

Abstract
Aβ42 peptide aggregation and deposition is an important component of the neuropathology of Alzheimer’s disease (AD). Gene-gun mediated gene vaccination targeting Aβ42 is a potential method to prevent and treat AD. APPswe/PS1ΔE9 transgenic (Tg) mice were immunized with an Aβ42 gene construct delivered by the gene gun. The vaccinated mice developed Th2 antibodies (IgG1) against Aβ42. The Aβ42 levels in brain were decreased by 41% and increased in plasma 43% in the vaccinated compared with control mice as assessed by ELISA analysis. Aβ42 plaque deposits in cerebral cortex and hippocampus were reduced by 51% and 52%, respectively, as shown by quantitative immunolabeling. Glial cell activation was also significantly attenuated in vaccinated compared with control mice. One rhesus monkey was vaccinated and developed anti-Aβ42 antibody. These new findings advance significantly our knowledge that gene-gun mediated Aβ42 gene immunization effectively induces a Th2 immune response and reduces the Aβ42 levels in brain in APPswe/PS1ΔE9 mice. Aβ42 gene vaccination may be safe and efficient immunotherapy for AD.
Keywords: AD Alzheimer’s disease, Abeta- Amyloid beta, APPswe/PS1deltaE9- Amyloid precursor protein(Swedish mutation)/presenilin 1 deltaE9 mutation, ELISPOT- Enzyme-linked immunosorbent spot, nm-nanometer, SPSS-Statistical Package for the Social Sciences
1. Introduction
Alzheimer’s disease (AD) is the most common cause of dementia and its pathogenesis has been associated with the accumulation, aggregation and deposition of amyloid beta (Aβ) peptides in cerebral cortex, hippocampus and other subcortical structures [1,2]. Aβ is derived from a larger beta-amyloid precursor protein [3,4] (APP) that is preferentially expressed in higher levels in central nervous system [5,6]. The aggregated form of Aβ42 has been identified as a major component of senile plaques of AD brain [7–9], and thus, a major target of therapy for AD. Aβ42 peptide vaccination has been shown to reduce the amyloid burden in brain and improve the cognitive function in transgenic mouse models as well as in AD patients. However, Aβ42 peptide vaccination was discontinued because of the occurrence of meningoencephalitis in 6% of immunized patients [10–13]. We are exploring genetic immunization as a method to treat or prevent Alzheimer’s disease.
We have previously demonstrated that prophylactic gene-gun mediated Aβ42 gene vaccination can break tolerance against mouse Aβ42 peptide in wild type mice and by efficiently producing anti-Aβ42 antibody to human Aβ42 peptide in APPswe/PS1ΔE9 transgenic mice with Th2 polarized antibody production [14]. We have also demonstrated that Aβ42 gene vaccination can efficiently prevent Aβ42 plaque formation in APPswe/PS1ΔE9 transgenic mice [15].
We advance and extend our knowledge significantly by demonstrating here in APPswe/PS1ΔE9 transgenic mice that gene-gun mediated Aβ42 gene vaccination can efficiently elicit Th2 biased anti-Aβ42 antibodies and the Aβ42 deposition in treated mouse brain is significantly reduced. It is demonstrated further that glial cell activation is also significantly reduced by vaccination. We show that gene-gun mediated Aβ42 gene vaccination can also efficiently induce a IgG1 (Th2) anti-Aβ42 antibody response in a monkey against Aβ42 peptide. The development of an Aβ42 gene vaccine for AD is supported by these new findings.
2. Results
2.1. Aβ42 constructs
As previously reported [14,15], the Aβ42 gene was cloned into a genetic immunization plasmid vector under the control of an synthetic mammalian cell-specific promoter named SP72 and fused upstream with a leader signal of adenovirus E3 gene (E3L) and downstream with the endosome targeting sequence in frame with the Aβ42 gene sequence (pSP72-E3L-Aβ42-ET) (Fig. 1). The construct was sequenced to confirm if the insert was in the correct open reading frame. We have previously demonstrated that SP72 with the E3 leader and endosome targeting sequences was the most efficient plasmid vector in eliciting an Aβ42 immune response. Therefore, in the present study, we used this plasmid DNA vector as the Aβ42 vaccine carrier for immunization of APPswe/PS1ΔE9 transgenic mice delivered by the gene-gun method.
Fig. 1.

Gene vaccine vector SP72-E3L-Aβ42-ET. DNA sequence encoding E3 leader, Aβ42 and endosome targeting gene were cloned in frame in EcoRI and XbaI restriction sites under the control of SP72 mammalian promoter gene. AMPr, ampicillin resistant gene. ColE1 Ori, E. coli DNA replication origin. PolyA, polyadenylation signal sequence.
2.2. Gene vaccine elicits Th2 immune response in APPswe/PS1ΔE9 transgenic AD mice
On the basis of our previous study, we used the pSP72-E3L-Aβ42-ET construct as the vaccine carrier for gene immunization in APPswe/PS1ΔE9 transgenic AD mice. These mice begin to develop amyloid plaques at 6 months of age. Twelve mice were equally divided into a control group which was transfected using the gene gun with a control plasmid pSP72-luc and a treated group vaccinated with the pSP72-E3L-Aβ42-ET beginning at 3 months of age. The humoral immune response was detected with the ELISA method after 4 vaccinations within 2 months. Fig. 2A shows the ELISA titration of anti-Aβ42 antibodies in mouse serum after 15 vaccinations with the final serum obtained at 15 months of age. The average antibody titer against Aβ42 was 1:10,000 in six treated Tg mice while the anti-Aβ42 antibody in 6 control mice was at a background level. Western blot analyses showed that all sera samples of the six vaccinated mice (sera taken from 15 month old mice after 15 vaccinations) recognized the N-terminal (Aβ1–16), middle part (Aβ17–28) and C-terminal (Aβ29–42) epitopes with slightly more reactivity against N-terminal (1.5×106 pixels, 35%) and C-Terminal (1.6×106 pixels, 36%) epitopes than the middle (1.1×106 pixels, 27%) epitopes (Fig. 2B). The additional band above the GST-Aβ29–42 peptide (about 50 kDa) might be a dimer of the peptide or coexist with a tightly associated protein.
Fig. 2.

Immune response against human Aβ42 in APPswe/PS1ΔE9 mice immunized with Aβ42 gene vaccine. A: Anti-Aβ42 antibody titers in Tg mice assayed by ELISA. The sera were from AD Tg mice 15 months old (N=6) and the titer of the antibody was measured against the GST fused Aβ42 peptide. A titer of 1:10,000 was obtained. B: The same serum (1:2000) tested by western blot analysis recognizes epitopes of Aβ42 in treated mice. The sera from control mice are negative by western blot. Lanes 1, 2 and 3 were loaded with GST fused to Aβ1–16, Aβ17–28 or Aβ29–42, respectively. Serum was then added from one control or treated mouse. All other control and treated mice showed similar results.
ELISA isotyping showed that anti-Aβ1–42 antibodies in the sera of vaccinated Tg mice were predominantly IgG1 type. The level of IgG2a was undetectable with ELISA except in one treated (1/6) mouse in which that IgG2a antibody was clearly detectable with 40% level of IgG1 antibody (Fig. 3A). The production of IgG1 type antibody is an indirect measure of the relative contribution of Th2-type cytokines, whereas IgG2a antibodies reflect the contribution of Th1 cytokines to the immune response. Thus, the data in antibody isotyping in the present study indicated that gene-gun mediated Aβ42 gene vaccination predominantly elicits a Th2-polarized immune response in APPswe/PS1ΔE9 transgenic AD mice.
Fig. 3.

Immune responses against Aβ42 in APPswe/PS1ΔE9 mice immunized with the human Aβ42 gene vaccine. A: Isotyping of anti-Aβ42 antibodies after 15 immunizations with (pSP72-E3L-Aβ42-ET). The sera were diluted 1:200 for detection of IgG1, IgG2a subclasses of anti-Aβ42 antibodies. All vaccinated mice exhibited high levels of IgG1 antibody IgG1: t(5.00)=−6.33 p=0.001; IgG2a: t(5.02)=−1.43, p=0.212. (N=6). B and C: ELISPOT assays for IFNγ (B) and for IL4 (C). ANOVA for Fig. 3B: The average number of IFNγ cells was significantly higher in the presence versus of the absence of Aβ42 peptide (32.54+6.96 versus 17.33+3.60, [F(1,10)=69.61, p<0.001]). ANOVA for Fig. 3C: The combination of group (vaccinated versus control) and Aβ42 peptide (present versus absence) was significant [F(1,10)=19.76, p<0.001] with the present Aβ42 peptide-vaccinated group significantly higher than the other 3 combinations. The average change in the number of IL4 T cells from Aβ42 peptide present to absent changed more than 27 in the vaccinated group compared to a change in only 6.5 in the control group. In addition, the average number of IL4 T cells was significantly higher in the presence of Aβ42 peptide (45.0+23.03) compared to the absence of Aβ42 peptide (28.17+14.52; (F(1,10)=52.45, p<0.001).
Consistent with the antibody isotyping data, enzyme-linked immunospot assay (ELISPOT assays) demonstrated that the vaccinated Tg mice exhibited insignificant cellular immune responses compared to control mice as tested by interferon gamma (IFNγ) release in spleen T cells stimulated with Aβ9–18, Aβ1–40 and Aβ1–42 synthetic peptides (n=6, p=0.59). Both control and vaccinated Tg mice also exhibited similar numbers of IFNγ positive T cells in the absence of Aβ42 peptide (n=6, p=0.28) (Fig. 3B). We further tested IL4 positive T cells with the ELISPOT method and showed that vaccinated mice have a significantly higher number of IL4 positive T cells in the presence of Aβ9–18, Aβ1–40 and Aβ1–42 peptide with in vitro cultures compared to the control mice (n=6, p<0.05). There was no significant increase of the IL4 positive T cells in control Tg mice in the presence of Aβ9–18, Aβ1–40 and Aβ1–42 peptide with in vitro cultures compared to absence of Aβ peptides (Fig. 3C). However, in treated mice, a higher number of IL4 positive cells were observed in the presence of Aβ peptide compared to the absence of Aβ peptide with in vitro cultures of the spleen T cells, indicating IL4 positive cells are Aβ1–42 peptide specific. (n=6, p<0.05).
2.3. Aβ42 levels were reduced in brain but increased in plasma in vaccinated Tg mice as measured by ELISA
After 15 Aβ42 gene vaccinations at 15 months of age, brain tissue and plasma of Tg mice were extracted with quinidine–tris buffer and Aβ42 levels were measured by ELISA analysis using the Aβ42 ELISA kit (BioSource, California). In control mice, the median level of Aβ42 in cerebral cortex was 910 ng per gram wet weight tissue. In contrast, Aβ42 gene vaccinated mice had 41% less Aβ42, 533 ng per gram wet weight tissue, than the control group in cerebral cortical homogenates (n=6, t(6.73) = 3.98, p=0.006) (Fig. 4A). The heparinized plasma was similarly extracted with quinidine–tris buffer and subjected to ELISA Aβ42 analysis and showed a 43% increase of plasma Aβ42 in the vaccinated mice (9.0 ng/ml) than the control mice (5.1 ng/ml) (n=6, t(8)=−3.15, p=0.014), t-test results, (Fig. 4B).
Fig. 4.

Levels of Aβ42 peptide in forebrain (A) and in plasma (B) of APPswe/PS1ΔE9 mice 15 months of age treated with the Aβ42 gene vaccine (T) (n=6) or control plasmid (C) (n=6). (A) The forebrain was extracted with 5 M quinidine and Aβ42 was quantified by the sandwich ELISA kit. There is a 41% reduction of total Aβ42 in forebrain of vaccinated mice compared to the controls (t(6.73)=3.98, p=0.006). (B) Similarly plasma samples were extracted with quinidine–tris buffer and Aβ42 levels were measured by sandwich ELISA. The Aβ42 levels in plasma increased 43% in vaccinated mice compared to controls (t(8)=−3.15, p=0.014).
2.4. Brain Aβ42 plaques are significantly decreased in vaccinated mice as demonstrated by immunolabeling
To evaluate the Aβ42 burden in vaccinated and control Tg mouse brain, we performed fluorescence immunolabeling for Aβ42. The paraffin sections of the brain were subjected to Aβ42-specific immunolabeling with antibody A1976 (Sigma). Aβ42 deposits were stained in both cortical and hippocampal regions in Tg mice 15 months of age in both the treated and control groups. To quantitatively measure the brain Aβ42 burden in these mice, five imaging areas at 10× magnification were measured in cortical and hippocampal regions in each mouse (representative image areas from 3 sections in cerebral cortex and representative image areas from 2 sections in hippocampus for each mouse). The fluorescence labeled plaque areas and intensity were quantitatively and automatically measured by Image J (NIH) software. The plaque areas and mean plaque intensity were registered in pixels. The density measured in density units was given by accumulated plaque area multiplied by average plaque intensity. The size of the plaques of all six vaccinated mice was smaller and the number of the plaques was significantly less than the control mice. The Aβ42 plaque burden in cortex was calculated as 8.6% (6.3–10.9%) in control mice (n=6) and 4.4% (3.2–6.2%) in vaccinated mice (n=6), and in hippocampus, the Aβ42 burden is 4.6% (3.8–5.6%) in control mice and 2.4% (2.3–2.7%) in vaccinated mice. The percentages represent plaque area to the total area measured by the Image J (NIH) software. By multiplying total plaque area with mean fluorescence intensity, the total density (density units) in the measured area was obtained. The mean brain plaque density in cerebral cortex of control mice was 4.9±0.9×106 (3.4–6.0×106) pixels (density units), while in treated mice it was 2.4±0.5×106 (2.0–3.3×106) pixels (density units). The reduction was calculated as 51% in cortex according to the density units in treated mice compared to the controls (t(10)=5.07, p<0.001). In hippocampus, 2.5±0.4×106 pixels (density units) were obtained in the control group and 1.2±0.2×106 pixels (density units) in the treated group, a 52% reduction (t(10)=7.77, p<0.001). Fig. 5A shows a low magnification (10×) of representative images of all six control and treated mice of cerebral cortex and hippocampus. Fig. 6 shows the Aβ42 plaque burden and the total density comparison between control and treated mice.
Fig. 5.
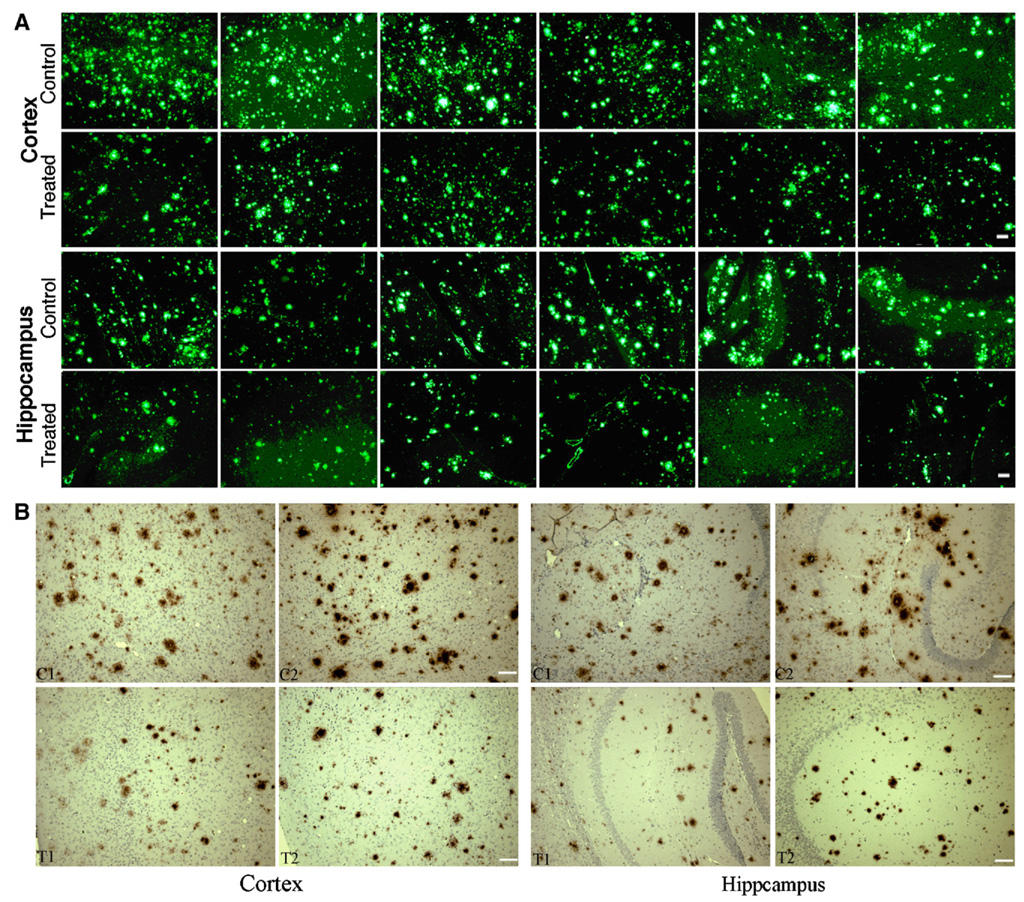
Aβ42 deposition in frontal cortex and in hippocampus of APPswe/PS1ΔE9 mice at 15 months of age immunized with the Aβ42 gene vaccine. Paraffin-embedded sections were labeled with anti-Aβ42 antibody followed by Alexa488-labeled second antibody (A). Shown are representative images at 10× magnification (6 control and 6 treated mice) of cerebral cortex and hippocampus. Plaques of increased size and number are seen in control compared to the treated mice. Amyloid angiopathy was a minor component of the total immunofluorescence and was seen in both control and treated brains. B, Representative sections for amyloid plaques using an immunohistochemical staining method from brains of Aβ42 gene vaccinated and control mice (two treated, T1, T2, and two control, C1, C2). Similar results can be seen as compared with the immunofluorescence method in both cortex and hippocampus. Scale bar=100 µm.
Fig. 6.

Aβ42 plaque burden (A and B, obtained by total plaque area in total measured area) and total density (C and D, obtained by total plaque area multiplied by mean intensity). (A) Aβ42 burden in cortex was 8.6% (6.3–10.9%) in control and 4.4% (3.2–6.2%) in vaccinated mice. (B) Aβ42 burden in hippocampus was 4.6% (3.8–5.6%) in control and 2.4% (2.3–2.7%) in vaccinated mice. (C) The total density in cortex was 4.9±0.9×106 pixels (density units) in control and 2.4±0.5×106 pixels (density units) in treated mice with a 51% plaque reduction (t(10)=5.02, p=0.001). (D) In hippocampus, a 52% plaque reduction was obtained with total density in control of 2.5±0.4×106 pixels (density units) and 1.2±0.2×106 pixels (density units) in treated mice (t(10)=7.77, p<0.001).
We also performed the immunohistochemical staining in paraffin-embedded brain sections using polyclonal anti-Aβ42 antibody (A1916, Sigma, St. Louis, MO). We observed similar results as with immunofluorescent labeling, in term of Aβ42 plaque numbers, plaque size and staining intensity in the hippocampus and frontal cortex (Fig. 5B). Compared with non-vaccinated APP/PS1 mice, Aβ1–42-stained plaques were shown to be smaller in size and less in numbers. Lightly stained plaques were more often seen in many brain areas of vaccinated mice.
2.5. Reduced glial fibrillary acidic protein staining in vaccinated mice
Glial fibrillary acidic protein (GFAP) is an intermediate-filament protein that is highly specific for cells of astroglial lineage and its increased expression has been associated with AD. We performed fluorescence immunolabeling for GFAP in paraffin sections of AD Tg mice. In cerebral cortex (representative image areas of 3 sections per mouse) and hippocampus ( representative image areas of 2 sections per mouse), intracellular GFAP positive staining was seen in glial cells. We quantitatively measured the GFAP staining burden and staining intensity. The control mice showed significantly higher levels of GFAP specific immunostaining in both cortex and in hippocampus compared to the treated mice. We quantitatively measured GFAP staining and the total density of the staining by Image J (NIH) software in representative areas with low magnification microscopy (10×). The cortical GFAP staining in control mice was 11.4% and in treated mice, it was 5.0%. The mean density in cortical in control mice (n=6) was 6.1±2.2×106 pixels (density units) per image area while in treated mice (n=6) it was 2.2±0.9×106 pixels (density units) (t(6.23)=3.435, p=0.013) (Fig. 7C). There is a 64% reduction in cortical levels of GFAP staining in gene-vaccinated mice compared to control mice (Fig. 7A–C). In hippocampus, GFAP staining in control mice was 14.2% and in vaccinated mice, it was 8.8%. The GFAP density of hippocampus in control mice (n=6) was 7.3±3.4×106 pixels (density units) and in treated mice (n=6) it was 3.6±1.9 × 106 pixels (density units) with a reduction of 51% in vaccinated mice (t(10) = 2.309, p=0.0444) (Fig. 7D–F). Immunohistochemical staining of the paraffin sections also showed similar result as with immunofluorescence method (Fig. 7G). Area covered by GFAP-positive astrocytes and the respective integral staining intensities were reduced in hippocampus and frontal cortex by Aβ42 gene vaccine treatment.
Fig. 7.
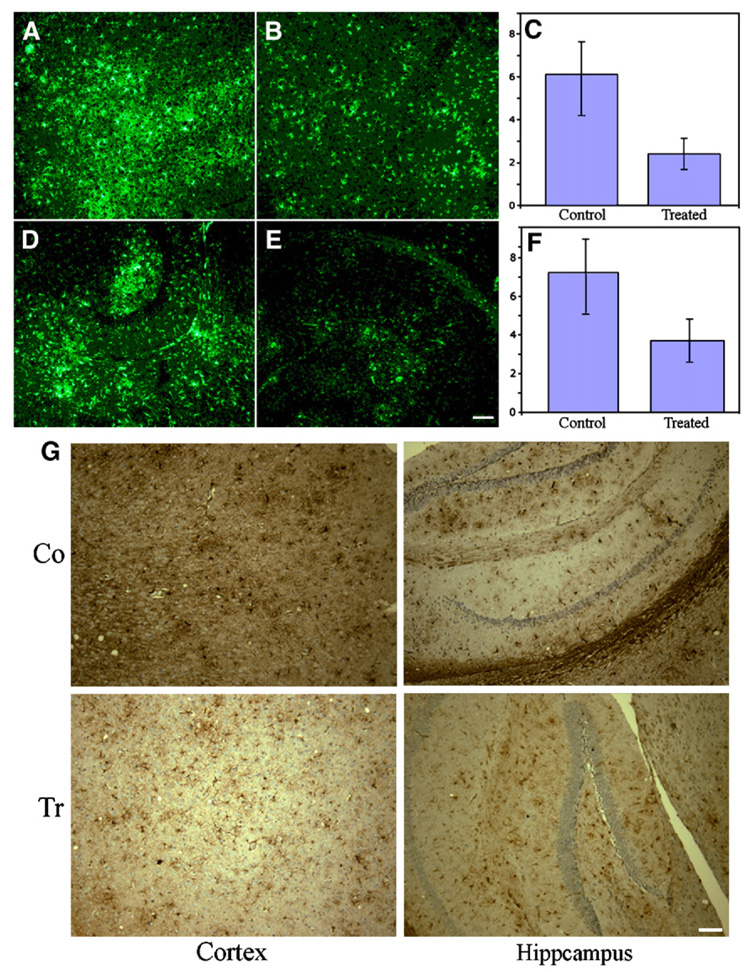
Fluorescence immunolabeling of glial fibrillary acidic protein in AD Tg mice. GFAP positive cells were stained with anti-GFAP antibody followed by G488 labeled second antibody. Shown are representative images of control (A, D) and treated (B, E) in cortex (A, B) and hippocampus (D, E). Shown in C and F are mean density (n=6) in cortex and in hippocampus of control and treated mice. C: The control mice had significantly higher cortical GFAP means than the treated mice (t(6.23)=3.44, p=0.013); control mice had significantly higher hippocampal GFAP means than the treated mice (t(10)=2.31, p=0.044). F: The control mice had significantly higher hippocampal GFAP means than the treated mice (t(10)=2.309, p=0.044). Representative image areas from 3 sections of cortex and representative image areas from 2 sections of hippocampus were analyzed from each control and treated mouse. G. Representative sections for GFAP staining using an immunohistochemical staining method from brains of Aβ42 gene vaccinated (Tr) and control mice (Co). Similar results can be seen as with the immunofluorescence method in both cortex and hippocampus. Scale bar=100 µm.
We also performed biotinylated BS-I isolectin B4 (Sigma) staining for macrophages. There was no significant difference in intensity for isolectin staining between control and treated mouse brain (data not shown). In addition, a careful histological examination of brain with hematoxylin and eosin staining showed no inflammatory cellular response. The immune response induced by gene vaccination to Aβ42 did not produce obvious signs of damage to neurons in Aβ42 gene-immunized mice. The weight gain was higher in vaccinated mice compared to the control mice with the mean weight in control mice at 15 months of age was 36±5.0 g and in treated mice, was 40±5.4 g (n=6).
2.6. Gene vaccine elicited antibodies against Aβ42 in an aged monkey
A 15 year old rhesus monkey was immunized using the same gene-gun delivered Aβ42 gene vaccine as used in mice. The monkey was injected in four monthly intervals with 8 µg DNA transfected into the abdominal skin each time. Serum was obtained 6 months after the first vaccination. ELISA analysis showed that the specific titer against Aβ42 was 1:20,000 (Fig. 8A) in comparison to the preimmune serum. Western blot analysis showed that the sera after Aβ42 gene immunization specifically recognized GST-Aβ42 with an antibody titer against Aβ1–16 and against Aβ29–42, residues similar to the mouse results (Fig. 8 B). The isotyping by ELISA showed that the vaccinated monkey produced an increased level of anti-Aβ42 IgG1 specific antibody (Th2 type response).
Fig. 8.

Aβ42 specific antibody from a vaccinated monkey. The monkey was vaccinated with the gene-gun delivered Aβ42 gene vaccine in 4 monthly intervals with 8 µg DNA and the serum obtained 2 months after the last immunization. A. ELISA assay shows anti-Aβ42 titer reached 1:20,000. B. Western blot shows Aβ42 gene vaccination in monkey elicits a specific antibody response against Aβ42 and also against Aβ1–16, Aβ17–28, and Aβ29–42. Lanes 1, 2 and 3 were loaded with GST fused to Aβ1–16, Aβ17–28 or Aβ29–42, respectively. Preimmune or immune serum was then added.
3. Discussion
In previous studies, we have demonstrated that prophylactic gene-gun mediated Aβ42 gene vaccine in APPswe/PS1ΔE9 transgenic mice induced a Th2 biased immune response and was effective in reducing Aβ42 plaque burden in brain [15]. The present studies significantly advance this analysis. Inducing a Th2 polarized immune response against Aβ42 peptide is critically important to minimize the risk of a cell mediated autoimmune encephalitis in vaccinating patients with Alzheimer’s disease. In the present study, we applied full length Aβ42 gene constructs as a vaccine carrier. Previous results have shown that a shorter epitope construct of Aβ1–16 was less effective in reducing Aβ42 burden in APPswe/PS1ΔE9 mice compared to the full length Aβ1–42 gene vaccine construct [15]. With full length Aβ42 gene constructs, a high titer of Th2 type antibodies (IgG1) was induced that recognized all segments of Aβ42 including: Aβ1–16, Aβ17–28, and Aβ29–42. The Th2 type IgG1 antibody has been shown to be effective in binding and clearing the Aβ plaques in AD mouse brain [16].
The Aβ42 antibody titer with the gene-gun method in the present study reached 1:10,000 (2.5 µg per ml serum) in APPswe/PS1ΔE9 mice and reached 1:20,000 (5 µg/ml) in a vaccinated monkey. In general, 1 µg of specific antibody per ml of serum is effective to neutralize a viral infection [17]. In the Aβ42 peptide vaccination protocol conducted on Alzheimer’s disease patients, only 20% of patients produced a significant anti-Aβ42 antibody titer (1:1200) [18,19]. The gene-gun delivered Aβ42 gene vaccine did induce anti-Aβ42 antibody in all six treated Tg mice and one monkey that provides an improved probability of being effective in reducing Aβ42 plaques in the brain of Alzheimer’s disease patients.
Avoiding a T cell mediated encephalitis is required for implementing a successful AD vaccination program. The ELISPOT method was used to discriminate Th1 and Th2 responses. The ELISPOT assay is a reliable method to test T cell responses to specific antigen stimulations [20]. IFNγ and IL4 are the two major primary cytokines to discriminate Th1 and Th2 T cells, respectively [21,22]. By quantitatively counting the cytokine containing spleen T cells in culture, the number of IL4 T cells was significantly increased after stimulation with Aβ42 peptide in vaccinated Tg mice compared to the absence of Aβ42 peptide in cell culture. Thus, increased IL4 T cells induced by Aβ42 peptide would increase a Th2 immune response in vaccinated Tg mice. There was no increase in the number of IL4 T cells from non-vaccinated APP/PS1 mice in the presence of Aβ42 peptide in cell culture compared to the absence of Aβ42 peptide. There was also no difference in the number of spleen IFNγ T cells in culture in the presence of Aβ42 between vaccinated and control mice. Interestingly, the number of IFNγ T cells was significantly increased in non-vaccinated Tg mice in the presence of Aβ42 peptide than in the absence of Aβ42 peptide as assayed in cell culture, indicating that these Tg mice might develop an autoimmune T cell response to the highly expressed transgenic Aβ42 peptide. This is consistent with human data that increased T cell reactivity to Aβ42 peptide increases with age [23].
Astrocytes may function as immunocompetent cells within the brain [24]. Activated astrocytes expressed an enhanced level of glial fibrillary acidic protein (GFAP) in control mouse brain that was significantly reduced by Aβ42 gene vaccination. This result can be explained by two possible mechanisms. First, activation of astrocytes is associated with Th1 autoimmunity against the highly expressed Aβ42 in these mice and the gene-gun mediated Aβ42 gene vaccine induced a Th2 immune response that inhibited the inflammatory immune response involved in astroglial cell activation. The second explanation may be that Aβ42 plaque deposition induced activation of glia cells and less deposition in vaccinated mice simultaneously reduced gliosis. Both mechanisms may be involved in the reduction of astroglial cell activation to express the GFAP [25–27].
Gene-gun mediated Aβ42 gene vaccination reduced Aβ42 deposition in brain by 50% as assessed by quantitative immunolabeling analysis. Analysis of brain homogenates by ELISA showed a 41% reduction, with a concomitant elevation of Aβ42 in the plasma by 43% in Aβ42 gene vaccinated compared with control mice. Brain homogenates assayed by ELISA assesses mainly insoluble Aβ42 peptide while immunolabeling mainly detects the plaque deposited Aβ42 peptide. These results and those of Okura et al. [28] and Ghochikyan et al [32] indicate that Aβ42 gene vaccination is efficient in inducing an immunoresponse against Aβ42 to reduce Aβ42 levels in brain. Ghochikyan et al. [32] used an Aβ42 gene fused to interleukin-4 gene (pAβ42-IL-4) to generate anti-Aβ antibodies and enhance the Th2-type of immune responses. They find that their gene-gun immunizations induced primarily IgG1 and IgG2b anti-Aβ antibodies. Okura et al. [28] similarly found that an Aβ DNA vaccine significantly reduced Aβ burden in transgenic mice. Both Okura et al. [28] and Ghochikyan et al. [32] point out that DNA (Aβ) gene vaccines produce lower titers of anti-Aβ42 antibodies compared to Aβ42 peptide vaccination [10,33]. Schultz et al. utilized both an Aβ42 gene vaccine together with low doses of preaggregated Aβ42 peptide and showed the presence of detectable titers of anti-Aβ42 within 2 weeks of vaccination [34]. They emphasize that the combination of gene and peptide vaccine results in higher anti-Aβ42 antibody titers than gene vaccination alone and that it results in a Th2 type of response.
One advantage to the Aβ42 gene vaccines is that despite their lower antibody titers, they reduce amyloid deposits, probably because DNA vaccination constantly induces the antibody production at a low titer for a long period. High anti-Aβ42 antibody titer levels may not be necessary for effective treatment with DNA vaccines [28].
Both mouse and monkey vaccinated with the gene-gun mediated Aβ42 gene vaccine produced anti-Aβ42 antibody of the Th2 type. The Th2 immune responses in mice and monkey induced by Aβ42 gene vaccination indicate the possibility of inducing a beneficial immune response in humans.
These new data indicate that gene-gun delivered Aβ42 gene vaccination may be an effective immunization method as therapy for AD.
4. Materials and methods
4.1. Mice
APPswe/PS1ΔE9 transgenic mice (7–10 week) carrying the human APP-Swedish and PS1ΔE9 mutations were purchased from Jackson Laboratory (Bar Harbor, ME). (Stock Number 004462). These animals develop numerous amyloid plaque deposits in the cerebral cortex and hippocampus by 6 months of age. The use of animals for this study was approved by the UT Southwestern Medical Center Animal Care and Use committee.
4.2. DNA constructs
The open reading frame of the human Aβ42 gene was chemically synthesized with optimal codons for mammalian cell expression and cloned into the genetic immunization vector as described previously. The Aβ42 gene was fused with the leader sequence of adenovirus E3 gene. An endosome-lysosome targeting sequence was fused downstream of the Aβ42 gene sequence. The synthetic SP72 promoter was used to drive the Aβ42 gene expression [15].
4.3. Gene-gun delivered vaccination
Immunizations of mice with plasmid DNA encoding the Aβ42 gene were performed on mouse ear skin using the Helios gene gun (Bio-Rad, Hercules, CA) as described. Briefly, DNA-coated gold particles were prepared by binding DNA to the gold beads (1–2 µm, Ferro Electronic Material Systems, South Plainfield, NJ) in 1 µg DNA per 1.2 mg gold beads in the presence of spermidine and calcium chloride. The DNA-gold was attached to the insides of the plastic tube and cut into 1.3 cm long as a bullet. The DNA coated gold particles were bombarded to both sides of the mouse ears using the gene gun with a helium gas pressure of 400 psi. The APPswe-PS1ΔE9 transgenic mice were immunized with the Aβ42 gene starting from the age of 3 months with 3 immunizations at one week intervals and then once per month for a total of 15 immunizations. Each vaccination consisted of 2 µg DNA injected with the gene gun into the skin of both external ears for a total of 4 µg DNA for each vaccination point. The control group received control plasmid pSP72-luc with the same immunization schedule. The blood was drawn from the tail vein and the serum sample was used to monitor the humoral immune responses. Similarly in a rhesus monkey, 8 µg of DNA binding to the gold-particles (2–4 µm) was injected into the abdominal skin monthly for 4 months using the gene gun. The pre-immune bleed and final bleed serum (2 weeks after final vaccine) was tested for anti-Aβ42 antibodies by ELISA and western blot.
4.4. Immunoassay for detection of anti-Aβ antibodies in serum
Enzyme-linked immunoabsorbent assay (ELISA) and western blot were used to monitor the humoral immune responses. Mouse blood was collected from the tail vein and serum was used to detect Aβ peptide by ELISA with a 96-microwell plate coated with GST-A β proteins. The antibody titer was determined by the dilution that produced a two fold higher OD value than the control sera. For western blot analysis, the GST-Aβ proteins in bacteria extract were separated by SDS-PAGE, blotted onto a PVDF membrane, incubated with the sera at 1:2000 dilutions. Antibodies against Aβ were detected using peroxidase-conjugated affinity-purified rabbit antiserum against mouse Ig. The blot was scanned and the reaction bands were measured and quantified in pixels. To determine the specific isotypes, sera from mice were diluted 1:200 and tested by ELISA as described above. To detect mouse IgG1, IgG2a, we used anti-mouse Ig-subclass-specific rabbit antibody (Pierce, Rockford, IL), followed by incubation with horseradish peroxidase (HRP)-conjugated Donkey anti-rabbit IgG. ELISA analysis was also applied for detection of Aβ42 specific antibodies produced in an Aβ42 gene vaccinated monkey. A panel of isotype-specific antibodies against monkey IgM and IgA (Rockland Inc, Bilbertsville, PA), and human IgG1, IgG2 and IgG4 (Sigma, St. Louis, MO) was used to discriminate the antibody isotype specific for Aβ42.
4.5. Enzyme-linked immunospot assay (ELISPOT)
The cell-mediated immune response was evaluated by ELISPOT for detection of spleen T cells to release interferon-gamma (IFN) and interleukin 4 (IL4) during in vitro re-stimulation with Aβ peptide [29]. Briefly, 96-well poly-vinylidene diflouride (PVDF) plates (Millipore, Bedford, MA) were coated with antibody to IFNs-γ or to IL4. The spleen T cells (Ficoll-Paque separated) were cultured at 2×105 per well in 0.2 ml of medium for re-stimulation with Aβ peptides Aβ9–18, Aβ1–40, Aβ1–42 (10 µg/ml) and Concanavalin A (5 µg/ml). After 48 h of incubation at 37 °C, the plates were washed, incubated with biotinylated anti-mouse IFNγ or anti-IL4, and further with Streptavidin-AP conjugate. After three washes, spots were developed with one-step NBT/BCIP reagent. Spots were counted using a stereomicroscope.
4.6. Enzyme linked-immunosorbent assay for detection of Aβ42 peptide in brain tissues and in plasma
The frontal lobe of brain was homogenized in 10 volumes of quinidine–tris buffer (5.0 M guanidine HCl/50 mM Tris–HCl, pH 8.0). The homogenates were mixed for 3 to 4 h at RT and stored at −80 °C until measured [30,31]. The Aβ42 in heparinized plasma was also extracted with 10 volumes of quinidine–tris buffer. For the sandwich ELISA assay, we used the Biosource Immunoassay kit for detection of Aβ42. In brief, 96-well plates were coated with monoclonal antibody that specifically recognize the N-terminal of Aβ42. Samples, including standards of known human Aβ1–42 content and brain or plasma extractions are added into these wells, followed by the addition of a rabbit antibody specific for the Aβ1–42 sequence of human Aβ. Bound rabbit antibody is detected by the use of an HRP labeled anti-rabbit antibody. The absorbance of the plates was read at 450 nm with a spectrophotometer.
4.7. Immunofluorescence and immunohistochemical staining and quantitation of amyloid burden and glia cells
For Aβ42 plaque detection, brain samples from APPswe/PS1ΔE9 transgenic mice were dissected, fixed with 4% paraformaldehyde for 4 h. The fixed samples were further processed for paraffin embedding and cut into 4 µm sections. After being deparaffinize, the section was treated with formic acid for 10 min, and further washed and blocked with blocking buffer (1% BSA in PBS with 0.05% Tween 20) for 30 min. After incubation with rabbit polyclonal anti serum (A1916, Sigma, St. Louis, MO) against Aβ42 (1:100, overnight at 4 °C), the sections were washed three times for 5 min each in PBS, again treated with blocking buffer (5 min, RT) before reacting with the secondary antibody (Alexa488-labeled goat anti-rabbit IgG, 1:400, 1 h, RT). Finally, the preparations were washed three times in PBS and observed with an Olympus fluorescence microscope. The amyloid plaque burden was analyzed with Image J (NIH) software and representative image areas of 3 (cortex) or 2 (hippocampus) sections were measured to calculate the percentage of plaque area against the total observed area. Anti-glial fibrillary acidic protein (GFAP) antibody (G9269, Sigma) was used in 1:200 dilution for immuno-labeling in paraffin sections. The rabbit IgG was further detected with goat anti-rabbit Alexa488 conjugated. The fluorescence was observed and quantity was measured by Image J software. In immunohistochemical staining, the same primary antibodies were applied to the paraffin sections and immunolabeling was detected by peroxidase labeled secondary antibody followed by development for 5 min with Metal-Enhanced diaminobenzidien substrate (Pierce, Rockford, IL).
4.8. Statistical analysis
Quantitative measurement of Aβ42, and GFAP burden in paraffin-embedded brain sections for control and treatment groups were compared using a two-way analysis of variance (ANOVA) with one between and one within factors and Student’s independent samples t-test. Assumptions for the statistical tests (normality and equal variances) were examined; when violated, non-equal variance formulas were used. SPSS was used to perform all analyses and the p value for statistical significance was set to 0.05.
Acknowledgments
This study was supported by P30AG12300 Alzheimer’s Disease Center grant from the National Institutes of Health, National Institute on Aging, Bethesda, MD; U01 AG16976 the National Alzheimer’s Coordinating Center grant, Seattle, WA.; Alzheimer’s Association Research Grant, and the Rudman Foundation, Dallas, Texas; The Luttrell Foundation, Belmont, California and unrestricted funds to SAJ in the Biodesign Institute.
Abbreviations
- AD
Alzheimer’s disease
- Aβ
Amyloid beta
- APPswe/PS1ΔE9
Amyloid precursor protein (Swedish mutation)/presenilin 1 delta E9 mutation
- ELISPOT
enzyme-linked immunosorbent spot
- nm
nanometer
- SPSS
Statistical Package for the Social Sciences
References
- 1.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg RN. The molecular and genetic basis of AD: the end of the beginning: the 2000 Wartenberg lecture. Neurology. 2000;54:2045–2054. doi: 10.1212/wnl.54.11.2045. [DOI] [PubMed] [Google Scholar]
- 3.Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, et al. Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 4.Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM, et al. Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science. 1992;258:126–129. doi: 10.1126/science.1439760. [DOI] [PubMed] [Google Scholar]
- 5.Lahiri DK, Ge YW. Role of the APP promoter in Alzheimer’s disease: cell type-specific expression of the beta-amyloid precursor protein. Ann NY Acad Sci. 2004;1030:310–316. doi: 10.1196/annals.1329.039. [DOI] [PubMed] [Google Scholar]
- 6.Fox NW, Johnstone EM, Ward KE, Schrementi J, Little SP. APP gene promoter constructs are preferentially expressed in the CNS and testis of transgenic mice. Biochem Biophys Res Commun. 1997;240:759–762. doi: 10.1006/bbrc.1997.7728. [DOI] [PubMed] [Google Scholar]
- 7.Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43) Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- 8.Iwatsubo T, Mann DM, Odaka A, Suzuki N, Ihara Y. Amyloid beta protein (A beta) deposition: A beta 42(43) precedes A beta 40 in Down syndrome. Ann Neurol. 1995;37:294–299. doi: 10.1002/ana.410370305. [DOI] [PubMed] [Google Scholar]
- 9.Yankner BA. Mechanisms of neuronal degeneration in Alzheimer’s disease. Neuron. 1996;16:921–932. doi: 10.1016/s0896-6273(00)80115-4. [DOI] [PubMed] [Google Scholar]
- 10.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 11.Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, et al. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- 12.Schenk D. Amyloid-beta immunotherapy for Alzheimer’s disease: the end of the beginning. Nat Rev Neurosci. 2002;3:824–828. doi: 10.1038/nrn938. [DOI] [PubMed] [Google Scholar]
- 13.Rosenberg RN. Immunization with β-Amyloid(1–42) protein for Alzheimer disease: genomics predicts the response. JAMA. 2005;294:2352–2353. [Google Scholar]
- 14.Qu B, Rosenberg RN, Li L, Boyer PJ, Johnston SA. Gene vaccination to bias the immune response to amyloid-beta peptide as therapy for Alzheimer disease. Arch Neurol. 2004;61:1859–1864. doi: 10.1001/archneur.61.12.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qu B, Boyer PJ, Johnston SA, Hynan LS, Rosenberg RN. Abeta42 gene vaccination reduces brain amyloid plaque burden in transgenic mice. J Neurol Sci. 2006;244:151–158. doi: 10.1016/j.jns.2006.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bard F, Barbour R, Cannon C, Carretto R, Fox M, Games D, et al. Epitope and isotype specificities of antibodies to beta-amyloid peptide for protection against Alzheimer’s disease-like neuropathology. Proc Natl Acad Sci U S A. 2003;100:2023–2028. doi: 10.1073/pnas.0436286100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bachmann MF, Kalinke U, Althage A, Freer G, Burkhart C, Roost H, et al. The role of antibody concentration and avidity in antiviral protection. Science. 1997;276:2024–2027. doi: 10.1126/science.276.5321.2024. [DOI] [PubMed] [Google Scholar]
- 18.Fox NC, Black RS, Gilman S, Rossor MN, Griffith SG, Jenkins L, et al. Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology. 2005;64:1563–1572. doi: 10.1212/01.WNL.0000159743.08996.99. [DOI] [PubMed] [Google Scholar]
- 19.Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- 20.Czerkinsky CC, Tarkowski A, Nilsson LA, Ouchterlony O, Nygren H, Gretzer C. Reverse enzyme-linked immunospot assay (RELISPOT) for the detection of cells secreting immunoreactive substances. J Immunol Methods. 1984;72:489–496. doi: 10.1016/0022-1759(84)90017-6. [DOI] [PubMed] [Google Scholar]
- 21.Torres KC, Dutra WO, Gollob KJ. Endogenous IL-4 and IFN-gamma are essential for expression of Th2, but not Th1 cytokine message during the early differentiation of human CD4+ T helper cells. Hum Immunol. 2004;65:1328–1335. doi: 10.1016/j.humimm.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 22.Nakamura T, Kamogawa Y, Bottomly K, Flavell RA. Polarization of IL-4- and IFN-gamma-producing CD4+ T cells following activation of naive CD4+ T cells. J Immunol. 1997;158:1085–1094. [PubMed] [Google Scholar]
- 23.Monsonego A, Zota V, Karni A, Krieger JI, Bar-Or A, Bitan G, et al. Increased T cell reactivity to amyloid beta protein in older humans and patients with Alzheimer disease. J Clin Invest. 2003;112:415–422. doi: 10.1172/JCI18104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- 25.Tani M, Glabinski AR, Tuohy VK, Stoler MH, Estes ML, Ransohoff RM. In situ hybridization analysis of glial fibrillary acidic protein mRNA reveals evidence of biphasic astrocyte activation during acute experimental autoimmune encephalomyelitis. Am J Pathol. 1996;148:889–896. [PMC free article] [PubMed] [Google Scholar]
- 26.Eng LF, Ghirnikar RS. GFAP and astrogliosis. Brain Pathol. 1994;4:229–237. doi: 10.1111/j.1750-3639.1994.tb00838.x. [DOI] [PubMed] [Google Scholar]
- 27.Marinkovic T, Garin A, Yokota Y, Fu YX, Ruddle NH, Furtado GC, et al. Interaction of mature CD3+ CD4+ T cells with dendritic cells triggers the development of tertiary lymphoid structures in the thyroid. J Clin Invest. 2006;116:2622–2632. doi: 10.1172/JCI28993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okura Y, Miyakoshi A, Kohyama K, Park K, Staufenbiel M, Matsumoto Y. Nonviral Aβ DNA vaccine therapy against Alzheimer’s disease: long term effects and safety. Proc Natl Acad Sci U S A. 2006;103:9619–9624. doi: 10.1073/pnas.0600966103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar A, Weiss W, Tine JA, Hoffman SL, Rogers WO. ELISPOT assay for detection of peptide specific interferon-gamma secreting cells in rhesus macaques. J Immunol Methods. 2001;247:49–60. doi: 10.1016/s0022-1759(00)00310-0. [DOI] [PubMed] [Google Scholar]
- 30.Vanderstichele H, Van Kerschaver E, Hesse C, Davidsson P, Buyse MA, Andreasen N, et al. Standardization of measurement of beta-amyloid(1–42) in cerebrospinal fluid and plasma. Amyloid. 2000;7:245–258. doi: 10.3109/13506120009146438. [DOI] [PubMed] [Google Scholar]
- 31.Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, et al. Amyloid precursor protein processing and A beta42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghochikyan A, Vasilevko V, Petrushina I, Movsesyan N, Davit B, Tian W, et al. Generation and characterization of the humoral immune response to DNA immunization with chimeric β-amyloid-interleukin-4 minigene. Eur J Immunol. 2003;33:3232–3241. doi: 10.1002/eji.200324000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Agadjanyan M, Ghochikyan A, Petrushina I, Vasilevko V, Movsesyan N, Mkrtichyan M, et al. Prototype Alzheimer’s disease vaccine using the immunodominant B cell epitope from β-amyloid and promiscuous T cell epitope pan HLA DR-binding peptide. J Immunol. 2005;174:1580–1586. doi: 10.4049/jimmunol.174.3.1580. [DOI] [PubMed] [Google Scholar]
- 34.Schultz J, Salzer U, Mohajeri M, Franke D, Heinrich J, Pavlovic J, et al. Antibodies form a DNA peptide vaccination decrease brain amyloid burden in a mouse model of Alzheimer’s disease. J Mol Med. 2004;82:706–714. doi: 10.1007/s00109-004-0570-z. [DOI] [PubMed] [Google Scholar]