

Abstract
Understanding the dynamics of redox elements in biologic systems remains a major challenge for redox signaling and oxidative stress research. Central redox elements include evolutionarily conserved subsets of cysteines and methionines of proteins which function as sulfur switches and labile reactive oxygen species (ROS) and reactive nitrogen species (RNS) which function in redox signaling. The sulfur switches depend upon redox environments in which rates of oxidation are balanced with rates of reduction through the thioredoxins, glutathione/glutathione disulfide and cysteine/cystine redox couples. These central couples, which we term redox control nodes, are maintained at stable but non-equilibrium steady states, are largely independently regulated in different subcellular compartments and are quasi-independent from each other within compartments. Disruption of the redox control nodes can differentially affect sulfur switches, thereby creating a diversity of oxidative stress responses. Systems biology provides approaches to address the complexity of these responses. In the present review, we summarize thiol/disulfide pathway, redox potential and rate information as a basis for kinetic modeling of sulfur switches. The summary identifies gaps in knowledge especially related to redox communication between compartments, definition of redox pathways and discrimination between types of sulfur switches. A formulation for kinetic modeling of GSH/GSSG redox control indicates that systems biology could encourage novel therapeutic approaches to protect against oxidative stress by identifying specific redox-sensitive sites which could be targeted for intervention.
Introduction
Cysteine (Cys) and methionine (Met) are the only amino acids in proteins which contain elements undergoing reversible oxidation under biologic conditions. These elements are aptly described as “sulfur switches” because the reversible oxidations provide means to control a broad range of activity and structure of proteins [1]. The sulfur atoms of both Cys and Met can undergo multiple oxidations, but the reversible oxidation of thiols to disulfides has been studied most extensively and serves as the basis for the current review. Changes in oxidation/reduction (redox) state of thiol/disulfide couples affects protein conformation, enzyme activity, transporter activity, ligand binding to receptors, protein-protein interactions, protein-DNA interactions, protein trafficking and protein degradation [2–6].
Considerable knowledge exists concerning specific redox-dependent components in redox signaling, transcriptional regulation, cell proliferation, apoptosis, hormonal signaling and other fundamental cell functions [7–10]. These redox-dependent components exist in redox signaling pathways [11]: the biological significance of these signaling pathways [12], the multiplicity of such pathways [13], and the sensitivity of these pathways to disruption by oxidative stress [14] has led to a refinement in the definition of oxidative stress as “an imbalance in prooxidants and antioxidants with associated disruption of redox circuitry and macromolecular damage” [15, 16]. This definition accommodates the multiple redox-dependent pathways (circuits) that exist and the heterogeneity in responses which can occur due to imbalances of oxidative and reductive processes [15–17]. At the same time, this definition suggests a need for integrated network maps to guide the search for redox-sensitive sites which can serve as targets for therapeutic interventions to prevent and treat oxidative stress. There has been limited progress in developing such maps and integrating the multiple redox pathways and compartments into common models.
The developing discipline of “systems biology” offers novel approaches for understanding the complex regulation of interacting redox networks. Established modeling formalisms, such as biochemical systems theory [18] and metabolic control analysis [19], which were once limited to metabolic pathways have been expanded as signal transduction and gene regulation regulatory networks have demonstrated similar control motifs [20]. Emerging principles in systems biology include the necessity of control points within a network (often referred as nodes) which serve as regulators of information or substrate flow. The realization that large biological networks can effectively be reduced into functional modules [21] linked together by control nodes has allowed researchers to tease apart the spaghetti diagrams into tractable pieces for validation and testing. The pace of scientific discovery and emphasis on cataloging and databasing [22–24] has increased the ability of descriptive kinetic modeling at every organizational level within the cell. Most importantly, the availability of high-throughput technologies for rapid, large-scale data acquisition has allowed novel data analysis tools to be developed for inferring network structures and extracting relationships between components. These computational and experimental tools can provide complementary approaches to traditional reductionist strategies for determining the interconnectivity between multiple redox couples that function as control nodes and their respective molecular targets.
The present review focuses on the redox states, stability and control mechanisms of the central thiol/disulfide redox couples (thioredoxins, GSH/GSSG and Cys/CySS) in mammalian cells with the intent to promote development of redox systems biology. These couples have previously been described as “redox control nodes” within electron-conducting circuits [15, 25]. These redox control nodes appear to regulate redox state of different sets of target proteins [26]. The purpose of the present review is to summarize the current knowledge of steady-state redox potentials of these redox control nodes and provide a framework for development of kinetic models which can describe the relatively stable non-equilibrium steady-state values of these redox couples and their functions in redox biology. A long-term goal of such an approach is to describe the steady-state redox potentials of individual redox couples as a function of all of the interacting electron transfer reactions within and between the cellular compartments.
The first section summarizes studies of the non-equilibrium redox states of the major thiol/disulfide redox couples in the subcellular and extracellular compartments. The second section contains an overview of some of the evolving principles which can be used to formulate models for redox systems biology. The third describes elements of kinetic models needed to account for non-equilibrium steady states of sulfur switches, using the GSH/GSSG couple as an example. This example illustrates the need for rate information for each step of biosynthesis, degradation/metabolism, transport, oxidation and reduction, to describe the steady-state relationships. The fourth section provides a framework to extend this approach to a more comprehensive systems biology description of redox signaling and control. This section also briefly considers the potential utility of such models for predictive health and development of new strategies to protect against disease.
Major thiol/disulfide couples are not at equilibrium in biologic systems
The redox potential (electromotive force, Eh), is a measure of the tendency of a chemical species to accept or donate electrons. This tendency is quantitatively expressed in millivolts relative to the standard hydrogen electrode reaction (H2/2H+ + 2e−). The Eh for an oxidation/reduction couple (e.g., GSH/GSSG) is dependent upon the inherent tendency of the chemical species to accept/donate electrons (Eo) and the concentrations of the respective acceptors and donors, defined by the Nernst equation (e.g., for the GSH/GSSG couple, Eh = Eo + RT/NF ln([GSSG]/[GSH]2).
The ΔEh between two couples is related to the free energy for the transfer of electrons between the couples (ΔG = −nFΔEh); however, the Eh values provide no information concerning whether a kinetically important pathway for electron transfer exists between the couples. The redox potential of a biologic system measured with a potentiometric electrode reflects the redox potential of the couples reacting most rapidly with the electrode, and not the Eh of components which do not interact with the electrode. Thus, establishing the electromotive force for redox couples in cells and tissues has created difficulties in defining a single metric for cellular redox environment; see [1, 27, 28] for examples of various proposed formalisms.
Two issues are problematic in trying to combine multiple redox couples together into a single definition of cellular redox state: 1) the potentials are not in equilibrium with one another and 2) the independent potentials vary among intracellular compartments. In the present review, we consider the redox control nodes as master control elements, which are independent and contribute to regulation of different sets of sulfur switches. They act as rheostats to control and integrate different cellular processes. Because there are multiple redox control nodes and a simple relation defining the cellular redox environment is ineffective and misleading, a systems modeling approach is needed to understand the relationships between redox couples.
In the following, we summarize the current knowledge of the steady-state Eh values for the thioredoxins, GSH/GSSG and Cys/CySS systems in mammalian systems to provide a foundation for such modeling. Values are calculated from the ratios of oxidized and reduced forms of thioredoxins and from concentrations of GSH, GSSG, Cys and CySS. The accuracy of comparisons of different couples is dependent upon the accuracy of the Eo values used for calculations. There is a considerable older literature on this subject, with a range of estimates for the GSH/GSSG and Cys/CySS couples [29]. For GSH/GSSG, the Eo′ value determined by Rost and Rapoport [29], −240 mV, is generally used. Experimental determinations of the Eo′ for Cys/CySS couple are more variable [30–32], and we use the value determined from the equilibrium of the 2 Cys + GSSG ↔ CySS + 2 GSH reaction, −226 mV [31]. The Eo values for human Trx1 (−230 mV) and Trx2 (−330 mV) were determined from the equilibria with the GSH/GSSG couple and the dithiothreitol/threitol disulfide couple under anaerobic conditions [33, 34].
Because the couples are not at equilibrium and differ among compartments, all biologic Eh values are operational, i.e., they are a function of the extraction, fractionation and assay methods. This includes both systematic errors, which can occur due to incorrect estimate of pH (5.9 mV/0.1 pH unit) and concentration (30 mV for 10-fold error in concentration estimates for GSH and Cys couples) [35], as well as variability, which can occur due to inefficient trapping of redox states. These issues were studied in detail to estimate cytoplasmic Eh values in HT29 cells [25, 36], with estimates of error/variability due to each as high as 10 mV.
GSH/GSSG Couple
Eh values for GSH/GSSG in extracts of cells cultured in vitro are typically in the range of −260 to −200 mV (Fig 1), with most values in the range of −230 to −220 mV (Table 1). Rapidly proliferating cells have a more negative (reduced) potential while non-dividing cells are more positive (oxidized). Cells undergoing apoptosis have a considerably more positive Eh value (−170 to −150 mV), due principally to a loss of GSH [37, 38]. More positive values also occur with depletion of GSH due to inhibition of GSH synthesis with buthionine sulfoximine (BSO) and due to removal of Cys and CySS from the cell culture media [39, 40].
Figure 1.
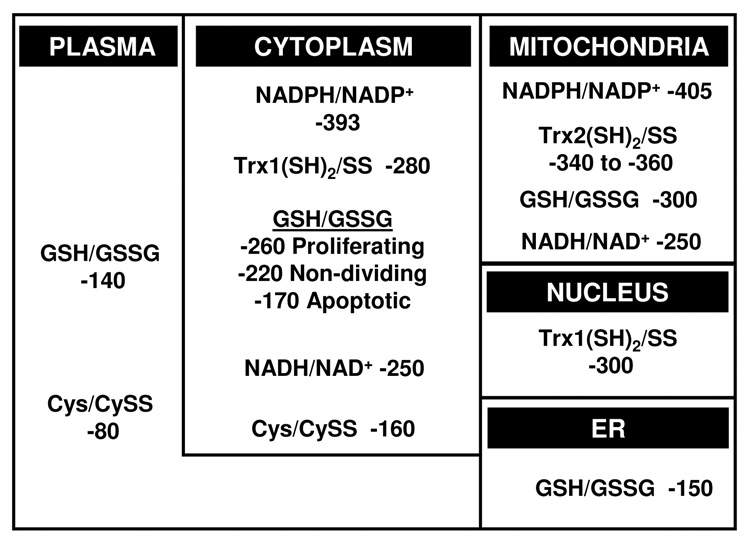
Steady-state redox potentials for thiol/disulfide control nodes. A variety of methods have been used to estimate redox states of thiol/disulfide systems in different compartments. Most information is available for total cellular GSH/GSSG, which appears to largely represent cytoplasm. Rapidly proliferating cells typically have values which are 30 to 60 mV more reducing than non-dividing cells. Cells undergoing apoptosis become considerably more oxidized, mostly due to a loss of GSH. Cytoplasmic Trx1 is more reduced than GSH and is not affected by cell proliferation and is not oxidized until terminal stages of apoptosis. Cytoplasmic Cys/CySS is more oxidized than GSH/GSSG and varies independently. Trx2 and GSH/GSSG have a more reduced redox state than cytoplasmic counterparts. Nuclear Trx1 is more reduced than cytoplasmic Trx1 and is much more resistant to oxidation. The redox state of nuclear GSH/GSSG is not known, but ratios of PrSH/PrSSG for nuclear and cytoplasmic proteins indicates that the nuclear GSH pool is more reduced than in the cytoplasm. The extracellular pools of GSH/GSSG and Cys/CySS are considerably oxidized compared to the respective cytoplasmic pools.
Table 1.
GSH/GSSG redox state (Eh) in rat intestines in vivo responds similarly to intestinal cell lines following experimental changes in growth stimulation, sulfur amino acid deficiency and inhibition of GSH synthesis.
| Control | Eh (mV) | Treatment | Eh (mV) | Reference |
|---|---|---|---|---|
| Differentiation and growth arrest | ||||
| HT29 cells | −258 | HT29 cells | −201 | [36] |
| Control media | Butyrate differentiation | |||
| Caco2 cells | −246 | Caco2 cells | −195 | [38] |
| Proliferating | Contact inhibited | |||
| Rat colon | −220 | Rat colon | −204 | [42] |
| ad libitum | 25% ad libitum | |||
| Growth stimulation by keratinocyte growth factor | ||||
| Caco2 cells | −230 | Caco2 cells | −240 | [7] |
| Control media | +KGF | |||
| Rat colon | −220 | Rat colon | −243 | [42] |
| Normal diet | +KGF | |||
| Sulfur amino acid deficiency | ||||
| HT29 cells | −224 | HT29 cells | −135 | [40] |
| Media with CySS | Media without CySS | |||
| Rat colon | −229 | Rat colon without | −215 | [137] |
| Normal diet | Sulfur amino acids | |||
| Inhibition of GSH synthesis with buthionine sulfoximine | ||||
| HT29 cells | −249 | HT29 cells | −220 | [25] |
| Without BSO | With BSO | |||
| Rat ileum | −215 | Rat ileum | −175 | [43] |
| Normal diet | With BSO | |||
Data from in vivo studies show values in the range of −255 mV (liver) to −193 mV (red blood cells) [41]. In rat colon, fasting and refeeding caused shifts in Eh from −220 mV (control), to −204 mV (fasted) to −243 mV (refeeding + KGF), in association with changes in cell proliferation (See Table 1) [42]. Similarly, following partial resection of rat ileum, a shift of the Eh from −215 mV (sham operated) to −225 mV (partial resection) was observed in association with increased proliferation [43]. Thus, the in vivo data are consistent with the in vitro studies establishing that Eh values for GSH/GSSG are within the range of −260 to −200 mV.
This range of values indicates that the GSH/GSSG couple is orders of magnitude out of equilibrium with the NADPH/NADP+ couple (Fig 1), which is maintained at about −400 mV [44]. The lack of equilibration of the GSH/GSSG couple with the NADPH/NADP+ couple was considered in detail by Gilbert [45], and can be partially explained by the kinetics of the GSSG reductase, which has a relatively high KM for GSSG. However, the range of GSH/GSSG values (from −260 to −200 mV) may also be important because this is sufficient to provide a 100-fold change in function of proteins with dithiol/disulfide motifs interacting with this couple [45].
GSH/GSSG couple in the cytoplasm
The redox state of the total cellular GSH/GSSG pool is the best estimate available for the cytoplasmic GSH/GSSG pool. In red blood cells, where there are no intracellular organelles, the Eh value was estimated to be −193 mV [41]. This value is more positive than estimates for cells with nuclei and mitochondria, which are typically <−200 mV, indicating that organelles with more negative values could introduce an error when using a cellular value to estimate the cytoplasmic value.
GSH/GSSG couple in the nucleus
Estimates of nuclear GSH have been controversial because of the potential artifacts due to perturbation during measurement [46–50]. Non-aqueous fractionation shows that GSH concentration in nuclei is similar to that in the cytoplasm, but redox potential values are not available [51]. Recent studies of the S-glutathionylation show that the ratio of protein thiol content (PrSH) to protein S-glutathione (PrSSG) is considerably higher in nuclear proteins than in cytosolic proteins [52]. While it is possible that evolution has resulted in nuclear proteins with a relative resistance to oxidation, i.e., Eo values which are more positive than for cytosolic proteins, the results are also consistent with nuclear GSH/GSSG being maintained at a more reduced steady state.
GSH/GSSG couple in the mitochondria
The mitochondrial GSH/GSSG pool has been estimated from GSH and GSSG concentrations following selective permeabilization of cells and also from isolated mitochondria incubated with respiratory substrates. Values from the former are contaminated by other organelles and tend to be somewhat more negative than the cytoplasmic values, i.e., about −280 mV [37, 53]. Isolated mouse liver mitochondria incubated with respiratory substrates can be substantially more negative, i.e., in the range of −330 to −300 mV (D. Cui, J.M. Johnson, D. P. Jones, unpublished). There is a possibility that the redox state in these isolated mitochondria is supra-physiologically reduced because of an artificially high concentration of respiratory substrates, but both approaches indicate that mitochondrial GSH/GSSG redox state is more negative (reduced) than the total cellular value.
GSH/GSSG couple in the plasma
The Eh for GSH/GSSG differs among extracellular compartments but is relatively well regulated within compartments. In human plasma, the mean value in young health adults was −138 ± 9 mV [54]. The difference between mean tissue values and plasma redox state is about 80 mV, indicating that the energy available from moving an electron from the cellular GSH/GSSG couple to the plasma GSH/GSSG couple is similar to the energy available from moving a Na+ ion down its electrochemical gradient. At present, potential functions of this redox gradient are not known.
The plasma GSH/GSSG redox state is oxidized in association with aging [41, 55], type 2 diabetes [41], cigarette smoking [56], chemotherapy [57] and risk of cardiovascular disease [58, 59]. Less information is available concerning other extracellular pools, but alcohol abuse has recently been shown to result in oxidation of the Eh in the lung lining fluid [60]. The lining fluid has a high GSH concentration and the GSH/GSSG redox state is similar to tissue redox values, i.e., −200 mV, perhaps reflecting the functional need to protect against oxidative toxicity and/or to avoid formation of protein disulfides. Fluidity of mucus is decreased by an oxidized redox state [61, 62], and crosslinking of proteins on the cell surfaces or in the lining fluid could also occur due to oxidation of the redox state. Thus, maintenance of a highly reduced redox state in the lining fluid would appear to be necessary to maintain deformability of the alveolar structures as well as signaling and other homeostatic functions.
GSH/GSSG couple in other organelles
The GSH/GSSG redox state in the secretory pathway has been estimated to be −172 to −188 mV [63]. This value is more oxidized than other subcellular compartments, and there is a possibility that the luminal redox state exists as a gradient in the vesicles communicating with extracellular thiol/disulfide pools. The endoplasmic reticulum contains an oxidase system which functions to introduce disulfides into proteins destined for export [64], and GSH is transported by isolated microsomes [65]. Thus, the endoplasmic reticulum contains both oxidative and reductive mechanisms which allow dynamic control of redox state in association with processing and secretory functions [66]. The lysosomal components of this membranal system are likely to differ considerably in redox control due to their function in protein degradation. Earlier studies showed that in cystinosis, a mutation results in loss of CySS transport and an accumulation of CySS within lysosomes [67, 68]. This suggests that lysosomes are a relatively oxidized compartment and are deficient in mechanisms to reduce disulfides to thiols.
Thioredoxins
Trx1 in the cytoplasm and nucleus
Thioredoxins are low molecular weight proteins containing a conserved dithiol motif which supports a range of biologic functions [69]. In mammalian systems, thioredoxin-1 (Trx1) is found in cytoplasm and nuclei, while Trx2 is found in mitochondria. The Eo value of the active site dithiol/disulfide couple of Trx1 was estimated to be −230 mV [34]. Using this value and measuring the percentage reduction with a redox western blot [34], cellular Trx1 has a steady-state redox potential of −280 mV. The value was more negative than the GSH/GSSG redox couple, and the couples were not in redox equilibrium [33, 38, 39]. The Trx1 and GSH/GSSG couples were found to vary independently during growth transitions [38], redox signaling [33, 39], and metal-induced toxicity [70]. Thus, Trx1 represents a distinct redox control node which can support a set of redox reactions which differ from those supported by GSH/GSSG.
Fractionation studies show that the nuclear Trx1 pool is about 20 mV more negative than cytosolic values and more resistant to oxidation induced by exogenous oxidant [33, 34]. The redox state of the nuclear Trx1 pool was preferentially preserved when cells were cultured in glucose- and glutamine free media, suggesting that nuclei contain special mechanisms to preserve Trx1 redox state [52]. This conclusion is reinforced by data showing that stimulation of H2O2 production in nuclei by a NLS-D-amino acid oxidase resulted in increased nuclear protein S-glutathionylation but not a detectable oxidation of nuclear Trx1 [71]. The available data show, therefore, that the redox state of nuclear Trx1 is maintained at least semi-autonomously from the nuclear GSH/GSSG pool and the cytoplasmic Trx1 pool.
Trx2 in the mitochondria
The mitochondrial Trx2 has a much more negative Eo value (−330 mV at pH 7.6) than cytoplasmic Trx1 (−230 mV at pH 7.0). The percentage of Trx2 present in the reduced form is variable in different cell types and under different metabolic conditions, with Eh values normally in the range of −360 to −340 mV (Fig 1). Trx2 is more susceptible to oxidation by exogenously added peroxides than either cellular Trx1 or GSH/GSSG [72]. Moreover, Trx2 is preferentially oxidized in a neuronal cell line treated with the respiratory inhibitor rotenone [73], in HeLa cells treated with TNF-α [70] and in HeLa cells treated with arsenic or cadmium [74]. In contrast, EGF signaling in keratinocytes oxidized cytosolic Trx1 but not Trx2 [33]. Together, these data show that the redox state of mitochondrial Trx2 is regulated independently of the cytoplasmic Trx1 pool. The substantial difference in redox potentials between the Trx2 couple and the mitochondrial GSH/GSSG couple also indicates that these couples are independently regulated in mitochondria [70, 74, 75].
Cys/CySS couple
Intracellular Cys/CySS
The Cys/CySS couple is considerably more oxidized than Trx1 and GSH/GSSG couples (Fig 1). In studies of total cellular Cys/CySS redox state, the digitonin-releasable fraction of Cys and CySS indicated that cytosolic Eh values were probably within about 10 mV of the values determined for whole cell extractions, i.e., about −160 mV [76]. This redox state is considerably oxidized relative to the GSH/GSSG and Trx1 couples, showing that reactions with the latter are not fast enough to equilibrate the pools within cells. The rate constant for reaction of low molecular weight thiols and disulfides can be conservatively estimated to be 20 Ms−1 [45]. Using this rate constant along with physiologic concentrations of cystine and GSH, the calculated thiol/disulfide exchange of CySS and GSH in cells is only 0.007 mM-min−1, a rate that is considerably slower that the estimated rate of CySS utilization by cells [77].
Experimental studies in which an inducer was used to stimulate GSH synthesis, an inhibitor was used to block GSH synthesis, and CySS-deficient media was used to deplete cellular thiol pools, showed that the cellular Cys/CySS couple is controlled independently of the GSH/GSSG couple [25, 40]. The Cys/CySS couple represents an important cellular redox control node which varies over a relatively narrow range and supports S-cysteinylation of proteins. Additional studies of the cellular Cys/CySS couple are needed because of the evidence that the extracellular Cys/CySS redox is associated with disease in humans and, as described above, is an important determinant of cell phenotype in cell culture.
Cys/CySS couple in plasma
The Cys/CySS pool is the major low-molecular weight thiol/disulfide couple in mammalian plasma. The redox state of this couple in human plasma averages −80 mV in young, healthy individuals; in rodents, the value is about −100 mV. Human studies show that the plasma Eh is oxidized in association with age [55], chemotherapy [57], cardiovascular disease [59] and smoking [56]. The value correlates with plasma protein S-glutathionylation [78, 79], and age-associated oxidation can be prevented by antioxidant supplements [80].
A number of studies have addressed the potential consequences of changes in plasma Cys/CySS redox state by systematically varying this parameter in cell culture. In colon carcinoma Caco2 cells and normal human retinal pigment epithelial (hRPE) cells, cell proliferation was greater at more negative Eh values [7, 81]. hRPE cells were more sensitive to oxidant-induced apoptosis at more positive Eh values [81]. ROS production was increased in bovine aortic endothelial cells exposed to a more positive Eh value [82]. Binding of human monocytic THP1 cells to endothelial cells was enhanced at more positive Eh values [82]. A lung fibroblast model relevant to pulmonary fibrosis also showed that positive Eh values stimulate fibroblast proliferation and matrix expression through upregulation of transforming growth factor-β [83]. Thus, the data show that the factors controlling the balance of plasma Cys/CySS are important in determining cell function and thereby can potentially contribute to disease risk.
Formulation of models for thiol/disulfide redox systems biology
Most proteins contain at least one Cys or Met which is subject to oxidation, and all aspects of life depend upon redox reactions. Consequently, there is considerable complexity to development of redox systems biology. Certain features are emerging which suggest that this complexity can be simplified by a set of rules or principles governing the biological redox reactions, which can be considered part of a biologic “redox code” [84–86]. For initial model development, these principles can be used as assumptions with the recognition that they are largely based upon observational data which can be strengthened by additional experimental validation.
In consideration of the available data, three interrelated features appear to be important for modeling, namely that 1) normal electron conducting pathways involving thiol/disulfide couples are insulated from each other so that it is possible to describe reactions in terms of discrete redox pathway maps, 2) pathway models can be simplified because there are common redox control nodes which control multiple sulfur switches and 3) specific functions can be defined for individual sulfur switches within these pathway maps so that accurate kinetic models can be developed.
Insulation of thiol/disulfide-dependent pathways
As summarized above, a range of experiments shows that the Trx1, GSH/GSSG and Cys/CySS couples are not rapidly interacting and not energetically coupled [25]. This lack of rapid interaction provides an important simplification for model development in that it allows description of redox processes in terms of pathways which are relatively insulated from each other. For instance, in Fig 2, examples of Trx and GSH are shown to support different reduction pathways in cytoplasm, nuclei and mitochondria. Although not explicitly proven, this interpretation is supported by studies in which Trx2 and GSH were found to have parallel functions in protection against TNF-α-induced cell death [87] and by a number of studies which establish separate control of specific proteins by either Trx1 or GSH, but not both [39, 74, 88].
Figure 2.
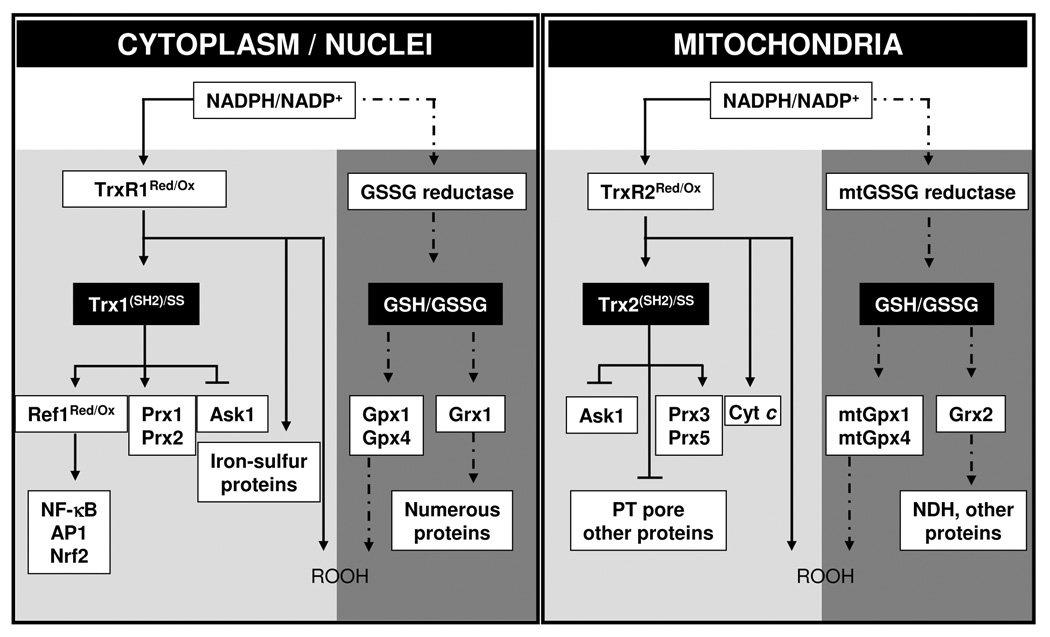
GSH and Trx-dependent systems support different protein-dependent systems in cytoplasm and nuclei (left), and in mitochondria (right). Model is based upon observations that 1) Trx (Trx1 for cytoplasm and nuclei, Trx2 for mitochondria) and GSH systems function in parallel in peroxide metabolism, 2) steady-state redox potentials of NADPH/NADP, Trx1 (SH)2/(SS), GSH/GSSG in cytoplasm and nuclei, Trx2(SH)2/(SS), GSH/GSSG in mitochondria are maintained at different values, 3) GSH and Trxs have multiple functions in protection against oxidative stress and in redox control mechanisms. Thioredoxin reductase-1 (TrxR1) in cytoplasm and nuclei and TrxR2 in mitochondria also have multiple activities which include peroxide metabolism. Redox factor-1 (Ref-1), peroxiredoxin-1 (Prx1), Prx2 and Ask1 for Trx1-dependent proteins and Ask1, Prx3, and Prx5 for Trx2-dependent proteins are represented, which function in parallel with the GSH/GSSG-dependent proteins [glutathione peroxidase-1 (Gpx1), Gpx4, glutaredoxin (Grx1) for cytoplasm, mtGpx1, mtGpx4, and Grx2 for mitochondria]. TrxR1 and TrxR2-dependent proteins other than Trx1 and Trx2 could also exist, such as iron-sulfur proteins and cytochrome c (Cyt c). In this model, the steady-state redox values are maintained separately for each couple by the balance of NADPH-dependent reduction and peroxide-dependent oxidation rates. Each couple can thereby be used for different redox-control processes, and in the presence of excess peroxide generation rates, each can contribute to detoxification. Arrowheads represent flow of electrons while blunt-end lines represent redox-dependent interactions which could occur without electron transfer.
The utility of conceptualizing redox events in terms of specific pathways is illustrated schematically in Fig 3. In this scheme, oxidative stress (Fig 3, A) is described by a pathway that is fundamentally the same as for redox signaling (Fig 3, B). In this scheme, upstream processes generate reactive species which oxidize downstream targets (Fig 3, A and B). Ordinarily, the high-flux electron transfer pathways that support ATP production in mitochondria, as well as other redox pathways in intermediary metabolism, are well insulated from other redox-sensitive components in cells. However, increased electron transfer between pathways can result in loss of the insulation and failure of systems by inappropriate transfer of electrons to a separate pathway [17]. A salient conclusion, therefore, is that kinetic models built upon well defined redox pathway maps could be very useful to identify sites and causes of transition between effectively insulated redox pathways and disrupted states of oxidative stress.
Figure 3.
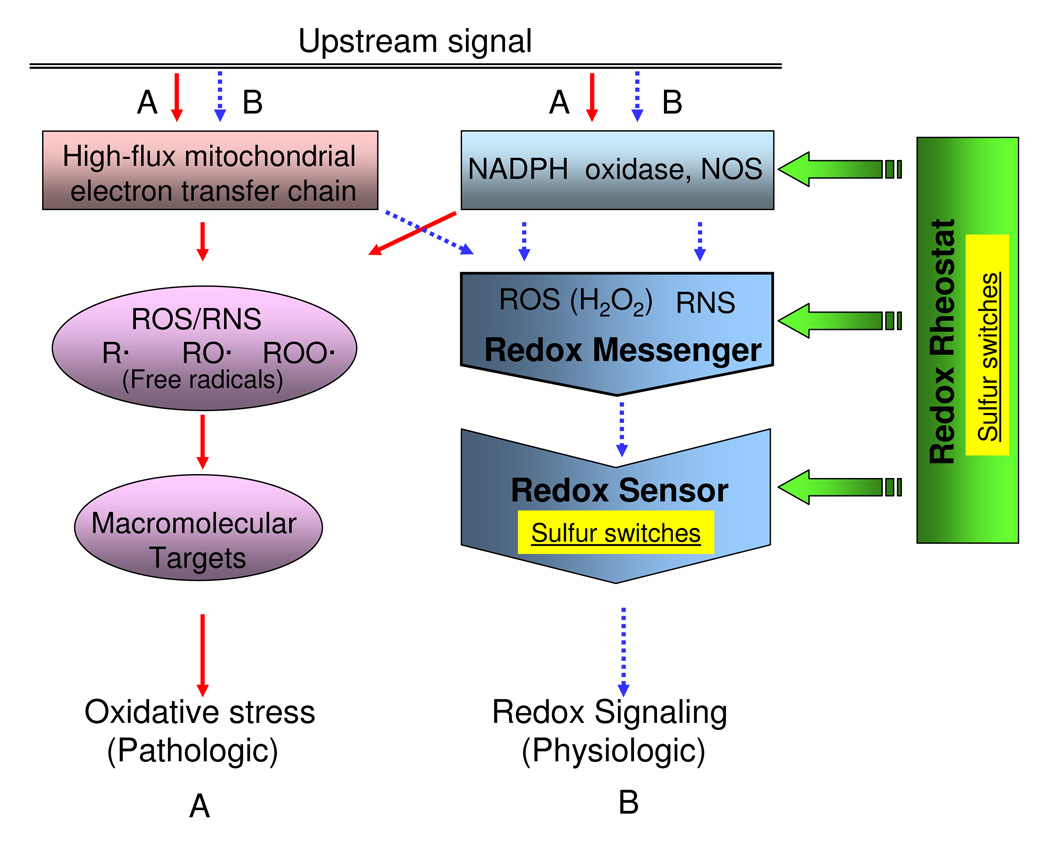
Sulfur switches can exist as "Redox Sensors" and as "Redox Rheostats" in generic redox circuitry models. Simplified models show (A) oxidative stress pathway (solid line) associated with high levels of ROS, RNS, and free radicals from mitochondrial dysfunction, NADPH oxidase activation, and nitric oxide synthase activation triggered by pathologic upstream signals. Redox signaling pathway (B, broken line) shows the process involving relatively low levels of diffusible reactive species associated with physiologic condition. In redox signaling, the small diffusible reactive species, termed "redox messengers" are generated by ROS/RNS sources and react with redox-sensitive macromolecules termed "redox sensors". These redox sensors are often sulfur switches, consisting of sulfur atoms within specific proteins. Sulfur switches also occur as regulatory elements, termed "redox rheostats", which are not needed to define the pathway but regulate its activity. The characteristics of these sulfur switches can differ because the redox sensors are integral components of pathways while the sulfur rheostats regulate activity.
The disequilibrium of Trx and GSH suggests that the rates of other thiol/disulfide couples in cells may also be slow, effectively insulating most sulfur switches from each other. This point is critical for development of kinetic models, but data are currently limited. A redox-sensitive GFP which changes fluorescence in response to thiol oxidation shows this characteristic of being relatively insulated because it is only slowly responsive to exogenously added oxidants [89]. Studies of the redox dynamics of the pathway NADPH → Trx reductase-1 → Trx-1 → Ref-1 → p65 (NF-κB) in cells exposed to glucose- and glutamine-deficient media also showed slow interaction in that components in the pathway were not fully reduced under steady-state conditions [52]. Steady-state levels of reduction in this pathway changed in association with redistribution of Trx1 to nuclei [52]. Similar rate limitations have recently been obtained for the mitochondrial pathway NADPH → TR2 → Trx2 → Prx3 → H2O2 under metabolic substrate deficiency (H. Zhang and D.P. Jones, unpublished). These rate limitations in kinetically active, well defined thiol/disulfide pathways suggests that non-equilibrium conditions may be a general feature of sulfur switching mechanisms which allows rapid and sensitive responses to perturbations in oxidant generation.
Specificity of thioredoxins and GSH in control of sulfur switches
Biochemical data show that thioredoxins and glutaredoxins catalyze different chemical reactions and have preferred substrates [90–93]. Thus, an important simplification for modeling will be possible if protein targets in cells can be classified as either targets of reduction by Trx or by GSH. In vivo data distinguishing Trx- and GSH-dependent activities are limited, but apoptosis signal-regulating kinase (Ask-1) activation has been associated with oxidation of Trx1 [74], while Nrf-2 translocation to nuclei [39] and oxidative regulation of NADH dehydrogenase [88] have been linked to oxidation of GSH/GSSG. Thus, for initial modeling, one can assume that Trx1, Trx2 and GSH largely support the reduction of different subsets of cellular proteins. Although considerable information is available, addition definition of these subsets represents an important subject area for future research.
A practical aspect of this pathway description emerges from the combination of the principle that sulfur switches can be classified in terms of control by Trx or GSH with the principle that the insulation of redox pathways is partly a consequence of differences in reactivities at prevailing concentrations in cells. In the non-equilibrium state, changes in abundance of thioredoxins and glutaredoxins can change the flux through pathways. This indicates that over-expression of redox-active components can, at least in principle, change the definition of the redox pathways by altering the abundance of key elements. Thus, a valuable contribution of pathway analysis and kinetic modeling will be to evaluate this possibility.
At present, it is not clear how many sulfur switches exist in cells or how many redox control nodes are present to support different subsets of these switches. In the discussion below, we address the redox control nodes including Trx1, Trx2, GSH/GSSG and Cys/CySS [25]. Evidence indicates that other redox control nodes may also exist because there are multiple protein targets for thioredoxin reductases [94, 95], Ref-1 [96–98], and thionein [99]. Additional research is needed to address the function of these redox couples to determine whether they should also be considered as central redox control nodes.
Sulfur switches: elements within redox signaling pathways (redox sensor) and elements for regulating redox signaling pathways (redox rheostat)
A third feature of sulfur switches which needs definition for modeling is the classification of switches according to function. Several lines of evidence suggest that sulfur switches have different functions which affect the way in which they are incorporated into kinetic models. In Fig 3 (center), a sulfur switch is depicted as a sensor within a pathway. Such an element can function as an on/off switch for a pathway, such as transmission of an NADPH oxidase-derived signal through a phosphatase. Alternatively, sulfur switches can be elements which function like rheostats to regulate the level of activity of a pathway but not be required for the pathway to operate (Fig 3, right). Hypothetical rheostat steps are indicated at upstream sites for generation of ROS/RNS, as well as subsequent sites regulating the concentrations reactive species and response sites (Fig 3, right). By being coupled to GSH/GSSG or thioredoxin, such rheostat elements could coordinate diverse processes within the context of the physiologic state [1, 36] without changing the fundamental way that a signaling pathway works.
Currently, there is no way to assess whether sulfur switches functioning as redox rheostats are rare or common in biologic systems. In consideration of this point, human Trx1 is a useful example. This protein has 5 Cys residues, two of which (C32,35) are present in the active site. Site-directed mutagenesis showed that C62, C69 and C73 are not necessary for function [100]. However, C73 functions in dimer formation [101] and also regulates activity by S-glutathionylation [102] and alkylation [103]. C62,69 is a redox-sensitive control for reduction by thioredoxin reductase-1 [34] and C69 and C73 are S-nitrosylation sites [104]. Thus, all of the Cys residues determine function even though only 2 are present in the active site and mutagenesis studies show the others are not required. Failure to detect redox rheostat elements can also because of the addition of GSH and dithiothreitol (DTT) for in vitro studies. Such reductants are often used to preserve activity of proteins which lose activity in purified form, and this could result in inability to detect redox rheostat mechanisms by eliminating redox-dependent changes. Thus, the available data point to a need for additional research to identify redox control elements which are not essential to the function of pathways but rather serve to regulate and integrate functions.
Kinetic models and potential mechanisms for control of non-equilibrium steady states
Electron transfer rates between specific components within biologic systems can vary considerably, but the overall rates are ultimately dictated by the rate of transfer to O2, the terminal electron acceptor for aerobic organisms. The maximal rate of O2 consumption by an athletically trained human is about 4 L/min, which is equivalent to about 2.5 mM-min−1 averaged throughout the body. This average equates to about 10 nmol/mg protein per min at the cellular level, which is about 40% of measured cellular rates [105, 106] and about 5–10% of maximal mitochondrial rates (100 to 200 nmol/mg protein per min). Calculated in terms of 2-electron transfer in cells, the mitochondrial rate is about 20–35 mM-min−1. In contrast, the rate estimated above for thiol-disulfide exchange between the Cys/CySS and GSH/GSSG couples at physiologic concentrations was 0.003 mM-min−1 (Table 2).
Table 2.
Comparison of glutathione fluxes across cellular compartments
| Flux | Abbrev. | mM/min | Source | |
|---|---|---|---|---|
| SLOW | GSSG transport | 0 | [138] | |
| cytosol → ER | ||||
| GSH + GSSG transport | νE_P | 4e-4* | [87] | |
| ER → plasma | ||||
| GSH disulfide exchange with Cys/CySS | 5e-4 | [139] | ||
| (plasma) | ||||
| GSH disulfide exchange with Cys/CySS | 0.003 | [140] | ||
| (cytosol) | ||||
| GSH +GSSG transport | νC_P | 0.007 | [141] | |
| cytosol → plasma | ||||
| MEDIUM | GSH transport | νC_E | 0.06 | [63] |
| cytosol → ER | ||||
| Cytosolic GSH synthesis | νs | 0.024-1.3 | [26,108] | |
| GSH transport | νC_M | 1.2 | [142] | |
| cytosol → mitochondria | ||||
| GSSG transport | νM_C | ? | ||
| mitochondria → cytosol | νM_C | ? | ||
| GSH + GSSG transport | νC_N | ? | ||
| cytosol → nucleus | ||||
| FAST | GSSG Reductase | 20 | [143] | |
| Thiol oxidase | 98 | [143] | ||
| Glutathione Peroxidase | 3000 | [143] | ||
upper bound
Where values were reported in other units, 106 cells were approximated as 1 mg protein and 5 µl intracellular water. Reports of transport flux under non-physiological conditions were ignored.
This comparison underscores the principle that redox signaling and control mechanisms require effective insulation from high-flux redox systems. The markedly slower rates suggest that sulfur switching may also require catalysis. Because of the relatively slow rates of thiol/disulfide reactions relative to other biologic oxidations, high catalytic efficiency is not needed. Localized generation of H2O2 can selectively oxidize thiols catalyzed by proximal amino acids or metals, with rates enhanced by conformational changes or binding interactions in which cationic amino acids are juxtaposed to thiols. Association of proteins into complexes can increase proximity of dithiols and decrease accessibility of the reductants such as GSH and thioredoxin. Insulation can be further achieved by compartmentation. High-flux electron transfer reactions are compartmented in the mitochondrial inner membrane, biosynthetic and degradative reactions dependent upon hydrogen peroxide are compartmented in the peroxisomes, oxidation of lipophilic xenobiotics is compartmented in the membrane of the endoplasmic reticulum, and controlled oxidations of proteins destined for secretion are compartmented in the cisternae of the endoplasmic reticulum. Such compartmentation allows relatively low flux redox signaling and control to occur separately in association with the plasma membrane, aqueous cytoplasm, nucleus and extracellular space.
Non-equilibrium thermodynamics
A systems modeling approach provides a means to study the relationships between sulfur switches and redox control nodes for improving the understanding of the critical sites of regulation and sensitivity to disruption in pathology and toxicity. In non-equilibrium systems, different mechanisms can cause systems to be out of equilibrium. For instance, introduction of a perturbation into an equilibrated system creates a disequilibrium followed by a decay back to a resting equilibrium state. In this scenario, the motion in the system is due to the perturbation. A second form of disequilibrium is a non-equilibrium steady state [107], in which a constant driving force in the system allows for a non-zero flux. The experimental data described above indicates that the multiple thiol/disulfide redox couples are maintained out of equilibrium and therefore are examples of the second scenario. Such disequilibrium can easily be mistaken for a system at rest because the concentrations are constant. Thus, a perturbation to the system in the form of oxidative stress (such as rapid introduction of ROS) is followed by a decay back to the non-zero steady-state fluxes in the same way that equilibrium systems decay back to equilibrium. A key difference, however, is that in the equilibrium state, the redox potentials are equilibrium values defined purely by the thermodynamics of the interacting components while in the nonequilibrium state, the redox potentials are steady-state values which are defined by the relative kinetics of the electron transfer reactions.
Modes of flux control in redox couples
In developing kinetic models for biologic redox systems with relatively slow electron transfer rates, the concentrations of electron carriers can change. Consequently, terms for synthesis and degradation as well as transport of the reduced and oxidized molecules across compartment membranes need to be included along with the oxidation/reduction reactions. To illustrate the role of various components as determinants of fluxes that give rise to the properties of a redox system, we use the GSH/GSSG couple as an example. Other redox couples - NADPH/NADP+ [108], NADH/NAD+, thioredoxin, Cys/CySS - can be analyzed in the same manner; however, there is much less information available.
For Eh of GSH/GSSG to vary from −140 in the plasma, −175 in the ER, −230 in the cytosol, and −270 in the mitochondria [75], there must be constant steady-state fluxes to maintain these non-equilibrated values. These fluxes are driven by the metabolic processes (mitochondrial respiration, NADPH oxidases, etc) that generate a constant flow of electrons to O2 to form superoxide anion radical, hydrogen peroxide, etc. in the cellular environment. The basal level of peroxide formation is on the order of 0.2–0.5 mM/min but can be stimulated to over 1.3 mM/min [109, 110]. We surveyed GSH sources and sinks from the cellular compartments to illustrate the primary contributors to the differences in GSH/GSSG potential by organelle. For many of the rates listed in Table 2, only unidirectional fluxes were available, generated under non-physiological conditions. Therefore, many of the values represent upper bounds of the actual basal level fluxes. Data missing from this Table (e.g., rates of transport of GSH or GSSG out of mitochondria and between cytoplasm and nuclei) identify key experimental data which are needed for functional kinetic models.
Table 2 reveals that transport rates in and out of cellular compartments vary between 10 µM/min and 1 mM/min. This is 10- to 1000-fold less than the metabolic rate of ATP synthesis, yet maximal rates are orders of magnitude greater than GSH synthesis. The synthesis of GSH is low (~ 0.024 mM/min in HT-29 cells [26], 0.1 mM/min for liver [111]) such that the distribution among cellular compartments can happen on a much faster time scale. From this, one can deduce that other factors are driving the fluxes of GSH and GSSG between compartments and not the synthesis rate of GSH. Because the GSH/GSSG couple in each compartment is also maintained out of equilibrium from other compartments, this indicates that the GSH/GSSG potential within compartments is driven by reactions unique to the organelles. The functional consequence is that the mitochondrial GSH/GSSG redox potential can be more negative (reducing) to control mitochondrial sulfur switches in the presence of local sources of ROS while the ER can be more positive (oxidizing) to regulate components for disulfide bond formation during protein folding by protein disulfide isomerase (for discussion see [112]).
Linear equations for modeling non-equilibrium systems
Using the values in Table 2, a system of linear equations can be generated to describe GSH movement in a compartmental model. Under steady-state conditions, a balance must exist between the fluxes of utilization/degradation for each compartment and the flow in and out of the compartment through transport, with the exception of the cytosol, where glutathione synthesis occurs. This balance can be represented by the following set of equations to mathematically formulate Figure 4:
GSHcytoplasm :−(νC_M + νC_N + νC_E + νC_P)=−(νs −νOC)
GSSGcytoplasm :−(νC_N + νC_P)= νOC
GSHmitochondria :νC_M = νOM
GSSGmitochondria :−νM_C = νOM
GSHnucleus :νC_N = νON
GSSGnucleus :νC_N = νON
GSHER :νC_E −νE_P = νOE
GSSGER :−νE_P = νOE
GSHplasma :νC_P + νE_P = νOP
GSSGplasma :νC_P + νE_P = νOP
In this formulation, the nomenclature designates the starting compartment followed by the target compartment, in which C for cytoplasm, M for mitochondria, N for nucleus, E for ER, and P for plasma. For example, νC_P represents the flux from the cytoplasm to the plasma. The utilization/degradation flux is symbolized by vO for “out” and the compartment abbreviation, such as νOM representing all utilization steps of mitochondrial GSH, including the conversion to GSSG. Thus, the GSH and GSSG fluxes are tied together by this overarching term which encompasses the biochemical oxidation/reduction reactions. The equations are structured to reflect the unknown effluxes on the right-hand side. The additional flux from the ER directly to the plasma is the only introduction of coupled variables in the system. One should note that in such formulations, fractional volumes must be used to convert data to molarity values to express the appropriate number of molecules in different subcellular volumes moving across membranes.
Figure 4.
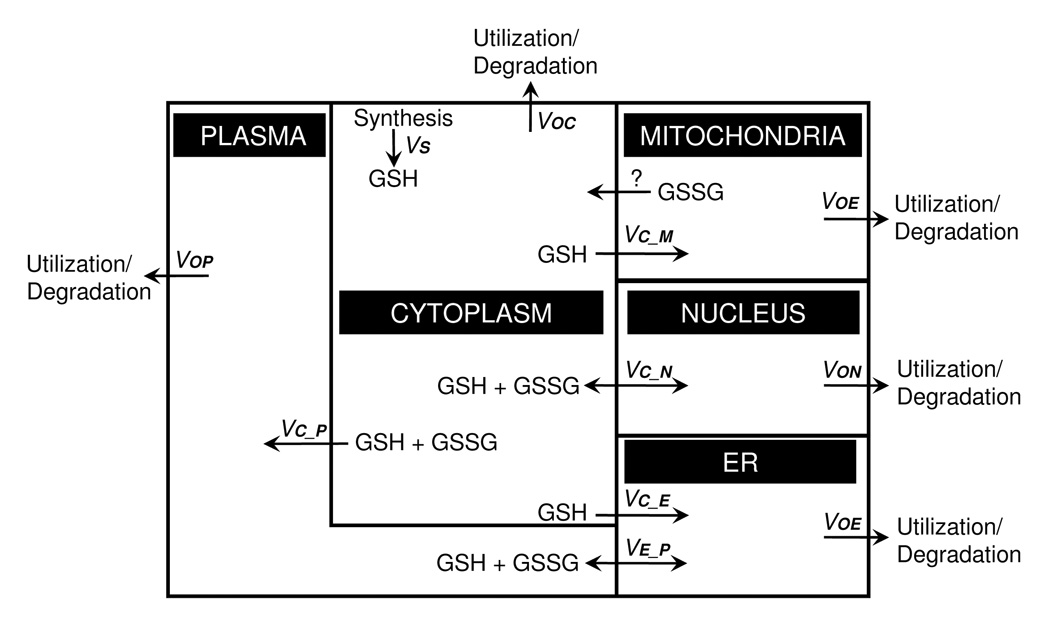
A compartmental model of steady-state glutathione fluxes. Under homeostatic conditions, GSH transport must be balanced by degradation/utilization to balance the rate of cytosolic GSH synthesis. In the diagram, GSH influx to the ER, mitochondria and plasma is considered to be unidirectional, while the exocytosis of metabolites through the ER will release GSH and GSSG into the plasma at the same rate. Mitochondrial loss of GSH is poorly understood but could occur by unidirectional efflux of GSSG. Passive transport to the nucleus will be bidirectional and include GSH + GSSG. Salient features revealed by this formulation include: 1) an uncharacterized mechanism for loss of GSH or GSSG from mitochondria is needed to balance mitochondrial GSH/GSSG redox state, 2) non-equal partitioning of reductases and oxidases between the cytoplasm and nucleus could maintain a disequilibrium between compartments, 3) differential regulation of GSH and GSSG transport between compartments can determine differences in steady-state redox potential between compartments, and 4) the secretory pathway could provide a kinetically important route for electron transfer between cytoplasm and plasma.
Summarizing from the section above, redox reactions within compartments are generally fast compared to movement between compartments, and movement between compartments is generally fast compared to entry (synthesis) and removal (degradation) from the system. Assumptions based upon these generalizations allow simplification of modeling. For instance, one can develop models for individual compartments assuming that there is no movement between compartments (See Figure 5, bottom left). These models can subsequently be incorporated into more complex models which address the interactions of the compartments (Figure 5, left panels). Additional relations can be included at the expense of a more complicated description.
Figure 5.
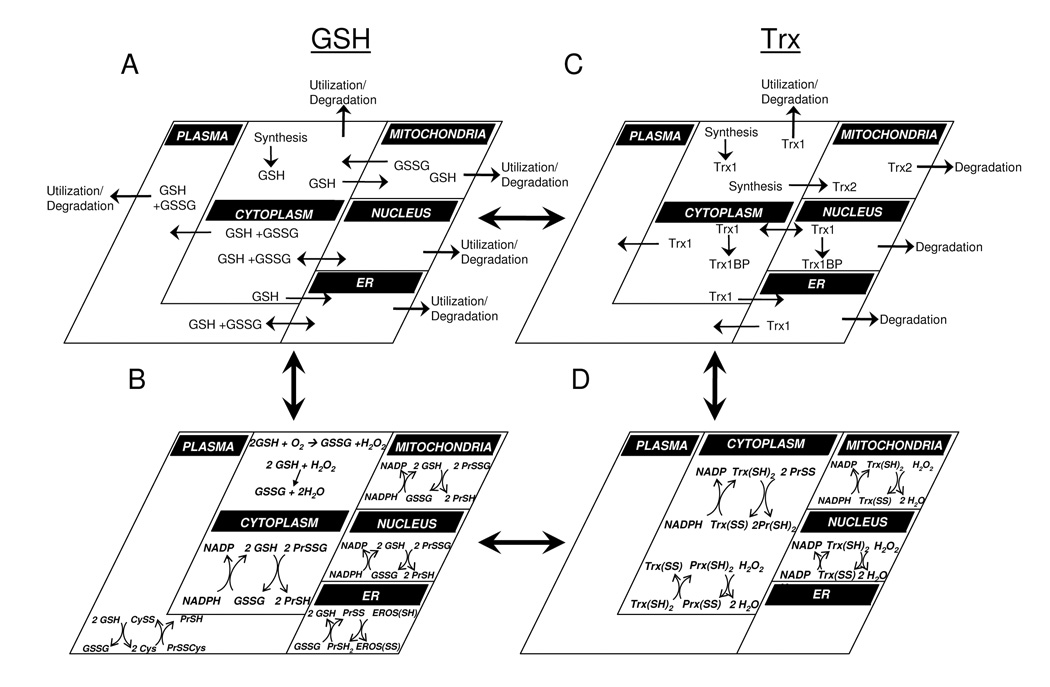
Complex redox systems maps can be assembled from models for redox couples in different compartments. The steady-state flux model for GSH and GSSG (Panel A) can be combined with compartmental models for oxidation/reduction of GSH/GSSG (Panel B) to provide a more complex model including synthesis/degradation and transport along with redox processes. Similar models can be developed for thioredoxins (Panels C, D), and these can be combined with A and B to provide more complex descriptions of interacting redox systems. Such models can then be used to analyze cellular control of sulfur switches, linking redox changes to specific sensor and rheostat functions. In this simplified model, only GSH and Trx couples are illustrated; however, these concepts can be extended to NADPH/NADP+, peroxiredoxins, Cys/CySS and other redox couples. For illustrative purposes, peroxiredoxin reactions are only depicted in the cytosol, but are present in the nucleus and mitochondria as well.
Because the reactions within compartments appear to be fast relative to the movement between compartments, it may also be possible to describe GSSG algebraically as a function of GSH so that transport processes can be studied in the presence of metabolism. In this modeling framework, oxidation of GSH to GSSG is included in the utilization term, reducing the number of variables from the above description. This consideration is potentially important because of the stability of steady-state thiol/disulfide systems. For instance, if one considers that autooxidation of thiols (plus possible sulfhydryl oxidases and thiol oxidases) results in continuous production of H2O2 and coupled (with glutaredoxin) generation of GSSG, one can write an overall balance for GSH oxidation and reduction described by the following equations:
With a constant flux of oxygen and NADPH regeneration, the glutathione couple acts as a balanced cycle of redox buffering under steady-state hydrogen peroxide synthesis. The first two reactions appear to be using a net consumption of four reducing equivalents for the conversion of O2 to water. However, in the context of a GSSG reductase, a balance occurs during basal metabolism as long as the NADPH regeneration and/or synthesis is held constant. Such a balance could explain the relatively stable GSH/GSSG redox potential in cells [113] so long as the rates are significantly faster than the transport fluxes. Limitations to this possibility are evident in the first step because there is little evidence for cellular thiol oxidases [114], and non-enzymatic thiol-disulfide exchange is slow [45]. An alternative is that an NADPH or NADH oxidase complements the mitochondria to maintain a relatively high constitutive rate of H2O2 generation which is balanced by the peroxidase and reductase reactions to maintain a relatively stable thiol/disulfide steady state. Such a possibility is suggested by the discovery an NADH oxidase coupled to peroxiredoxin in anaerobic bacteria (Amphibacillus xylanus and Sporolactobacillus inulinus) [115]. Although the function of this complex is not known, the organism is aerobic and lacks a respiratory chain so that a possible function could be to maintain a non-equilibrium thiol/disulfide redox potential of sulfur switches.
Regardless of simplifications used to evaluate individual processes, the ultimate goal is to develop a model which can be used to develop testable hypotheses concerning critical sites of oxidative stress. For a non-equilibrium homeostasis to exist, all fluxes must ultimately be balanced such that synthesis (νs in Figure 4 and the above equations) matches utilization of GSH in each compartment (through the glutathionylation of proteins, for example) or degradation (i.e. to Cys) because the GSH synthesis rate is the only introduction of material to the system. For high import rates across a membrane such as in the mitochondria, this forced balance means that either the mitochondria must utilize GSH at a high rate, or bidirectional transport exists to reduce the net rate into the mitochondria. From the diagram, one can observe that with a low synthesis rate (24 µM/min) [26, 108], the transport balance must be very fast because a high utilization/degradation rate would rapidly deplete cytoplasmic GSH. The discrimination between the two options may be compartment-specific. In the ER, the measured transport is primarily uni-directional into the cisternae of the ER; thus, it is not clear whether GSH is recycled back to the cytoplasm or lost to the plasma. In the mitochondria, lack of recycling for GSH or GSSG would result in mitochondria being an irreversible sink for GSH. Thus, the model predicts that mechanisms for movement of GSH or GSSG back to cytoplasm must occur to maintain cellular balance, and additional research is needed to understand these mechanisms.
Extending this point, the model includes the contribution of GSSG influx/efflux to the cytosol/mitochondria, ER/plasma, and cytosol/nucleus exchanges, but several of these rates are poorly characterized. Additionally, some compartments are not even considered. Consequently, this formulation identifies an important need for redox system biology is to obtain more complete information concerning transmembrane transfer rates and function of specific subcellular compartments in thiol/disulfide redox control.
Challenges to development of redox systems biology
Maintaining the redox couples out of equilibrium poses serious challenges in computational modeling when one wants to introduce oxidative stress conditions into the simulations. Specifically, the model must accommodate the maintenance of a steady-state system with proper redox potentials prior to the onset of ROS formation and then adjust rates, which can include transport, synthesis and degradation, into the overall model of electron flow. Although challenging, a strength of the approach is that it can address more complex issues such as the indirect linkages between control nodes. For instance, the GSH/GSSG and Trx systems are linked despite their disequilibrium due to the use of a common substrate, NADPH, between Trx reductase and GSSG reductase and the glutathionylation of thioredoxin to enhance activity [102]. The independent behavior of the individual couples can be addressed by extending the GSH/GSSG model to include the Trx systems.
The description of transport between subcellular compartments that relies on a concentration gradient and/or membrane potential creates additional difficulties. In such a system, components will flow toward equilibrium prior to the stimulus of oxidative stress conditions. The steady-state flux contributions to GSH disequilibrium across membranes can be primarily attributed to movement of the GSH molecule rather than reduction of GSSG to GSH. During dynamic simulation of an oxidative stress response, one could consider these fluxes of molecules static in order to focus on faster changes occurring at the level of sulfur switches within compartments; however, the disequilibrium set by the differences in potentials across compartments will make it difficult to discern which switches are initiated by an oxidative stimulus versus the change in distribution of the molecules within the system. One solution is to include all of the pertinent influx and effluxes (such as in Figure 4) to maintain the non-equilibrium steady-state values; however, this is not always feasible and adds complexity to a given model. The lack of appropriate steady-state flux values for all compartments also introduces error into the model parameters.
Another modeling approach is to define the driving force of electron transfer reactions as a function of the difference between the homeostatic potential and the actual potential, such that under steady-state conditions the driving force is non-existent. Transport across membranes similarly could be defined as a function of the difference between the homeostatic concentration gradient and the oxidative stress concentration gradient. Although not mechanistically correct, it does provide a work-around to a complex issue without explicitly defining the multiple contributors to the net driving force. A final complication in modeling redox potentials in cellular systems is the 80 mV gradient across the plasma membrane for ΔEGSH, which could possible result in a driving force for solute transport.
Challenges to the modeling of Trx systems
The fluxes in the Trx1 system include transcription, translation and diffusion/transport (Figure 5, top right) in addition to oxidation-reduction reactions within compartments (Figure 5, bottom right). On the time scales of the sulfur switch systems, most of these fluxes between compartments can be considered zero. In describing the responses to oxidative stress or physiologic challenge, however, increased synthesis alters abundance of Trx1. In addition, Trx1 distribution between the cytoplasm and nuclei is altered by oxidative stress [116, 117]. The rates and regulation for this diffusion/transport across the cytosolic-nuclear membrane to maintain a non-equilibrium state [−280 mV (cytoplasm), −300 mV (nuclei)] are unknown. Furthermore, it is unclear how much Trx1 exists as sequestered protein (Trx1-BP, Fig. 5 top right) under basal conditions, such as its relationship with Vitamin D3-Upregulated Protein [also named as Trx-interacting protein (TxnIP)] where it is bound until a change in redox environment releases it for catalytic activity [118, 119]. The fraction of Trx1 bound to ASK1 is small relative to total Trx1 [120]. If a significant fraction of thioredoxin is bound to other proteins, then the modeler will have to consider this as a rapid source of protein on the same timescale as the switch initiation events.
Challenges to modeling the Cys/CySS system
The Cys/CySS couple is the predominant low molecular weight thiol/disulfide system in most extracellular fluids and has been studied extensively in the plasma. Plasma Cys is derived from the diet, from cell export, from GSH hydrolysis and from reduction of disulfide forms (e.g., CySS, protein-SS-Cys). Cys is cleared from the plasma by numerous amino acid transporters, by oxidation to CySS and by loss in other extracellular fluids (e.g., urine). Plasma CySS is present at a higher concentration than Cys and is derived principally from oxidation of Cys and hydrolysis of GSH-related disulfides [121, 122]. CySS is cleared from the plasma by numerous amino acid transporters [67, 123, 124], by reduction and by loss in urine and other fluids. Isotopic tracer methods have been used to estimate whole body turnover of plasma Cys and CySS in humans [125] and rodents [126], but relatively little information is available concerning the contributions of the different pathways in vivo.
Eh values for the Cys/CySS couple have been estimated in HT29 cells and found to vary independently of the GSH/GSSG and Trx1 couples [38]. Little knowledge is available concerning transport or redox regulation of the Cys/CySS redox state in different subcellular compartments. Transport into mitochondria must occur because of the presence of protein synthesis within this organelle, and transport out of lysosomes must occur because of the degradation processes. However, very few of the rate parameters which are necessary for modeling are available and the extent to which the Cys/CySS couple is regulated in different compartments is unknown. This represents an important area for future research.
Perspectives for Redox Systems Biology
Figure 4, which is focused on the GSH/GSSG couple, delineates only one aspect of the overall cellular redox system including Trx couples and Cys/CySS. The description can be expanded to include the Trx and Cys/CySS couples by incorporating relevant fluxes and distributions across cellular compartments such as shown in Figure 5. These redox control nodes, in turn, provide the set points by which different sulfur switches can operate independently. Because systems biology is driven to explore the emergent properties that results from complex multivariate relationships, the multiple redox couples that exist out of equilibrium with each other and within different cellular locations is an excellent example of a biological system suited for the emerging analytical techniques associated with this field. The critical importance of these redox processes in health and disease suggest that this should be a priority for systems biology development.
Redox systems biology is in a nascent stage of development as researchers shift from empirical observation and component identification to a quantitative understanding of the interrelationships between components. As evident from the lack of critical rate information, systems biologists will need to work in concert with experimentalists in this development. For example, the cloning of mammalian Trx2 occurred as late as 1997 [127], providing new insight in the compartmentalization of the Trx2 in mitochondria (mitochondria, Figure 5 right). Similarly, the Cys/CySS couple in the plasma has only slowly surfaced as a significant independent player in protein thiol oxidation, with relevance to clinical conditions such as cardiovascular disease [58, 59, 82, 128] and alcoholic lung disease [129]. Furthermore, identification of novel protein targets that are regulated by redox state has been increasing as redox proteomic techniques improve [130–132].
To develop effective kinetic models to understand the relationships between redox control nodes and sulfur switches, there is a need for multivariate data acquisition across cellular compartments and in response to physiologic challenge. This will require improved high-throughput sample analysis, such as being developed in mass spectrometry applications that detect protein modifications through oxidation and glutathionylation [133, 134]. Ultimately, nanoscale tools may be needed to allow dynamic quantification of redox states of sulfur switches in the pertinent cellular compartments.
Implications for Predictive Health and Personalized Medicine
Many disease states are associated with either changes in redox states or redox regulation of protein thiol modification. The development of models that establish the interconnectedness of these redox changes and their responses during oxidative stress will aid in understanding and managing the complex regulation of redox process under pathological conditions. For example, attempts to use dietary antioxidants such as Vitamin E in the treatment of cancer, heart disease [135] and Alzheimer’s disease [136] have been disappointing in terms of clinical benefit despite clear evidence for protective activities in controlled experiments. As evident from the complexity of the redox pathways, redox-active chemicals used to abrogate pathologic processes by scavenging ROS may also alter the normal redox circuitry (Figure 3). A framework in which the interconnected nature of non-equilibrium redox control nodes can be analyzed concertedly with the switch mechanisms of their regulatory targets will help elucidate how therapeutics can be used to limit the pathologic events while preserving the physiological signaling pathways.
Many challenges exist for the development of useful kinetic models; however, the formulation of a conceptual framework incorporating the key compartments, redox control nodes and mathematical formulations identifies needed experimental data and supplies the context for such pursuits. There are many unmeasured contributions to control of sulfur switches that must be determined for modeling accuracy. Until the magnitudes of relevant fluxes are established, it is difficult to discern how the cell exploits these fluxes to generate new set points in response to physiologic and pathologic events. Additional information is needed about transport systems for reduced versus oxidized components, vis-à-vis, whether preferential transport is a critical mechanism for maintenance of non-equilibrium conditions across compartments. It is likely that many unidentified sulfur switches remain to be discovered; addition of such features within a model will increase complexity but also improve accuracy. Finally, the role of different redox control nodes for control of different switches is also not clearly defined. The micromolar concentrations of Trx1 and Trx2 compared to the millimolar GSH concentrations suggest that Trx may used for specificity of signaling or other highly critical functions while GSH is used more globally for detoxification and redox buffering. On the other hand, the redox changes of GSH/GSSG in association with cell proliferation, differentiation and apoptosis, may indicate that GSH is used enzymatically for S-thiylation of critical master switches, thereby providing a context-dependent control of cell functions. How this specificity is conferred and the extent of overlap between GSH- and Trx-dependent sulfur switches remains to be investigated.
In summary, sulfur switches functions as sensors in redox signaling pathways and as redox rheostats which control and integrate metabolic pathways. Three major redox control nodes, consisting of thioredoxins, GSH/GSSG and Cys/CySS, play an important role in regulation of these sulfur switches. The redox control nodes are maintained independently under non-equilibrium steady-states in different subcellular compartments, thereby supporting diverse redox regulatory behavior of the cell as distinct functional modules. An initial framework for mathematical modeling has begun to provide form to the thiol/disulfide redox code governing these complex interactions. This framework identifies key needs for inter-compartment flux measurements. The formulation also illustrates a considerable gap in knowledge of the number, identity and mechanistic control of sulfur switches. Future elaboration of the multivariate approaches of systems biology can be expected to improve understanding of critical sites of disruption during oxidative stress and provide a foundation for new approaches for therapeutic intervention.
Acknowledgments
Research by the authors upon which this review was based was supported by NIH grants ES011195, ES009047, ES012929, and by support from the Whitaker Foundation.
List of Abbreviations
- BSO
buthionine sulfoximine
- Cys
cysteine
- CySS
cystine
- DTT
dithiothreitol
- ER
endoplasmic reticulum
- GSH
glutathione
- GSSG
glutathione disulfide
- hRPE
human retinal pigment epithelial
- ROS
reactive oxygen species
- Trx
thioredoxin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free radical biology & medicine. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 2.Hamdane D, Kiger L, Dewilde S, Green BN, Pesce A, Uzan J, Burmester T, Hankeln T, Bolognesi M, Moens L, Marden MC. The redox state of the cell regulates the ligand binding affinity of human neuroglobin and cytoglobin. The Journal of biological chemistry. 2003;278:51713–51721. doi: 10.1074/jbc.M309396200. [DOI] [PubMed] [Google Scholar]
- 3.Jordan PA, Gibbins JM. Extracellular disulfide exchange and the regulation of cellular function. Antioxidants & redox signaling. 2006;8:312–324. doi: 10.1089/ars.2006.8.312. [DOI] [PubMed] [Google Scholar]
- 4.Kerblat I, Drouet C, Chesne S, Marche PN. Importance of thioredoxin in the proteolysis of an immunoglobulin G as antigen by lysosomal Cys-proteases. Immunology. 1999;97:62–68. doi: 10.1046/j.1365-2567.1999.00748.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reeves JP, Bailey CA, Hale CC. Redox modification of sodium-calcium exchange activity in cardiac sarcolemmal vesicles. The Journal of biological chemistry. 1986;261:4948–4955. [PubMed] [Google Scholar]
- 6.Yang J, Chen H, Vlahov IR, Cheng JX, Low PS. Evaluation of disulfide reduction during receptor-mediated endocytosis by using FRET imaging. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:13872–13877. doi: 10.1073/pnas.0601455103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jonas CR, Gu LH, Nkabyo YS, Mannery YO, Avissar NE, Sax HC, Jones DP, Ziegler TR. Glutamine and KGF each regulate extracellular thiol/disulfide redox and enhance proliferation in Caco-2 cells. American journal of physiology. 2003;285:R1421–R1429. doi: 10.1152/ajpregu.00702.2002. [DOI] [PubMed] [Google Scholar]
- 8.Pignocchi C, Kiddle G, Hernandez I, Foster SJ, Asensi A, Taybi T, Barnes J, Foyer CH. Ascorbate oxidase-dependent changes in the redox state of the apoplast modulate gene transcript accumulation leading to modified hormone signaling and orchestration of defense processes in tobacco. Plant physiology. 2006;141:423–435. doi: 10.1104/pp.106.078469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sen CK, Packer L. Antioxidant and redox regulation of gene transcription. Faseb J. 1996;10:709–720. doi: 10.1096/fasebj.10.7.8635688. [DOI] [PubMed] [Google Scholar]
- 10.Ueda S, Masutani H, Nakamura H, Tanaka T, Ueno M, Yodoi J. Redox control of cell death. Antioxidants & redox signaling. 2002;4:405–414. doi: 10.1089/15230860260196209. [DOI] [PubMed] [Google Scholar]
- 11.Forman HJ, Fukuto JM, Torres M. Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am J Physiol Cell Physiol. 2004;287:C246–C256. doi: 10.1152/ajpcell.00516.2003. [DOI] [PubMed] [Google Scholar]
- 12.Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arteriosclerosis, thrombosis, and vascular biology. 2000;20:2175–2183. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- 13.Allen RG, Tresini M. Oxidative stress and gene regulation. Free radical biology & medicine. 2000;28:463–499. doi: 10.1016/s0891-5849(99)00242-7. [DOI] [PubMed] [Google Scholar]
- 14.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 15.Jones DP. Redefining oxidative stress. Antioxidants & redox signaling. 2006;8:1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- 16.Sies H, Jones DP. Oxidative Stress. Elsevier; 2006. [Google Scholar]
- 17.Jones DP. Disruption of mitochondrial redox circuitry in oxidative stress. Chemico-biological interactions. 2006;163:38–53. doi: 10.1016/j.cbi.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 18.Voit EO, Savageau MA. Accuracy of alternative representations for integrated biochemical systems. Biochemistry. 1987;26:6869–6880. doi: 10.1021/bi00395a042. [DOI] [PubMed] [Google Scholar]
- 19.Kacser H, Burns JA. The control of flux. Symposia of the Society for Experimental Biology. 1973;27:65–104. [PubMed] [Google Scholar]
- 20.Milo R, Shen-Orr S, Itzkovitz S, Kashtan N, Chklovskii D, Alon U. Network motifs: simple building blocks of complex networks. Science (New York, N.Y. 2002;298:824–827. doi: 10.1126/science.298.5594.824. [DOI] [PubMed] [Google Scholar]
- 21.Hartwell LH, Hopfield JJ, Leibler S, Murray AW. From molecular to modular cell biology. Nature. 1999;402:C47–C52. doi: 10.1038/35011540. [DOI] [PubMed] [Google Scholar]
- 22.Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M. From genomics to chemical genomics: new developments in KEGG. Nucleic acids research. 2006;34:D354–D357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schomburg I, Chang A, Schomburg D. BRENDA, enzyme data and metabolic information. Nucleic acids research. 2002;30:47–49. doi: 10.1093/nar/30.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M. BioGRID: a general repository for interaction datasets. Nucleic acids research. 2006;34:D535–D539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones DP, Go YM, Anderson CL, Ziegler TR, Kinkade JM, Jr, Kirlin WG. Cysteine/cystine couple is a newly recognized node in the circuitry for biologic redox signaling and control. Faseb J. 2004;18:1246–1248. doi: 10.1096/fj.03-0971fje. [DOI] [PubMed] [Google Scholar]
- 26.Anderson CL, Iyer SS, Ziegler TR, Jones DP. Control of extracellular cysteine/cystine redox state by HT-29 cells is independent of cellular glutathione. American journal of physiology. 2007;293:R1069–R1075. doi: 10.1152/ajpregu.00195.2007. [DOI] [PubMed] [Google Scholar]
- 27.Hancock JT, Desikan R, Neill SJ, Cross AR. New equations for redox and nano-signal transduction. Journal of theoretical biology. 2004;266:60–68. doi: 10.1016/j.jtbi.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 28.Martinovich GG, Cherenkevich SN, Sauer H. Intracellular redox state: towards quantitative description. Eur Biophys J. 2005;34:937–942. doi: 10.1007/s00249-005-0470-3. [DOI] [PubMed] [Google Scholar]
- 29.Rost J, Rapoport S. Reduction-Potential of Glutathione. Nature. 1964;201:185. doi: 10.1038/201185a0. [DOI] [PubMed] [Google Scholar]
- 30.Gillin FD, Diamond LS. Inhibition of clonal growth of Giardia lamblia and Entamoeba histolytica by metronidazole, quinacrine, and other antimicrobial agents. J Antimicrob Chemother. 1981;8:305–316. doi: 10.1093/jac/8.4.305. [DOI] [PubMed] [Google Scholar]
- 31.Jocelyn PC. The standard redox potential of cysteine-cystine from the thioldisulphide exchange reaction with glutathione and lipoic acid. European journal of biochemistry / FEBS. 1967;2:327–331. doi: 10.1111/j.1432-1033.1967.tb00142.x. [DOI] [PubMed] [Google Scholar]
- 32.Segal IH. Oxidation-reduction reactions. New York: John Wiley and Son; 1976. [Google Scholar]
- 33.Halvey PJ, Watson WH, Hansen JM, Go YM, Samali A, Jones DP. Compartmental oxidation of thiol-disulphide redox couples during epidermal growth factor signalling. The Biochemical journal. 2005;386:215–219. doi: 10.1042/BJ20041829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watson WH, Pohl J, Montfort WR, Stuchlik O, Reed MS, Powis G, Jones DP. Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. The Journal of biological chemistry. 2003;278:33408–33415. doi: 10.1074/jbc.M211107200. [DOI] [PubMed] [Google Scholar]
- 35.Clarke WM. The standard hydrogen half-cell and the standardization of oxidation-reduction potentials and pH numbers. Oxidation-reduction potentials of organic systems. Baltimore. 1960:248–247. [Google Scholar]
- 36.Kirlin WG, Cai J, Thompson SA, Diaz D, Kavanagh TJ, Jones DP. Glutathione redox potential in response to differentiation and enzyme inducers. Free radical biology & medicine. 1999;27:1208–1218. doi: 10.1016/s0891-5849(99)00145-8. [DOI] [PubMed] [Google Scholar]
- 37.Cai J, Jones DP. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. The Journal of biological chemistry. 1998;273:11401–11404. doi: 10.1074/jbc.273.19.11401. [DOI] [PubMed] [Google Scholar]
- 38.Nkabyo YS, Ziegler TR, Gu LH, Watson WH, Jones DP. Glutathione and thioredoxin redox during differentiation in human colon epithelial (Caco-2) cells. Am J Physiol Gastrointest Liver Physiol. 2002;283:G1352–G1359. doi: 10.1152/ajpgi.00183.2002. [DOI] [PubMed] [Google Scholar]
- 39.Hansen JM, Watson WH, Jones DP. Compartmentation of Nrf-2 redox control: regulation of cytoplasmic activation by glutathione and DNA binding by thioredoxin-1. Toxicol Sci. 2004;82:308–317. doi: 10.1093/toxsci/kfh231. [DOI] [PubMed] [Google Scholar]
- 40.Miller LT, Watson WH, Kirlin WG, Ziegler TR, Jones DP. Oxidation of the glutathione/glutathione disulfide redox state is induced by cysteine deficiency in human colon carcinoma HT29 cells. The Journal of nutrition. 2002;132:2303–2306. doi: 10.1093/jn/132.8.2303. [DOI] [PubMed] [Google Scholar]
- 41.Samiec PS, Drews-Botsch C, Flagg EW, Kurtz JC, Sternberg P, Jr, Reed RL, Jones DP. Glutathione in human plasma: decline in association with aging, age-related macular degeneration, and diabetes. Free radical biology & medicine. 1998;24:699–704. doi: 10.1016/s0891-5849(97)00286-4. [DOI] [PubMed] [Google Scholar]
- 42.Jonas CR, Estivariz CF, Jones DP, Gu LH, Wallace TM, Diaz EE, Pascal RR, Cotsonis GA, Ziegler TR. Keratinocyte growth factor enhances glutathione redox state in rat intestinal mucosa during nutritional repletion. The Journal of nutrition. 1999;129:1278–1284. doi: 10.1093/jn/129.7.1278. [DOI] [PubMed] [Google Scholar]
- 43.Tian J, Washizawa N, Gu LH, Levin MS, Wang L, Rubin DC, Mwangi S, Srinivasan S, Gao Y, Jones DP, Ziegler TR. Stimulation of colonic mucosal growth associated with oxidized redox status in rats. American journal of physiology. 2007;292:R1081–R1091. doi: 10.1152/ajpregu.00050.2006. [DOI] [PubMed] [Google Scholar]
- 44.Sies H. Nicotinamide nucleotide compartmentation. London: A Subsidiary of Harcourt Brace Jovanovic; 1982. [Google Scholar]
- 45.Gilbert HF. Molecular and cellular aspects of thiol-disulfide exchange. Advances in enzymology and related areas of molecular biology. 1990;63:69–172. doi: 10.1002/9780470123096.ch2. [DOI] [PubMed] [Google Scholar]
- 46.Bellomo G, Vairetti M, Stivala L, Mirabelli F, Richelmi P, Orrenius S. Demonstration of nuclear compartmentalization of glutathione in hepatocytes. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:4412–4416. doi: 10.1073/pnas.89.10.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Briviba K, Fraser G, Sies H, Ketterer B. Distribution of the monochlorobimane-glutathione conjugate between nucleus and cytosol in isolated hepatocytes. The Biochemical journal. 1993;294(Pt 3):631–633. doi: 10.1042/bj2940631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cotgreave IA. Analytical developments in the assay of intra- and extracellular GSH homeostasis: specific protein S-glutathionylation, cellular GSH and mixed disulphide compartmentalisation and interstitial GSH redox balance. BioFactors (Oxford, England) 2003;17:269–277. doi: 10.1002/biof.5520170126. [DOI] [PubMed] [Google Scholar]
- 49.Jevtovic-Todorovic V, Guenthner TM. Depletion of a discrete nuclear glutathione pool by oxidative stress, but not by buthionine sulfoximine. Correlation with enhanced alkylating agent cytotoxicity to human melanoma cells in vitro. Biochemical pharmacology. 1992;44:1383–1393. doi: 10.1016/0006-2952(92)90540-y. [DOI] [PubMed] [Google Scholar]
- 50.Voehringer DW, McConkey DJ, McDonnell TJ, Brisbay S, Meyn RE. Bcl-2 expression causes redistribution of glutathione to the nucleus. Proceedings of the National Academy of Sciences of the United States of America. 1998:2959–2960. doi: 10.1073/pnas.95.6.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soboll S, Grundel S, Harris J, Kolb-Bachofen V, Ketterer B, Sies H. The content of glutathione and glutathione S-transferases and the glutathione peroxidase activity in rat liver nuclei determined by a non-aqueous technique of cell fractionation. The Biochemical journal. 1995;311(Pt 3):889–894. doi: 10.1042/bj3110889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Go YM, Ziegler TR, Johnson JM, Gu V, Hansen JM, Jones DP. Selective protection of nuclear thioredoxin-1 and glutathione redox systems against oxidation during glucose and glutamine deficiency in human colonic epithelial cells. Free radical biology & medicine. 2007;42:363–370. doi: 10.1016/j.freeradbiomed.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rebrin I, Sohal RS. Comparison of thiol redox state of mitochondria and homogenates of various tissues between two strains of mice with different longevities. Experimental gerontology. 2004;39:1513–1519. doi: 10.1016/j.exger.2004.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jones DP, Carlson JL, Mody VC, Cai J, Lynn MJ, Sternberg P. Redox state of glutathione in human plasma. Free radical biology & medicine. 2000;28:625–635. doi: 10.1016/s0891-5849(99)00275-0. [DOI] [PubMed] [Google Scholar]
- 55.Jones DP, Mody VC, Jr, Carlson JL, Lynn MJ, Sternberg P., Jr Redox analysis of human plasma allows separation of pro-oxidant events of aging from decline in antioxidant defenses. Free radical biology & medicine. 2002;33:1290–1300. doi: 10.1016/s0891-5849(02)01040-7. [DOI] [PubMed] [Google Scholar]
- 56.Moriarty SE, Shah JH, Lynn M, Jiang S, Openo K, Jones DP, Sternberg P. Oxidation of glutathione and cysteine in human plasma associated with moking. Free radical biology & medicine. 2003;35:1582–1588. doi: 10.1016/j.freeradbiomed.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 57.Jonas CR, Puckett AB, Jones DP, Griffith DP, Szeszycki EE, Bergman GF, Furr CE, Tyre C, Carlson JL, Galloway JR, Blumberg JB, Ziegler TR. Plasma antioxidant status after high-dose chemotherapy: a randomized trial of parenteral nutrition in bone marrow transplantation patients. The American journal of clinical nutrition. 2000;72:181–189. doi: 10.1093/ajcn/72.1.181. [DOI] [PubMed] [Google Scholar]
- 58.Abramson JL, Hooper WC, Jones DP, Ashfaq S, Rhodes SD, Weintraub WS, Harrison DG, Quyyumi AA, Vaccarino V. Association between novel oxidative stress markers and C-reactive protein among adults without clinical coronary heart disease. Atherosclerosis. 2005;178:115–121. doi: 10.1016/j.atherosclerosis.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 59.Ashfaq S, Abramson JL, Jones DP, Rhodes SD, Weintraub WS, Hooper WC, Vaccarino V, Harrison DG, Quyyumi AA. The relationship between plasma levels of oxidized and reduced thiols and early atherosclerosis in healthy adults. Journal of the American College of Cardiology. 2006;47:1005–1011. doi: 10.1016/j.jacc.2005.09.063. [DOI] [PubMed] [Google Scholar]
- 60.Brown LA, Ping XD, Harris FL, Gauthier TW. Glutathione availability modulates alveolar macrophage function in the chronic ethanol-fed rat. Am J Physiol Lung Cell Mol Physiol. 2007;292:L824–L832. doi: 10.1152/ajplung.00346.2006. [DOI] [PubMed] [Google Scholar]
- 61.Bai C, Jones DP. GSH transport and GSH-dependent detoxication in small intestine of rats exposed in vivo to hypoxia. The American journal of physiology. 1996;271:G701–G706. doi: 10.1152/ajpgi.1996.271.4.G701. [DOI] [PubMed] [Google Scholar]
- 62.Neu J, Roig JC, Meetze WH, Veerman M, Carter C, Millsaps M, Bowling D, Dallas MJ, Sleasman J, Knight T, Auestad N. Enteral glutamine supplementation for very low birth weight infants decreases morbidity. The Journal of pediatrics. 1997;131:691–699. doi: 10.1016/s0022-3476(97)70095-7. [DOI] [PubMed] [Google Scholar]
- 63.Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science (New York, N.Y. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 64.Gross E, Sevier CS, Heldman N, Vitu E, Bentzur M, Kaiser CA, Thorpe C, Fass D. Generating disulfides enzymatically: reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:299–304. doi: 10.1073/pnas.0506448103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chakravarthi S, Jessop CE, Bulleid NJ. The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO reports. 2006;7:271–275. doi: 10.1038/sj.embor.7400645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gorlach A, Klappa P, Kietzmann T. The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxidants & redox signaling. 2006;8:1391–1418. doi: 10.1089/ars.2006.8.1391. [DOI] [PubMed] [Google Scholar]
- 67.Cherqui S, Kalatzis V, Trugnan G, Antignac C. The targeting of cystinosin to the lysosomal membrane requires a tyrosine-based signal and a novel sorting motif. The Journal of biological chemistry. 2001;276:13314–13321. doi: 10.1074/jbc.M010562200. [DOI] [PubMed] [Google Scholar]
- 68.Forestier L, Jean G, Attard M, Cherqui S, Lewis C, van't Hoff W, Broyer M, Town M, Antignac C. Molecular characterization of CTNS deletions in nephropathic cystinosis: development of a PCR-based detection assay. American journal of human genetics. 1999;65:353–359. doi: 10.1086/302509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Holmgren A. Reduction of disulfides by thioredoxin. Exceptional reactivity of insulin and suggested functions of thioredoxin in mechanism of hormone action. The Journal of biological chemistry. 1979;254:9113–9119. [PubMed] [Google Scholar]
- 70.Hansen JM, Zhang H, Jones DP. Mitochondrial thioredoxin-2 has a key role in determining tumor necrosis factor-alpha-induced reactive oxygen species generation, NF-kappaB activation, and apoptosis. Toxicol Sci. 2006;91:643–650. doi: 10.1093/toxsci/kfj175. [DOI] [PubMed] [Google Scholar]
- 71.Halvey PJ, Hansen JM, Johnson JM, Go YM, Samali A, Jones DP. Selective Oxidative Stress in Cell Nuclei by Nuclear-Targeted D-Amino Acid Oxidase. Antioxidants & redox signaling. 2007;9:807–816. doi: 10.1089/ars.2007.1526. [DOI] [PubMed] [Google Scholar]
- 72.Chen Y, Cai J, Jones DP. Mitochondrial thioredoxin in regulation of oxidant-induced cell death. FEBS letters. 2006;580:6596–6602. doi: 10.1016/j.febslet.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ramachandiran S, Hansen JM, Jones DP, Richardson JR, Miller GW. Divergent mechanisms of paraquat, MPP+, and rotenone toxicity: oxidation of thioredoxin and caspase-3 activation. Toxicol Sci. 2007;95:163–171. doi: 10.1093/toxsci/kfl125. [DOI] [PubMed] [Google Scholar]
- 74.Hansen JM, Zhang H, Jones DP. Differential oxidation of thioredoxin-1, thioredoxin-2, and glutathione by metal ions. Free radical biology & medicine. 2006;40:138–145. doi: 10.1016/j.freeradbiomed.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 75.Hansen JM, Go YM, Jones DP. Nuclear and mitochondrial compartmentation of oxidative stress and redox signaling. Annual review of pharmacology and toxicology. 2006;46:215–234. doi: 10.1146/annurev.pharmtox.46.120604.141122. [DOI] [PubMed] [Google Scholar]
- 76.Fayos BE, Wattenberg BW. Regulated exocytosis in vascular endothelial cells can be triggered by intracellular guanine nucleotides and requires a hydrophobic, thiol-sensitive component. Studies of regulated von Willebrand factor secretion from digitonin permeabilized endothelial cells. Endothelium. 1997;5:339–350. doi: 10.3109/10623329709052598. [DOI] [PubMed] [Google Scholar]
- 77.Mannervik B, Axelsson K, Sundewall AC, Holmgren A. Relative contributions of thioltransferase-and thioredoxin-dependent systems in reduction of low-molecular-mass and protein disulphides. The Biochemical journal. 1983;213:519–523. doi: 10.1042/bj2130519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pastore A, Tozzi G, Gaeta LM, Bertini E, Serafini V, Di Cesare S, Bonetto V, Casoni F, Carrozzo R, Federici G, Piemonte F. Actin glutathionylation increases in fibroblasts of patients with Friedreich's ataxia: a potential role in the pathogenesis of the disease. The Journal of biological chemistry. 2003;278:42588–42595. doi: 10.1074/jbc.M301872200. [DOI] [PubMed] [Google Scholar]
- 79.Piemonte F, Pastore A, Tozzi G, Tagliacozzi D, Santorelli FM, Carrozzo R, Casali C, Damiano M, Federici G, Bertini E. Glutathione in blood of patients with Friedreich's ataxia. European journal of clinical investigation. 2001;31:1007–1011. doi: 10.1046/j.1365-2362.2001.00922.x. [DOI] [PubMed] [Google Scholar]
- 80.Moriarty-Craige SE, Ha KN, Sternberg P, Jr, Lynn M, Bressler S, Gensler G, Jones DP. Effects of long-term zinc supplementation on plasma thiol metabolites and redox status in patients with age-related macular degeneration. American journal of ophthalmology. 2007;143:206–211. doi: 10.1016/j.ajo.2006.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jiang S, Moriarty-Craige SE, Orr M, Cai J, Sternberg P, Jr, Jones DP. Oxidant-induced apoptosis in human retinal pigment epithelial cells: dependence on extracellular redox state. Investigative ophthalmology & visual science. 2005;46:1054–1061. doi: 10.1167/iovs.04-0949. [DOI] [PubMed] [Google Scholar]
- 82.Go YM, Jones DP. Intracellular proatherogenic events and cell adhesion modulated by extracellular thiol/disulfide redox state. Circulation. 2005;111:2973–2980. doi: 10.1161/CIRCULATIONAHA.104.515155. [DOI] [PubMed] [Google Scholar]
- 83.Ramirez A, Ramadan B, Ritzenthaler JD, Rivera HN, Jones DP, Roman J. Extracellular cysteine/cystine redox potential controls lung fibroblast proliferation and matrix expression through upregulation of transforming growth facto-{rbeta} Am J Physiol Lung Cell Mol Physiol. 2007 doi: 10.1152/ajplung.00010.2007. [DOI] [PubMed] [Google Scholar]
- 84.Delaunay A, Pflieger D, Barrault MB, Vinh J, Toledano MB. A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell. 2002;111:471–481. doi: 10.1016/s0092-8674(02)01048-6. [DOI] [PubMed] [Google Scholar]
- 85.Helmann JD. OxyR: a molecular code for redox sensing. Sci STKE. 2002;2002:PE46. doi: 10.1126/stke.2002.157.pe46. [DOI] [PubMed] [Google Scholar]
- 86.Lockwood TD. The transfer of reductive energy and pace of proteome turnover: a theory of integrated catabolic control. Antioxidants & redox signaling. 2005;7:982–998. doi: 10.1089/ars.2005.7.982. [DOI] [PubMed] [Google Scholar]
- 87.Zhang H, Go YM, Jones DP. Mitochondrial thioredoxin-2/peroxiredoxin-3 system functions in parallel with mitochondrial GSH system in protection against oxidative stress. Archives of biochemistry and biophysics. 2007;465:119–126. doi: 10.1016/j.abb.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 88.Taylor ER, Hurrell F, Shannon RJ, Lin TK, Hirst J, Murphy MP. Reversible glutathionylation of complex I increases mitochondrial superoxide formation. The Journal of biological chemistry. 2003;278:19603–19610. doi: 10.1074/jbc.M209359200. [DOI] [PubMed] [Google Scholar]
- 89.Dooley CT, Dore TM, Hanson GT, Jackson WC, Remington SJ, Tsien RY. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. The Journal of biological chemistry. 2004;279:22284–22293. doi: 10.1074/jbc.M312847200. [DOI] [PubMed] [Google Scholar]
- 90.Giustarini D, Rossi R, Milzani A, Colombo R, Dalle-Donne I. S-glutathionylation: from redox regulation of protein functions to human diseases. Journal of cellular and molecular medicine. 2004;8:201–212. doi: 10.1111/j.1582-4934.2004.tb00275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jung CH, Thomas JA. S-glutathiolated hepatocyte proteins and insulin disulfides as substrates for reduction by glutaredoxin, thioredoxin, protein disulfide isomerase, and glutathione. Archives of biochemistry and biophysics. 1996;335:61–72. doi: 10.1006/abbi.1996.0482. [DOI] [PubMed] [Google Scholar]
- 92.Klatt P, Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. European journal of biochemistry / FEBS. 2000;267:4928–4944. doi: 10.1046/j.1432-1327.2000.01601.x. [DOI] [PubMed] [Google Scholar]
- 93.Marchand C, Le Marechal P, Meyer Y, Decottignies P. Comparative proteomic approaches for the isolation of proteins interacting with thioredoxin. Proteomics. 2006;6:6528–6537. doi: 10.1002/pmic.200600443. [DOI] [PubMed] [Google Scholar]
- 94.Chang EY, Son SK, Ko HS, Baek SH, Kim JH, Kim JR. Induction of apoptosis by the overexpression of an alternative splicing variant of mitochondrial thioredoxin reductase. Free radical biology & medicine. 2005;39:1666–1675. doi: 10.1016/j.freeradbiomed.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 95.Turanov AA, Su D, Gladyshev VN. Characterization of alternative cytosolic forms and cellular targets of mouse mitochondrial thioredoxin reductase. The Journal of biological chemistry. 2006;281:22953–22963. doi: 10.1074/jbc.M604326200. [DOI] [PubMed] [Google Scholar]
- 96.Diamond DA, Parsian A, Hunt CR, Lofgren S, Spitz DR, Goswami PC, Gius D. Redox factor-1 (Ref-1) mediates the activation of AP-1 in HeLa and NIH 3T3 cells in response to heat shock. The Journal of biological chemistry. 1999;274:16959–16964. doi: 10.1074/jbc.274.24.16959. [DOI] [PubMed] [Google Scholar]
- 97.Iwasaki K, Mackenzie EL, Hailemariam K, Sakamoto K, Tsuji Y. Hemin-mediated regulation of an antioxidant-responsive element of the human ferritin H gene and role of Ref-1 during erythroid differentiation of K562 cells. Molecular and cellular biology. 2006;26:2845–2856. doi: 10.1128/MCB.26.7.2845-2856.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nishi T, Shimizu N, Hiramoto M, Sato I, Yamaguchi Y, Hasegawa M, Aizawa S, Tanaka H, Kataoka K, Watanabe H, Handa H. Spatial redox regulation f a critical cysteine residue of NF-kappa B in vivo. The Journal of biological chemistry. 2002;277:44548–44556. doi: 10.1074/jbc.M202970200. [DOI] [PubMed] [Google Scholar]
- 99.Sagher D, Brunell D, Hejtmancik JF, Kantorow M, Brot N, Weissbach H. Thionein can serve as a reducing agent for the methionine sulfoxide reductases. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:8656–8661. doi: 10.1073/pnas.0602826103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gasdaska JR, Berggren M, Powis G. Cell growth stimulation by the redox protein thioredoxin occurs by a novel helper mechanism. Cell Growth Differ. 1995;6:1643–1650. [PubMed] [Google Scholar]
- 101.Gasdaska JR, Kirkpatrick DL, Montfort W, Kuperus M, Hill SR, Berggren M, Powis G. Oxidative inactivation of thioredoxin as a cellular growth factor and protection by a Cys73-->Ser mutation. Biochemical pharmacology. 1996;52:1741–1747. doi: 10.1016/s0006-2952(96)00595-3. [DOI] [PubMed] [Google Scholar]
- 102.Casagrande S, Bonetto V, Fratelli M, Gianazza E, Eberini I, Massignan T, Salmona M, Chang G, Holmgren A, Ghezzi P. Glutathionylation of human thioredoxin: a possible crosstalk between the glutathione and thioredoxin systems. Proceedings of the National Academy of Sciences of the United States of America. 2002;9:9745–9749. doi: 10.1073/pnas.152168599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Debarbieux L, Beckwith J. The reductive enzyme thioredoxin 1 acts as an oxidant when it is exported to the Escherichia coli periplasm. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:10751–10756. doi: 10.1073/pnas.95.18.10751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Haendeler J. Thioredoxin-1 and posttranslational modifications. Antioxidants & redox signaling. 2006;8:1723–1728. doi: 10.1089/ars.2006.8.1723. [DOI] [PubMed] [Google Scholar]
- 105.Jones DP. Effect of mitochondrial clustering on O2 supply in hepatocytes. The American journal of physiology. 1984;247:C83–C89. doi: 10.1152/ajpcell.1984.247.1.C83. [DOI] [PubMed] [Google Scholar]
- 106.Jones DP. Intracellular diffusion gradients of O2 and ATP. The American journal of physiology. 1986;250:C663–C675. doi: 10.1152/ajpcell.1986.250.5.C663. [DOI] [PubMed] [Google Scholar]
- 107.Qian H, Beard DA, Liang SD. Stoichiometric network theory for nonequilibrium biochemical systems. European journal of biochemistry / FEBS. 2003;270:415–421. doi: 10.1046/j.1432-1033.2003.03357.x. [DOI] [PubMed] [Google Scholar]
- 108.Tribble DL, Jones DP. Oxygen dependence of oxidative stress. Rate of NADPH supply for maintaining the GSH pool during hypoxia. Biochemical pharmacology. 1990;39:729–736. doi: 10.1016/0006-2952(90)90152-b. [DOI] [PubMed] [Google Scholar]
- 109.Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. The Biochemical journal. 1972;128:617–630. doi: 10.1042/bj1280617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tretter L, Takacs K, Hegedus V, Adam-Vizi V. Characteristics of alpha-glycerophosphate- evoked H2O2 generation in brain mitochondria. Journal of neurochemistry. 2007;100:650–663. doi: 10.1111/j.1471-4159.2006.04223.x. [DOI] [PubMed] [Google Scholar]
- 111.Shan XQ, Aw TY, Jones DP. Glutathione-dependent protection against oxidative injury. Pharmacology & therapeutics. 1990;47:61–71. doi: 10.1016/0163-7258(90)90045-4. [DOI] [PubMed] [Google Scholar]
- 112.Gilbert HF. Protein disulfide isomerase and assisted protein folding. The Journal of biological chemistry. 1997;272:29399–29402. doi: 10.1074/jbc.272.47.29399. [DOI] [PubMed] [Google Scholar]
- 113.Jones DP. Redox potential of GSH/GSSG couple: assay and biological significance. Methods in enzymology. 2002;348:93–112. doi: 10.1016/s0076-6879(02)48630-2. [DOI] [PubMed] [Google Scholar]
- 114.Ziegler DM. Role of reversible oxidation-reduction of enzyme thiols-disulfides in metabolic regulation. Annual review of biochemistry. 1985;54:305–329. doi: 10.1146/annurev.bi.54.070185.001513. [DOI] [PubMed] [Google Scholar]
- 115.Nishiyama Y, Massey V, Takeda K, Kawasaki S, Sato J, Watanabe T, Niimura Y. Hydrogen peroxide-forming NADH oxidase belonging to the peroxiredoxin oxidoreductase family: existence and physiological role in bacteria. Journal of bacteriology. 2001;183:2431–2438. doi: 10.1128/JB.183.8.2431-2438.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Malik G, Gorbounov N, Das S, Gurusamy N, Otani H, Maulik N, Goswami S, Das DK. Ischemic preconditioning triggers nuclear translocation of thioredoxin and its interaction with Ref-1 potentiating a survival signal through the PI-3-kinase-Akt pathway. Antioxidants & redox signaling. 2006;8:2101–2109. doi: 10.1089/ars.2006.8.2101. [DOI] [PubMed] [Google Scholar]
- 117.Wei SJ, Botero A, Hirota K, Bradbury CM, Markovina S, Laszlo A, Spitz DR, Goswami PC, Yodoi J, Gius D. Thioredoxin nuclear translocation and interaction with redox factor-1 activates the activator protein-1 transcription factor in response to ionizing radiation. Cancer research. 2000;60:6688–6695. [PubMed] [Google Scholar]
- 118.Schulze PC, De Keulenaer GW, Yoshioka J, Kassik KA, Lee RT. Vitamin D3-upregulated protein-1 (VDUP-1) regulates redox-dependent vascular smooth muscle cell proliferation through interaction with thioredoxin. Circulation research. 2002;91:689–695. doi: 10.1161/01.res.0000037982.55074.f6. [DOI] [PubMed] [Google Scholar]
- 119.Yamawaki H, Pan S, Lee RT, Berk BC. Fluid shear stress inhibits vascular inflammation by decreasing thioredoxin-interacting protein in endothelial cells. The Journal of clinical investigation. 2005;115:733–738. doi: 10.1172/JCI200523001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liu H, Nishitoh H, Ichijo H, Kyriakis JM. Activation of apoptosis signal-regulating kinase 1 (ASK1) by tumor necrosis factor receptor-associated factor 2 requires prior dissociation of the ASK1 inhibitor thioredoxin. Molecular and cellular biology. 2000;20:2198–2208. doi: 10.1128/mcb.20.6.2198-2208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Butler JD, Spielberg SP. Accumulation of cystine from glutathione-cysteine mixed disulfide in cystinotic fibroblasts; blockade by an inhibitor of gamma-glutamyl transpeptidase. Life sciences. 1982;31:2563–2570. doi: 10.1016/0024-3205(82)90729-9. [DOI] [PubMed] [Google Scholar]
- 122.Hildebrandt W, Kinscherf R, Hauer K, Holm E, Droge W. Plasma cystine concentration and redox state in aging and physical exercise. Mechanisms of ageing and development. 2002;123:1269–1281. doi: 10.1016/s0047-6374(02)00013-1. [DOI] [PubMed] [Google Scholar]
- 123.Bannai S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. The Journal of biological chemistry. 1986;261:2256–2563. [PubMed] [Google Scholar]
- 124.Deora AB, Ghosh RN, Tate SS. Progressive C-terminal deletions of the renal cystine transporter, NBAT, reveal a novel bimodal pattern of functional expression. The Journal of biological chemistry. 1998;273:32980–32987. doi: 10.1074/jbc.273.49.32980. [DOI] [PubMed] [Google Scholar]
- 125.Raguso CA, Regan MM, Young VR. Cysteine kinetics and oxidation at different intakes of methionine and cystine in young adults. The American journal of clinical nutrition. 2000;71:491–499. doi: 10.1093/ajcn/71.2.491. [DOI] [PubMed] [Google Scholar]
- 126.Ensunsa JL, Hirschberger LL, Stipanuk MH. Catabolism of cysteine, cystine, cysteinesulfinate, and OTC by isolated perfused rat hindquarter. The American Journal of physiology. 1993;264:E782–E789. doi: 10.1152/ajpendo.1993.264.5.E782. [DOI] [PubMed] [Google Scholar]
- 127.Spyrou G, Enmark E, Miranda-Vizuete A, Gustafsson J. Cloning and expression of a novel mammalian thioredoxin. The Journal of biological chemistry. 1997;72:2936–2941. doi: 10.1074/jbc.272.5.2936. [DOI] [PubMed] [Google Scholar]
- 128.Neuman RB, Bloom HL, Shukrullah I, Darrow LA, Kleinbaum D, Jones DP, Dudley SC., Jr Oxidative stress markers are associated with persistent trial fibrillation. Clinical chemistry. 2007;53:1652–1657. doi: 10.1373/clinchem.2006.083923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yeh MY-P. Medicine. Atlanta: Emory University; 2007. Determining and monitoring systemic and pulmonary redox states in chronic alcohol abusers; p. 154. [Google Scholar]
- 130.Butterfield DA, Perluigi M, Sultana R. Oxidative stress in Alzheimer's disease brain: new insights from redox proteomics. European journal of pharmacology. 2006;545:39–50. doi: 10.1016/j.ejphar.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 131.Dalle-Donne I, Scaloni A, Giustarini D, Cavarra E, Tell G, Lungarella G, Colombo R, Rossi R, Milzani A. Proteins as biomarkers of oxidative/nitrosative stress in diseases: the contribution of redox proteomics. Mass spectrometry reviews. 2005;24:55–99. doi: 10.1002/mas.20006. [DOI] [PubMed] [Google Scholar]
- 132.Fratelli M, Gianazza E, Ghezzi P. Redox proteomics: identification and functional role of glutathionylated proteins. Expert review of proteomics. 2004;1:365–376. doi: 10.1586/14789450.1.3.365. [DOI] [PubMed] [Google Scholar]
- 133.Brennan JP, Wait R, Begum S, Bell JR, Dunn MJ, Eaton P. Detection and mapping of widespread intermolecular protein disulfide formation during cardiac oxidative stress using proteomics with diagonal electrophoresis. The Journal of biological chemistry. 2004;279:41352–41360. doi: 10.1074/jbc.M403827200. [DOI] [PubMed] [Google Scholar]
- 134.Fratelli M, Demol H, Puype M, Casagrande S, Eberini I, Salmona M, Bonetto V, Mengozzi M, Duffieux F, Miclet E, Bachi A, Vandekerckhove J, Gianazza E, Ghezzi P. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:3505–3510. doi: 10.1073/pnas.052592699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pham DQ, Plakogiannis R. Vitamin E supplementation in cardiovascular disease and cancer prevention: Part 1. Ann Pharmacother. 2005;39:1807–1878. doi: 10.1345/aph.1G211. [DOI] [PubMed] [Google Scholar]
- 136.Boothby LA, Doering PL. Vitamin C and vitamin E for Alzheimer's disease. Ann Pharmacother. 2005;39:2073–2080. doi: 10.1345/aph.1E495. [DOI] [PubMed] [Google Scholar]
- 137.Nkabyo YS, Gu LH, Jones DP, Ziegler TR. Thiol/disulfide redox status is oxidized in plasma and small intestinal and colonic mucosa of rats with inadequate sulfur amino acid intake. The Journal of nutrition. 2006;136:1242–1248. doi: 10.1093/jn/136.5.1242. [DOI] [PubMed] [Google Scholar]
- 138.Banhegyi G, Lusini L, Puskas F, Rossi R, Fulceri R, Braun L, Mile V, di Simplicio P, Mandl J, Benedetti A. Preferential transport of glutathione versus glutathione disulfide in rat liver microsomal vesicles. The Journal of biological chemistry. 1999;274:12213–12216. doi: 10.1074/jbc.274.18.12213. [DOI] [PubMed] [Google Scholar]
- 139.Kleinman WA, Richie JP., Jr Status of glutathione and other thiols and disulfides in human plasma. Biochemical pharmacology. 2000;60:19–29. doi: 10.1016/s0006-2952(00)00293-8. [DOI] [PubMed] [Google Scholar]
- 140.Stipanuk MH, Coloso RM, Garcia RA, Banks MF. Cysteine concentration regulates cysteine metabolism to glutathione, sulfate and taurine in rat hepatocytes. The Journal of nutrition. 1992;122:420–427. doi: 10.1093/jn/122.3.420. [DOI] [PubMed] [Google Scholar]
- 141.Lee TK, Hammond CL, Ballatori N. Intracellular glutathione regulates taurocholate transport in HepG2 cells. Toxicology and applied pharmacology. 2001;174:207–215. doi: 10.1006/taap.2001.9208. [DOI] [PubMed] [Google Scholar]
- 142.Xu F, Putt DA, Matherly LH, Lash LH. Modulation of expression of rat mitochondrial 2-oxoglutarate carrier in NRK-52E cells alters mitochondrial transport and accumulation of glutathione and susceptibility to chemically induced apoptosis. The Journal of pharmacology and experimental therapeutics. 2006;316:1175–1186. doi: 10.1124/jpet.105.094599. [DOI] [PubMed] [Google Scholar]
- 143.Kowalski DP, Aw TY, Park Y, Jones DP. Postanoxic oxidative injury in at hepatocytes: lactate-dependent protection against tert-butylhydroperoxide. Free radical biology & medicine. 1992;12:205–212. doi: 10.1016/0891-5849(92)90028-f. [DOI] [PubMed] [Google Scholar]