

Abstract
β-arrestins have important roles in the regulation of seven-transmembrane receptors (7TMRs). Smoothened (Smo) is a 7TMR that mediates effects of Hedgehog on developmental processes and whose dysregulation may cause tumorigenesis. β-arrestins are required for endocytosis of Smo and signaling to Gli transcription factors. In mammalian cells, Smo-dependent signaling requires translocation to primary cilia. We demonstrate that β-arrestins mediate the activity-dependent interaction of Smo and the kinesin motor protein Kif3A. This multimeric complex localized to primary cilia and was disrupted in cells transfected with β-arrestin siRNA. β-arrestin 1 or β-arrestin 2 depletion prevented localization of Smo to primary cilia and Smo-dependent activation of Gli. These results support roles for β-arrestins in mediating the intracellular transport of a 7TMR to its obligate subcellular location for signaling.
β-arrestins (βarrs) are mediators of 7TMR desensitization and internalization (1, 2), and also function as scaffolding proteins that mediate distinct receptor-activated signaling events (3). The roles of the 7TMR Smoothened (Smo), a component of the Hedgehog (Hh) signaling pathway, have been well-defined in multiple organisms (4), yet regulatory mechanisms of Smo signaling are only now being elucidated. Smo activity is repressed by the twelve-membrane spanning receptor Patched (Ptc). Hh binding to Ptc, relieves repression of Smo, allowing Smo to act as a positive mediator of Hh signaling by activating the Gli family of transcription factors. Hh signaling regulates patterning and cell fate, thus loss of Hh pathway activity causes birth defects in several organisms (5), whereas inappropriate activation of the pathway leads to tumorigenic phenotypes (6). βarrs are important for Smo-mediated signaling and Sonic Hedgehog (Shh) responsiveness in mammalian and zebrafish model systems (7-9). Activation of Smo causes increased association between β-arrestin 2 (βarr2) and Smo in a heterotrimeric guanine nucleotide-binding protein (G-protein) coupled receptor kinase 2 (Grk2)-dependent manner (7). Furthermore, morpholino-mediated silencing of βarr2 recapitulates Shh signaling defects, and causes a lack of Gli responsiveness in zebrafish (8). Smo requires translocation to the primary cilium to function properly (10-12) and to activate Gli (5, 13). We therefore tested whether β-arrestin 1 (βarr1) and βarr2 might function in mediating activity-dependent translocation of Smo to the primary cilium.
We checked for localization of βarrs to the primary cilium. Primary cilia were visualized by confocal microscopy in mouse NIH-3T3 cells (Fig. 1A). βarrs were localized and concentrated in the primary cilium when stained alone, stained together with acetylated tubulin, a common marker for primary cilia (Fig. 1A) or in cells also stained for pericentrin (Fig. 1B), which marks the basal bodies of the primary cilia. We also found that βarrs had no effect on ciliogenesis (Fig. S1). The βarrs join a list of Smo-related signaling molecules that localize to the primary cilium (14, 15).
Fig. 1. Localization of β-arrestins, Smoothened and Kif3A to the primary cilia.
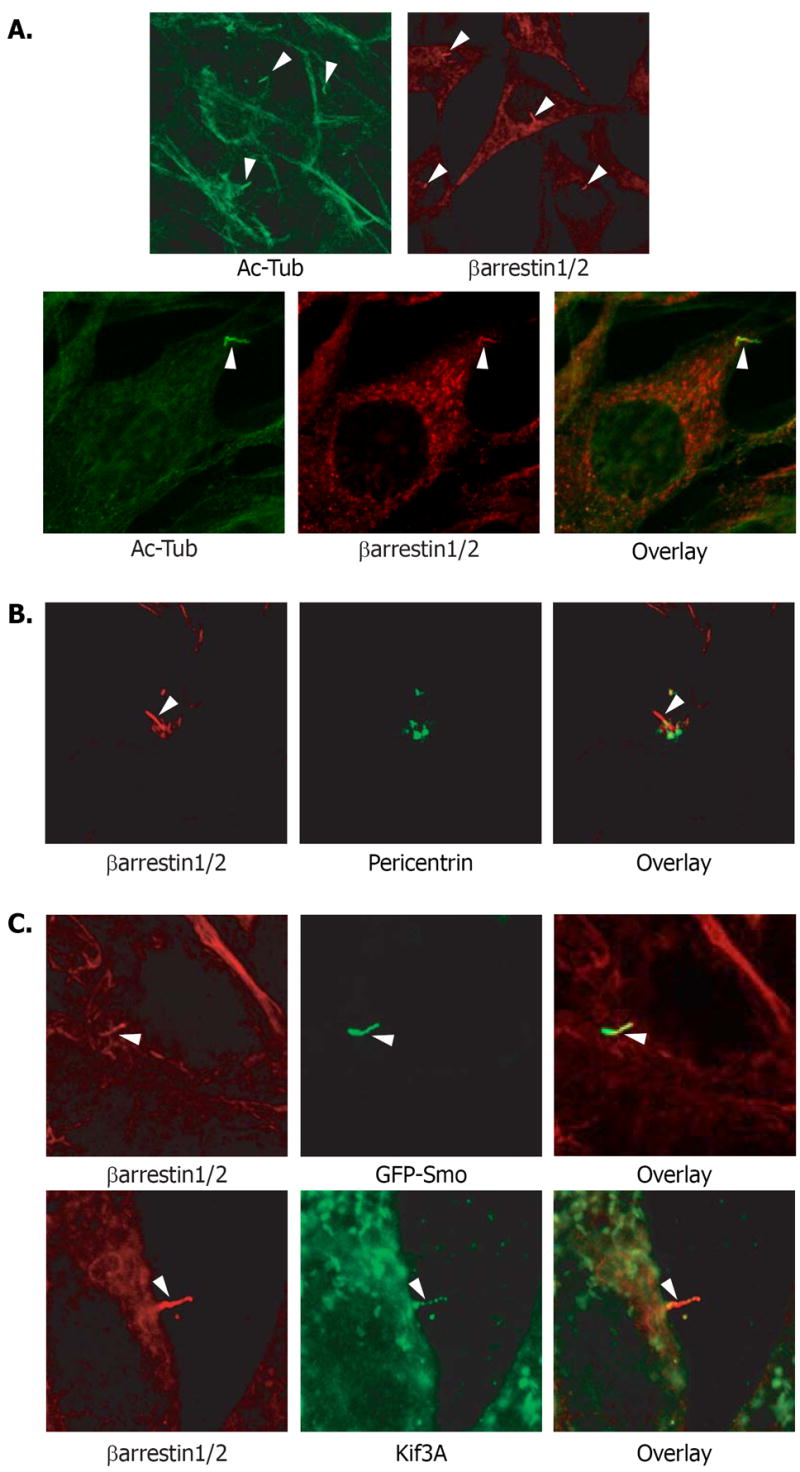
(A) NIH-3T3 cells were immunostained using antibodies to acetylated tubulin or βarr1 and βarr2 (top right panel – mouse monoclonal, bottom middle – rabbit polyclonal), to visualize primary cilia (arrowheads). Colocalization is shown in yellow (overlay). (B) NIH-3T3 cells were stained with βarr antibody (red) and pericentrin antibody (green) which marks the basal bodies of primary cilia. (C) (Top panels) NIH-3T3 cells overexpressing GFP-Smo (green) were immunostained with βarr antibody (red). Colocalization of GFP-Smo and endogenous βarr1 and βarr2 in the primary cilia (arrowhead) can be visualized (yellow). (Bottom panels) NIH-3T3 cells were immunostained antibodies to βarr (red) and Kif3A (green). Colocalization in the primary cilia (arrowhead) can be observed (yellow).
The translocation of Smo to primary cilia is mediated by members of the intraflagellar transport (IFT) complex (16). One IFT family member that has been implicated in Shh-mediated signaling, Kif3A, is a subunit of the kinesin-2 motor complex (14), an anterograde molecular motor that transports protein complexes to and within cilia and flagella (17). Kif3A is essential for Shh-mediated signaling in mammalian systems (5), and we identified Kif3A as a βarr1 binding partner in a proteomics screen (18). To test whether βarrs, Smo, and Kif3A might work in concert we examined cells for localization of these proteins together at the primary cilia. Both βarr1 and βarr2 localize to the primary cilia along with an overexpressed green fluorescent protein (GFP)-Smo fusion protein in NIH-3T3 cells (Fig. 1C). The Smo in these cells was constitutively active as endogenous amounts of the Smo suppressor, Patched (Ptc) are insufficient for inactivation of overexpressed Smo (7). Thus, as expected, GFP-Smo was localized to the cilia without Shh stimulation as were βarr1, βarr2, and Kif3A (Fig. 1C).
To assess the interaction of endogenous βarrs , Smo, and Kif3A, we examined NIH-3T3 cells left untreated or treated with Shh. Stimulation of cells with Shh increased the amount of endogenous Smo and Kif3A that coimmunoprecipitated with βarrs (Fig. 2A). Treatment of cells with cyclopamine prevented this Shh-induced association (Fig. 2A). Thus, activation of Smo in response to Shh-mediated inhibition of Ptc activity causes the formation of a complex between endogenous Smo, βarrs and Kif3A.
Fig. 2. Multimeric complex formation between β-arrestins, Kif3A, and Smoothened.
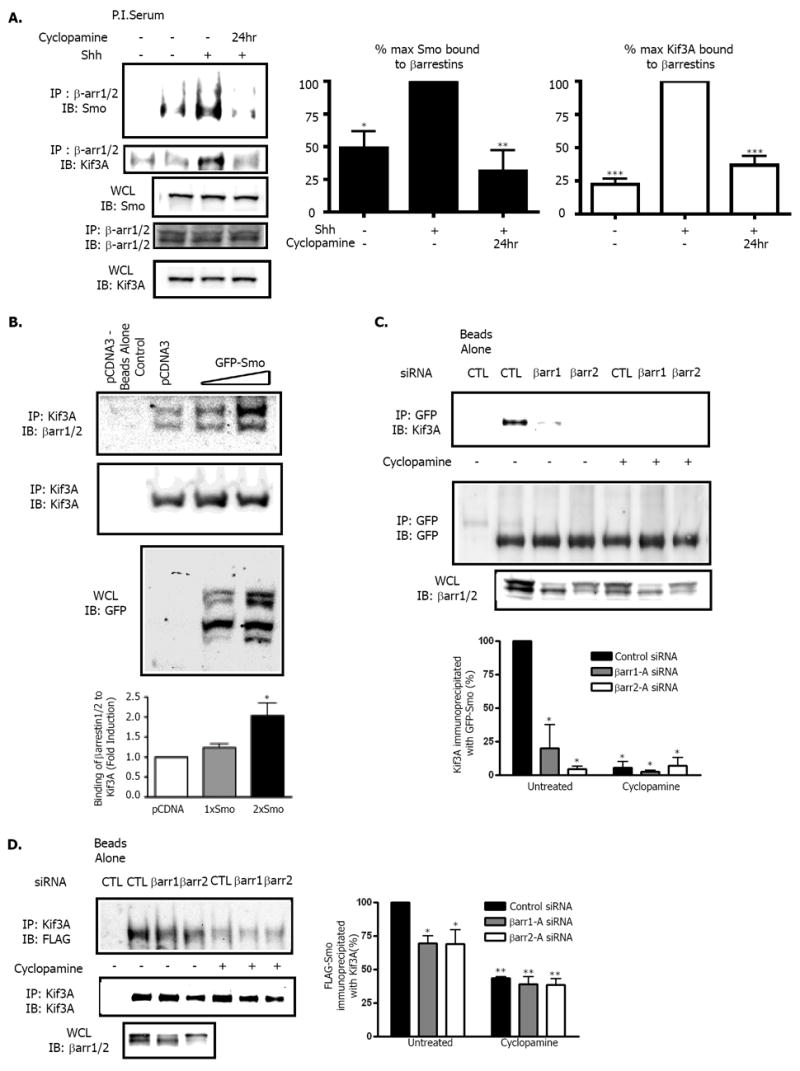
(A) Effects of Shh on association of Smo and Kif3A with βarrs. NIH 3T3 cells treated with Shh-conditioned media +/- cyclopamine were lysed and immunoprecipitated with an antibody that recognizes both βarr1 and βarr2. Samples were subjected to SDS-polyacrylamide gel electrophoresis (PAGE) analysis and immunoblotted for the presence of endogenous Smo or Kif3A. (WCL = whole cell lysate) *p< 0.05, **p<0.005, and ***p<0.001 compared to Shh treated maximum, n=4. (B) Effect of overexpressed Smo on association of Kif3A and βarrs. NIH-3T3 cells were transiently transfected with pCDNA control vector or various amounts of GFP-Smo. Proteins from lysates were immunoprecipitated with Kif3A antibody and immunoblotted for the presence of endogenous βarr or Kif3A. In the βarr blot, the top band is endogenous βarr1, and the lower band is endogenous βarr2. *p< 0.05 compared to pCDNA, n=3. (C) Effect of depletion of βarrs on association of Kif3A with GFP-Smo. NIH-3T3 cells stably overexpressing GFP-Smo were transfected with control (CTL), βarr1 or βarr2 siRNA oligonucleotides. Samples were left untreated (DMSO) or treated with 6μM Cyclopamine for 1 hour. Proteins from lysates were immunoprecipitated with GFP antibody followed by immunoblotting for endogenous Kif3A and GFP-Smo. *p< 0.001 compared to control siRNA untreated sample, n=4. (D) Effect of depletion of βarrs on association of FLAG-Smo with Kif3A. NIH-3T3 cells stably overexpressing FLAG-Smo were transfected with control (CTL), βarr1 or βarr2 siRNA oligonucleotides. Samples were left untreated (DMSO) or treated with 6μM cyclopamine for 1 hour. Cells were lysed and proteins were immunoprecipitated with antibody to Kif3A and immunoblotted for FLAG-Smo and endogenous Kif3A. *p< 0.05, **p< 0.001 when compared to control siRNA, n=3. All p values analyzed by one-way ANOVA with Bonferoni correction. Data are shown as mean +/- SEM.
We next examined the nature of this complex with constiuitively active Smo in the overexpression system used to localize Smo, βarrs, and Kif3A to the primary cilium. Increasing the amount of Smo overexpression led to an increased association between endogenous Kif3A and endogenous βarr1 and βarr2 (Fig. 2B). This increased binding supports the idea that βarrs act as facilitators of Smo binding to Kif3A, thus regulating subsequent Smo translocation to the primary cilia. If this is correct, siRNA-mediated depletion of the βarrs should affect the binding of Smo to Kif3A (Fig. S2 for siRNA sequences). In cells treated with control non-silencing siRNA oligonucleotide, endogenous Kif3A coimmunoprecipitated with GFP-Smo. However, this interaction was lost upon depletion of βarr1 or βarr2 (Fig. 2C). Inhibition of constitutively active GFP-Smo by treatment of cells with cyclopamine also abolished the interaction between endogenous Kif3A and overexpressed Smo (Fig. 2C). Our previous results suggest that cyclopamine treatment inhibits the association of overexpressed βarr2 with Smo (7), and here endogenous βarr1 and βarr2 dissociated from the receptor after cyclopamine treatment (Fig. S3). Furthermore, we examined the amount of FLAG-Smo present in endogenous Kif3A immunoprecipitates under various conditions. In cells treated with a control siRNA oligonucleotide, stably overexpressed FLAG-Smo coimmunoprecipitated with endogenous Kif3A. However, after depletion of βarr1 or βarr2, the interaction between motor and receptor was reduced (Fig. 2D). Similarly, the interaction between Smo and endogenous Kif3A was also abolished by cyclopamine treatment in this context. These results clearly demonstrate that active Smo and Kif3A form a complex in a βarr1 and βarr2–dependent manner.
We interrogated whether the βarr-mediated association of Smo and Kif3A affects localization of Smo to primary cilia. NIH-3T3 cells overexpressing a FLAG-Smo fusion protein were immunostained with antibody to FLAG and examined by confocal microscopy. Fields were scored for the percentage of Smo-containing cilia per total number of cells (Fig. 3A). A significant decrease in the percentage of Smo-containing cilia was seen after depletion of either βarr1, or βarr2 (Fig. 3B). We also observed a significant decrease in Smo-containing primary cilia in cells treated with Kif3A siRNA (Fig. 3B). To ensure that knockdown of βarr1, βarr 2 or Kif3A did not alter cilia formation in this assay, we assessed the total percentage of cilia that contained Smo by staining FLAG-Smo and acetylated tubulin. We then counted the total number of cilia that were marked by acetylated tubulin, and counted the number of those cilia that also contained FLAG-Smo (Fig. 3C). A significant decrease in the percentage of cilia containing Smo was seen after depletion of βarr1 by four different βarr1 siRNA oligonucleotides, after depletion of βarr2 by three different βarr2 siRNA oligonucleotides and after depletion of Kif3A by 3 different Kif3A siRNA olgonucleotides (Fig. 3D). Visualization of cilia with acetylated tubulin antibody demonstrates that functional cilia were formed in these cells, and that Smo was not properly localized after siRNA treatment (Fig. 3C). The use of numerous specific siRNA oligonucleotides also confirms that these are on-target effects. Furthermore, the loss of Smo localization to the primary cilia could be rescued by the overexpression of constructs containing silent mutations that overcame the depletion of endogenous βarr1 and βarr 2 (Fig. 3E and F). Using these rescue constructs, we counted the number of transfected cells in which overexpressed Smo could be detected in cilia (Fig. 3E). Cells transfected with either of the rescue constructs showed similar percentages of Smo-containing cilia as control cells (Fig. 3G). Thus, βarr1, βarr2 and Kif3A appear to be required for localization of Smo to the primary cilia.
Fig. 3. Disrupted localization of Smo after siRNA mediated depletion of β – arrestin 1, 2, or Kif3A.
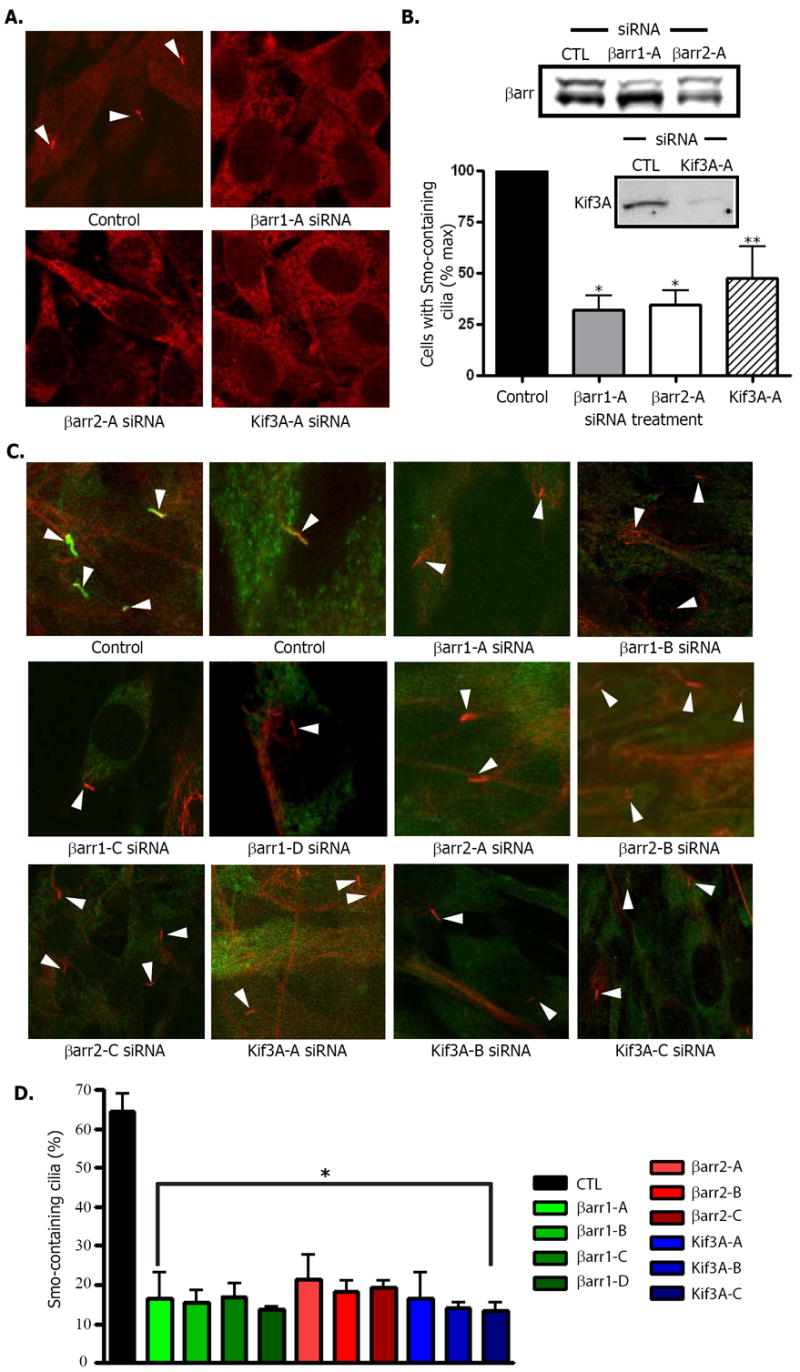
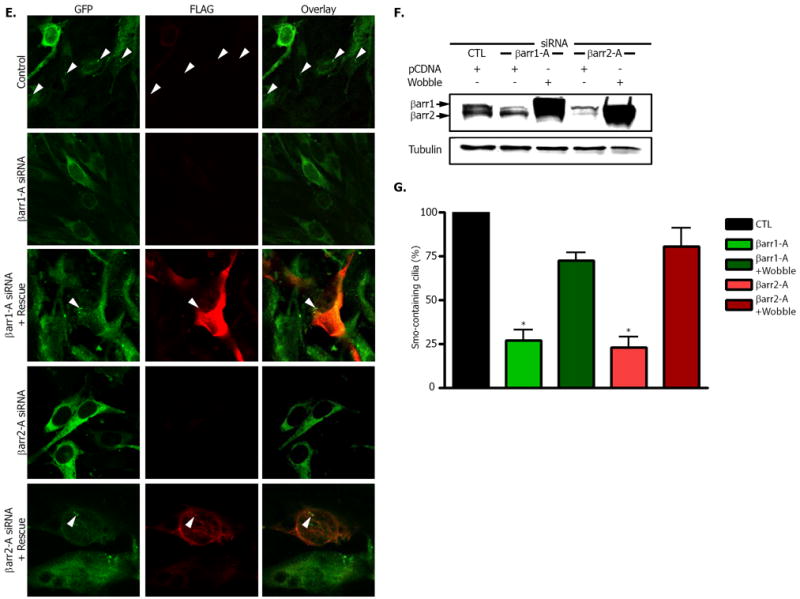
(A) Smo was visualized with FLAG antibody in NIH-3T3 cells overexpressing FLAG-Smo and transfected with control (CTL), βarr1 (βarr1-A) or βarr2 (βarr2-A) siRNA oligonucleotides. Representative fields were examined for each and the total number of cilia (arrowheads) was counted along with total number of nuclei to delineate total cell number. (B) The percentage of cells that had FLAG-Smo positive cilia was calculated and normalized to control samples. * p < .001, **p < 0.01 vs. control, n=4. Inset: Western blot of representative samples for βarr1 (top band) and βarr2 (bottom band). (C) Samples were prepared as in (A) with multiple siRNA oligos for each protein (βarr1-A - D, βarr2-A - C, and Kif3A-A - C) as indicated, and immunostained with both antibody to FLAG (green) and acetylated tubulin (red). (D) Percentage of total cilia in which Smo was detected. FLAG-Smo containing cilia and acetylated tubulin-stained cilia were counted. *p < .001 vs. control, n=3. (E and F) Effects of rescue constructs containing silent mutations for the region targeted by the indicated siRNA. Cells were stained for GFP-Smo (green) or FLAG (red) to indicate expression of the rescue construct (E) with βarr1 or βarr2 siRNA as indicated. After SDS-PAGE analysis, membranes were blotted for the presence of βarr1 and βarr2 (F). (G) Percentage of cells with Smo–containing cilia. For rescued samples, one hundred cells in which FLAG expression was detected were counted and examined for the presence of Smo in cilia. *p < .001 vs. control, n=3. All p values analyzed by one-way ANOVA with Bonferoni correction. Data are shown as mean +/- SEM.
Next we examined if the altered localization of Smo to primary cilia is correlated with a change in Gli activity. We monitored Shh-mediated gene induction in NIH-3T3 cells that endogenously expressed Patched (Ptc), Smo, βarr1 and βarr2 and Kif3A. βarr1, βarr2 or Kif3A were depleted, cells were then treated with Shh for 18 hours, and Gli-reporter activity was assayed. Silencing of βarr1 or βarr2 through use of multiple oligonucleotides caused a complete loss of Shh-mediated Gli reporter activation (Fig. 4A). Several siRNA oligos to Kif3A showed a similar effect (Fig. 4B). Gli-reporter activity could be rescued in this system by overexpression of β-arrestin mutants not subject to siRNA mediated silencing (Fig. 4C). The rescue constructs increased basal activity in the Gli-reporter system (Fig. 4C), but the total change in Gli activity in response to Shh in such cells was similar to that observed in control samples (Fig. 4D). Thus, βarrs appear to be required for efficient downstream signaling through Gli in these cells, probably due to their effects on localization of Smo to the primary cilium.
Figure 4. Inhibition of Shh signaling after siRNA-mediated depletion of β-arrestin 1, β-arrestin 2 or Kif3A.
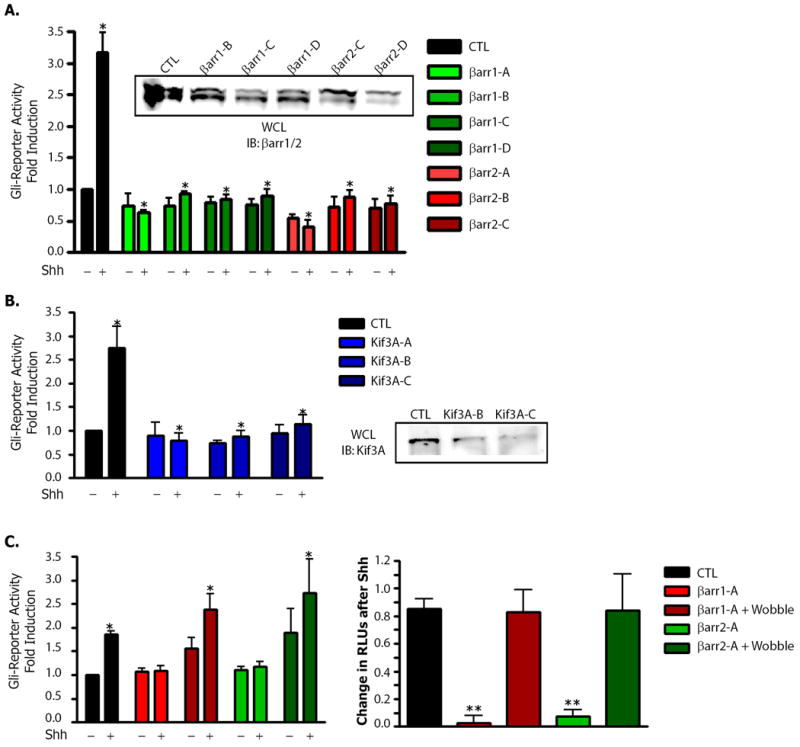
(A) Gli-luciferase reporter activity was assayed in NIH-3T3 cells transfected with either control (CTL), βarr1 (βarr1-A - D) or βarr2 (βarr2-A - C) siRNA oligonucleotides, a Gli-luciferase reporter and a firefly renilla plasmid. Samples were plated, grown and then treated with mock- or Shh-containing medium. Dual luciferase assay data were pooled from 4 separate experiments with each sample repeated in triplicate. *p < 0.001 vs. control / Shh treated, n=5. Inset: western blot for βarr knockdown. (B) Effect of depletion of Kif3A in NIH-3T3 cells transfected with either control (CTL), or Kif3A (Kif3A-A - C) siRNA oligonucleotides, a Gli-luciferase reporter and a firefly renilla plasmid. Lysates were examined as above. *p < 0.001 vs. control / Shh treated, n=3. Inset: Western blot for Kif3A knockdown. (C) Rescue of siRNA treated NIH-3T3 cells transfected with either control (CTL), βarr1 (βarr1-A) or βarr2 (βarr2-A) siRNA oligonucleotides alone or with the corresponding rescue construct, a Gli-luciferase reporter and a firefly renilla plasmid as an internal control. Samples were plated, grown and then treated with mock- or Shh-containing media. For the left panel data were plotted as in (A) and (B). *p < 0.05 vs. control / untreated as analyzed by one-way ANOVA with Bonferoni correction, n=4. For the right panel, values in each individual experiment were analyzed for the change in RLUs after Shh treatment as a relative measure of induction over basal for the indicated condition. **p < 0.001 vs. control / Shh treated, n=4. All p analyzed by one-way ANOVA with Bonferoni correction. Data are shown as mean +/- SEM.
Our findings indicate that together β-arrestins and Kif3A may mediate the transport of Smo to the primary cilium, where it must reside to activate Gli transcription factors. While Kif3A functions downstream of Ptc in response to Shh stimulation, it may both positively and negatively regulate Smo activity (19, 20). Interestingly, Kif3A also mediates the transport of another ciliary protein, von-Hippel Lindau (21), and is responsible for transporting visual arrestin between the inner and outer segments of the mammalian photoreceptor, which is a specialized and modified primary cilium (22). Furthermore, in addition to Smo, several other 7TMRs localize to the primary cilium including the 5-HT(6) serotonin receptor (23), the somatostatin receptor (sst(3)) (24) and angiotensin II receptor (25). Consequently, it seems plausible that β-arrestins may regulate localization of 7TMRs and other signaling proteins to the primary cilia, and function at the primary cilia to regulate signaling networks.
Supplementary Material
Acknowledgments
We thank S. Dewire, J. Violin, S. Ahn, and E. Reiter for critical commentary, J. Reiter for scientific discussions, C. Hubbert and A. Guardiola for technical assistance, and D. Addison, E. Hall and D. Sawyer for administrative assistance. Supported in part by National Institutes of Health grants HL16037 and HL70631. Additional funding was provided by NIH grant 5RO1 CA113656-02. J.J.K. is supported by NIH grant 5T32 AI007217-25. W.C. is a V Foundation Scholar. R.J.L. is an Investigator with the Howard Hughes Medical Institute.
References
- 1.Lefkowitz RJ, Whalen EJ. Curr Opin Cell Biol. 2004 Apr;16:162. doi: 10.1016/j.ceb.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 2.Shenoy SK, Lefkowitz RJ. J Biol Chem. 2003 Apr 18;278:14498. doi: 10.1074/jbc.M209626200. [DOI] [PubMed] [Google Scholar]
- 3.Lefkowitz RJ, Rajagopal K, Whalen EJ. Mol Cell. 2006 Dec 8;24:643. doi: 10.1016/j.molcel.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, McMahon AP, Allen BL. Curr Opin Cell Biol. 2007 Apr;19:159. doi: 10.1016/j.ceb.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Huangfu D, Anderson KV. Development. 2006 Jan;133:3. doi: 10.1242/dev.02169. [DOI] [PubMed] [Google Scholar]
- 6.Evangelista M, Tian H, de Sauvage FJ. Clin Cancer Res. 2006 Oct 15;12:5924. doi: 10.1158/1078-0432.CCR-06-1736. [DOI] [PubMed] [Google Scholar]
- 7.Chen W, et al. Science. 2004 Dec 24;306:2257. doi: 10.1126/science.1104135. [DOI] [PubMed] [Google Scholar]
- 8.Meloni AR, et al. Mol Cell Biol. 2006 Oct;26:7550. doi: 10.1128/MCB.00546-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilbanks AM, et al. Science. 2004 Dec 24;306:2264. doi: 10.1126/science.1104193. [DOI] [PubMed] [Google Scholar]
- 10.Corbit KC, et al. Nature. 2005 Oct 13;437:1018. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- 11.Huangfu D, Anderson KV. Proc Natl Acad Sci U S A. 2005 Aug 9;102:11325. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Satir P, Christensen ST. Annu Rev Physiol. 2007;69:377. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- 13.Haycraft CJ, et al. PLoS Genet. 2005 Oct;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huangfu D, et al. Nature. 2003 Nov 6;426:83. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- 15.Rohatgi R, Milenkovic L, Scott MP. Science. 2007 Jul 20;317:372. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- 16.May SR, et al. Dev Biol. 2005 Nov 15;287:378. doi: 10.1016/j.ydbio.2005.08.050. [DOI] [PubMed] [Google Scholar]
- 17.Hirokawa N. Traffic. 2000 Jan;1:29. doi: 10.1034/j.1600-0854.2000.010105.x. [DOI] [PubMed] [Google Scholar]
- 18.Xiao K, et al. Proc Natl Acad Sci U S A. 2007 Jul 17;104:12011. doi: 10.1073/pnas.0704849104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kolpakova-Hart E, Jinnin M, Hou B, Fukai N, Olsen BR. Dev Biol. 2007 Sep 15;309:273. doi: 10.1016/j.ydbio.2007.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koyama E, et al. Development. 2007 Jun;134:2159. doi: 10.1242/dev.001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mans DA, et al. Exp Cell Res. 2008 Apr 1;314:1229. doi: 10.1016/j.yexcr.2007.12.020. [DOI] [PubMed] [Google Scholar]
- 22.Marszalek JR, et al. Cell. 2000 Jul 21;102:175. doi: 10.1016/s0092-8674(00)00023-4. [DOI] [PubMed] [Google Scholar]
- 23.Brailov I, et al. Brain Res. 2000 Jul 28;872:271. doi: 10.1016/s0006-8993(00)02519-1. [DOI] [PubMed] [Google Scholar]
- 24.Handel M, et al. Neuroscience. 1999 Mar;89:909. doi: 10.1016/s0306-4522(98)00354-6. [DOI] [PubMed] [Google Scholar]
- 25.Woost PG, et al. In Vitro Cell Dev Biol Anim. 2006 Jul-Aug;42:189. doi: 10.1290/0511076.1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.