

Abstract
The functional differentiation of T cells is mediated by lineage-specific transcription factors. TH17 has been recently identified as a distinct TH lineage that mediates tissue inflammation. RORγ was previously shown to regulate TH17 differentiation; RORγ deficiency, however, did not completely abolish TH17 cytokine expression. Here we report that TH17 cells also highly express another related nuclear receptor RORα, which is induced by TGFβ and IL-6 in a STAT3-dependent manner. Over-expression of RORα promotes TH17 differentiation and significantly upregulates IL-17 and IL-17F expression, possibly through the CNS2 element in the IL-17-IL-17F gene locus. RORα deficiency results in reduced IL-17 expression in vitro and in vivo. Furthermore, we found that RORα and RORγ co-expression synergistically leads to greater TH17 differentiation. Double deficiencies in RORα and RORγ globally impair TH17 generation and completely protect mice against experimental autoimmune encephalomyelitis. Therefore, TH17 lineage differentiation is mediated by both RORα and RORγ.
Introduction
Upon activation by antigen-presenting cells (APC), naïve TH cells undergo clonal expansion and functional differentiation into cytokine-secreting effector cells. Effector TH cells have been historically classified into TH1 and TH2 subsets (Dong and Flavell, 2000; Glimcher and Murphy, 2000). TH1 cells make interferonγ (IFNγ) and regulate antigen presentation and cellular immunity. TH2 cells, on the other hand, secrete IL-4, -5 and -13, which together regulate humoral and anti-parasite immunity. The cytokine environment during TH activation determines TH effector differentiation, through selective signal transducer and activator of transcription (STAT) proteins leading to expression of lineage-specific master transcription factors (Dong, 2006; Glimcher and Murphy, 2000). TH1 differentiation and IFNγ production is promoted by IL-12, a heterodimeric cytokine produced by activated antigen-presenting cells (APCs) that signals through STAT4. IFNγ-STAT1 pathway in turn sustains TH1 development leading to the induction of transcription factor T-bet. On the other hand, IL-4, secreted by activated T cells, drives TH2 polarization in a STAT6-dependent manner, resulting in activation of transcription factor GATA3.
A novel TH subset, termed THIL-17, TH17 or inflammatory TH (THi), has been recently identified as a distinct TH lineage (Bettelli et al., 2007; Dong, 2006; Reiner, 2007; Weaver et al., 2006). TH17 differentiation is initiated by TGFβ and IL-6 in mouse (Bettelli et al., 2006; Mangan et al., 2006; Veldhoen et al., 2006), possibly via regulating the chromatin remodeling of the IL-17-IL-17-F locus (Akimzhanov et al., 2007), which is further reinforced by IL-23 (Yang et al., 2007; Zhou et al., 2007). TH17 cells produce IL-17, IL-17F and IL-22, all of which regulate inflammatory responses by tissue cells (Chung et al., 2006; Langrish et al., 2005; Liang et al., 2006; Zheng et al., 2007). Recently, IL-21 was reported as an autocrine factor induced by IL-6 to regulate TH17 differentiation (Korn et al., 2007; Nurieva et al., 2007; Zhou et al., 2007). TH17 differentiation is negatively regulated by IFN-γ, IL-4, IL-27 and IL-2 (Batten et al., 2006; Harrington et al., 2005; Laurence et al., 2007; Park et al., 2005; Stumhofer et al., 2006).
Analogous to STAT4 and STAT1 in TH1 and STAT6 in TH2 differentiation, STAT3 was found to selectively mediate TH17 differentiation (Laurence et al., 2007; Yang et al., 2007). Overexpression of a hyperactive STAT3 enhanced TH17 differentiation while STAT3 deficiency impairs TH17 differentiation in vitro (Laurence et al., 2007; Yang et al., 2007) and in vivo (Zhou et al., 2007). STAT3 is necessary for IL-6 induction of IL-21 expression and is required for IL-21-mediated TH17 differentiation (Nurieva et al., 2007). The precise biochemical function of STAT3 is unclear at this point. Although STAT3 has been shown to bind to IL-17 gene promoter (Chen et al., 2006), STAT3 appears to control more than just IL-17 gene expression (Yang et al., 2007). Likely, STAT3, similar to STAT1 and STAT6, may function in regulation of lineage-specific master transcription factor expression.
One such TH17-specific transcription factor is an orphan nuclear receptor RORγ. Its specific isoform RORγt was recently shown as the first transcription factor to be selectively expressed in TH17 cells (Ivanov et al., 2006) and is regulated by STAT3 (Laurence et al., 2007; Yang et al., 2007). Overexpression of RORγt promoted TH17 differentiation when TH1 and TH2 development was inhibited (Ivanov et al., 2006). Conversely, RORγt deficiency resulted in profound TH17 deficiency and was reported to protect mice from EAE. However, RORγt defect did not completely abolish TH17 differentiation or totally inhibit EAE, suggestive of additional factors involved.
In the current study, we report that TH17 cells also highly express another orphan nuclear receptor RORα, which is induced by TGF-β and IL-6 in a STAT3-dependent manner. Overexpression of RORα promotes TH17 differentiation and significantly upregulates IL-17 and IL-17F expression. RORα deficiency results in reduced IL-17 expression in vitro and in vivo. Furthermore, we found that RORα and RORγt co-expression synergistically drive greater TH17 differentiation especially under non-favorable conditions. Double deficiencies in RORα and RORγ entirely impair TH17 generation in vitro and completely inhibit EAE disease. Therefore, RORα is another TH17-specific factor that together with RORγ directs TH17 lineage differentiation.
Results
RORα is highly expressed in TH17
In a gene expression profiling analysis of in-vitro differentiated TH1, TH2 and TH17 cells, we found that the expression of two RAR-related orphan receptor (ROR), RORα and RORγ, was significantly elevated in TH17 cells compared to TH1 or TH2 cells (data not shown). The ROR family comprises 3 members, RORα, RORβ and RORγ (Jetten, 2004). While RORβ is not expressed in TH17 cells (data not shown), RORγ, in particular the RORγt isoform, was previously identified as a critical transcription factor regulating TH17 differentiation (Ivanov et al., 2006). Whether RORα, specifically the isoform 4 that is expressed in TH17 cells (data not shown), is also involved in TH17 regulation has not yet been established. To examine the potential role of RORα, we first assessed the expression levels of RORα in TH1, TH2 and TH17 subsets. CD4+ T cells purified from OT-II mice were differentiated under neutral condition or into TH1, TH2 and TH17 cells and the expression levels of RORαand RORγt mRNA were measured on day 5 by quantitative real-time RT-PCR. As demonstrated for RORγt, RORα mRNA was found highly expressed in TH17 compared with TH1 and TH2 (Fig. 1A).
Figure 1. RORα is highly expressed in TH17 cells.
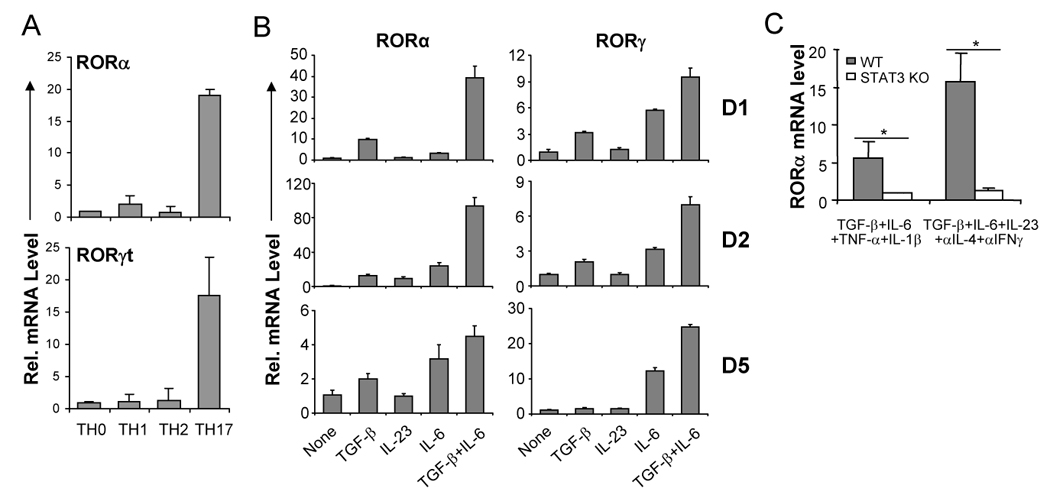
(A) CD4+ T cells from OTII mice were differentiated with OVA323–339 peptide and splenic antigen presenting cells (APCs) under neutral (TH0), TH1, TH2 and TH17 conditions for 5 days, and RORα and RORγt mRNA expression levels were assessed by real-time RT-PCR after restimulation of T cells by anti-CD3 for 4 hours. Data shown are derived from 2 independent experiments with consistent results and normalized using expression levels of Actb. Expression levels in TH0 were referred as 1. (B) FACS-sorted naïve CD4+CD25−CD62LhiCD44lo T cells from C57BL/6 mice were activated with anti-CD3 and anti-CD28 in the presence of indicated cytokine stimuli for 1, 2 or 5 days, and RORα and RORγ mRNA expression levels were assessed by real-time RT-PCR. Expression levels in the condition without exogenous cytokine stimulus were referred as 1. (C) Naïve CD4+ T cells from STAT3 KO or wild-type (WT) mice were activated with plate-bound anti-CD3 and anti-CD28 under the indicated stimuli for 4–5 days. RORα mRNA expression levels were assessed by real-time RTPCR. The expression levels in STAT3 KO cells were referred as 1. The experiments were repeated at least 3 times with consistent results. *, student t test p≤0.05.
TH17 differentiation is initiated by TGF-β and IL-6 (Bettelli et al., 2006; Mangan et al., 2006; Veldhoen et al., 2006) and reinforced by IL-23 (Yang et al., 2007). We next asked whether these cytokines regulate RORα expression. FACS-sorted CD4+CD25−CD62LhiCD44lo naïve T cells were activated with plate-bound anti-CD3 and anti-CD28 in the presence of various cytokines for 1, 2 and 5 days. On days 1 and 2, IL-23 alone did not have a significant effect on RORα mRNA levels; the combination of TGF-β and IL-6 synergistically upregulated RORα mRNA levels (Fig. 1B), which correlates with the induction of IL-17 and IL-17F gene transcription (Akimzhanov et al., 2007). On day 5, either IL-6 or TGF-β increased RORα mRNA and the combination of TGFβ and IL-6 further enhanced RORα mRNA expression (Fig. 1B).
We and others previously showed that STAT3 is an essential factor regulating cytokine-mediated TH17 differentiation and that RORγt is regulated by STAT3 (Laurence et al., 2007; Yang et al., 2007). To determine whether RORα expression is STAT3-dependent, naïve T cells were purified from STAT3f/Δ Tie2-Cre+ (STAT3 KO) or wild-type (WT) mice and differentiated into TH17 cells using the following two conditions: TGF-β, IL-6, TNF-α plus IL-1β or TGF-β, IL-6, IL-23 and blocking antibodies against IL-4 and IFN-γ. Real-time RT-PCR revealed that under both conditions, RORα mRNA expression was defective in STAT3 KO cells in comparison with control cells (Fig. 1C). Therefore, like RORγt, RORα is highly expressed in TH17 cells in a STAT3-dependent manner.
RORα overexpression drives TH17 differentiation
In order to assess RORα function in TH17 differentiation, we over-expressed RORα by retroviral transduction. Naïve CD4+ T cells from OT-II mice were FACS-purified and activated with Ova peptide and splenic APC in the presence of blocking antibodies to IL-4 and IFN-γ. On day 2, the activated cells were infected with bicistronic retroviruses containing an IRES-GFP. 3 days after infection, IL-17- and IFN-γ-expressing cells were measured by intracellular staining. Compared to cells infected with a control virus, RORα over-expression substantially increased the percentage of IL-17-secreting cells even in the absence of TGF-β and IL-6 and reduced the frequency of IFN-γ-producing cells (Fig. 2A). Anti-IL-17 staining correlated with high levels of GFP expression and GFP negative cells did not exhibit any significant increase in IL-17 expression (Fig. S1), indicating a cell-intrinsic effect by RORα. In the presence of TGF-β or IL-6, RORα facilitated TH17 differentiation and further enhanced this process in the presence of both factors (Fig. 2A). To further understand the role of RORα in directing the TH17 program, we sorted GFP+ cells from the retrovirus-transduced cells differentiated under neutral conditions and their gene expression profiles were assessed using real-time RT-PCR. In comparison with cells infected with the control virus, RORα over-expression greatly enhanced IL-17, IL-17F and IL-23R mRNA expression, while IL-22 expression level was moderately increased (Fig. 2B). Furthermore, RORα overexpression facilitated TH17 differentiation in RORγ-deficient cells (Fig. S1). Thus, RORα promotes TH17 differentiation and is sufficient to upregulate most of TH17-specific genes.
Figure 2. RORα overexpression promotes TH17 differentiation.

(A) FACS-sorted naïve OT-II CD4+ T cells were activated with splenic APCs pulsed with OVA peptide in the presence of blocking antibodies for IL-4 and IFN-γ and the indicated stimuli and infected with an IRES-GFP-containing bicistronic retrovirus expressing RORα or a vector control virus. IL-17- and IFN-γ-expressing cells were measured by intracellular staining on a GFP+ gate. The experiments were repeated at least for three times with consistent results. (B) GFP+ cells were sorted from the above neutral culture conditions and mRNA expression was measured by real-time RT-PCR. Expression levels in cells transduced with the vector control virus were referred as 1. (C) Naïve CD4+ T cells were activated with anti-CD3 and anti-CD28 in the presence of indicated stimuli for 2 days and histone H3 acetylation in CNS2 of IL-17-IL-17F locus was assessed by chromatin immuno-precipitation (ChIP) and PCR using two different pairs of primers (2a and 2b). Data were normalized to levels of histone H3 acetylation at the Actb promoter. (D) ROR binding to RORE at CNS2 was assessed by Electrophoretic Mobility Shift Assay. Nuclear proteins were prepared from TH17 cells differentiated from naïve OT-II T cells using splenic APC pulsed with OVA peptide in the presence of polarizing cytokines. A CNS2 RORE was used as probe. Competitors were a consensus element and a mutant of the CNS2 RORE. (E) RORα or RORγ overexpression enhances transcription from an IL-17 minimal promoter in the presence of CNS2. EL-4 cells were transfected with RORα, RORγ or both o0r vector alone with an IL-17 promoter (promoter) or IL-17-promoter-CNS2 (promoter+CNS2) luciferase reporter vector. Renilla luciferase was used to normalize transfection efficiency. Background luciferase activities in cells transfected with promoter reporter and empty vectors were referred as 1. Data shown represent two independent experiments with similar results.
How RORγt regulates TH17 differentiation is poorly understood. Since RORα and RORγ are highly homologous in their DNA binding motif, we search for putative ROR response elements (RORE) in the IL-17-IL-17F gene locus. While IL-17 and IL-17F gene promoters do not have perfect RORE, we noticed that CNS2 site contains 2 RORE that are also conserved in human. We previously found CNS2 to be evolutionarily conserved and associated with acetylated histone H3 in TH17 but not TH1 or TH2 cells (Akimzhanov et al., 2007). Furthermore, we found that this chromatin change at CNS2 was initiated by both TGFβ and IL-6 (Fig. 2C). To further characterize the potential regulation of CNS2 by ROR factors, we performed electrophoresis mobility shift assay (EMSA) with a CNS2 oligonucleotide probe containing RORE. Nuclear factors from TH17 cells were found to bind to this CNS2 probe (Fig. 2D) but not to a CNS2 probe containing a mutation in RORE (data not shown). Furthermore, unlabelled consensus probe but not the mutant CNS2 probe diminished this binding in a dose-dependent manner (Fig. 2D). ROR factor binding to CNS2 was detected by chromatin immunoprecipitation assay in TH cells overexpressing RORα or RORγt (Fig. S2). To determine whether RORα and RORγt regulate the activity of CNS2, we cloned this element together with a minimal promoter of the IL-17 gene in front of a luciferase reporter. In EL-4 cells, the minimal IL-17 promoter alone was not activated by RORα and/or RORγt (Fig. 2E). In the presence of CNS2, either RORα or RORγt significantly enhanced the transcription of IL-17 promoter (Fig. 2E). There was no synergistic effect when both were co-expressed (Fig. 2E).
RORα deficiency reduced IL-17 expression
Therefore, RORα, like RORγt, is sufficient in inducing TH17 program. We next tested the function of RORα using Staggerer mice which bear a spontaneous deletion in RORα ligand-binding domain and exhibit the same phenotype as the ROR α knockout mice (Hamilton et al., 1996; Steinmayr et al., 1998). When naïve CD4+ T cells isolated from homozygous Staggerer (referred to as ROR αsg/sg hereafter) or wild-type (WT) splenocytes were differentiated into TH17 cells, an approximately 3-fold reduction in IL-17-producing cells was observed in RORαsg/sg versus WT cells under the stimuli of TGF-β, IL-6, TNF-α and IL-1β, but this reduction was less pronounced when cells were differentiated in the presence of TGF-β, IL-6, IL-23, anti-IL-4 and anti-IFN-γ (Fig. 3A). When analyzed by real-time RT-PCR, in addition to reduced IL-17 mRNA expression, IL-23R mRNA was also consistently decreased in RORαsg/sg cells as well (Fig. 3B). In contrast, T-bet mRNA was increased in RORαsg/sg samples, suggesting that RORα not only promotes the TH17 program but also may inhibit the TH1 program. Unlike IL-17 and IL-23R, the expression levels of IL-22, IL-21 and IL-17F mRNA were not significantly affected by RORα deficiency (Fig. 3B), indicating differential dependency of TH17 cytokines on RORα.
Figure 3. RORα deficiency reduces IL-17 production in vitro.

(A) FACS-sorted naïve CD4+ T cells from WT or homozygous Staggerer (RORαsg/sg) were activated under the indicated conditions for 4 d. IL-17- or IFN-γ-expressing cells were measured by intracellular staining. The experiments were repeated at least for 3 times. (B) Total RNA was isolated from the above culture and mRNA expression for indicated genes was assessed by real-time RT-PCR. The lowest expression levels in each PCR were referred as 1. *, p≤0.05.
To understand the function of RORα in vivo, we employed experimental autoimmune encephalomyelitis (EAE), a TH17-mediated central nerve system (CNS) inflammation disease model (Langrish et al., 2005; Park et al., 2005). Since RORαsg/sg mice have developmental defects in brain (Hamilton et al., 1996), we reconstituted Rag1 knockout mice on C57BL/6 background with bone marrow cells from RORαsg/sg or WT mice. RORαsg/sg hematopoietic stem cells were previously shown to reconstitute lymphoid compartments in Rag-deficient mice (Dzhagalov et al., 2004; Ivanov et al., 2006). EAE was induced in the chimeric mice using MOG peptide. In comparison with mice reconstituted with WT bone marrow cells, RORαsg/sg constituted mice exhibited the same disease incidence but fewer mice in this group developed limb paralysis (score≥3) (Fig. 4A). To further understand the impact of RORα mutation on EAE pathogenesis, we sacrificed both groups of mice on day 14 after the 2nd immunization and examined IL-17- and IFN-γ-expressing cells in the CNS and spleen by intracellular staining. The CNS infiltrates in RORαsg/sg chimeras contained reduced numbers of IL-17-secreting cells than WT chimeras (Fig. 4B). The number of IL-17-producing CD4+ cells in spleen was decreased upon restimulation, while the number of IFN-γ-expressing cells was increased in Rag1−/− mice reconstituted with RORαsg/sg cells versus those with WT cells (Fig. 4B). Moreover, after restimulation with MOG peptide, RORαsg/sg splenocytes exhibited reduced IL-17 and increased IFN-γ expression compared to WT cells, while IL-22 production was not affected (Fig. 4C). Thus, our in vitro and in vivo analysis indicates that IL-17 expression is selectively dependent on RORα.
Figure 4. RORα deficiency reduces IL-17 production in vivo.

(A) Rag1 KO mice were reconstituted with bone marrow from RORαsg/sg or WT mice. 6–8 weeks later, EAE was induced in the chimeric mice. Data shown are a combination of two independent experiments. WT, n=10; RORαsg/sg, n=9. Max disease, score ≥ 3. (B) Infiltrates in central nerve system or splenocytes from the EAE mice were isolated on day 14 after the 2nd immunization and restimulated with PMA/Ionomycin and MOG35–55 peptide, respectively. IL-17- or IFN-γ-expressing cells were measured by intracellular staining. Data shown are on gated CD4+ cells. CNS, central nerve system; Sp, spleen. (C) Splenocytes from the above mice were stimulated with MOG peptide and cytokine expression levels were measured by ELISA. Mean values were shown as horizontal bars. Data shown represent two independent experiments with consistent results.
RORα and RORγt co-expression synergistically promotes TH17 differentiation in vitro
RORα exhibits a similar expression pattern as RORγt and both factors appear to regulate TH17 differentiation. To understand why both receptors are co-expressed in TH17 cells, we first examined whether RORα synergizes with RORγt in programming the TH17 lineage using a retroviral co-infection strategy. Naïve OT-II T cells were activated as above and co-infected with two viruses expressing RORα-GFP or GFP vector and RORγt-hCD2 or hCD2 vector. IL-17- and IFN-γ-secreting cells were assessed by intracellular staining on a GFP+hCD2+ gate. Under neutral condition, either RORα or RORγt but not the control virus was sufficient to promote the generation of IL-17-producing cells (Fig. 5). Remarkably, in the absence of exogenous cytokine, co-expression of RORα and RORγt led to greatly enhanced IL-17 production. No effect was observed on GFP- non-transduced cells (Fig. S3A), indicating a cell-intrinsic regulation. RORα was found to enhance the numbers of IL-17-expressing T cells in a dose-dependent manner in cells expressing zero, low or high levels of RORγt-hCD2 (Fig. S3A). To further understand the relationship between RORα and RORγt in directing TH17 differentiation, we sorted out the GFP+hCD2+ cells from the culture above and examined the expression of TH17-specific genes by RT-PCR. Compared with cells infected with control viruses, RORα or RORγt alone increased IL-17, IL-17F, and IL-23R mRNA expression while co-expression of RORα and RORγt synergistically upregulated IL-17, IL-17F, IL-23R and IL-22 mRNA levels (Fig. S3B). RORα or RORγt expression also downregulated T-bet mRNA expression (Fig. S3C). Associated with activation of TH17-specific genes, we found that histone H3 acetylation was enhanced by RORα and RORαt at CNS2 but not IL-17 gene promoter (Fig. S3D), further supporting CNS2 as a potential direct target of ROR factors.
Figure 5. RORα and RORγt synergizes in promoting TH17 differentiation.

Naïve OT-II CD4+ T cells were activated with OVA peptide-pulsed splenic APCs under the Neutral (anti-IL-4 and anti-IFN-γ); iTreg (inducible regulatory T cells, TGF-β, anti-IL-4 and anti-IFN-γ), TH1 or TH2 conditions and co-infected with two bicistronic retroviruses expressing RORα-GFP or GFP vector and RORγt-hCD2 or hCD2 vector. Cytokine- or Foxp3-expressing cells were assessed by intracellular staining. Data shown are gated on GFP+hCD2+ cells. The experiments were repeated at least twice with consistent results.
We next tested whether RORα and RORγt combination could induce TH17 cells under non-favorable conditions. Naïve OT-II T cells were activated with TGF-β to induce regulatory T cells (iTreg) or under TH1 and TH2 polarized conditions and infected with RORα, RORγt and/or empty vector viruses. IL-17- and Foxp3- (iTreg), IFN-γ- (TH1) or IL-5- (TH2 condition) expressing cells were assessed by intracellular staining in a GFP+hCD2+ gate. Under iTreg generation condition, RORα appears to downregulate Foxp3 expression; RORα and RORγt synergistically induced IL-17 expression (Fig. 5). Under the TH1 condition, neither RORα nor RORγt over-expression alone resulted in significant numbers of TH17 cells, whereas co-expression of RORα and RORγt promoted TH17 differentiation and partially inhibited TH1 development (Fig. 5). Under the TH2 condition, interestingly, either RORα or RORγt led to TH17 differentiation and their co-expression greatly increased TH17 cell number, or cytokine protein and mRNA expression (Fig. 5, S3E). Thus, RORα and RORαt together promote TH17 development and are required to antagonize the TH1 program.
RORα and RORα double deficiencies completely inhibit TH17 differentiation
Since RORα or RORγt single deficiency did not completely abrogate TH17 differentiation, we analyzed RORα and RORγ double deficient cells (RORαsg/sg/α−/−) in TH17 differentiation. Naïve T cells were isolated from RORαsg/sg/γ−/−, RORγ−/− and WT mice and differentiated under TH17 conditions in the presence of IL-6 or IL-21 with or without anti-IL-2. In comparison to WT T cells, IL-17-secreting cell numbers were greatly reduced in RORγ−/− cells; however, the generation of IL-17-producing cells was further reduced by more than 5 fold and completely impaired in double deficient cells (Fig. 6A, S4, S5). Using real-time RT-PCR, RORγ deficiency resulted in reduction of IL-17, IL-17F, IL-22 and IL-23R mRNA levels compared to WT, while RORα/α double deficiencies completely abrogated the expression of all these genes (Fig. 6B). Moreover, while IL-21 expression was not affected in T cells lacking RORγ (Fig. 6B), we found that double deficiencies greatly reduced the expression of IL-21 mRNA (Fig. 6B).
Figure 6. RORα and RORγ double deficiencies completely abrogate TH17 differentiation in vitro.

Naïve CD4+CD25−CD62LhiCD44lo T cells from spleens of RORγ−/−, RORα-RORγ double deficient (RORαsg/sg/γ−/− or WT mice were activated with plated-bound anti-CD3 and anti-CD28 under the indicated conditions. (A) IL-17- and IFN-γ-secreting cells were assessed by intracellular staining. Protein levels of IL-17 were measured by ELISA after anti-CD3 restimulation. (B) Expression levels of indicated genes were measured by real-time RT-PCR. The lowest expression levels for each gene were referred as 1.
To further understand the contribution of RORα and RORγ in vivo, we reconstituted Rag1−/− mice with bone marrow cells from WT, RORα−/− or RORαsg/sgγ−/− animals. RORγ−/− hematopoietic stem cells were previously shown to reconstitute lymphoid compartments in Rag-deficient mice (Dzhagalov et al., 2004; Ivanov et al., 2006). Similar to singly defective cells, reconstitution with RORαsg/sg/γ−/− bone marrow cells gave rise to normal CD4 T cell numbers in blood or spleen, while in peripheral and mesenteric lymph nodes, RORαsg/sg/γ−/− exhibited reduced CD4 T numbers compared to WT or RORα/sg/sg chimera but were comparable to RORγ−/− chimeras (Fig. S6 and data not shown). Similar to CD4+ T cells from RORαsg/sg/γ−/− animals, those from Rag1−/− mice with RORαsg/sgγ/−/− lymphocytes also exhibited profound TH17 defects during in vitro differentiation while their induction of Foxp3 expression in response to TGFβ was normal (Fig. S7). CD4+ T cells in lamina propria were previously reported to express IL-17 constitutively, which is partially dependent on RORγ (Ivanov et al., 2006). We found a modest reduction of IL-17-expressing cells in RORαsg/sg chimeras when compared to RORγ−/− chimeras (Fig. 7A). In Rag1-RORαsg/sg/γ−/− animals, the total number of cells recovered from lamina propria was consistently lower than the other groups of chimeric mice (data not shown) and these cells were most profoundly impaired in IL-17 expression (Figure 7A).
Figure 7. RORα and RORγ compound mutations completely inhibit TH17 differentiation in vivo.

(A) Lamina propria cells were isolated from Rag1−/− mice reconstituted with RORαsg/sg, RORγ−/− RORαsg/sg/γ−/− or WT bone marrow cells and IL-17 expression was assessed by intracellular staining. Student t test, *, p<0.05; **, p<0.005. (B) EAE was induced in the indicated Rag1-deficient mice reconstituted with indicated bone marrow cells. WT, n=5; RORγ−/−, n=4; RORαsg/sg/γ−/−, n=5. Max disease, score ≥ 3. *, student t test RORαsg/sg/γ−/− vs WT, p<0.001; RORαsg/sg/γ−/− vs RORγ−/−, p<0.05. Infiltrates in central nerve system or splenocytes from the EAE mice were isolated on day 13 after the 2nd immunization and IL-17- or IFN-γ- expressing cells were measured by intracellular staining. Data shown are on gated CD4+ cells.
Next, we subjected Rag1−/− chimeric mice containing WT, RORγ−/− or RORαsg/sg/−/− lymphocytes to EAE. Using our EAE protocol, the onset and severity of disease were only moderately reduced in Rag1−/− mice containing RORγ−/− lymphocytes (Fig. 7B). However, in sharp contrast, all mice containing doubly deficient cells were completely protected against this disease with no sign of neurological impairments (Fig. 7B). When we examined infiltrating mononuclear cells in the central nervous system and splenic CD4 T cells, RORγ−/− animals had severely reduced but still detectable IL-17+ cells, whereas RORαsg/sg/γ−/− had virtually none (Fig. 7B). IFNγ production was not defective in these animals (Fig. 7B), suggesting that there was a selective defect in TH17 differentiation in the absence of RORγ or both RORα and RORγ. To further confirm the role of RORα/γ in initial differentiation of IL-17-producing TH17 cells in vivo, we immunized Rag1−/− mice constituted with WT and doubly deficient bone marrow cells with KLH in CFA. One week later, spleen cells from immunized mice were analyzed for IL-17 and IFNγ expression. Although CD4+ T cells from Rag1-RORαsg/sg/γ−/− mice exhibited IFNγ expression, IL-17-producing cells was almost completely absent (Fig. S8). Together, compound mutation of both RORα and RORγ resulted in impaired TH17 differentiation in vivo and complete protection against EAE.
Discussion
T cell functional differentiation is regulated not only by cytokines from the environment but also their intrinsic programs. TH17 as a novel lineage of T cells has now been shown to be positively and negatively regulated by many cytokines. However, the genetic programming of their lineage differentiation in response to these cytokines has not been well understood. In the current study, we found that RORα, downstream of STAT3, functions together with RORγ in directing TH17 differentiation and cytokine expression.
RORα, regulated by STAT3, is selectively expressed in TH17 cells. RORα overexpression promotes IL-17 and IL-17F but not IL-22 expression. However, RORα deficiency only selectively impairs IL-17 but not IL-17F expression. Only in the absence of RORγ, RORα accounts for the remaining IL-17F production. RORα thus appears only to be important for IL-17 expression among all TH17 cytokines. The past work has indicated that TH cell cytokine expression is not only determined by master regulators but also regulated by other lineage-specific transcription factors that fine tune specific gene expression. For example, in TH1 cells, Hlx genetically interacts with T-bet to promote TH1 response (Mullen et al., 2002; Zheng et al., 2004). In TH2 cells, c-MAF and JunB selectively regulate IL-4 expression (Kim et al., 1999; Li et al., 1999). We previously found ICOS-c-Maf pathway only controls IL-4 but not IL-5 or IL-10 expression in effector TH2 cells and ICOS deficiency selectively abrogates IL-4- dependent IgE production but not IL-5-mediated airway eosinophilia (Dong et al., 2001; Nurieva et al., 2003). Interestingly, recently we observed different ratios of IL-17 and IL-17F expression in different T cell populations in vitro and in vivo (unpublished data). This suggests differential cytokine expression in differentiated TH17 cells. Whether RORα regulates this differential regulation is unknown at this point. Nor was the biological or pathologic significance of this regulation. In addition to IL-17 regulation, RORα also appears to upregulate IL-23R and downregulate IFNγ and T-bet expression. The molecular basis of this regulation also requires further investigation.
Since RORα or RORγ on their own is sufficient to induce TH17 differentiation under neutral conditions, it is curious why both are co-expressed in TH17 cells. Single ROR over-expression or deficiency did not substantially alter the expression of the other (data not shown), suggesting that these are two parallel but not sequential or inter-dependent pathways induced by STAT3. In support of this idea, RORα expression in RORγ-deficient cells drove TH17 differentiation, indicating that RORα could function independent of RORγ. Our retroviral co-expression has revealed a synergistic function by RORα and RORγ during TH differentiation. The molecular basis for this synergy is unclear at this point. ROR factors do not typically form heterodimeric complexes, which we confirmed via over-expression and co-immunoprecipitation of RORα and RORγt (data not shown). Moreover, we did not observe any synergy of the two factors in activating CNS2-IL-17 reporter expression. Although it is possible that RORα and RORγ synergistically activate some other elements in the locus, our data also strongly suggest a dose-dependent function of RORs in activation of IL-17 production. Low expression levels of RORα or RORγt did not effectively induce IL-17 expression (Fig. S1). Moreover, RORα enhanced IL-17 production in a dose-dependent manner in cells expressing variable concentrations of RORγt (Fig. S3A). These observations from retrovirus overexpression experiments suggest that when RORαt is expressed at physiological concentration, it requires the presence of RORα to induce TH17 differentiation effectively. Alternatively, both receptors are activated by similar but different ligands which can be regulated differentially to control the amount of pro-inflammatory TH17 cells. Co-expression and co-activation of both factors is necessary to counteract the inhibitory mechanisms to reach the threshold levels for TH17 differentiation. In support of this idea, we observe that during polarized differentiation of T cells into TH1, TH2 or iTreg cells, RORs had different capacity of supporting TH17 development-TH2 was the easiest while iTreg was the most difficult. Consistent with these results, we found that TH17 differentiation was strongly inhibited by T-bet and Foxp3 but not by GATA-3 (unpublished results). RORα and RORγ can both be induced to some extent by TGFβ and IL-6; however, this level of expression may not be sufficient to drive TH17 differentiation.
In addition, our results also indicate redundancy of ROR factors. Although RORγ deficiency substantially inhibits TH17 differentiation, TH17 development was not completely abrogated (Ivanov et al., 2006). The combination with RORα mutation further diminished the expression of TH17-specific genes by at least several folds. The redundancy is best seen in terms of IL-21 expression. While single mutation did not affect, the compound mutation greatly inhibited IL-21 production. This redundancy may have a biological significance, as in our EAE model involving two times of immunization, whereas RORγ−/− cell-reconstituted mice only exhibited moderately reduced EAE, in contrast to an earlier report using RORγt knockout mice (Ivanov et al., 2006). Importantly, RORα and RORγ double deficiencies led to complete protection. Since IFNγ production is not diminished in the absence of both ROR factors, our current results, in agreement with our data on IL-21 −/− (Nurieva et al., 2007) and IL-17−/− (our unpublished results) mice, support TH17 cells as pathogenic mediators in this disease. Our data also suggest that simultaneous inhibition of RORα and RORγ would be most beneficial for treatment of autoimmune diseases, although it is not clear at this stage whether they are required for maintaining TH17 programs.
In summary, our study has elucidated the function of RORα, another TH17 lineage-specific transcription factor and its synergy and redundancy with RORγ. This work not only demonstrates genetic interaction between two nuclear receptors in TH17 differentiation but also reveals the complexity in the regulation of TH17 program and function.
Methods
Mice
Heterozygous Staggerer (ROR αsg/+) mice on C57BL/6 background were obtained from Jackson Laboratory and interbred to generate RORαsg/sg mice. Some RORαsg/sg mice die after birth but a few survive to adulthood and used for some experiments. Their CD4+ cells in spleen exhibited normal naïve and regulatory T cell population (data not shown). RORγ−/− mice, described previously (Kurebayashi et al., 2000), were backcrossed 6–7 generations onto C57BL/6 background and their CD4 T cells in spleen were used for some experiments in the study. RORαsg/sg/γ−/− mice were generated by crossing heterozygous RORαsg/+ mice with RORγ−/− and subsequent mating of RORαsg/+/γ−/− mice Kang et al., 2007). Some RORαsg/sg/γ−/− mice survived to adulthood and were used in our studies. Their splenic CD4+ T cells exhibited normal regulatory T cell compartment (data not shown). Littermates were used as WT controls. Stat3 fl and Tie2-Cre mice were bred to yield fl/Δ Cre+ and Cre− littermates as described (Yang et al., 2007), and their spleen and lymph node cells were used for in vitro differentiation. RORαsg/sg, RORγ−/−, RORαsg/sg/γ−/− and WT bone marrow chimeras were generated by reconstitution of 5 × 106 bone marrow cells from above mice into sublethally irradiated (500 rad) Rag1 KO mice on the same background. The animal experiments were performed at the age of 6–10 weeks using protocols approved by Institutional Animal Care and Use Committee.
T cell differentiation
Differentiation of OT-II cells in Figure 1A was performed using Ova peptide, splenic APC from C57BL/6 mice in the absence (for neutral differentiation) or presence of polarizing cytokines (10 µg/ml anti-IL-4 and 2 ng/ml IL-12 for TH1, 10 µg/ml anti-IFNγ and 10 ng/ml IL-4 for TH2, 10 ng/ml IL-6, 5 ng/ml TGFβ, 5 ng/ml IL-23 and anti-IL-4/anti- IFNγ for TH17 differentiation) as previously described (Chung et al., 2006). For naïve T cell differentiation in other experiments, CD4+CD25−CD62LhiCD44lo cells were FACS-sorted as described (Nurieva et al., 2007; Yang et al., 2007). Naïve CD4+ T cells were activated with plate-bound 2 µg/ml anti-CD3 and 2 µg/ml anti-CD28 and in the presence of 50 units/ml IL-2 (in some experiments, exogenous IL-2 was not added or with 10 µg/ml anti-IL2 was added instead of IL-2), 2.5 ng/ml TGF-β (Peprotech), 30 ng/ml IL-6 (Peprotech), 50 ng/ml IL-23 (R&D system), 10 µg/ml anti-IL-4 (11B11), 10 µg/ml anti-IFN-γ (XMG 1.2), 10 ng/ml TNF-α, 10 ng/ml IL-1β or combination of these stimuli. Four to five days after activation, cells were washed and restimulated with PMA and ionomycin in the presence of Golgi-stop for 5 h, after which IL-17- and IFNγ-producing cells were analyzed using intracellular staining. Intracellular staining for Foxp3 was performed by using a Foxp3 staining kit (eBioscience).
Quantitative real-time PCR
Total RNA was prepared from T cells using TriZol reagent (Invitrogen). cDNA were synthesized using Superscript reverse transcriptase and oligo(dT) primers (Invitrogen) and gene expression was examined with a Bio-Rad iCycler Optical System using iQ™ SYBR green real-time PCR kit (Bio-Rad Laboratories, Inc.). The data were normalized to Actb reference. RORα primers were forward, 5′-tctccctgcgctctccgcac, and reverse, 5′-tccacagatcttgcatgga, detecting the predominant isoform 4 expressed in TH17. The primers for IL-17, IL-17F, IL-23R, IL-22, IL-21, RORγt, T-bet, and Actb were previously described (Nurieva et al., 2007; Yang et al., 2007).
Retroviral transduction
RORα (Genbank Acc. XM_903197) and RORγt (Genbank Acc.AJ132394) were cloned into bicistronic retroviral vector pGFP-RV (Ouyang et al., 1998) or pMIG-hCD2 (Deftos et al., 1998) containing IRES-regulated GFP and human CD2, respectively. Naïve CD4+CD25−CD62LhiCD44lo T cells from OT-II mice were FACS-sorted and activated with Ova peptide and irradiated wild-type splenic APCs in the presence or absence of 2.5 ng/ml TGF-β, 30 ng/ml IL-6, IL-4 (Peprotech), IL-12 (Peprotech), 10 µg/ml anti-IL-4, 10 µg/ml anti-IFN-γ or combination of these stimuli as indicated. 24 hours after activation, the cells were infected by retroviruses expressing RORα-GFP or control empty vector (containing only IRES-GFP). 3 days after infection, the cells were restimulated with PMA and ionomycin in the presence of Golgi-stop for 5 h, after which IL-17- and IFNγ-producing cells were analyzed using intracellular staining on a GFP+ gate. Co-infection was performed with two bicistronic retroviruses expressing RORα or empty vector (both have IRES-GFP) and RORγt or control vector (both with IRES-hCD2). The analysis was performed on a GFP+hCD2+ gate.
EAE induction
For the induction of EAE, female bone marrow chimeric mice were immunized with the MOG peptide emulsified in CFA. Mice were immunized subcutaneously at the dorsal flanks with 150 µg of MOG peptide in CFA at day 0 and day 7. Pertussis toxin was given intraperitoneally at day 1 and day 8 with the dosage of 500 ng per mouse. Signs of EAE were assigned scores on a scale of 1–5 as follows: 0, none; 1, limp tail or waddling gait with tail tonicity; 2, wobbly gait; 3, hind limb paralysis; 4, hind limb and forelimb paralysis; 5, death. In Figure 2C–E, disease incidence and max scores from two independent experiments were combined and p values calculated using the student t test by comparing the disease scores. To analyze central nervous system infiltrates, both brain and spinal cord were collected from perfused mice and mononuclear cells were prepared by percoll gradient.
Transcription reporter assay
Expression vectors encoding RORα or RORγ were transfected into EL-4 cells with luciferase constructs containing IL-17 minimal promoter (−1131 to +1 relative to translation start site) with or without CNS2 element (2.3 kb, the cloning primers were 5’-TGTTTGTGGGTGTATGAG and 5’-GAATCCCGTTCTAATGTGAC). The dual-luciferase reporter system (Promega) was used to assay firefly and Renilla luciferase activity in each sample. Renilla luciferase was used to normalize transfection efficiency and luciferase activity.
Chromatin immuno-precipitation (ChIP) assays
ChIP assays were carried out as described (Akimzhanov et al., 2007). In brief, cells were cross-linked using formaldehyde, and after the nuclei were isolated and sonicated, DNA-protein complexes were immunoprecipitated with protein-A sepharose pre-blocked with salmon sperm DNA using anti-acetylated histone H3 Ab (Upstate, Lake Placid, NY; # 06-599), anti-Flag (M2, Sigma) or anti-RORγ (H-190, Santa Cruz Biotechnology, Inc.). After washing, elution and reversion of cross-links, the DNA was isolated and used in radioactive PCR reactions. The primers used for Actb, CNS2a were as described before (Akimzhanov et al., 2007). The primers for IL-17 promoter were, 5′-GCAGCAGCTTCAGATATGTCC-3′ and 5′-GGGGTGACACCATTTGAGTAA-3′.
Electrophoretic Mobility Shift Assays (EMSA)
Nuclear proteins from TH17 cells were prepared and used in EMSAs as described (Klein-Hessling et al., 1996). The following oligonucleotides were used as probes and competitors and to introduce mutations: ROR element (RORE) in IL-17 CNS2 (CNS2), 5′-GAAAGTTTTCTGACCCACTTTAAATCAATTT-3′; CNS2 RORE mutant (mutant): 5′- GAAAGTTTTCTGACACACTTTAAATCAATTT-3′; RORE consensus in Pcp2 promoter (consensus), 5′-GGAGTCCCCTGACCCAGTTACTATAACACA-3′.
Supplementary Material
Acknowledgements
We thank Bruz Marzolf in Institute for Systems Biology for his assistance in microarray analysis, Dr. Ken Murphy for RV-GFP vector, Dr. Steve Reiner for advice on dual retroviral transduction and the Dong lab members for their help. The work is supported by research grants from NIH (AR050772to CD), an Intramural Research Program of the NIEHS, NIH (to AMJ) and MD Anderson Cancer Center (to SSW). RN received a postdoctoral fellowship from the Arthritis Foundation and is a recipient of a Scientist Development Grant from the American Heart Association. BP receives an Odyssey Fellowship of MD Anderson Cancer Center. ADP receives an NIH predoctoral training grant and an American Legion Auxiliary Award. CD and KS are MD Anderson Cancer Center Trust Fellows and CD is a Cancer Research Institute Investigator and an American Lung Association Career Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akimzhanov AM, Yang XO, Dong C. Chromatin remodeling of interleukin-17 (IL-17)-IL-17F cytokine gene locus during inflammatory helper T cell differentiation. J Biol Chem. 2007;282:5969–5972. doi: 10.1074/jbc.C600322200. [DOI] [PubMed] [Google Scholar]
- Batten M, Li J, Yi S, Kljavin NM, Danilenko DM, Lucas S, Lee J, de Sauvage FJ, Ghilardi N. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat Immunol. 2006;7:929–936. doi: 10.1038/ni1375. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Oukka M, Kuchroo VK. TH-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu BM, Tato C, Yoshimura A, Hennighausen L, O'Shea JJ. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci U S A. 2006;103:8137–8142. doi: 10.1073/pnas.0600666103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung Y, Yang X, Chang SH, Ma L, Tian Q, Dong C. Expression and regulation of IL-22 in the IL-17-producing CD4+ T lymphocytes. Cell Res. 2006;16:902–907. doi: 10.1038/sj.cr.7310106. [DOI] [PubMed] [Google Scholar]
- Deftos ML, He YW, Ojala EW, Bevan MJ. Correlating notch signaling with thymocyte maturation. Immunity. 1998;9:777–786. doi: 10.1016/s1074-7613(00)80643-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C. Diversification of T-helper-cell lineages: finding the family root of IL-17-producing cells. Nat Rev Immunol. 2006;6:329–334. doi: 10.1038/nri1807. [DOI] [PubMed] [Google Scholar]
- Dong C, Flavell RA. Cell fate decision: T-helper 1 and 2 subsets in immune responses. Arthritis Res. 2000;2:179–188. doi: 10.1186/ar85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Juedes AE, Temann UA, Shresta S, Allison JP, Ruddle NH, Flavell RA. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature. 2001;409:97–101. doi: 10.1038/35051100. [DOI] [PubMed] [Google Scholar]
- Dzhagalov I, Giguere V, He Y-W. Lymphocyte Development and Function in the Absence of Retinoic Acid-Related Orphan Receptor {alpha} J Immunol. 2004;173:2952–2959. doi: 10.4049/jimmunol.173.5.2952. [DOI] [PubMed] [Google Scholar]
- Glimcher LH, Murphy KM. Lineage commitment in the immune system: the T helper lymphocyte grows up. Genes Dev. 2000;14:1693–1711. [PubMed] [Google Scholar]
- Hamilton BA, Frankel WN, Kerrebrock AW, Hawkins TL, FitzHugh W, Kusumi K, Russell LB, Mueller KL, van Berkel V, Birren BW, et al. Disruption of the nuclear hormone receptor RORalpha in staggerer mice. Nature. 1996;379:736–739. doi: 10.1038/379736a0. [DOI] [PubMed] [Google Scholar]
- Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Jetten AM. Recent advances in the mechanisms of action and physiological functions of the retinoid-related orphan receptors (RORs) Curr Drug Targets Inflamm Allergy. 2004;3:395–412. doi: 10.2174/1568010042634497. [DOI] [PubMed] [Google Scholar]
- Kang HS, Angers M, Beak JY, Wu X, Gimble JM, Wada T, Xie W, Collins JB, Grissom SF, Jetten AM. Physiol. Genom. 2007. Gene expression profiling reveals a regulatory role for RORα and RORγ in Phase I and Phase II Metabolism. In press. [DOI] [PubMed] [Google Scholar]
- Kim JI, Ho IC, Grusby MJ, Glimcher LH. The Transcription Factor c-Maf Controls the Production of Interleukin-4 but Not Other Th2 Cytokines. Immunity. 1999;10:745–751. doi: 10.1016/s1074-7613(00)80073-4. [DOI] [PubMed] [Google Scholar]
- Klein-Hessling S, Schneider G, Heinfling A, Chuvpilo S, Serfling E. HMG I(Y) interferes with the DNA binding of NF-AT factors and the induction of the interleukin 4 promoter in T cells. Proc Natl Acad Sci U S A. 1996;93:15311–15316. doi: 10.1073/pnas.93.26.15311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory TH17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurebayashi S, Ueda E, Sakaue M, Patel DD, Medvedev A, Zhang F, Jetten AM. Retinoid-related orphan receptor gamma (RORgamma) is essential for lymphoid organogenesis and controls apoptosis during thymopoiesis. Proc Natl Acad Sci U S A. 2000;97:10132–10137. doi: 10.1073/pnas.97.18.10132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, et al. Interleukin-2 Signaling via STAT5 Constrains T Helper 17 Cell Generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Li B, Tournier C, Davis RJ, Flavell RA. Regulation of IL-4 expression by the transcription factor JunB during T helper cell differentiation. Embo J. 1999;18:420–432. doi: 10.1093/emboj/18.2.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- Mullen AC, Hutchins AS, High FA, Lee HW, Sykes KJ, Chodosh LA, Reiner SL. Hlx is induced by and genetically interacts with T-bet to promote heritable TH1 gene induction. Nat Immunol. 2002;3:652–658. doi: 10.1038/ni807. [DOI] [PubMed] [Google Scholar]
- Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, Dong C. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- Nurieva RI, Duong J, Kishikawa H, Dianzani U, Rojo JM, Ho I, Flavell RA, Dong C. Transcriptional regulation of th2 differentiation by inducible costimulator. Immunity. 2003;18:801–811. doi: 10.1016/s1074-7613(03)00144-4. [DOI] [PubMed] [Google Scholar]
- Ouyang W, Ranganath SH, Weindel K, Bhattacharya D, Murphy TL, Sha WC, Murphy KM. Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity. 1998;9:745–755. doi: 10.1016/s1074-7613(00)80671-8. [DOI] [PubMed] [Google Scholar]
- Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner SL. Development in motion: helper T cells at work. Cell. 2007;129:33–36. doi: 10.1016/j.cell.2007.03.019. [DOI] [PubMed] [Google Scholar]
- Steinmayr M, Andre E, Conquet F, Rondi-Reig L, Delhaye-Bouchaud N, Auclair N, Daniel H, Crepel F, Mariani J, Sotelo C, Becker-Andre M. staggerer phenotype in retinoid-related orphan receptor alpha-deficient mice. Proc Natl Acad Sci U S A. 1998;95:3960–3965. doi: 10.1073/pnas.95.7.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumhofer JS, Laurence A, Wilson EH, Huang E, Tato CM, Johnson LM, Villarino AV, Huang Q, Yoshimura A, Sehy D, et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol. 2006;7:937–945. doi: 10.1038/ni1376. [DOI] [PubMed] [Google Scholar]
- Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–688. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- Zheng W-p, Zhao Q, Zhao X, Li B, Hubank M, Schatz DG, Flavell RA. Up-Regulation of Hlx in Immature Th Cells Induces IFN-{gamma} Expression. 2004:114–122. doi: 10.4049/jimmunol.172.1.114. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, Ouyang W. Interleukin-22, a TH17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.