expressed as percent of untreated control ± SEM; * indicates significant difference compared to control (p < 0.001) based on one-way ANOVA. Scalebars = 10 μm.
To more closely evaluate the effects of ML-9 on EYFP-Stim1 localization, we imaged EYFP-Stim1 expressing HEK293 cells by confocal microscopy. As shown in Figure 5, store depletion with thapsigargin in nominally Ca2+-free extracellular solution caused rearrangement of EYFP-Stim1 from tubular into discrete punctate structures. Addition of ML-9 (100 μM) in the continued presence of thapsigargin and nominal extracellular Ca2+ completely reversed the punctate EYFP-Stim1 localization, and EYFP-Stim1 returned to tubular structures. With this protocol, Ca2+ stores remained empty after ML-9 addition as assessed by experiments examining the size of ionomycin-releasable Ca2+ pools (data not shown, (Bird and Putney, Jr. 2005)). Notably, these tubular structures seen following ML-9 addition were nearly identical to those seen in the initial store-replete state. Consistent with the return of EYFP-Stim1 into tubular structures by ML-9, we also found in time-lapse TIRFM imaging that constitutive EYFP-Stim1 movements reinitiated when EYFP-Stim1 localization was reversed with 100 μM ML-9 (Supplemental Movie 2). Thus, by two independent measures, the reversal of EYFP-Stim1 localization by ML-9 appears to be complete.
Figure 5. Reversal of Stim1 localization by ML-9 is complete.

Shown are confocal images of cells in the presence of 1.8 mM extracellular Ca2+ (left panel), 15 minutes following store depletion with thapsigargin (Tg; 2 μM) in nominally Ca2+-free extracellular solution (center panel), and 5 minutes following addition of 100 μM ML-9 in the continued presence of Tg and Ca2+-free extracellular solution. Scalebars = 10 μm.
Inhibition of Ca2+ Entry is Due to Inhibition of Stim1 Rearrangement
It was apparent that the concentration dependence of inhibition of SOCE by ML-9 in wildtype (unconjugated EYFP transfected) cells was more sensitive than that of overexpressed EYFP-Stim1 rearrangement. One possibility for this discrepancy is that overexpression of EYFP-Stim1 in TIRFM experiments may shift the concentration dependence of inhibition. To test this more directly, we compared the concentration dependence of inhibition of SOCE by ML-9 in wildtype HEK293 cells overexpressing unconjugated EYFP to that in HEK293 cells overexpressing EYFP-Stim1. Data for this analysis were obtained by performing experiments as described in Figure 3C; i.e., ML-9 was added after the initiation of SOCE. The concentration-dependent responses for EYFP alone and EYFP-Stim1-expressing cells are shown in Figure 6A. It is apparent that overexpression of EYFP-Stim1 caused a rightward shift in the concentration dependence, and the IC50 for inhibition of SOCE by ML-9 in EYFP-Stim1-expressing cells was 66 μM, compared to 16 μM in unconjugated EYFP-expressing cells. Furthermore, the IC50 for inhibition of SOCE in EYFP-Stim1 expressing cells was close to that for inhibition of EYFP-Stim1 rearrangement from TIRFM experiments. The fact that overexpression of EYFP-Stim1 has a rightward-shifting effect on the concentration dependence of SOCE inhibition by ML-9 provides strong evidence that the inhibition of SOCE is related to effects on Stim1 function.
We also analyzed the kinetics of inhibition of SOCE and EYFP-Stim1 rearrangement by simultaneously measuring TIRFM fluorescence intensity and intracellular Ca2+ concentrations (Fig. 6B). In these experiments thapsigargin was added to the cells in the presence of 1.8 mM extracellular Ca2+; thus, the Ca2+ response that is seen is representative of both an initial release from the ER as well as subsequent activation of SOCE. When 100 μM ML-9 was added during the sustained SOCE phase, both the intracellular Ca2+ concentration and the TIRFM fluorescence intensity rapidly decreased to near their original, basal levels (Fig. 6B). When both the Ca2+ and TIRFM responses to ML-9 were normalized to the same minimum and maximum scale, it was apparent that the decrease in TIRFM intensity preceded the decrease in Ca2+ concentration (Fig. 6C). This further supports the conclusion that the reversal of Stim1 localization by ML-9 is the cause of inhibition of SOCE.
ML-9 Inhibits a Constitutively Active Mutant of Stim1
Mutations of Ca2+-binding residues within the EF-hand domain of Stim1 render the mutated Stim1 constitutively active; i.e., EF-hand mutated Stim1 localizes in near-plasma membrane punctae even when intracellular Ca2+ stores are full, and constitutive SOCE is observed. As shown in Figure 7A, cells that overexpress an EYFP-tagged human Stim1 in which the aspartic acids at positions 76 and 78 were mutated to asparagines (EYFP- D76N/D78N-Stim1) exhibit constitutive SOCE, as indicated by high intracellular Ca2+ levels that significantly decreased upon removal of extracellular Ca2+, despite full intracellular Ca2+ stores. ML-9 inhibited this constitutive SOCE activity, since the addition of 100 μM ML-9 in the presence of 1.8 mM extracellular Ca2+ caused a rapid decrease in the intracellular Ca2+ concentration, and removal of extracellular Ca2+ in the continued presence of ML-9 resulted in only a small additional decrease. In TIRFM imaging, cells overexpressing EYFP-D76N/D78N-Stim1 exhibited intense near-plasma membrane punctae in the absence of store depletion, and consistent with the Ca2+ data, the EYFP-D76N/D78N-Stim1 fluorescence intensity rapidly decreased upon addition of 100 μM ML-9 (Fig. 7B). Remarkably, confocal imaging demonstrated that when the constitutively punctate distribution of EYFP-D76N/D78N-Stim1 was reversed by ML-9, the construct adopted a tubular distribution that is indistinguishable from the configuration of wildtype EYFP-Stim1 in the presence of replete Ca2+ stores (Fig. 7C). This striking relocalization of EYFP-D76N/D78N-Stim1 by ML-9 was further exemplified by the fact that EYFP-D76N/D78N-Stim1 began to exhibit constitutive movements when cells were treated with 100 μM ML-9 in time-lapse TIRFM imaging (Supplemental Movie 3).
Effects of ML-9 on Stim1 Are Likely not Due to Inhibition of MLCK
We determined whether other methods of decreasing MLCK activity could recapitulate the effects on Stim1 function that we have observed with ML-9. As shown in Figures 8A and B, transfection of HEK293 cells with two out of three siRNA constructs targeted to MLCK resulted in a significant reduction in expression of MLCK protein, whereas a control siRNA construct had no significant effect. However, the TIRFM responses to store depletion by thapsigargin were not inhibited in cells that were co-transfected with EYFP-Stim1 and the MLCK#3 siRNA construct (Fig. 8D), which reduced MLCK expression by approximately 80%, compared to the responses in cells transfected with the control siRNA (Fig. 8C). Expression of the other two MLCK siRNA constructs also had no inhibitory effect on the TIRFM response (data not shown). We also attempted to pharmacologically replicate the effects of ML-9. However, pre-treatment of cells with 20 μM wortmannin, which inhibits MLCK activity (Nakanishi et al. 1992), had no effect on the TIRFM response of EYFP-Stim1 to store depletion (data not shown).
Figure 8. Knockdown of MLCK protein expression by siRNA does not inhibit EYFP-Stim1 rearrangement.

A) HEK293 cells left untransfected (WT; lane 1), or transfected with control siRNA (lane 2) or one of three different MLCK siRNA constructs (lanes 3−5) were subjected to Western blotting with an antibody against MLCK. B) Three independent experiments were performed as described in (A), and the average band intensities for each condition are expressed as a percentage of WT ± SEM; * indicates significant difference compared to WT (p < 0.01) based on one-way ANOVA. C) Cells were transfected with control siRNA or (D) with MLCK #3, and TIRFM experiments were performed in which stores were depleted with 2 μM thapsigargin in nominally Ca2+-free extracellular solution. Each trace represents a single cell, and the responses of all the cells measured from a single coverslip are shown.
EYFP-Stim1 Punctae Form in Pre-determined Locations
The ability to reversibly block the punctate localization of EYFP-Stim1 provided us with the opportunity to explore whether EYFP-Stim1 punctae form in similar locations in the same cell each time their formation is induced. If this is the case, then it would imply that cellular structures or molecules other than Stim1 itself determine the localization of Stim1 punctae formation. For this analysis we used ML-9 to reverse EYFP-Stim1 localization since reversal with ML-9 was consistently more effective than that seen with any other strategy. As shown in the TIRFM fluorescence intensity profile in Figure 9A and the representative images in Figure 9B, store depletion with thapsigargin induced EYFP-Stim1 punctae formation that was rapidly and completely reversed by 100 μM ML-9. Upon washout of the ML-9, punctae rapidly reformed with a fluorescence intensity similar to that seen prior to ML-9 treatment. To compare the locations of EYFP- Stim1 punctae pre-reversal with ML-9 to those post-reversal (i.e., following washout of ML-9), a pre-reversal TIRFM image was pseudocolored red and merged with a post-reversal image that was pseudocolored green (Figure 9C). These image manipulations revealed that many punctae were located in the same, or near to the same position in the images taken pre- and post-reversal. Furthermore, evaluation of several merged images in sequence (Figure 9C and Supplemental Movie 5) revealed that these closely apposed pre- and post-reversal punctae remained closely apposed over time, indicating that this correlation was not stochastic or specific to one pair of merged images. It should be noted that EYFP-Stim1 punctae do not remain static over time, but instead exhibit small lateral movements as can be seen in the complete time-lapse movie of the experiment shown in Figure 9 (Supplemental Movie 4). Thus, it is not expected that EYFP-Stim1 punctae should form in the exact same locations pre- and post-reversal, and is it reasonable to expect that there may be some regions of the cell where there is little correlation between the pre- and post-reversal punctae. More importantly, in three out of three experiments performed in this manner, we were able to find regions of the cells where clear spatial coincidence between pre- and post-reversal punctae was apparent.
Figure 9. Single EYFP-Stim1 punctae form in similar locations upon multiple rearrangement stimuli.

A) Two EYFP-Stim1-expressing HEK293 cells were imaged by time-lapse TIRFM, during which Ca2+ stores were depleted with 2 μM thapsigargin in the presence of nominal extracellular Ca2+, 100 μM ML-9 was added to reverse punctae formation, and ML-9 was removed to restimulate punctae formation. Shown is the fluorescence intensity profile, with each trace representing a single cell. B) Representative TIRFM images taken at the times indicated (i-iv) in the intensity profile in panel A. The complete image series of this experiment is shown in Supplemental Movie 4. C) The TIRFM image taken at 380 seconds (store-depleted with thapsigargin prior to ML-9 treatment) was pseudocolored red. This image was then merged with the image taken at 710 seconds (after ML-9 washout), which was pseudocolored green. Image b on the right is a close-up of the region denoted by the white rectangle in the full-size image on the left. Image a was generated from the merge of the images 60 seconds prior to the images used to generate the full-size image on the left, and image c was generated from the merge of the images 60 seconds after the images used to generate the full-size image on the left. The arrows in image b point to pairs of red and green punctae that remain consistent throughout the series of merged images. Scalebars = 10 μm, except in images a-c in panel C in which the Scalebar = 1.0 μm
DISCUSSION
Physiological Reversal of Stim1 and Orai1 Localization by Store Refilling
Since the discovery of the role of Stim1 in SOCE, the majority of studies have focused on the mechanisms by which Stim1 rearranges into near-PM punctae and activates Ca2+ entry. It is equally important, however, that we also understand the mechanisms by which SOCE is terminated. Unregulated Ca2+ entry could lead to overload of cytoplasmic and/or ER Ca2+, both of which can be detrimental to cellular physiology. Furthermore, discrete Ca2+ signals, such as Ca2+ oscillations, may depend on tightly controlled SOCE events (Bird and Putney, Jr. 2005; Wedel et al. 2007). The reversibility of Stim1 rearrangement has previously been established (Liou et al. 2005; Varnai et al. 2007), and we have now clearly demonstrated that this reversibility absolutely requires Ca2+ store refilling through store-operated channels. It is also apparent that upon reversal, Stim1 localizes into tubular structures that are analogous to those seen prior to store depletion. Consistent with this, we also observed constitutive, comet-like movements of reversed EYFP-Stim1 that are typical of those exhibited by EYFP-Stim1 prior to store depletion (Baba et al. 2006). Thus, by all measures, EYFP-Stim1 reverses to its basal state upon store refilling, allowing it to respond to subsequent store depletion events, as evidenced by the ability of store depletion with thapsigargin to trigger rearrangement of EYFP-Stim1 into punctae after store refilling-induced reversal. We also report for the first time that the rearrangement of Orai1 into punctate structures as a result of Ca2+ store depletion is similarly reversed by store refilling. Thus, the two basic components of the SOCE machinery are completely reversed by store refilling through the SOC channels. The fact that overexpressed Orai1 does not rearrange to a significant degree in response to store depletion in cells that do not also overexpress Stim1 implies that rearrangement of Stim1 directs the rearrangement of Orai1, although this has yet to be proven. Our data are consistent with this in that reversal of Stim1 localization occurs concomitantly with reversal of Orai1 localization.
Reversal of Stim1 Localization by ML-9
We have, for the first time, demonstrated inhibition of store depletion-induced rearrangement of EYFP-Stim1 into near-PM punctae by a method that does not involve replenishment of Ca2+ stores, namely by use of the pharmacological agent, ML-9. We have also established that the ability of ML-9 to inhibit SOCE and Icrac is due to its inhibition of Stim1 localization into punctae. This finding is highly significant, given the lack of SOCE inhibitors for which the molecular basis of inhibition within the SOCE pathway has been established.
In SOCE experiments, the IC50 for inhibition by ML-9 is similar whether the compound is added prior to or after activation of SOCE. Thus, it appears that ML-9 is equally as effective at reversing SOCE as it is at preventing its activation. We obtained similar results in TIRFM experiments in that ML-9 effectively prevented store depletion-induced rearrangement of EYFP-Stim1 into near-PM punctae as well as reversed this PM localization after it was established. The IC50 for reversal of the TIRFM response in EYFP-Stim1-expressing cells was significantly greater than that for reversal of SOCE in wildtype cells, but was similar to that for reversal of SOCE in cells that overexpressed EYFP-Stim1. Unfortunately, a lack of Stim1 antibodies suitable for immunofluorescence in wild-type cells has prevented us from evaluating the effect of ML-9 on rearrangement of endogenous Stim1. However, it appears from our data that in the case of overexpressed EFYP-Stim1, the amount of EYFP-Stim1 that is localized in punctae as a function of ML-9 inhibition directly correlates with the extent of SOCE, based on the similarities between the inhibition curves. This is, to our knowledge, the first demonstration of a quantitative correspondence between the amount of Stim1 localized in near-PM punctae and the magnitude of SOCE. It also provides strong evidence indicating that the inhibition of SOCE by ML-9 is a result of inhibition of Stim1 localization. Why overexpression of EYFP-Stim1 reduces the efficacy of ML-9's inhibition of EYFP-Stim1 rearrangement is unclear. Further evidence supporting the conclusion that ML-9 inhibits SOCE by reversing Stim1 localization comes from simultaneous TIRFM and intracellular Ca2+ measurements, in which the reversal of EYFP-Stim1 localization precedes that of SOCE. This is, in effect, a functional corollary of the recent finding that EYFP-Stim1 rearrangement to the PM precedes the initiation of Icrac (Wu et al. 2006).
Closer examination of the reversal of EYFP-Stim1 localization by confocal microscopy revealed that EYFP-Stim1 reformed tubular structures upon ML-9 treatment that were nearly identical to those observed prior to store depletion. Furthermore, constitutive movements of EYFP-Stim1 were also observed following reversal by ML-9. Thus, similar to the reversal achieved by means of store refilling, reversal by ML-9 reestablished the basal, store-replete localization and behavior of EYFP-Stim1. This is quite remarkable given that in the case of ML-9 treatment, Ca2+ stores remained empty and yet EYFP-Stim1 still localized to the same structures typical of the store-replete state. Thus, ER Ca2+ is not required per se for EYFP-Stim1 to localize in tubular structures. Consistent with this is the fact that EYFP-D76N/D78N-Stim1, which is constitutively arranged in near-PM punctae regardless of Ca2+ store content, becomes localized to tubular structures and exhibits constitutive movements in response to ML-9 treatment. In this case, EYFP-D76N/D78N-Stim1 is able to localize to tubular structures in the same manner as wild-type Stim1, despite the fact that it harbors a mutation that renders it insensitive to ER Ca2+ concentrations. This finding also demonstrates that ML-9 does not simply substitute for Ca2+ by binding to the N-terminal Ca2+ binding site.
The mechanism by which ML-9 inhibits EYFP-Stim1 rearrangement is unclear at this point. Previous studies that have demonstrated inhibition of SOCE by ML-9 have generally attributed this effect to inhibition of MLCK, the characterized target of ML-9 (Watanabe et al. 1996; Takahashi et al. 1997; Norwood et al. 2000). However, this conclusion has only been corroborated with molecular data in one study, in which an antisense oligonucleotide targeted to the MLCK gene inhibited SOCE in human monocytes/macrophages (Tran et al. 2001). In our hands, significant knockdown of MLCK protein expression by siRNA did not have any measurable effect on store depletion-induced rearrangement of EYFP-Stim1. Furthermore, wortmannin, which is also known to inhibit MLCK (Nakanishi et al. 1992), had no effect on EYFP-Stim1 rearrangement. A related point is that Icrac develops optimally when intracellular Ca2+ is buffered to extremely low levels, in which case MLCK, a Ca2+/calmodulin-activated enzyme (Somlyo and Somlyo 2003), would be essentially inactive. It therefore appears unlikely that MLCK plays a significant role in the activation mechanism for SOC channels, and thus inhibition of MLCK by ML-9 is unlikely to underlie the inhibition of Stim1 rearrangement and SOCE. Notably, ML-9 did not significantly affect the structure of the ER when monitored by confocal microscopy or TIRFM (data not shown). Thus, it also seems unlikely that the effects of ML-9 on Stim1 function are due to effects on ER structure, although changes at the ultra-structural level cannot be ruled out. Alternatively, it is possible that ML-9 may directly influence the conformation of Stim1, or may alter Stim1 phosphorylation, given that Stim1 has been demonstrated to be a phosphoprotein (Manji et al. 2000). The mechanism by which ML-9 affects Stim1 function may reveal important information on the activation mechanism of this important signaling protein, and will be a topic of further research.
EYFP-Stim1 Forms Similarly Localized Punctae Upon Multiple Stimulations
We have taken advantage of the reversibility of EYFP-Stim1 by store refilling and ML-9 treatment to demonstrate that EYFP-Stim1 forms punctae in similar locations upon multiple stimulations. This indicates that the locations of Stim1 punctae formation are governed by cellular components other than Stim1 itself, and are not random in nature. In these experiments the reversal of EYFP-Stim1 punctae was complete in as much as punctae were no longer visible by TIRFM; therefore, it is unlikely that small amounts of EYFP-Stim1 remained at the PM and directed the reversed EYFP-Stim1 back to the same sites. Furthermore, our demonstration that Orai1 also reverses upon store refilling suggests that Orai1 does not direct EYFP-Stim1 into the same punctae upon subsequent stimulations. Thus, molecules other than Stim1 and Orai1 may be involved in localizing Stim1 and Orai1 in punctae, as has recently been suggested (Varnai et al. 2007). It has also been demonstrated that when stores are depleted Stim1 localizes in punctae at sites of close apposition between the ER and the PM (Wu et al. 2006; Varnai et al. 2007); therefore, it is likely that these areas of ER to PM contact ultimately define the sites of Stim1 and Orai1 interaction.
In conclusion, Ca2+ store depletion-induced Stim1 and Orai1 rearrangements are completely reversed by store refilling, a finding that provides a molecular basis for the self-regulating nature of the SOCE process. Stim1 rearrangement can also be reversed experimentally with ML-9, independently of Ca2+ store content. Elucidation of the mechanism by which ML-9 affects Stim1 function could help reveal the mechanisms by which Stim1 localization is regulated. Finally, the reversibility of Stim1 rearrangement has allowed us to determine that the sites of Stim1 punctae formation are governed by additional, as yet unidentified molecular determinants.
MATERIALS AND METHODS
Cell Culture and Transfections
HEK293 cells were obtained from ATCC and cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 2 mM glutamine (DMEM) in a 37°C, 5% CO2 humidified incubator. Transfections of cDNA were performed using the Lipofectamine 2000 reagent (Invitrogen). EYFP-Stim1 was obtained from Dr. Tobias Meyer (Stanford University), CFP-Orai1 was made in our laboratory by fusion of CFP to Orai1, purchased from Invitrogen, as described (DeHaven et al. 2007), and the m5 muscarinic receptor plasmid was obtained from Dr. Lutz Birnbaumer, NIEHS, Research Triangle Park, NC, USA. EYFP-D76N/D78N-Stim1 was generated in our laboratory by site-directed mutagenesis of the construct obtained from Dr. Tobias Meyer as previously described (Mercer et al. 2006). For transfections, 0.5 μg of the EYFP-Stim1, EYFP-D76N/D78N-Stim1, and CFP-Orai1 plasmids were used, and 1.0 μg of the m5 muscarinic receptor plasmid was used. In indicated experiments, cells were transfected with the m5 muscarinic receptor plasmid to enhance the responsiveness of cells to the muscarinic receptor agonist carbachol.
Intracellular Ca2+ Measurements
Single-cell Ca2+ concentrations were measured in live cells plated on glass coverslips and mounted in Teflon chambers. Prior to experiments, cells were incubated in 1 μM Fura-5F/AM (Invitrogen) for 25 minutes in DMEM at 37°C. Cells were then bathed in Hepes-buffered saline solution (HBSS; in mM: 120 NaCl, 5.4 KCl, 1.8 CaCl2, 0.8 MgCl2, 11 glucose, and 20 Hepes, pH 7.4) throughout the course of the experiments. Fura-5F fluorescence was measured when cells were excited consecutively at 340 nm and 380 nm using a microscope-based digital fluorescence imaging system (InCyt Im2; Intracellular Imaging Inc.), and relative Ca2+ concentrations are reported as the ratio of fluorescence emission at the two excitation wavelengths. At the end of each experiment, Fura-5F/AM fluorescence was quenched by treating cells with 10 μM ionomycin and 20 mM MnCl2 to obtain background fluorescence values; these background values were subtracted from each experimental measurement. Cells transfected with unconjugated EYFP or EYFP-Stim1 were identified based on their fluorescence emission at 530 nm when excited at 477 nm.
Live-cell Confocal and TIRFM Imaging
Confocal and TIRFM imaging were performed on cells bathed in HBSS. All confocal images were obtained with a pinhole setting of 1 Airy Unit using a Zeiss LSM 510 laser scanning system and a 63× oil-immersion (N.A. 1.4). For EYFP-Stim1, 488 nm or 514 nm illumination was provided by an Argon laser and emission was selected with a 530−600 nm bandpass filter. The excitation for CFP-Orai1 was 458 nm from an Argon laser and emission was selected with a 470−510 bandpass filter. When cells expressing multiple probes were imaged, lack of bleed-through between channels was verified by imaging cells that expressed each of the probes individually. TIRFM was performed using an Olympus IX2 illumination system mounted on an Olympus IX71 inverted microscope. Excitation light was provided by a 488 nm argon ion laser (Melles Griot) directed through a fiber optic cable. The angle of illumination incident on the interface between the glass coverslip and the aqueous medium was controlled by adjusting the lateral position of the laser beam prior to passing through a 60× oil-immersion objective (N.A. 1.45). The emitted fluorescence passed through a D525/50 nm filter (Chroma) and was captured by a Photometrics Cascade 512F cooled CCD (Roper Scientific). Acquisition and image analysis were performed using MetaFluor software (Molecular Devices). For fluorescence intensity profiles, data are represented as the fluorescence intensity at each time point divided by the fluorescence intensity at the start of the experiment (F/F0). Fluorescence intensities were collected from regions of interest encompassing the visible footprints of single cells and were background subtracted. Time-lapse movies of TIRFM images were generated using ImageJ software. For simultaneous TIRFM and intracellular Ca2+ measurements, cells were first loaded with Fura-5F as described, and each TIRFM image acquisition was immediately followed by a pair of acquisitions with excitation at 360 nm and 380 nm and emission at 525 nm. The 360 nm emission was chosen because the TIRFM objective did not effectively transmit 340 nm light. Excitation for Fura-5F measurements was provided by a 75W Xenon Arc lamp (Zeiss) and excitation and emission filters were controlled by a Lambda 10−2 filter wheel (Sutter Instruments Co.). Intensity measurements at 360 nm and 380 nm were independently background subtracted, and data are presented as 360/380 ratios.
Patch-clamp Electrophysiology
Whole-cell currents were measured as previously described (DeHaven et al. 2007). The standard extracellular Hepes buffered saline solution contained (mM): 145 NaCl, 3 KCl, 10 CsCl, 1.2 MgCl2, 10.0 CaCl2, 10 glucose, and 10 Hepes (pH to 7.4 with NaOH). The standard divalent free solution (DVF) was prepared by removing the CaCl2 and MgCl2 from the Hepes solution and adding 0.1 mM EGTA. The intracellular pipette solution contained (in mM) 145 Cs-methanesulfonate, 20 BAPTA, 10 HEPES, and 8 MgCl2 (pH to 7.2 with CsOH) with the addition of 25 μM inositol 1,4,5-trisphosphate (IP3, hexasodium salt). Currents were acquired with pCLAMP-10 (Axon Instruments) and analyzed with Clampfit (Axon Instruments).
siRNA Knockdown of MLCK Expression
siRNA constructs targeted to human MLCK were obtained from Invitrogen (Stealth RNAi); these constructs are predicted to suppress all MLCK isoforms with equal efficacy. The MLCK siRNA constructs had the following sequences: #1, auagaggacagucuuccaauggcuc; #2, caaucuugcagucaaaucuagcagc; #3, uagucuaucuggaaguggcgggacu. The control siRNA construct was the Stealth RNAi Negative Control Medium GC (Invitrogen). siRNA was transfected into cells using Metafectine reagent (Biontex) at a final concentration of 100 nM; cells were co-transfected with EYFP cDNA to identify transfected cells. Cells were assayed 48−72 hours following siRNA transfection.
Western Blotting
Cells were lysed in RIPA buffer (in mM: 50 Tris HCL, 150 NaCl, 1 EDTA, 1% v/v Nonidet P-40, 0.25% w/v sodium deoxycholate, 1 phenylmethylsulfonyl fluoride, pH 7.5) containing 1 Complete Mini Protease Inhibitor Tablet (Roche Applied Sciences) per 10 ml, and total protein content in the lysates was determined using the DC Protein Assay Kit (BioRad). Lysates were normalized based on protein content and were electrophoresed in 6% polyacrylamide gels. Proteins were then transferred electrophoretically to PVDF membranes. Membranes were blocked for 1 hour at room temperature in TBS-T (in mM: 24.7 Tris base, 137 NaCl, 2.7 KCl, 0.1% Tween-20, pH 7.4) containing 2% BSA, incubated in primary antibody (mouse monoclonal anti-MLCK clone K36, Sigma) in TBS-T with BSA overnight at 4°C, and in secondary antibody (horseradish peroxidase-linked anti-mouse IgG, Amersham) in TBS-T with BSA for 1 hour at room temperature. Membranes were washed for 10 minutes in TBS-T three times following each antibody incubation. Membranes were developed using ECL reagent (Amersham) and were exposed to film (Hyperfilm, Amersham). Band intensities were analyzed using PhotoShop software.
Reagents
ML-9 and wortmannin were obtained from Calbiochem; both compounds were dissolved in DMSO. Thapsigargin was obtained from Alexis Biochemicals. All other reagents were from Sigma unless stated otherwise.
Supplemental Materials
CFP-Orai1 does not rearrange upon store depletion when Stim1 is not overexpressed. Shown are confocal images of cells overexpressing CFP-Orai1 alone in the presence of 1.8 mM extracellular Ca2+ (left panel), following store depletion with thapsigargin (2 μM) in nominally Ca2+-free extracellular solution (center panel), and following store refilling with atropine and 1.8 mM extracellular Ca2+ (right panel). Scalebar = 10 μm.
{kind=link}
ML-9 reverses EYFP-Stim1 punctae when Ca2+ stores are depleted with ionomycin. TIRFM was performed on EYFP-Stim1-expressing HEK293 cells that were treated at the times indicated with 400 nM ionomycin in nominal extracellular Ca2+, followed by restoration of 1.8 mM extracellular Ca2+, and finally addition of 100 μM ML-9. Shown is the fluorescence intensity profile averaged from 7 cells measured on a single coverslip; representative of 3 identical experiments (n = 12 cells total).
EYFP-Stim1 exhibits constitutive movements when punctate localization is reversed by store refilling. A single EYFP-Stim1 overexpressing HEK293 cell was imaged by TIRFM beginning in extracellular solution containing 1.8 mM Ca2+. At the times indicated in the upper left, the extracellular solution was changed to 300 μM carbachol (Carb) in nominal Ca2+, then to 50 μM atropine (Atn) with 1.8 mM Ca2+, and finally to 2 μM thapsigargin (Tg) in nominal Ca2+. Images were captured every 10 seconds, and the frame rate of the movie is 10 frames per second. The Scalebar is 10 μm.
Reversal with ML-9 re-establishes constitutive movements of EYFP-Stim1. A single EYFP-Stim1 overexpressing HEK293 cell was imaged by TIRFM beginning in extracellular solution containing 1.8 mM Ca2+. At the times indicated in the upper left, the extracellular solution was changed to 2 μM thapsigargin (Tg) in nominal Ca2+ and then to 100 μM ML-9 in the continued presence of Tg and nominal Ca2+. Images were captured every 5 seconds, and the frame rate of the movie is 10 frames per second. The Scalebar is 10 μm.
ML-9 induces constitutive movements of EYFP-D76N/D78N-Stim1. A single HEK293 cell over-expressing EYFP-D76N/D78N-Stim1 was imaged by TIRFM beginning in extracellular solution containing 1.8 mM Ca2+. At the time indicated in the upper left, the extracellular solution was changed to 100 μM ML-9 in the continued presence of 1.8 mM Ca2+. Images were captured every 5 seconds, and the frame rate of the movie is 10 frames per second. The Scalebar is 10 μm.
EYFP-Stim1 punctae are similar before and after reversal and reformation. HEK293 cells overexpressing EYFP-Stim1 were imaged by TIRFM beginning in extracellular solution containing 1.8 mM Ca2+. At the times indicated in the upper left, the extracellular solution was changed to 2 μM thapsigargin (Tg) in nominal Ca2+, then to 100 μM ML-9 in the continued presence of Tg and nominal Ca2+, and lastly to Tg in nominal Ca2+ without ML-9 (Wash). Images were captured every 10 seconds, the frame rate of the movie is 10 frames per second. The Scalebar is 10 μm. Images correspond to the experiment presented in Figure 9.
Pre- and post-reversal EYFP-Stim1 punctae exhibit close spatial correlation over time. As described in Figure 9C, images prior to reversal with ML-9 were pseudocolored red and merged with images taken following ML-9 washout (pseudocolored green). This movie shows a time-lapse series of merged images, zoomed into a region of the cell to demonstrate spatial correlation between red and green punctae. The frame rate of the movie is 5 frames per second. The total experiment time represented by this movie is 2 minutes.
Acknowledgements
Drs. Steven Shears and David Miller read the manuscript and provided useful comments. This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
Reference List
- Baba Y, Hayashi K, Fujii Y, Mizushima A, Watarai H, Wakamori M, Numaga T, Mori Y, Iino M, Hikida M, Kurosaki T. Coupling of STIM1 to store-operated Ca2+ entry through its constitutive and inducible movement in the endoplasmic reticulum. Proc.Nat.Acad.Sci.USA. 2006;103:16704–16709. doi: 10.1073/pnas.0608358103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird GS, Putney JW., Jr. Capacitative calcium entry supports calcium oscillations in human embryonic kidney cells. J.Physiol. 2005;562:697–706. doi: 10.1113/jphysiol.2004.077289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeHaven WI, Smyth JT, Boyles RR, Putney JW., Jr. Calcium inhibition and calcium potentiation of Orai1, Orai2, and Orai3 calcium release-activated calcium channels. J Biol Chem. 2007;282:17548–17556. doi: 10.1074/jbc.M611374200. [DOI] [PubMed] [Google Scholar]
- Dziadek MA, Johnstone LS. Biochemical properties and cellular localisation of STIM proteins. Cell Calcium. 2007;42:123–132. doi: 10.1016/j.ceca.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr., Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr.Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luik RM, Wu MM, Buchanan J, Lewis RS. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J.Cell Biol. 2006;174:815–825. doi: 10.1083/jcb.200604015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manji SS, Parker NJ, Williams RT, Van SL, Pearson RB, Dziadek M, Smith PJ. STIM1: a novel phosphoprotein located at the cell surface. Biochim.Biophys.Acta. 2000;1481:147–155. doi: 10.1016/s0167-4838(00)00105-9. [DOI] [PubMed] [Google Scholar]
- Mercer JC, DeHaven WI, Smyth JT, Wedel B, Boyles RR, Bird GS, Putney JW., Jr. Large store-operated calcium-selected currents due to co-expression of orai1 or orai2 with the intracellular calcium sensor, stim1. J.Biol.Chem. 2006;281:24979–24990. doi: 10.1074/jbc.M604589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi S, Kakita S, Takahashi I, Kawahara K, Tsukuda E, Sano T, Yamada K, Yoshida M, Kase H, Matsuda Y. Wortmannin, a microbial product inhibitor of myosin light chain kinase. J Biol Chem. 1992;267:2157–2163. [PubMed] [Google Scholar]
- Norwood N, Moore TM, Dean DA, Bhattacharjee R, Li M, Stevens T. Store-operated calcium entry and increased endothelial cell permeability. Am J Physiol Lung Cell Mol Physiol. 2000;279:L815–L824. doi: 10.1152/ajplung.2000.279.5.L815. [DOI] [PubMed] [Google Scholar]
- Parekh AB, Putney JW., Jr. Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- Saitoh M, Ishikawa T, Matsushima S, Naka M, Hidaka H. Selective inhibition of catalytic activity of smooth muscle myosin light chain kinase. J Biol Chem. 1987;262:7796–7801. [PubMed] [Google Scholar]
- Saitoh M, Naka M, Hidaka H. The modulatory role of myosin light chain phosphorylation in human platelet activation. Biochem Biophys.Res Commun. 1986;140:280–287. doi: 10.1016/0006-291x(86)91087-9. [DOI] [PubMed] [Google Scholar]
- Smyth JT, DeHaven WI, Bird GS, Putney JW., Jr. Role of the microtubule cytoskeleton in the function of the store-operated Ca2+ channel activator, Stim1. J.Cell Sci. 2007 doi: 10.1242/jcs.015735. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth JT, DeHaven WI, Jones BF, Mercer JC, Trebak M, Vazquez G, Putney JW., Jr. Emerging perspectives in store-operated Ca2+ entry: roles of Orai, Stim and TRP. Biochim.Biophys.Acta. 2006;1763:1147–1160. doi: 10.1016/j.bbamcr.2006.08.050. [DOI] [PubMed] [Google Scholar]
- Soboloff J, Spassova MA, Tang XD, Hewavitharana T, Xu W, Gill DL. Orai1 and STIM Reconstitute Store-operated Calcium Channel Function. J.Biol.Chem. 2006;281:20661–20665. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- Stathopulos PB, Li GY, Plevin MJ, Ames JB, Ikura M. Stored Ca2+ Depletion-induced Oligomerization of Stromal Interaction Molecule 1 (STIM1) via the EF-SAM Region: AN INITIATION MECHANISM FOR CAPACITIVE Ca2+ ENTRY. J.Biol.Chem. 2006;281:35855–35862. doi: 10.1074/jbc.M608247200. [DOI] [PubMed] [Google Scholar]
- Takahashi R, Watanabe H, Zhang XX, Kakizawa H, Hayashi H, Ohno R. Roles of inhibitors of myosin light chain kinase and tyrosine kinase on cation influx in agonist-stimulated endothelial cells. Biochem Biophys.Res Commun. 1997;235:657–662. doi: 10.1006/bbrc.1997.6856. [DOI] [PubMed] [Google Scholar]
- Tran QK, Watanabe H, Le HY, Pan L, Seto M, Takeuchi K, Ohashi K. Myosin light chain kinase regulates capacitative ca(2+) entry in human monocytes/macrophages. Arterioscler.Thromb.Vasc.Biol. 2001;21:509–515. doi: 10.1161/01.atv.21.4.509. [DOI] [PubMed] [Google Scholar]
- Varnai P, Toth B, Toth DJ, Hunyady L, Balla T. Visualization and Manipulation of Plasma Membrane-Endoplasmic Reticulum Contact Sites Indicates the Presence of Additional Molecular Components within the STIM1-Orai1 Complex. J.Biol.Chem. 2007;282:29678–29690. doi: 10.1074/jbc.M704339200. [DOI] [PubMed] [Google Scholar]
- Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, Kinet JP. CRACM1 Is a Plasma Membrane Protein Essential for Store-Operated Ca2+ Entry. Science. 2006;312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Takahashi R, Zhang XX, Kakizawa H, Hayashi H, Ohno R. Inhibition of agonist-induced Ca2+ entry in endothelial cells by myosin light-chain kinase inhibitor. Biochem Biophys.Res Commun. 1996;225:777–784. doi: 10.1006/bbrc.1996.1250. [DOI] [PubMed] [Google Scholar]
- Wedel B, Boyles RR, Putney JW, Bird GS. Role of the Store-operated Calcium Entry Proteins, Stim1 and Orai1, in Muscarinic-Cholinergic Receptor Stimulated Calcium Oscillations in Human Embryonic Kidney Cells. J Physiol. 2007;579:679–689. doi: 10.1113/jphysiol.2006.125641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MM, Buchanan J, Luik RM, Lewis RS. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. The Journal of Cell Biology. 2006;174:803–813. doi: 10.1083/jcb.200604014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Lu J, Li Z, Yu X, Chen L, Xu T. Aggregation of STIM1 underneath the plasma membrane induces clustering of Orai1. Biochem.Biophys.Res Commun. 2006;350:969–976. doi: 10.1016/j.bbrc.2006.09.134. [DOI] [PubMed] [Google Scholar]
- Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc.Natl.Acad.Sci U.S.A. 2006;103:9357–9362. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
CFP-Orai1 does not rearrange upon store depletion when Stim1 is not overexpressed. Shown are confocal images of cells overexpressing CFP-Orai1 alone in the presence of 1.8 mM extracellular Ca2+ (left panel), following store depletion with thapsigargin (2 μM) in nominally Ca2+-free extracellular solution (center panel), and following store refilling with atropine and 1.8 mM extracellular Ca2+ (right panel). Scalebar = 10 μm.
ML-9 reverses EYFP-Stim1 punctae when Ca2+ stores are depleted with ionomycin. TIRFM was performed on EYFP-Stim1-expressing HEK293 cells that were treated at the times indicated with 400 nM ionomycin in nominal extracellular Ca2+, followed by restoration of 1.8 mM extracellular Ca2+, and finally addition of 100 μM ML-9. Shown is the fluorescence intensity profile averaged from 7 cells measured on a single coverslip; representative of 3 identical experiments (n = 12 cells total).
EYFP-Stim1 exhibits constitutive movements when punctate localization is reversed by store refilling. A single EYFP-Stim1 overexpressing HEK293 cell was imaged by TIRFM beginning in extracellular solution containing 1.8 mM Ca2+. At the times indicated in the upper left, the extracellular solution was changed to 300 μM carbachol (Carb) in nominal Ca2+, then to 50 μM atropine (Atn) with 1.8 mM Ca2+, and finally to 2 μM thapsigargin (Tg) in nominal Ca2+. Images were captured every 10 seconds, and the frame rate of the movie is 10 frames per second. The Scalebar is 10 μm.
Reversal with ML-9 re-establishes constitutive movements of EYFP-Stim1. A single EYFP-Stim1 overexpressing HEK293 cell was imaged by TIRFM beginning in extracellular solution containing 1.8 mM Ca2+. At the times indicated in the upper left, the extracellular solution was changed to 2 μM thapsigargin (Tg) in nominal Ca2+ and then to 100 μM ML-9 in the continued presence of Tg and nominal Ca2+. Images were captured every 5 seconds, and the frame rate of the movie is 10 frames per second. The Scalebar is 10 μm.
ML-9 induces constitutive movements of EYFP-D76N/D78N-Stim1. A single HEK293 cell over-expressing EYFP-D76N/D78N-Stim1 was imaged by TIRFM beginning in extracellular solution containing 1.8 mM Ca2+. At the time indicated in the upper left, the extracellular solution was changed to 100 μM ML-9 in the continued presence of 1.8 mM Ca2+. Images were captured every 5 seconds, and the frame rate of the movie is 10 frames per second. The Scalebar is 10 μm.
EYFP-Stim1 punctae are similar before and after reversal and reformation. HEK293 cells overexpressing EYFP-Stim1 were imaged by TIRFM beginning in extracellular solution containing 1.8 mM Ca2+. At the times indicated in the upper left, the extracellular solution was changed to 2 μM thapsigargin (Tg) in nominal Ca2+, then to 100 μM ML-9 in the continued presence of Tg and nominal Ca2+, and lastly to Tg in nominal Ca2+ without ML-9 (Wash). Images were captured every 10 seconds, the frame rate of the movie is 10 frames per second. The Scalebar is 10 μm. Images correspond to the experiment presented in Figure 9.
Pre- and post-reversal EYFP-Stim1 punctae exhibit close spatial correlation over time. As described in Figure 9C, images prior to reversal with ML-9 were pseudocolored red and merged with images taken following ML-9 washout (pseudocolored green). This movie shows a time-lapse series of merged images, zoomed into a region of the cell to demonstrate spatial correlation between red and green punctae. The frame rate of the movie is 5 frames per second. The total experiment time represented by this movie is 2 minutes.