

Summary
Background
The proinflammatory cytokine Tumor Necrosis Factor–α (TNF) elicits cellular responses by signaling through a receptor complex that includes the essential adaptor molecule RIP. One important consequence of signaling is activation of the transcription factor NF-κB, and failure to downregulate TNF-induced NF-κB transcriptional activity results in chronic inflammation and death. Internalization of the receptor complex plays an important regulatory role in TNF signaling.
Results
We report that CARP-2, a RING domain-containing ubiquitin protein ligase (E3), is a negative regulator of TNF-induced NF-κB activation. By virtue of its phospholipid-binding FYVE domain, CARP-2 localized to endocytic vesicles where it interacted with internalized TNF-receptor complex, resulting in RIP ubiquitination and degradation. Knockdown of CARP-2 stabilized TNFR1-associated polyubiquitinated RIP levels after TNF simulation and enhanced activation of NF-κB.
Conclusions
CARP-2 acts at the level of endocytic vesicles to limit the intensity of TNF-induced NF-κB activation by the regulated elimination of a necessary signaling component within the receptor complex.
Keywords: RIP, FYVE domain, ubiquitination, endocytic vesicles, signal transduction
Introduction
TNF is a multifunctional cytokine that elicits many biological responses that are critical for cellular homeostasis [1, 2]. Deregulation of TNF signaling has been implicated in a variety of pathological conditions such as cachexia, inflammation, osteoporosis, and rheumatoid arthritis. TNF orchestrates most of its cellular functions, either cell survival and proliferation or apoptosis, via TNF-receptor-1 (TNF-R1) [3, 4].
TNF-induced NF-κB activation is crucial for immune responses. Binding of TNF triggers recruitment of many cytosolic proteins to TNF-R1 including TNF receptor associated death domain protein (TRADD), receptor interacting kinase (RIP), and TNF-receptor associated factor 2 (TRAF-2) [5]. The assembly of this complex (also known as complex I) takes place immediately after stimulation at the plasma membrane, where posttranslational modifications like polyubiquitination of several complex-associated proteins occur, resulting in the activation of the IκB kinase (IKK) complex and subsequently NF-κB [6-9]. This receptor-associated complex containing RIP internalizes within minutes of formation and fuse with the trans-golgi network to form multivesicular endosomes and this event is believed to be critical to attenuate inflammatory responses [5, 10]. However, the underlying mechanisms that regulate NF-κB signaling during endocytosis of the activated TNF-receptor complexes remain unknown.
RIP, a death domain-containing serine/threonine kinase, is heavily ubiquitinated upon recruitment to early cell surface TNF-R1 complexes in TNF-stimulated cells. RIP is essential for TNF pathway, because RIP-deficient Jurkat human T lymphoma cells and mouse fibroblasts have no TNF-inducible NF-κB response [11-13]. NEMO, the regulatory subunit of the IKK complex, specifically recognizes K63-linked polyubiquitinated chains on RIP, triggering activation of IKK and NF-κB [6, 8, 14]. The domain structure of RIP is organized into three distinct regions, a C-terminal death domain (DD), an intermediate domain (ID) and an N-terminal kinase domain (KD). While the DD mediates RIP recruitment to the early TNF-R1 complex, Lysine 377 in the ID is the site of K63-linked polyubiquitination, probably mediated by TRAF-2 [6, 11, 14]. Both DD and ID are essential for TNF-induced IKK and NF-κB activation. Whereas no functional role for the RIP KD has been established, it is thought to be dispensable for downstream NF-κB signaling [12, 13].
Recent reports indicate that a number of peripheral (as opposed to integral membrane) proteins, that localize to specific organelles through lipid-protein and protein-protein interactions, target the internalized receptor complexes and provide necessary spatial and temporal regulation for signaling [15, 16]. Because internalized TNF-R1 complexes are known to associate with endocytic membrane vesicles, we hypothesized that proteins in these intracellular compartments might affect TNF-induced NF-κB activation by targeting signaling complexes. Here, we present evidence that CARP-2, a phospholipid-binding domain containing protein with ubiquitin protein ligase activity, localizes to endocytic vesicles, recruits to internalized early TNF-R1 complex, targets internalized RIP for proteasome-mediated degradation, and limits TNF-induced NF-κB activation.
Results
CARP-2 is a novel regulator of TNF-induced NF-κB activation
To identify proteins that regulate TNF signaling in the endocytic pathway, we focused on proteins that associate with the endosomal and Golgi membrane vesicles with which fusion of internalized TNF receptor complexes occurs. Besides trans-membrane proteins, proteins with a particular motif known as FYVE (conserved in Fab1p/YOTB/Vac1p /EEA1) are known to localize to these vesicles [17, 18]. Therefore, we searched the NCBI gene bank for FYVE motif-containing proteins. Interestingly, two of the 45 FYVE domain-containing proteins found also contain RING domains, a sequence that confers ubiquitin protein ligase activity to many molecules [19, 20]. Reasoning that molecules containing such properties might be especially well-suited to interact with and modify components of early TNF-R1 associated proteins, we tested their ability to affect TNF-induced NF-κB activation. One of the molecules, CARP-2 (caspase-8 and –10-associated RING protein-2) (Fig. 1A), is known to bind and negatively regulate DED caspases, but has not been reported to regulate TNF signaling [21]. CARP-2 (also known as Rififylin/SAKURA) belongs to a minor subgroup of FYVE-domain containing proteins with no conserved phosphoinositide-binding motif [22-24]. In spite of the missing PI3P binding property of the FYVE motif, this sequence has been shown in overexpression studies to target the protein to endocytic membrane vesicles [23]. The RING domain is also functional and exhibits ubiquitin protein ligase (E3) activity in vitro and ex vivo [21, 22].
Fig. 1. CARP-2 negatively regulates TNF-induced NF-κB activation.

(A). Schematic diagram shows the domain structure and position of conserved Histidine 333 in the RING domain of CARP-2.
(B) and (C). CARP-2 inhibits TNF-induced IKK activation and IκB α degradation. Extracts of 293T cells transfected with indicated plasmids and treated with TNF for 5 min (B), or as indicated (C) were subjected to in vitro IKK kinase assay (B), or probed for expression of indicated proteins (B and C).
(D) and (F). CARP-2 inhibits TNF-induced NF-κB reporter activity. Extracts of TNF-stimulated 293T cells expressing reporter plasmids along with different amounts of FLAG-CARP-2 variants (D), or ShRNA constructs (F) were assayed for reporter activities and probed for CARP-2.
(E). CARP-2 inhibits TNF-induced IL-6 production. IL-6 secreted from mouse embryonic fibroblasts (MEFs) expressing CARP-2 variants was measured by ELISA and expression of CARP-2 and β-actin in the extracts is shown.
(G) and (H). Knockdown of CARP-2 prolongs TNF-induced IKK activation and IκB αdegradation. Extracts of TNF-treated 293T cells expressing ShRNA constructs were subjected to in vitro IKK kinase assay (G), or probed for expression of indicated proteins (G and H).
To assess the effect of CARP-2 expression on TNF-induced NF-κB activation, we analyzed the endogenous IKK activity and IκBα degradation (Fig. 1B and C). As expected, treatment of vector only expressing cells with TNF resulted in increased IKK activation (Fig. 1B). Expression of CARP-2 wild type decreased IKK activity both at basal level and upon TNF stimulation. A substitution of alanine for a histidine in the RING domain (H333A) that abrogates E3 activity failed to reduce IKK activity suggesting that E3 activity is required for CARP-2 inhibitory function (Fig. 1B). Consistent with these results expression of CARP-2 wild type (Fig. 1C) that did not affect the level of IκBα in unstimulated cells prevented its TNF-induced degradation. In contrast, the RING-mutant had shown no such effect (Fig. 1C).
Additionally, NF-κB reporter assays were used to assess the effect of increased CARP-2 expression on NF-κB activation. CARP-2 reduced TNF-induced NF-κB reporter activity in a dose-responsive fashion (Fig. 1D). At high concentrations the RING mutant also exhibited some inhibition, which may result from the ability of the RING mutant to bind to target protein(s) and affect its (their) function in a subtle way. This downregulation of NF-κB activity by CARP-2 was observed in all cell lines such as HT1080 (human fibrosarcoma), HeLa (human cervical carcinoma) and C2C12 (mouse myoblast) tested (data not shown). To investigate the effect of CARP-2 expression on NF-κB mediated cytokine production, we examined IL-6 secretion in response to TNF stimulation in mouse embryonic fibroblasts that express CARP-2 variants. Treatment of vector–only expressing cells with TNF resulted in increased production of IL-6, but cells that express wild type CARP-2 produced very little IL-6 (Fig. 1E). In agreement with the requirement of E3 activity for CARP-2 inhibitory function, MEFs that express the RING mutant (H333A) exhibited increased IL-6 production both at the basal levels and upon TNF stimulation (Fig. 1E). Therefore, in TNF stimulated cells CARP-2 inhibits activation of NF-κB in a largely RING dependent manner.
To investigate the physiologic function of endogenous CARP-2, we designed small hairpin RNA (ShRNA) specific for two different regions of CARP-2. Transfection of the siRNA hairpins in 293T cells resulted in a large reduction in the level of endogenous CARP-2 protein (Fig. 1F). Knockdown of endogenous CARP-2 expression enhanced TNF-induced NF-κB reporter activity by approximately two fold (Fig. 1F). Consistent with this, knockdown of CARP-2 prolonged the IKK activation to as late as 60 min (Fig. 1G) and delayed the recovery of IκBα (beginning at 30-45 min in control cells but occurring at 60-90 min in ShRNA-treated cells) (Fig. 1H). The observed effects of ShRNAs are specific because co-expression of ShRNA-resistant CARP-2 but not wild type rescued TNF-induced NF-κB reporter activity (Fig. S1A-B). The increase in TNF signaling in cells with reduced CARP-2 suggests that the physiological function of this molecule is to limit the intensity or duration of signaling.
CARP-2 localizes to membrane compartments and recruits to vesicles containing endocytosed TNF-receptor
Previous studies have shown that overexpressed mouse CARP-2 associates with membrane compartments in the perinuclear region that are positive for the endosomal markers, Rab5 and Rab11[23]. Therefore, to determine if endogenous CARP-2 constitutively associates with endocytic membrane vesicles, we developed a monoclonal antibody that specifically recognizes CARP-2 (Fig. S2A and B). Confocal microscopic analysis of cells stained with this antibody showed punctate staining for endogenous CARP-2, mostly in the cytosol (Fig. 2A). A subset of these cytosolic vesicles also stained positive with antibody specific for Rab5 (Fig. 2B). No significant co-localization between CARP-2 and either Golgi marker GM130, or the lysosomal marker Lamp1 was observed (Fig. S2C). Collectively these results suggest that endogenous CARP-2 associates constitutively with, albeit undefined, membrane vesicles that fuse with internalized TNF-receptor complex (see below) and are referred as CARP-2 positive vesicles in this manuscript.
Fig. 2. CARP-2 localizes to endocytic vesicles.

(A) and (B). CARP-2 localizes to endocytic membrane vesicles. Confocal images of HeLa cells immunostained with control or CARP-2 (Red) (A), or CARP-2 (Red) and Rab 5 (Green) antibodies (B). Scale bar is 10 μm. Areas of colocalization was shown by line intensity profile.
(C). CARP-2 is recruited to TNF-R1 receptosomes. Magnetic TNF-R1 fractions and post-nuclear extracts (lysate) from HeLa cells were probed for indicated proteins.
(D). Internalized TNF-R1 vesicles associate with CARP-2 positive vesicles. Confocal images of HeLa cells labeled with Biotin-TNF/FITC (Green) conjugated streptavidine and anti-CARP-2 (Red). Colocalization analyses were performed with Zeiss AIM software v3.2 to give the overlay image of colocalized pixels, scatter plots and Pearson's coefficients. Ch 3 is FITC (green) and Ch4 is CY3 (Red). Scale bar is10 μm.
Given that internalized TNF-receptor complex fuse with endocytic vesicles to form multi vesicular endosomes (also known as TNF receptosomes), we examined whether these isolated TNF receptosomes include CARP-2 [10, 25]. As expected all the receptosome preparations from both HeLa and U937 cells contain TNF-R1 (Fig. 2C and Fig. S2D). After 5 minutes of internalization significant amounts of CARP-2 were found along with markers of other endocytic vesicles such as Rab 5 and Rab 7. Of note, barely detectable in the cell lysate, considerable enrichment of these proteins in isolated receptosomes indicate that either CARP-2 is directly recruited to endocytosed TNF complex, or CARP-2 vesicles along with that of Rab 5 and 7 fuse with internalized receptor complex. While the recruitment of CARP-2 occurred in as early as 5 minutes, Lamp-1 (a lysosomal protein) is recruited around 30-60 minutes suggesting that the association of CARP-2 is an early event that occurs much before the fusion of the internalized TNF-receptor with lysosomal compartments.
Additionally, confocal microscopy was used to investigate the association between CARP-2 and endocytosed TNF-receptor complex. The intracellular trafficking of TNF/TNF-R1 was followed by first, incubating HeLa cells with biotin TNF/streptavidin FITC at a low temperature and then by increasing the temperature to 37°C. Confocal analysis of these cells stained with anti-CARP-2 antibody showed (30 minutes after internalization) significant co-localization of the receptor with CARP-2 (Fig. 2D). The co-localization coefficient (Pearson's coefficient) for TNF receptor after 30 minutes of internalization ranged from 0.314 to 0.956. Thus, these results demonstrate that internalized TNF-R1 complex indeed associates with CARP-2.
CARP-2 promotes endogenous RIP degradation in TNF-stimulated cells
The association of the internalized TNF-R complexes with CARP-2 suggests that CARP-2 inhibits NF-κB activation in TNF-stimulated cells possibly by targeting one or more components of the early TNF-R1 complex. To test this possibility CARP-2 was co-expressed with a number of gene products found in the early TNF-R1 complex whose overexpression in cells is known to activate NF-κB. Co-expression of CARP-2 substantially inhibited the NF-κB activation induced by all the three proteins and the RING mutant was only slightly inhibitory (Fig. 3A). The NF-κB activity induced by IKKα was minimally inhibited by CARP-2, independent of its RING function. Therefore, CARP-2 likely inhibits NF-κB activation at the level of the early signaling complex.
Fig. 3. CARP-2 functions at the level of the early TNF-R1 complex.
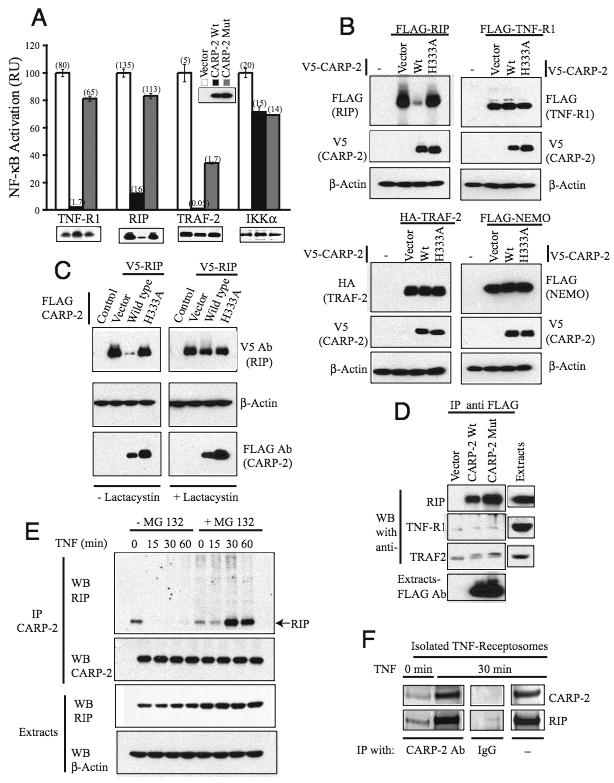
(A). CARP-2 inhibits NF-κB activation induced by early TNF-R1 complex components. Extracts from 293T cells transfected with indicated constructs along with reporter plasmids were assayed for reporter activities. The fold increase in the activity relative to vector background is shown in parenthesis. Expression of signaling molecules and one representative set of CARP-2 are shown.
(B) and (C). CARP-2 downregulates, and lactacystin rescues RIP expression. Expression of exogenous proteins in the extracts of 293T cells transfected with indicated constructs and treated with proteasomal inhibitor lactacystin are shown.
(D). CARP-2 binds to endogenous RIP. Extracts from 293T cells expressing FLAG-CARP-2 variants were immunoprecipitated with anti-FLAG antibody and probed for indicated proteins.
(E). Endogenous CARP-2 binds and promotes endogenous RIP degradation in TNF-stimulated cells. CARP-2 from extracts of TNF-stimulated HeLa cells was immunoprecipitated and probed for RIP and CARP-2.
(F). Endogenous CARP-2 binds endogenous RIP in TNF receptosomes. TNF receptosomes isolated from MG-132 treated HeLa cells were subjected to immunoprecipitation with anti-CARP-2 antibody coupled to magnetic MACS microbeads or with anti-rabbit IgG magnetic microbeads. RIP and CARP-2 in total receptosomes are shown.
Given that ubiquitin ligase (E3) activity of CARP-2 is essential for its function, we asked if CARP-2 might affect the expression of the above signaling molecules. RIP protein level was downregulated by co-expression of CARP-2 (Fig. 3B). The downregulation was specific for RIP, as CARP-2 had no effect on the level of expression of either TNF-R1, or TRAF-2, or NEMO, another critical regulator of NF-κB activation, under the same experimental conditions (Fig. 3B). The function of CARP-2 appears to be specific for TNF, as co-expression of CARP-2 had no effect on the expression of IRAK (Interleukin-1 receptor-associated kinase) a functional homologue of RIP in IL-1 signaling and an essential modulator of IL-1-induced NF-κb activation (Fig. S3). The RING mutant did not affect RIP protein expression (Fig. 3B), demonstrating that the protein ubiquitin ligase activity of CARP-2 is critical for this effect. This was strongly supported by the finding that the proteasome inhibitor lactacystin reverses the loss of RIP protein in the presence of CARP-2 (Fig. 3C). In order to determine whether CARP-2 binds directly to RIP, immunoprecipitates of FLAG-tagged CARP-2 from 293T cells were probed for endogenous proteins with antibodies against TNF-R1, RIP, and TRAF-2 (Fig. 3D). Consistent with the specific downregulation of RIP protein, immunoprecipitates from cell extracts expressing wild-type CARP-2 but not vector contained substantial amounts of RIP protein and only a very little amount of TNF-R1 and TRAF-2. Interestingly the RING-domain mutant, which was unable to affect RIP expression or inhibit TNF signaling, also co-immunoprecipitated these proteins equally well as wild-type CARP-2 indicating that the RING-domain is dispensable for RIP-binding. These interactions were further confirmed as extracts of cells expressing FLAG-tagged RIP and V5-tagged CARP-2 contained RIP/CARP-2 complexes (Fig. S4).
Because CARP-2 binds and downregulates RIP, we asked if stimulation with TNF promotes interaction between these proteins at endogenous level. To answer this endogenous CARP-2 was immunoprecipitated from cells stimulated with TNF in the presence or absence of MG-132, and the immunoprecipitated material was probed with anti-RIP (Fig. 3E). Some interaction between RIP and CARP-2 was detected in unstimulated cells. But in stimulated cells we found no interaction in the absence of MG-132 between CARP-2 and RIP. However, when proteasomal degradation is inhibited, anti-CARP-2 immunoprecipitates RIP from cells stimulated by TNF for 30 min or more, suggesting that after TNF stimulation CARP-2 binds and rapidly degrades RIP (Fig. 3E). In order to evaluate the direct interaction of endogenous CARP-2 with endogenous RIP in internalized TNF receptosomes, CARP-2 was immunoprecipitated from TNF receptosomes isolated from MG-132-treated HeLa cells after 0 and 30 min of internalization and probed for CARP-2 and RIP (Fig. 3F). TNF receptosomes in 30 min contained both RIP and CARP-2 and RIP from lysed TNF receptosomes was found to coimmunoprecipitate specifically with CARP-2 antibody. These data demonstrate that CARP-2 directly interacts with RIP at internalized TNF receptosomes.
CARP-2 targets the internalized receptor associated RIP for degradation
To test the hypothesis that RIP degradation at endocytic vesicles is the mechanism to terminate NF-κB signaling, we investigated the effect of internalization on RIP degradation in TNF-stimulated cells. Plasma membrane TNF-R1 is reported to internalize through two distinct endocytic routes, namely, clathrin mediated endocytosis (CME) and raft/caveolar endocytosis (RCE) [26-28] . Results from a previous study that show reduced TNF-induced NF-κB target gene expression upon inhibition of endocytosis indicate that active signaling continues during TNF-receptor endocytosis [29]. While most cell types upon TNF stimulation exhibit rapid degradation of receptor associated RIP, only NEMO deficient cells show significant decrease in total cellular RIP [8]. For this reason, to investigate the effect of internalization on RIP ubiquitination and degradation we have also used NEMO deficient cells in our studies.
As reported earlier, TNF stimulation significantly reduced total cellular RIP in NEMO −/− MEFs (Fig. 4A). This decrease in RIP level is not the result of apoptosis, as pan caspase inhibitor Z-VAD-FMK had failed to rescue the loss (Fig. 4B). Treatment with chlorpromazine (CPZ), a specific CME inhibitor, effectively reduced the loss in a dose dependent manner (Fig.4A). Inhibition of TNF-R1 endocytosis by monodansyl cadaverine (MDC) also prevented RIP loss (data not shown). However, the presence of another inhibitor Filipin at a concentration that specifically inhibits RCE, had very little effect suggesting that CME promotes the degradation of RIP in these cells. Next, the effect of internalization on RIP ubiquitination was assessed from cells treated with or without CPZ in the presence of proteasome inhibitor MG-132, and stimulated with TNF. In agreement with earlier published reports proteasomal inhibition efficiently blocked TNF-induced RIP loss (Fig. 4B). While TNF stimulation in the presence of MG-132 promoted RIP ubiquitination, the presence of CPZ blocked RIP modification suggesting that the observed loss of RIP might be the consequence of internalization and subsequent ubiquitination (Fig. 4C).
Fig. 4. Endogenous CARP-2 targets internalized receptor-associated RIP for degradation.

(A) and (B). Inhibitors of internalization and MG-132 but not Z-VAD FMK rescue TNF–induced RIP degradation. Expression of endogenous RIP in TNF-stimulated NEMO −/− MEFs, pretreated with medium or FILIPIN (2 μg/ml) or chlorpromazine (CPZ) (1.6 μg/ml, 3.2 μg/ml, 6.4 μg/ml) (A), or with indicated compounds (B) is shown.
(C). CPZ inhibits TNF–induced RIP ubiquitination. RIP from extracts of TNF-stimulated NEMO −/− MEFs pretreated with MG-132 and medium (UT) or CPZ (10 μg/ml) was immunoprecipitated under denaturing conditions and probed for ubiquitin and RIP.
(D). Knockdown of CARP-2 rescues TNF-induced RIP loss. Extracts of TNF-stimulated NEMO−/− MEFs expressing ShRNA constructs were probed for indicated proteins.
(E). Ubiquitinated RIP accumulates on TNF-R1 endocytic vesicles in the presence of MG-132. Magnetic TNF-R1 fractions from U937 cells were probed for indicated proteins.
(F). CARP-2 targets TNF receptor-associated RIP for degradation. Extracts from TNF-stimulated HeLa cells expressing ShRNA constructs were precipitated with hamster anti TNF-R1 antibody and probed for indicated proteins.
To investigate the role of CARP-2 in RIP degradation, we transfected NEMO−/−cells with ShRNA specific for mouse CARP-2. While the knockdown of CARP-2 protein expression had minimal effect on RIP level in unstimulated cells, it partially rescued the loss of RIP in stimulated cells (Fig. 4D) suggesting a possible role for CARP-2 in the degradation of RIP.
This finding prompted us to examine whether inhibition of RIP degradation with MG-132 results in accumulation of ubiquitinated RIP at endocytic vesicles. To address this we purified TNF-R1 associated endocytic vesicles from U937 cells treated with or without MG-132 and analyzed the vesicles for RIP by immunoblotting. MG-132 treatment did not alter the TNF-R1 complex fusion to endocytic vesicles (Fig. 4E). Compared to control (- MG-132) cells, inhibition of proteasomal degradation resulted in increased amounts of both polyubiquitinated and unubiquitinated RIP (30 and 60 min samples). Not surprisingly, the presence of MG-132 also stabilized TNF-R1 protein (Fig. 4E). Nevertheless, these results suggest that, in TNF stimulated cells, internalized RIP ubiquitination and degradation broadly occur at endocytic vesicles. While the initial ubiquitination of RIP that occurs immediately after stimulation at the plasma membrane (presumably mediated by TRAF-2) promotes NF-κB activation, the later ubiquitination at endocytic vesicles might be required for its degradation resulting in termination of NF-κB activation. If CARP-2, indeed, promotes RIP degradation we reasoned that the knockdown of CARP-2 expression should result in stabilization of this receptor associated RIP.
To test this, we immunoprecipitated TNF-R1 from HeLa cells expressing CARP-2 ShRNA and treated with TNF, and immunoblotted for associated endogenous RIP. Consistent with earlier reports, ubiquitinated and unubiquitinated RIP was found to coimmunoprecipitate with TNF-R1 from TNF-stimulated cells (Fig. 4F). Knockdown of CARP-2 resulted in a substantial increase in receptor-associated RIP, both polyubiquitinated and unubiquitinated, indicating that CARP-2 targets RIP within the early TNF-R1 complex for degradation. Hence, an increase in TNF-induced IKK/NF-κB activation in CARP-2 ShRNA expressing cells (Fig. 1F - H) could be the result of stabilization of receptor-associated RIP.
CARP-2 is an ubiquitin protein ligase for RIP
Given that CARP-2 promotes RIP degradation, we investigated whether CARP-2 functions as an ubiquitin protein ligase (E3) for RIP. We first assessed the effect of CARP-2 on endogenous RIP ubiquitination in TNF-stimulated cells transfected with CARP-2 and His-ubiquitin. While endogenous RIP was ubiquitinated on CARP-2 overexpression, this was significantly increased upon TNF-stimulation (Fig. 5A left panel). Furthermore, in co-transfection experiments V5-CARP-2 wild type but not RING mutant markedly enhanced the ubiquitination of FLAG RIP (Fig. 5A right panel). In these experiments overexpression of FLAG-RIP alone resulted in moderate ubiquitination. Since, CARP-2 ubiquitination of RIP effectively results in its degradation, we hypothesized that CARP-2 promoted ubiquitination of RIP was K48-linked. To test this, we co-transfected cells with CARP-2 variants along with HA-ubiquitin mutants in which only K-48 or K-63 are available for polymerization and analyzed ubiquitination status of endogenous RIP from TNF stimulated cells. As expected, CARP-2 wild type markedly promoted the K48-linked ubiquitination of RIP and very little K63-linked RIP ubiquitination was observed under these conditions (Fig. 5B). In agreement with this expression of mutant RIP that is unable to undergo K63-linked polyubiquitination (K377F) was reduced in RING-dependent manner by CARP-2 (Fig. S5). Additionally, the effect of CARP-2 variants on the turnover of FLAG-RIP was assessed by pulse chase analysis. Consistent with its function as an E3 ligase, the degradation of transfected FLAG-RIP was rapid in the presence CARP-2 wild type (Fig. 5C). To determine if CARP-2 can directly ubiquitinate RIP, in vitro ubiquitination assays were performed using methylated (blocked) ubiquitin. In the presence of ATP, wild-type but not the RING-domain mutant of CARP-2 was found to ubiquitinate RIP protein (Fig. 5D). Taken together, the results demonstrate that CARP-2 binds and ubiquitinates RIP, leading to its degradation.
Fig 5. CARP2 is an ubiquitin ligase (E3) for RIP.

(A). CARP-2 promotes TNF-induced endogenous RIP ubiquitination. Purified Hisubiquitinated proteins from TNF-stimulated 293T cells transfected with indicated plasmids and treated with MG-132 were analyzed with anti-RIP antibody (left panel). 293T cells expressing pcDNA3-HA-ubiquitin and pCMV-tag2B-RIP-His along with indicated plasmids were pretreated with MG-132. His-RIP was purified and analyzed for ubiquitination (right panel).
(B). CARP-2 mediates K48-linked polyubiquitination of endogenous RIP. Endogenous RIP from TNF-stimulated 293T cells expressing indicated plasmids, preincubated with MG-132, was immunoprecipitated under denaturing conditions and probed for ubiquitin and RIP.
(C). CARP-2 promotes RIP degradation. 293T cells transfected with pFLAG-CMV-RIP and pcDNA3-nV5-CARP2 variants were incubated with DMEM containing [35S] ProMix for 30 minutes and chased by growing in complete serum medium. RIP from the extracts was immunoprecipitated and analyzed by autoradiography and PhosphorImager.
(D). In vitro ubiquitination of RIP by CARP-2. Modified RIP band is indicated.
RIP kinase domain is essential for CARP-2 mediated RIP degradation
Earlier reports have shown that the RIP intermediate domain and death domain are essential but the kinase domain is dispensable for NF-κB activation [12, 13]. To determine which portion of RIP interacts with CARP-2, we tested its binding to individual regions of RIP. Because the different constructs were not expressed in transiently transfected cells to the same level, we expressed FLAG-tagged RIP constructs and V5-tagged CARP-2 constructs individually in 293T cells and used different amounts of the extracts from these cells for immunoprecipitation experiments. Interestingly the RIP kinase domain, but not RIP lacking the KD, was found to specifically co-immunoprecipitate with CARP-2 (Fig. 6A), suggesting a requirement of RIP KD for CARP-2 mediated RIP degradation. In order to test this possibility we examined the effect of CARP-2 on the degradation of RIP without KD in co-transfection and pulse-chase experiments. In agreement with its inability to bind to RIP without its kinase domain, CARP-2 did not affect RIPΔKD expression but reduced the full length RIP protein level (Fig. 6B). Consistent with this, in pulse-chase experiments the degradation of FLAG-RIPΔKD remained unchanged in the presence of wild type or RING mutant CARP-2 (Fig. 6C). Thus, these results prove that the kinase domain of RIP is essential for its binding and subsequent degradation by CARP-2.
Fig. 6. CARP-2 interacts with RIP kinase domain.

(A). CARP-2 binds RIP kinase domain. Extracts from 293T cells expressing FLAG tagged RIP kinase domain (KD) or without kinase domain (ΔKD) were incubated with extracts from cells expressing vector or wild type V5-CARP-2. RIP was immuno-precipitated and probed for CARP-2.
(B). Kinase domain is essential for CARP-2 mediated RIP loss. Extracts from HeLa cells transfected with pcDNA3-nV5-RIP Full length (FL) or ΔKD together with CARP-2 were probed for indicated proteins.
(C). Kinase domain is essential for CARP-2 mediated RIP degradation. 293T cells transfected with pcDNA3-nV5 -RIPΔKD and pFLAG-CMV-CARP2 variants were analyzed by procedure described in figure 5C.
(D). RIP kinase domain negatively regulates TNF-induced NF-κB activation. Extracts of medium (Med) or TNF treated RIP−/− MEFs transfected with indicated constructs in pMSCV vector along with reporter plasmids were assayed for the reporter activities.
Given this apparent paradox on the role of the kinase domain in NF-κB signaling we hypothesized that this domain might regulate the termination of NF-κB activation by targeting internalized RIP to CARP-2 in intracellular compartments. In such a scenario, cells expressing only RIP without the kinase domain should exhibit increased or prolonged NF-κB activation. In fact, earlier it has been shown that expression of a construct containing the ID-DD region of RIP in RIP-deficient Jurkat cells has increased NF-κB activity as compared to full-length RIP-expressing cells [13]. Indeed, TNF stimulation of RIP −/− MEFs expressing RIP lacking the kinase domain are consistently found to have an increase in basal as well as TNF-inducible NF-κB activity (Fig. 6D). Thus CARP-2 negatively regulates NF-κB signaling by binding to RIP of the internalized early complex via the kinase domain leading to RIP ubiquitination and degradation.
Discussion
Endocytic organelles are organized as a transport network consisting of physically and biochemically distinct membranous domains, which serve as intracellular stations that integrate diverse signals to provide a high order of specificity and regulation to signaling [16, 30-32]. The end result of a signaling process is determined by factors, presumably associated with membrane vesicles that regulate the duration and intensity of the signal. While duration of signaling is an important parameter that ultimately determines the biological outcome, to our knowledge, no factor(s) at the vesicles that terminate TNF signaling have been reported.
In this report, we propose that degradation of internalized RIP by CARP-2 is the mechanism to dampening TNF-induced IKK/NF-κB activation (Fig. 7). Endocytosed TNF-receptor complex that includes RIP is known to fuse with intracellular compartments to form TNF-receptosomes [10]. We now present evidence that CARP-2 localizes to compartments that associate with receptosomes and targets endocytosed RIP for K-48 linked ubiquitination and proteosomal degradation. Whereas, results from pulse-chase experiments showed marginal differences between wild-type and mutant CARP-2, collective evidence from several experiments including in vivo and in vitro ubiquitination assays proves that CARP-2 is an ubiquitin protein ligase (E3) for RIP. In knockdown experiments, we observed only small effect in RIP ubiquitination/ IKK/NF-κB activation possibly owing to the presence of some residual protein in our shRNA experiments, and/or other molecules that could functionally offset the effect of loss of CARP-2. A20, an ubiquitin-editing enzyme that negatively regulates TNF-induced NF-κB activation targets, like CARP-2, TNFR1-associated ubiquitinated RIP for degradation [7]. Knockdown of either CARP-2 or A20 stabilized RIP in a similar manner (Fig. S6A), and suppression of A20 expression together with CARP-2 further increased RIP ubiquitination and slightly decreased the level of IκBα compared to reduction of A20 alone (Fig. S6B). These results indicate that CARP-2 might function co-operatively with A20 in regulating TNF-induced NF-κB activation with CARP-2 acting at the endocytic compartments. Notwithstanding of these similarities the level of CARP-2, unlike A20 whose expression is substantially upregulated by NF-κB, remained unchanged upon TNF treatment (Fig. S6C). More experiments are needed to understand the relative contribution of A20 and CARP-2 in the regulation of TNF signaling.
Fig. 7.

Model for regulation of TNF mediated NF-κB activation by CARP-2
An unexpected outcome of our studies is the requirement of the kinase domain of RIP for CARP-2 function. The dispensability of the KD for TNF-induced NF-κB activation has long been known [12, 13, 33]. Our data suggests, however, that the kinase domain may have an inhibitory effect on RIP function. Increased NF-κB activity observed in our experiments in RIP −/− MEFs expressing RIP KD deletion mutant is in agreement with earlier reports using RIP deficient Jurkat cells [13]. In principle it is possible that kinase activity might negatively regulate TNF-induced NF-κB activity by affecting RIP/CARP-2 interaction. Such a notion is supported by earlier data that showed increased NF-κB transcriptional activity in TNF-stimulated RIP deficient cells expressing kinase inactive version of RIP (K45R or D138N) [13, 33].
Unlike other FYVE proteins that localize to Rab 5 or EEA-1 positive endosomes endogenous CARP-2 mostly associates with non-conventional endocytic vesicles. This could partly be because of the differences in FYVE domains' affinity to phospholipids [24]. We speculate that the FYVE domain restricts the function of CARP-2 to a specific set of endocytic vesicles along the endocytic pathway of TNF signaling. Knowledge of pathways such as this may have important implications in understanding the constitutive activation of NF-κB in tumor cells.
Materials and methods
Details of materials and methods can be found in supplementary information.
Supplementary Material
Acknowledgements
The research was supported by the Intramural Research Program of the NIH, Center for Cancer research, National Cancer Institute. This work was supported in part by NIH grant CA97100 to W.S.E-D and by DFG grands SCHU 733/7-1 and SFB 415, project A11 to S.S. We thank Jonathan Ashwell, Remy Bosselut and Stanley Lipkowitz for helpful discussions, Michelle A. Kelliher for RIP −/− MEFs and MARC Schmidt-Supprian for NEMO −/− MEFs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001;11:372–377. doi: 10.1016/s0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- 2.Dempsey PW, Doyle SE, He JQ, Cheng G. The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev. 2003;14:193–209. doi: 10.1016/s1359-6101(03)00021-2. [DOI] [PubMed] [Google Scholar]
- 3.MacEwan DJ. TNF receptor subtype signalling: differences and cellular consequences. Cell Signal. 2002;14:477–492. doi: 10.1016/s0898-6568(01)00262-5. [DOI] [PubMed] [Google Scholar]
- 4.Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. 2002;296:1634–1635. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- 5.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 6.Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 7.Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, Ma A, Koonin EV, Dixit VM. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 8.Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected] Nat Cell Biol. 2006;8:398–406. doi: 10.1038/ncb1384. [DOI] [PubMed] [Google Scholar]
- 9.Zhang SQ, Kovalenko A, Cantarella G, Wallach D. Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKgamma) upon receptor stimulation. Immunity. 2000;12:301–311. doi: 10.1016/s1074-7613(00)80183-1. [DOI] [PubMed] [Google Scholar]
- 10.Schneider-Brachert W, Tchikov V, Neumeyer J, Jakob M, Winoto-Morbach S, Held-Feindt J, Heinrich M, Merkel O, Ehrenschwender M, Adam D, Mentlein R, Kabelitz D, Schutze S. Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity. 2004;21:415–428. doi: 10.1016/j.immuni.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 11.Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 1998;8:297–303. doi: 10.1016/s1074-7613(00)80535-x. [DOI] [PubMed] [Google Scholar]
- 12.Meylan E, Tschopp J. The RIP kinases: crucial integrators of cellular stress. Trends Biochem Sci. 2005;30:151–159. doi: 10.1016/j.tibs.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 13.Ting AT, Pimentel-Muinos FX, Seed B. RIP mediates tumor necrosis factor receptor 1 activation of NF-kappaB but not Fas/APO-1-initiated apoptosis. Embo J. 1996;15:6189–6196. [PMC free article] [PubMed] [Google Scholar]
- 14.Li H, Kobayashi M, Blonska M, You Y, Lin X. Ubiquitination of RIP is required for tumor necrosis factor alpha-induced NF-kappaB activation. J Biol Chem. 2006;281:13636–13643. doi: 10.1074/jbc.M600620200. [DOI] [PubMed] [Google Scholar]
- 15.Cho W. Building signaling complexes at the membrane. Sci STKE. 2006;2006:pe7. doi: 10.1126/stke.3212006pe7. [DOI] [PubMed] [Google Scholar]
- 16.Cho W, Stahelin RV. Membrane-protein interactions in cell signaling and membrane trafficking. Annu Rev Biophys Biomol Struct. 2005;34:119–151. doi: 10.1146/annurev.biophys.33.110502.133337. [DOI] [PubMed] [Google Scholar]
- 17.Blatner NR, Stahelin RV, Diraviyam K, Hawkins PT, Hong W, Murray D, Cho W. The molecular basis of the differential subcellular localization of FYVE domains. J Biol Chem. 2004;279:53818–53827. doi: 10.1074/jbc.M408408200. [DOI] [PubMed] [Google Scholar]
- 18.Lindmo K, Stenmark H. Regulation of membrane traffic by phosphoinositide 3-kinases. J Cell Sci. 2006;119:605–614. doi: 10.1242/jcs.02855. [DOI] [PubMed] [Google Scholar]
- 19.Gao M, Karin M. Regulating the regulators: control of protein ubiquitination and ubiquitin-like modifications by extracellular stimuli. Mol Cell. 2005;19:581–593. doi: 10.1016/j.molcel.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 20.Haglund K, Dikic I. Ubiquitylation and cell signaling. Embo J. 2005;24:3353–3359. doi: 10.1038/sj.emboj.7600808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McDonald ER, 3rd, El-Deiry WS. Suppression of caspase-8- and -10-associated RING proteins results in sensitization to death ligands and inhibition of tumor cell growth. Proc Natl Acad Sci U S A. 2004;101:6170–6175. doi: 10.1073/pnas.0307459101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Araki K, Kawamura M, Suzuki T, Matsuda N, Kanbe D, Ishii K, Ichikawa T, Kumanishi T, Chiba T, Tanaka K, Nawa H. A palmitoylated RING finger ubiquitin ligase and its homologue in the brain membranes. J Neurochem. 2003;86:749–762. doi: 10.1046/j.1471-4159.2003.01875.x. [DOI] [PubMed] [Google Scholar]
- 23.Coumailleau F, Das V, Alcover A, Raposo G, Vandormael-Pournin S, Le Bras S, Baldacci P, Dautry-Varsat A, Babinet C, Cohen-Tannoudji M. Over-expression of Rififylin, a new RING finger and FYVE-like domain-containing protein, inhibits recycling from the endocytic recycling compartment. Mol Biol Cell. 2004;15:4444–4456. doi: 10.1091/mbc.E04-04-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tibbetts MD, Shiozaki EN, Gu L, McDonald ER, 3rd, El-Deiry WS, Shi Y. Crystal structure of a FYVE-type zinc finger domain from the caspase regulator CARP2. Structure. 2004;12:2257–2263. doi: 10.1016/j.str.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 25.Schneider-Brachert W, Tchikov V, Merkel O, Jakob M, Hallas C, Kruse ML, Groitl P, Lehn A, Hildt E, Held-Feindt J, Dobner T, Kabelitz D, Kronke M, Schutze S. Inhibition of TNF receptor 1 internalization by adenovirus 14.7K as a novel immune escape mechanism. J Clin Invest. 2006;116:2901–2913. doi: 10.1172/JCI23771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mosselmans R, Hepburn A, Dumont JE, Fiers W, Galand P. Endocytic pathway of recombinant murine tumor necrosis factor in L-929 cells. J Immunol. 1988;141:3096–3100. [PubMed] [Google Scholar]
- 27.D'Alessio A, Al-Lamki RS, Bradley JR, Pober JS. Caveolae participate in tumor necrosis factor receptor 1 signaling and internalization in a human endothelial cell line. Am J Pathol. 2005;166:1273–1282. doi: 10.1016/S0002-9440(10)62346-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee KH, Feig C, Tchikov V, Schickel R, Hallas C, Schutze S, Peter ME, Chan AC. The role of receptor internalization in CD95 signaling. Embo J. 2006;25:1009–1023. doi: 10.1038/sj.emboj.7601016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bradley JR, Johnson DR, Pober JS. Four different classes of inhibitors of receptor-mediated endocytosis decrease tumor necrosis factor-induced gene expression in human endothelial cells. J Immunol. 1993;150:5544–5555. [PubMed] [Google Scholar]
- 30.Di Fiore PP, De Camilli P. Endocytosis and signaling. an inseparable partnership. Cell. 2001;106:1–4. doi: 10.1016/s0092-8674(01)00428-7. [DOI] [PubMed] [Google Scholar]
- 31.Polo S, Di Fiore PP. Endocytosis conducts the cell signaling orchestra. Cell. 2006;124:897–900. doi: 10.1016/j.cell.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 32.Raiborg C, Rusten TE, Stenmark H. Protein sorting into multivesicular endosomes. Curr Opin Cell Biol. 2003;15:446–455. doi: 10.1016/s0955-0674(03)00080-2. [DOI] [PubMed] [Google Scholar]
- 33.Lee TH, Shank J, Cusson N, Kelliher MA. The kinase activity of Rip1 is not required for tumor necrosis factor-alpha-induced IkappaB kinase or p38 MAP kinase activation or for the ubiquitination of Rip1 by Traf2. J Biol Chem. 2004;279:33185–33191. doi: 10.1074/jbc.M404206200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.