

Abstract
In otherwise non-autoimmune-prone C57BL/6 (B6) mice rendered genetically deficient in CD152 (CTLA-4), polyclonal hypergammaglobulinemia with increased levels of SLE-associated IgG autoantibodies, glomerular IgG and C3 deposition, and interstitial nephritis all developed by 3-5 weeks of age. Remarkably, superimposing genetic deficiency of BAFF onto CD152 deficiency did not substantially attenuate humoral autoimmunity and immunopathology in these mice, despite the resulting marked reduction in B-lineage cells. Although superimposing a BAFF transgene (resulting in constitutive BAFF overexpression) onto CD152-deficient mice did lead to increases in B-lineage cells and serum levels of certain SLE-associated IgG autoantibodies, renal immunopathology remained largely unaffected. Taken together, these results demonstrate that global T cell dysregulation, even in an otherwise non-autoimmune-prone host, can promote systemic humoral autoimmunity and immunopathology in a BAFF-independent manner. Moreover, supra-physiologic expression of BAFF in the setting of ongoing autoimmunity does not necessarily lead to greater immunopathology. These findings may help explain the limited clinical efficacy appreciated to date of BAFF antagonists in human SLE.
Keywords: rodent, autoimmunity, cytokines
Introduction
B cell activating factor belonging to the TNF family (BAFF3; also known as BLyS, TALL-1, THANK, TNFSF13B, and zTNF4) is a 285-amino acid type II transmembrane protein member of the TNF ligand superfamily (1-6). Myeloid lineage cells (monocytes, dendritic cells, macrophages, neutrophils) and bone marrow (BM)-derived radiation-resistant stromal cells are the major systemic sources of BAFF (1-3, 5, 7-9). Three BAFF receptors have been identified (BCMA, TACI, and BAFFR [also called BR3]), and their expression is largely (albeit not exclusively) limited to B cells (10-13). Cleavage of BAFF by a furin protease from the cell surface results in release of a soluble, biologically active 17-kD molecule (1, 7) which serves as a vital survival factor for B cells (14-20). BAFF also serves as a B cell differentiation factor, promoting the differentiation of immature B cells to mature B cells (21, 22) and promoting Ig class switching and Ig production by B cells via engagement of TACI and BAFFR (23-25).
The importance of BAFF to in vivo humoral immunity is highlighted by states of either excessive BAFF or absent BAFF. Administration of exogenous BAFF to mice at the time of immunization enhances antigen-specific antibody production (15), and repeated administration of BAFF to mice even without specific antigenic immunization results in B cell expansion and polyclonal hypergammaglobulinemia (2). Moreover, constitutive overexpression of BAFF in BAFF-transgenic (Tg) mice that otherwise are not autoimmune-prone leads not just to B cell expansion and polyclonal hypergammaglobulinemia but to features of systemic lupus erythematosus (SLE), including elevated circulating titers of multiple autoantibodies and immune-complex glomerulonephritis (GN) (6, 26, 27). As long as MyD88-mediated signaling is intact, SLE-like features develop in BAFF-Tg mice even in the complete absence of T cells (28). Conversely, non-autoimmune-prone mice genetically rendered deficient in BAFF exhibit marked global reductions in B cells beyond the transitional 1 (T1) maturational state and in baseline serum Ig levels and Ig responses to T cell-dependent and T cell-independent antigens (29, 30). Given the profound reductions in mature B cells and circulating Ig levels in BAFF-deficient non-autoimmune-prone mice, the expectation was that BAFF deficiency would greatly attenuate autoimmunity in hosts that otherwise are autoimmune-prone. Surprisingly, BAFF-deficient SLE-prone NZM 2328 mice, despite reductions in mature B cell numbers as severe as those observed in BAFF-deficient non-autoimmune-prone mice, developed hypergammaglobulinemia, serological autoimmunity (including nephrophilic autoantibodies), and end-organ (kidney) pathology as they aged (31). This clearly demonstrated that B cell-based autoimmunity could emerge despite the life-long absence of BAFF, but the underlying driving force(s) remained uncertain.
Since T cell dysregulation is a feature of SLE, one plausible explanation for development of autoimmunity in BAFF-deficient NZM 2328 mice is that dysregulated T cell activation promotes differentiation of, and Ig production by, the limited numbers of B cells extant in the BAFF-deficient, but otherwise SLE-prone, hosts. However, a myriad of T cell-independent abnormalities, including those involving B cells and/or the innate immune system, in these mice could potentially contribute in a meaningful way to development of autoimmunity. That is, development of BAFF-independent autoimmunity in NZM 2328 mice may not necessarily be (solely)T cell-driven. To unambiguously address development of T cell-driven BAFF-independent autoimmunity and immunopathology, we turned to a model system based on deficiency of CD152 (CTLA-4) in an otherwise non-autoimmune-prone host.
CD152 is a vital homeostatic regulator of T cell activation. The suppressor effects of CD4+CD25+ regulatory T cells are mediated, at least in part, via CD152 (32, 33), and engagement of CD152 is crucial to development and/or maintenance of tolerance (34, 35). C57BL/6 (B6) or BALB/c mice genetically deficient in CD152 (cd152-/- mice) spontaneously develop massive systemic T cell expansion and infiltration into vital organs which is lethal by as early as 3 weeks of age (36-38). Negative and positive selection in the thymus are normal in cd152-/- mice (39, 40), indicating that the physiologic defect is in control of peripheral T cell activation rather than in central T cell development. Since the accelerated T cell activation, T cell expansion, and mortality are markedly attenuated in TCR-Tg cd152-/- mice that express highly limited T cell repertoires (40-43), it is likely that the proliferating T cells in non-TCR Tg cd152-/- mice respond to highly prevalent environmental antigens and/or self antigens. The diverse and unbiased TCR repertoire in these non-TCR Tg cd152-/- mice (44) indicates that no individual self (or environmental) antigen is uniquely driving the pathologic response, but it does make it likely that autoreactive T cells are represented among the activated and proliferating T cells.
Development of humoral autoimmunity in cd152-/- mice has, to date, not been reported. In this report, we demonstrate that cd152-/- mice bearing a non-autoimmune-prone B6 genetic background (B6.cd152-/- mice) develop, by 3-5 weeks of age,high circulating levels of SLE-associated IgG autoantibodies along with renal deposition of Ig and complement and interstitial nephritis. Remarkably, BAFF-deficient B6.cd152-/- (B6.cd152-/-.baff-/-) mice develop similarly robust autoimmunity and immunpathology, demonstrating that dysregulated T cells, even in the context of an otherwise non-autoimmune-prone environment, can promote humoral autoimmunity and immunopathology in a BAFF-independent manner. These findings may have profound ramifications for BAFF-targeted therapeutic approaches in human autoimmune diseases.
Materials and methods
General
All reported studies were approved by the University of Southern California (USC) Institutional Animal Care and Use Committee. All assays and tissue evaluations described below were performed by individuals who were blinded to the genotypes of the mice.
Mice
All mice were housed at USC in a single specific pathogen-free room. To generate BAFF-deficient (baff-/-) cd152-/- mice, baff-/- mice (30) that had been backcrossed to B6 mice for > 9 generations (B6.baff-/- mice) were first crossed with (baff+/+) B6.cd152+/- mice (45), and the pups were screened by PCR for the cd152+/- genotype (45) and the baff+/- genotype. (31) The resulting B6.baff+/-.cd152+/- mice were backcrossed to B6.baff-/- mice, and the pups were screened for the cd152+/- and baff-/- genotypes. The resulting male and female B6.baff-/-.cd152+/- mice were intercrossed, giving rise to B6.baff-/-.cd152+/+, B6.baff-/-.cd152+/-, and B6.baff-/-.cd152-/- mice.
To generate BAFF-Tg cd152-/- mice, B6.cd152+/- mice were first crossed with (cd152+/+) BAFF-Tg B6 (B6.BTg) mice (26, 46), and the pups were screened by PCR for the BAFF Tg (46) and for the cd152+/- genotype (45). The resulting B6.BTg.cd152+/- mice were then crossed with B6.cd152+/- mice, giving rise to B6.cd152+/+, B6.cd152+/-, B6.cd152-/-, B6.BTg.cd152+/+, B6.BTg.cd152+/-, and B6.BTg.cd152-/- mice.
Cell surface staining
Single-cell suspensions from spleen or BM were stained with combinations of the following conjugated mAb: FITC-conjugated anti-CD43 (BD Biosciences, San Jose, CA), anti-Igλ (Southern Biotechnology Associates, Birmingham, AL),and anti-Igκ (Southern Biotechnology); APC-conjugated anti-CD19 (BD Biosciences) and anti-CD93 (eBioscience, San Diego, CA); APC Cy7-conjugated anti-B220 (eBioscience); PE-conjugated anti-Igλ (Southern Biotechnology), anti-Igκ (Southern Biotechnology), anti-CD23 (BD Biosciences), and anti-CD138 (BD Biosciences); PE Cy5-conjugated anti-GR1 (BD Biosciences), anti-CD4 (BD Biosciences), anti-CD8 (BD Biosciences), and anti-F4/80 (BD Biosciences); PE Cy5.5-conjugated anti-CD21/35 (Allman lab, University of Pennsylvania); PE Cy7-conjugated anti-IgM (eBiosciences); biotin-conjugated anti-CD138 (BD Biosciences), anti-CD23 (BD Biosciences), and anti-IgD (Southern Biotechnology); Streptavidin-conjugated PerCP Cy5.5 (BD Biosciences) and Pacblue (Invitrogen, Carlsbad, CA). For intracellular antigen discrimination, samples were fixed and rendered permeable with Solutions A and B (Invitrogen) prior to addition of the antibodies toward intracellular antigens. Dead cells were excluded from analyses with Aqua Fixable Live/Dead (Invitrogen). Analyses were performed on a 17-color 4-laser LSRII (Becton Dickinson). Flow cytometry data were analyzed using FlowJo 8.6 software (Tree Star, Ashland, OR).
Spleen immunofluorescence
OCT-embedded frozen spleen sections were stained with PE-conjugated anti-CD45R/B220 mAb (BD Biosciences) and FITC-conjugated anti-MOMA-1 mAb (Serotec, Raleigh, NC) for 1 hr at room temperature and mounted with Fluoromount G (Electron Microscopy Sciences, Hatfield, PA). Stained sections were examined by fluorescence microscopy (Nikon E600).
Serum Ig and autoantibody determinations
For quantification of individual total Ig class or subclass concentrations, serial dilutions of mouse sera were added to ELISA plates that had been coated with capture antibodies against the indicated specific Ig class or subclass. This was followed by alkaline phosphatase-conjugated detection antibodies to the specific Ig class or subclass (Southern Biotechnology) (47). The Ig concentrations were calculated from standard curves concurrently generated with purified mouse myeloma proteins (Sigma, St. Louis, MO).
For quantification of IgG autoantibody concentrations, mouse sera (1:250 dilution) were added to ELISA plates that had been coated with chromatin (5 μg/ml), histone (20 μg/ml), ssDNA (100 μg/ml), or dsDNA (100 μg/ml) followed by alkaline phosphatase-conjugated goat anti-mouse IgG (Southern Biotechnology) (48). Each sample was normalized to the mean OD of serum from 5-month-old MRL-lpr/lpr mice, the latter being arbitrarily assigned a value of 100 U/ml.
Kidney immunofluorescence
OCT-embedded frozen kidney sections were incubated with FITC-conjugated F(ab)2 fragments of goat anti-mouse IgM or IgG γ chain, FITC-conjugated goat anti-mouse IgG1, IgG2b, or IgG3 isotypes (Jackson ImmunoResearch Laboratories, West Grove, PA), or goat anti-mouse C3 (MP Biomedicals, Aurora, OH) followed by FITC-conjugated anti-goat IgG (Jackson). Stained sections were examined by fluorescence microscopy (Nikon).
Kidney histology
Individual 5-μ sections of formalin-fixed kidneys were stained with hematoxylin and eosin, PAS, and Masson’s trichrome stain and were examined by light microscopy. Each case was assessed for GN using a modification of the World Health Organization classification for lupus nephritis (46) as previously described (31) and for interstitial nephritis.
Statistical analysis
All analyses were performed using SigmaStat software (SPSS, Chicago, IL). If the raw (untransformed) data did not follow a normal distribution, they were log-transformed to achieve normality. Parametric testing between two matched or unmatched groups was performed by the paired or unpaired t test, respectively. Parametric testing among three or more groups was performed by one-way ANOVA. When the data were not normally distributed even after log-transformation or the equal variance test was not satisfied, non-parametric testing was performed by the Mann-Whitney rank sum test between two groups and by Kruskal-Wallis one-way ANOVA on ranks among three or more groups. Correlations were determined by Pearson product moment correlation for interval data and by Spearman rank order correlation for ordinal data or for interval data which did not follow a normal distribution.
Results
Effects of CD152 deficiency on B cell phenotype in B6.baff+/+, B6.BTg, and B6.baff-/- mice
We chose CD152-deficient mice as a model of robust global T cell dysregulation, inasmuch as such mice spontaneously undergo massive systemic T cell expansion and activation. A large percentage dies by 6 weeks of age from inflammatory infiltrates in multiple vital organs (e.g., heart, liver) (36-39, and unpublished data), so we focused our studies on 3- to 5-week-old mice.
CD152 deficiency in B6.baff+/+ mice led to modest increases in the percentages of BM pre/pro B cells and to substantial reductions in percentages of BM immature B cells (Figure 1A a, d). These changes were mirrored in B6.baff-/- mice but were blunted in B6.BTg mice (Figure 1A b, c, e, f). (In all staining combinations, results for cd152+/- mice were identical to those for cd152+/+ mice, so results only for the latter are shown.) Importantly, CD152 deficiency in B6.baff+/+ mice did not lead to a reduction in the percentages of BM mature recirculating B cells (Figure 1A g, j). Although the percentages of these cells in CD152-sufficient B6.BTg and B6.baff-/- mice were, respectively, greater than and less than that in CD152-sufficient B6.baff+/+ mice, CD152 deficiency failed to lead to any relative reductions in the percentages of BM mature recirculating B cells in either B6.BTg or B6.baff-/- mice (Figure 1A h, i, k, l).
Figure 1. BM and spleen B cell subsets in CD152-intact and CD152-deficient B6.baff+/+, B6.BTg, and B6.baff-/- mice.

Panel A: Contour plots of surface CD23 and IgM staining of gated BM B220+AA4.1+ cells (a-f) and B220+AA4.1- cells (g-l) from the indicated mice are illustrated. The numbers in panels a-f indicate the percentages of pro/pre (lower left), CD23- immature (upper left), and CD23+ immature (upper right) B cells. The numbers adjacent to the rectangles in panels g-l indicate the percentages of mature recirculating B cells. Panel B: Contour plots of surface CD23 and IgM staining of gated spleen B220+AA4.1+ cells (a-f) and contour plots of surface IgM and CD21 staining of sorted spleen B220+AA4.1- cells (g-l) from the indicated mice are illustrated. The numbers adjacent to the rectangles in panels a-f indicate the percentages of T1 (upper left), T2 (upper right), and T3 (lower right) B cells. The numbers adjacent to the ovals and quadrangles in panels g-l respectively indicate the percentages of FO and MZ B cells. Panel C: Contour plots of surface CD138 and intracellular κ/λ staining of gated BM (a-f) or spleen (g-l) CD4-CD8-GR-1-B220lo/-sIgM- cells from the indicated mice are illustrated. The numbers inside the rectangles indicate the percentages of plasma cells. Panel D: Sections of fresh-frozen spleens from the indicated mice (a-d) were stained with anti-B220-PE (red) + anti-MOMA-1-FITC (green). Original magnification is 10X.
Among spleen transitional B cells, CD152-sufficient B6.BTg mice harbored greater percentages of late transitional (T3) B cells and lesser percentages of early transitional (T1) B cells than did CD152-sufficient B6.baff+/+ mice, consistent with the ability of BAFF to serve as a B cell differentiation factor (21, 22).CD152 deficiency in both B6. baff+/+ and B6.BTg mice led to increases in the percentages of T3 B cells and decreases in the percentages of T1 B cells (Figure 1B a, b, d, e). Strikingly, transitional B cell progression beyond the T1 stage was largely arrested in CD152-sufficient B6.baff-/- mice, consistent with previous reports (29, 30). Nevertheless, there was an unmistakable increase in the percentages of the more mature T2 and T3 B cells in B6.baff-/-.cd152-/- mice (Figure 1B c, f), demonstrating that the consequences of BAFF deficiency on transitional B cell maturation could be at least partly overcome.
In addition to the substantial reduction in T2 and T3 B cells in BAFF-deficient (CD152-sufficient) mice, FO and MZ B cells are also substantially reduced in them (29, 30). In contrast, MZ B cells are expanded in BAFF-Tg (CD152-sufficient) mice (16, 26). Analysis of the FO and MZ B cell subsets in CD152-sufficient B6.baff-/- and B6.BTg mice confirmed their respective reductions and expansions relative to their B6.baff+/+ counterparts (Figure 1B g-i). Regardless of the baff genotype, CD152-deficient mice harbored substantially lower percentages of both FO and MZ B cells than did the corresponding CD152-sufficient mice (Figure 1B j-l), although the effect on MZ B cells was difficult to appreciate in B6.baff-/-.cd152-/- mice due to the very low percentages of MZ B cells harbored by B6.baff-/-.cd152+/+ and B6.baff-/-.cd152+/- mice.
Preservation of plasma cells (PC) and development of hypergammaglobulinemia and SLE-associated autoantibodies in CD152-deficient B6.baff+/+, B6.BTg, and B6.baff-/- mice
In contrast to the reductions in percentages of T2, T3, FO, and MZ B cells in CD152-deficient mice relative to their CD152-sufficient counterparts, CD152 deficiency in B6.baff+/+, B6.BTg, and B6.baff-/- mice led to no change or modest increases in the percentages of BM and/or spleen PC (Figure 1C a-l), populations which include both CD138+ and CD138- cells (49). Moreover, the collapsed spleen follicles seen in CD152-sufficient B6.baff-/- mice were substantially restored in their CD152-deficient counterparts (Figure 1D a-b). (The paucity of MOMA-1+ rings surrounding the spleen follicles in B6.baff-/-.cd152-/- mice is likely a feature of CD152 deficiency per se, inasmuch as a similar paucity of MOMA-1+ rings was observed in CD152-deficient, but not CD152-sufficient, B6.baff+/+ mice [Figure 1D c-d].)
Consistent with the relative increase in PC and, in the case of B6. baff-/- mice, the restoration of follicular structure, serum levels of total IgM, IgG1, and IgG2b were significantly greater in CD152-deficient mice than in corresponding CD152-sufficient mice, regardless of the baff genotype (p ≤ 0.006 for total IgM; p ≤ 0.001 for total IgG1; p ≤ 0.005 for total IgG2b; Figure 2A a-c). Although serum total IgM and IgG2b levels in B6.baff-/-.cd152-/- mice were significantly lower than those in B6.baff+/+.cd152-/- or B6.BTg.cd152-/- mice (p < 0.001 for each comparison), there were no significant differences in serum total IgG1 levels among these discrete mouse cohorts.
Figure 2. Serum total Ig and IgG autoantibody levels in CD152-sufficient and CD152-deficient B6.baff+/+, B6.BTg, and B6.baff-/- mice.
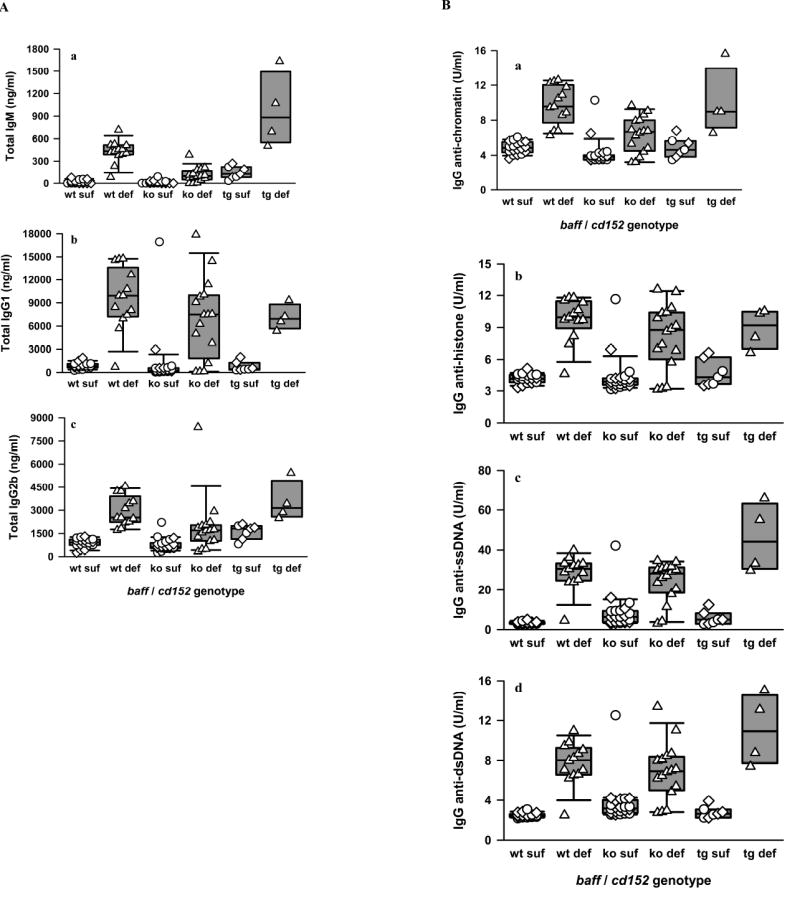
Panel A: Sera from CD152-sufficient B6.baff+/+ (wt suf; n = 9 cd152+/+ + 9 cd152+/-), CD152-deficient B6.baff+/+ (wt def; n = 13), CD152-sufficient B6.baff-/- (ko suf; n = 11 cd152+/+ + 11 cd152+/-), CD152-deficient B6.baff-/- (ko def; n = 16), CD152-sufficient B6.BTg (tg suf; n = 3 cd152+/+ + 4 cd152+/-), and CD152-deficient B6.BTg (tg def; n = 4) mice were assayed for levels of total IgM (a), total IgG1 (b), and total IgG2b (c). Circles, diamonds, and triangles respectively represent individual baff+/+, baff+/-, and baff-/- mice. The composite results are plotted as box plots. The lines inside the boxes indicate the medians; the outer borders of the boxes indicate the 25th and 75th percentiles; and the bars extending from the boxes indicate the 10th and 90th percentiles. Panel B: Sera from the same mice were assayed for levels of IgG antibodies against chromatin (a), histone (b), ssDNA (c), and dsDNA (d). Results are presented as in panel A.
Not only did hypergammaglobulinemia develop in CD152-deficient mice regardless of their baff genotype, but these mice also harbored considerable levels of serum SLE-associated IgG autoantibodies, including autoantibodies directed against chromatin, histone, ssDNA, and dsDNA (Figure 2B a-d). In contrast, serum levels of these autoantibodies were low in CD152-sufficient hosts, with no significant differences being appreciated between cd152+/+ and cd152+/- mice. Of note, serum autoantibody levels were no greater in CD152-sufficient B6.BTg mice than in CD152-sufficient B6.baff+/+ mice, consistent with previous observations that elevated circulating IgG anti-chromatin and IgG anti-dsDNA autoantibodies were not detected in 3-month-old B6.BTg (cd152+/+) mice (46). For any baff genotype, autoantibody levels against each of the 4 tested specificities were significantly greater in CD152-deficient mice than in corresponding CD152-sufficient mice (p ≤ 0.008 for IgG anti-chromatin; p ≤ 0.003 for IgG anti-histone; p ≤ 0.006 for IgG anti-dsDNA; p ≤ 0.006 for IgG anti-ssDNA).
The only IgG autoantibodies whose serum levels were statistically greater in CD152-deficient B6.BTg mice than in the other tested CD152-deficient mice were anti-ssDNA (p = 0.003) and anti-dsDNA (p = 0.032). That is, constitutive overexpression of BAFF in CD152-deficient B6 mice did not discernibly promote greater IgG anti-chromatin or anti-histone antibody responses. Strikingly, serum IgG anti-histone, anti-ssDNA, and anti-dsDNA levels were no different in CD152-deficient B6.baff-/- mice than in CD152-deficient B6.baff+/+ mice. The only IgG autoantibody whose serum levels were statistically lower in the former than in the latter was anti-chromatin (p < 0.001). Although modest quantitative differences were detected among the individual CD152-deficient mouse cohorts in IgG subclass distribution of the SLE-associated IgG autoantibodies (data not shown), the serologic studies collectively indicate that the generation and maintenance of elevated levels of at least some of these autoantibodies in CD152-deficient B6 mice do not require BAFF. Morever, when levels of such autoantibodies are already high, they frequently are not meaningfully augmented further by BAFF overexpression.
Development of renal immunopathology in CD152-deficient B6.baff+/+, B6.BTg, and B6.baff-/- mice
The development of serologic autoimmunity in CD152-deficient mice by 3-5 weeks of age raised the possibility that development of target-organ (kidney) immunopathology might also be rapid in these mice. Immunofluorescence studies of kidney sections revealed discrete patterns of Ig deposition among the tested groups of mice. Renal IgM deposition was detected in CD152-sufficient B6.baff+/+ and B6.BTg mice (Figure 3A a, c), with staining intensity being similar to that in 2-month-old (prior to development of clinical autoimmunity) NZM 2328 mice (Figure 3A g). In contrast, no renal IgM deposition was appreciated in CD152-sufficient B6.baff-/- mice (Figure 3A e). However, renal IgM deposition was strong in all CD152-deficient mice, including B6.baff-/-.cd152-/- mice, with staining intensity approaching that in 6-month-old (clinically sick) NZM 2328 mice (Figure 3A b, d, f, h).
Figure 3. Renal Ig and C3 deposition in CD152-sufficient and CD152-deficient B6.baff+/+, B6.BTg, and B6.baff-/- mice.
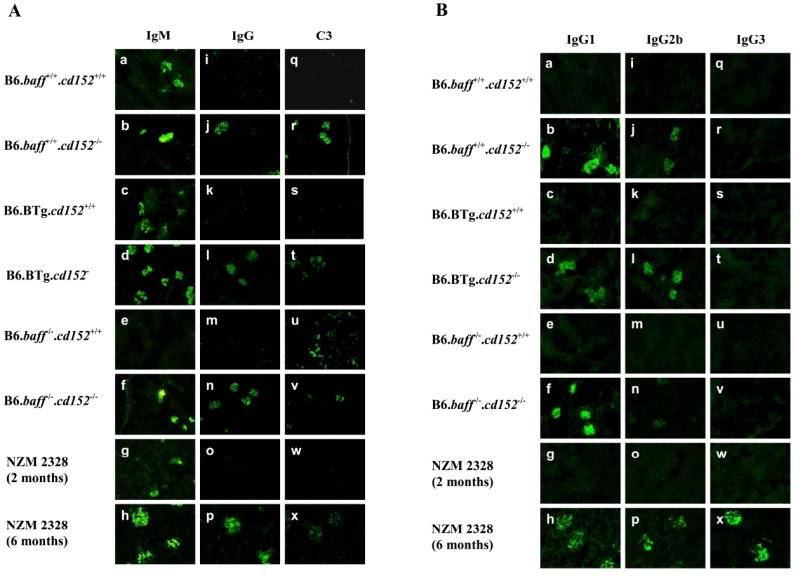
Panel A: Sections of fresh-frozen kidneys from the indicated mice were stained for total IgM (a-h), total IgG (i-p), and C3 (q-x). Original magnification is 40X. Panel B: Kidneys sections were stained for IgG1 (a-h), IgG2b (i-p), and IgG3 (q-x). Original magnification for all photomicrographs is 40X.
Deposition of IgG in the kidneys of the tested mice was striking in its prevalence. Although renal IgG deposition was not appreciated in any of the CD152-sufficient mice (Figure 3A i, k, m), all the CD152-deficient mice, including B6.baff-/-.cd152-/- mice, developed considerable renal IgG deposition (Figure 3A j, l, n). This renal IgG deposition by 3-5 weeks of age in the CD152-deficient mice is especially remarkable, inasmuch as no renal IgG deposition was yet appreciated in 2-month-old (clinically healthy)NZM 2328 mice despite their development of considerable renal IgG deposition by 6 months of age (when clinically sick) (Figure 3A o-p). In general, renal deposition of C3 paralleled deposition of IgG (Figure 3A q-x), although occasionally, some interstitial C3 deposition was also observed, irrespective of the baff or cd152 genotypes of the mice (Figure 3A u).
In BAFF-deficient NZM 2328 mice, deposition of IgG in the kidneys is substantial, albeit delayed in time relative to that in corresponding BAFF-sufficient mice, but the distribution of IgG subclasses deposited in the kidneys of BAFF-deficient NZM 2328 mice differs from that in BAFF-sufficient NZM 2328 mice. In the former, IgG1 deposition predominates with limited IgG2a or IgG2b deposition. In contrast, all three subclasses are amply represented in the latter (31).
Given the dramatic deposition of IgG in the kidneys of both B6.baff+/+.cd152-/- mice and B6.baff-/-.cd152-/- mice, we assessed the distribution of IgG subclasses in the renal deposits. Consistent with the lack of detectable deposition of total IgG in CD152-sufficient mice of any baff genotype, no renal IgG1, IgG2b, or IgG3 deposition was detected in any CD152-sufficient mouse (Figure 3B a, c, e, i, k, m, q, s, u). In contrast, renal IgG1 deposition was abundant in all CD152-deficient mice of any baff genotype (Figure 3B b, d, f). Renal IgG2b deposition was also detected in all CD152-deficient mice, but among these mice, the staining appeared to be less intense in B6.baff-/-.cd152-/- mice (Figure 3B j, l, n). Of note, renal IgG3 deposition was not detected in any CD152-deficient mouse tested (Figure 3B r, t, v), which contrasts with the substantial renal deposition of IgG1, IgG2b, and IgG3 in 6-month-old, but not 2-month-old, NZM 2328 mice (Figure 3B g, h, o, p, w, x).
In contrast to the severe and widespread GN that develops with age in either BAFF-sufficient or BAFF-deficient NZM 2328 mice (31), GN in 3- to 5-week-old CD152-deficient (or CD152-sufficient) mice was quite limited, with endocapillary proliferation and mesangial hypercellularity only occasionally seen. In contrast, a very different picture emerged with regard to interstitial nephritis. Whereas interstitial nephritis was largely absent in B6.baff+/+.cd152+/+ mice at 3-5 weeks of age, it was prominent in B6.baff+/+.cd152-/- and in B6.BTg.cd152-/- mice of the same age (Figure 4 a, c, and unpublished data). Strikingly and unexpectedly, interstitial nephritis was also a feature of CD152-sufficient B6.baff-/- mice and was further intensified in CD152-deficient B6.baff-/- mice (Figure 4 b, d). Taken together, not only were the immunopathological consequences of CD152 deficiency for the kidney not inhibited by BAFF deficiency, but they may actually have been aggravated by BAFF deficiency.
Figure 4. Renal histology in CD152-sufficient and CD152-deficient B6.baff+/+ and B6.baff-/- mice.
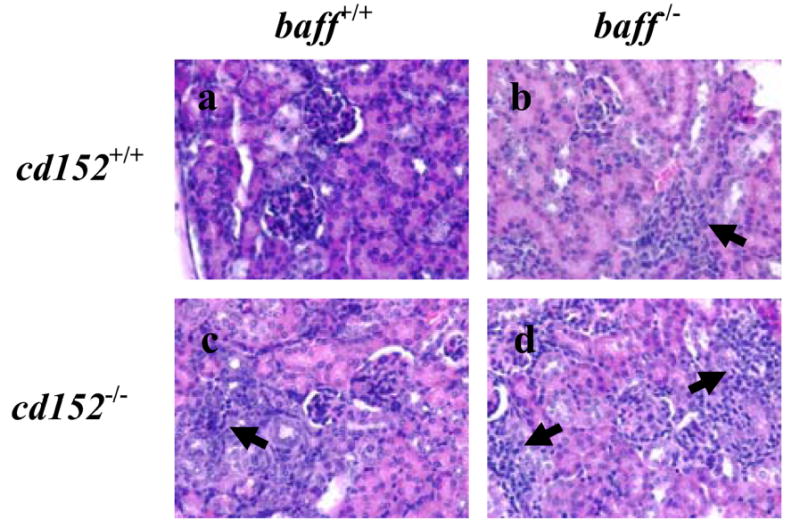
Representative sections of formalin-fixed kidneys from B6.baff+/+.cd152+/+ (a), B6.baff-/-.cd152+/+ (b), B6.baff+/+.cd152-/- (c), and B6.baff-/-.cd152-/- (d) mice were stained with hematoxylin and eosin. The arrows point to overt areas of interstitial nephritis. Original magnification is 400X.
Discussion
Two striking conclusions can be drawn from the present study. First, the complete absence of CD152 leads to development of hypergammaglobulinemia and considerable humoral autoimmunity, including the production of nephrophilic autoantibodies and/or autoantibodies commonly associated with SLE (IgG anti-chromatin, IgG anti-histone, IgG anti-dsDNA, and IgG anti-ssDNA antibodies). The development of such humoral autoimmunity in CD152-deficient B6 mice (which harbor ostensibly “normal” B cells, a “normal” innate immune system, and an otherwise “non-autoimmune-prone” environment) by 3-5 weeks of age is far more rapid than that which occurs in “bona fide” SLE mice (e.g., NZM 2328 mice). Deposition of IgG and C3 in the glomeruli of CD152-deficient B6 mice is readily demonstrable, and such mice develop interstitial nephritis to a much greater degree than do their CD152-sufficient counterparts. Of interest, dramatic glomerular histologic changes are not associated with the renal IgG and C3 deposition. The paucity and early stage of histologic GN may reflect the very young age of the mice at the time of their sacrifice. It is possible that widespread GN would develop were the mice able to live longer. This speculation, however, is inherently indeterminate due to the uniformly early mortality of CD152-deficient mice from failure of non-renal vital organs (e.g., heart, liver).
The humoral autoimmunity and immunopathology that develop in CD152-deficient are almost certainly due to global T cell dysregulation rather than due to any intrinsic B cell abnormality. Experiments with chimeric mice harboring CD152-sufficient T cells and CD152-deficient B cells have demonstrated that the frequencies of peripheral B cells, total circulating IgM and IgG levels, and primary and secondary antigen-specific IgG responses are no different than those in chimeric mice harboring CD152-sufficient B cells rather than CD152-deficient B cells (50).
Although complete absence of CD152 has never been associated with human autoimmune disease, polymorphisms within the Cd152 gene have been associated with several human disorders characterized by autoantibody production, including SLE (51-53). This raises the possibility that alterations in CD152 function may importantly affect development of autoantibodies, including potentially pathogenic ones, not just in mice but in humans as well. Moreover, autoantibodies against CD152 circulate in some patients with systemic immune-based rheumatic diseases (54) and, thereby, may further compromise the regulatory function of CD152 and aggravate the autoimmune process.
The second and, perhaps, more striking conclusion from the present study is that the entire spectrum of autoimmune features observed in CD152-deficient mice can develop in the complete absence of BAFF. Regardless of their genetic background (non-autoimmune-prone or autoimmune-prone), there is a ~90% reduction in spleen B cells (including transitional, FO, and MZ B cells) among baff-/- mice relative to their baff+/+ counterparts (30, 31). Although CD152 deficiency, regardless of the baff genotype, considerably affected the distribution of B cells among phenotypically-defined B cell subsets, CD152 deficiency did not result in appreciable expansion of B cells and, of great importance, did not restore B cell numbers in B6.baff-/- mice to levels observed in B6.baff+/+ mice. That is, CD152-deficient B6.baff-/- mice displayed a marked B cell deficiency, similar in degree to that displayed by CD152-sufficient B6.baff-/- mice.
This B cell deficiency notwithstanding, hypergammaglobulinemia developed in CD152-deficient mice by 3-5 weeks of age. Although serum levels of total IgM and total IgG2b were significantly lower in B6.baff-/-.cd152-/- mice than in B6. baff+/+.cd152-/- mice, serum total IgG1 levels in these respective mice were similar. Since BAFF can preferentially promote Th1 responses (55), the preferential global production of IgG1 in BAFF-deficient hosts may be a reflection of a shift from Th1 responses to Th2 responses (which would favor production of IgG1 over IgG2b).
In any case, there was only limited difference among B6.baff-/-.cd152-/- mice, B6.BTg.cd152-/- mice, and B6.baff+/+.cd152-/- mice when it came to production of SLE-associated autoantibodies. Although serum levels of IgG anti-chromatin antibodies in B6.baff-/-.cd152-/- mice were modestly (albeit significantly) lower than those in the other two cohorts, serum levels of IgG anti-histone, IgG anti-ssDNA, and IgG anti-dsDNA antibodies were essentially identical in B6.baff-/-.cd152-/- and B6.baff+/+.cd152-/- mice. Importantly, despite serum levels of IgG anti-ssDNA and IgG anti-dsDNA being greater in B6.BTg.cd152-/- mice than in the other CD152-deficient cohorts, glomerular deposition of IgG1 and IgG2b in B6.baff-/-.cd152-/- mice, B6.BTg.cd152-/- mice, and B6.baff+/+.cd152-/- mice was comparable and was associated with concurrent deposition of C3. Thus, global T cell dysregulation arising from CD152 deficiency drove considerable serologic autoimmunity and renal IgG and C3 deposition in non-autoimmune-prone B6 mice by 3-5 weeks of age. This was not substantially affected either by the complete absence of BAFF or by constitutive supra-physiologic overexpression of BAFF. The fact that some differences exist between B6.baff-/-.cd152-/- (and B6. baff+/+.cd152-/- or B6.BTg.cd152-/-) and bona fide SLE-prone NZM 2328 mice, such as the renal deposition of IgG3 or the intact MOMA-1+ rings surrounding spleen follicles in the latter but not in the former, likely demonstrates that the numerous genetic differences between NZM 2328 and CD152-deficient B6 mice do lead to phenotypic differences.
Although we have tacitly assumed that the B cells in B6.baff-/- mice are “normal”, this may not necessarily be strictly correct. Since all the B cells in BAFF-deficient hosts will have matured and differentiated along a BAFF-independent pathway, they may be more sensitive and/or responsive to alternate maturation/differentiation factors than are B cells that mature and differentiate in a BAFF-sufficient environment. Future studies that assess the effects of pharmacologic inhibition of BAFF on autoimmunity in CD152-deficient BAFF-sufficient hosts should lend insight into this matter.
Given that widespread T cell expansion and T cell infiltration into vital organs are cardinal features of CD152-deficient mice (36-38), it is not surprising that we documented the development of substantial interstitial nephritis in our CD152-deficient mice. What was unexpected was the considerable “baseline” interstitial nephritis that developed in CD152-sufficient B6.baff-/- mice by 3-5 weeks of age. Previous reports of BAFF-deficient (non-autoimmune-prone) mice have not included descriptions of kidney histology (29, 30, 56, 57), so the clinical importance of interstitial nephritis in these mice and the underlying mechanisms warrant further investigation.
The unexpected finding of interstitial nephritis in B6.baff-/- mice notwithstanding, the development of autoimmunity in the absence of BAFF should not be taken to mean that BAFF plays no contributory role in disease pathogenesis. Indeed, BAFF overexpression can promote SLE-like autoimmunity even in the absence of T cells (28). That is, abnormal T cell help may be dispensable for SLE-like features. Disease that develops in hosts bearing certain intrinsic “abnormalities” of B cells and/or the innate immune system might be highly responsive to BAFF elimination/neutralization.Nevertheless, the rapid T cell-driven development of substantial serological autoimmunity and end-organ (kidney) immunopathology in a host that harbors an otherwise “non-autoimmune-prone” environment despite the complete absence of BAFF does raise some doubt regarding the ultimate utility of BAFF antagonists in clinical practice. Although treatment of SLE-prone (NZB × NZW)F1 or MRL-lpr/lpr mice with the BAFF antagonist, TACI-Ig, did attenuate disease and enhance survival (6), at least some of the clinical efficacy may have come from the ability of TACI-Ig to neutralize not just BAFF but APRIL as well. Although BAFFR-Ig, a BAFF-specific antagonist without any APRIL-neutralizing activity, was able to attenuate end-organ pathological changes in murine SLE (58, 59), only short-term effects were assessed. It remains plausible that in the long-term, disease would have emerged in these mice despite the neutralization of BAFF. Indeed, the development of serological and pathological features of SLE in BAFF-deficient NZM 2328 mice (31) and the development of full-blown serological, pathological, and clinical disease in MRL-lpr/lpr mice bearing a mutant BAFFR incapable of transducing BAFF-triggered signals (60) support this premise.
Human experience to date also raises concerns regarding the efficacy of BAFF antagonism. Although clinical trials in human SLE with a neutralizing anti-BAFF mAb have documented a highly favorable safety profile, they have demonstrated, at most, only modest clinical benefit (61, 62). Although these observations should not be interpreted as showing the absence of any benefit from solely targeting BAFF, they do strongly suggest that therapeutic targeting of BAFF alone is inadequate. Optimal therapeutic utility of BAFF antagonism may require combination with other agents. Further investigation in both murine models and in human subjects will be needed to delineate the precise therapeutic niche for BAFF antagonists.
Footnotes
This work was supported in part by NIH grants R01 AR050193 (WS), R01 AI054488 (MPC), R01 AI073939 (MPC), R01 AR049765 (LP), R01 AR048692 (CP), and P01 AI051392 (CP); a grant from the Mary Kirkland Center for Lupus Research (LP), a Target Identification in Lupus Award from the Alliance for Lupus Research/Arthritis Foundation (CP), and a Hulda Irene Duggan Arthritis Investigator Award from the Arthritis Foundation (CP). The authors have no conflicting financial interests.
Abbreviations: B6, C57BL/6; BAFF, B cell activating factor belonging to the TNF family; BTg, BAFF-transgenic; FO, follicular; GN, glomerulonephritis; MZ, marginal zone; SLE, systemic lupus erythematosus; T1, transitional 1; Tg, transgenic; USC, University of Southern California
References
- 1.Schneider P, MacKay F, Steiner V, Hofmann K, Bodmer J-L, Holler N, Ambrose C, Lawton P, Bixler S, Acha-Orbea H, Valmori D, Romero P, Werner-Favre C, Zubler RH, Browning JL, Tschopp J. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med. 1999;189:1747–1756. doi: 10.1084/jem.189.11.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore PA, Belvedere O, Orr A, Pieri K, LaFleur DW, Feng P, Soppet D, Charters M, Gentz R, Parmelee D, Li Y, Galperina O, Giri J, Roschke V, Nardelli B, Carrell J, Sosnovtseva S, Greenfield W, Ruben SM, Olsen HS, Fikes J, Hilbert DM. BLyS: member of the tumor necrosis factor family and B lymphocyte stimulator. Science. 1999;285:260–263. doi: 10.1126/science.285.5425.260. [DOI] [PubMed] [Google Scholar]
- 3.Shu H-B, Hu W-H, Johnson H. TALL-1 is a novel member of the TNF family that is down-regulated by mitogens. J Leukocyte Biol. 1999;65:680–683. [PubMed] [Google Scholar]
- 4.Mukhopadhyay A, Ni J, Zhai Y, Yu G-L, Aggarwal BB. Identification and characterization of a novel cytokine, THANK, a TNF homologue that activates apoptosis, nuclear factor-κB, and c-Jun NH2-terminal kinase. J Biol Chem. 1999;274:15978–15981. doi: 10.1074/jbc.274.23.15978. [DOI] [PubMed] [Google Scholar]
- 5.Tribouley C, Wallroth M, Chan V, Paliard X, Fang E, Lamson G, Pot D, Escobedo J, Williams LT. Characterization of a new member of the TNF family expressed on antigen presenting cells. Biol Chem. 1999;380:1443–1447. doi: 10.1515/BC.1999.186. [DOI] [PubMed] [Google Scholar]
- 6.Gross JA, Johnston J, Mudri S, Enselman R, Dillon SR, Madden K, Xu W, Parrish-Novak J, Foster D, Lofton-Day C, Moore M, Littau A, Grossman A, Haugen H, Foley K, Blumberg H, Harrison K, Kindsvogel W, Clegg CH. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. 2000;404:995–999. doi: 10.1038/35010115. [DOI] [PubMed] [Google Scholar]
- 7.Nardelli B, Belvedere O, Roschke V, Moore PA, Olsen HS, Migone TS, Sosnovtseva S, Carrell JA, Feng P, Giri JG, Hilbert DM. Synthesis and release of B-lymphocyte stimulator from myeloid cells. Blood. 2001;97:198–204. doi: 10.1182/blood.v97.1.198. [DOI] [PubMed] [Google Scholar]
- 8.Scapini P, Nardelli B, Nadali G, Calzetti F, Pizzolo G, Montecucco C, Cassatella MA. G-CSF-stimulated neutrophils are a prominent source of functional BLyS. J Exp Med. 2003;197:297–302. doi: 10.1084/jem.20021343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gorelik L, Gilbride K, Dobles M, Kalled SL, Zandman D, Scott ML. Normal B cell homeostasis requires B cell activation factor production by radiation-resistant cells. J Exp Med. 2003;198:937–945. doi: 10.1084/jem.20030789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laabi Y, Gras M-P, Brouet J-C, Berger R, Larsen C-J, Tsapis A. The BCMA gene, preferentially expressed during B lymphoid maturation, is bidirectionally transcribed. Nucleic Acids Res. 1994;22:1147–1154. doi: 10.1093/nar/22.7.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.von Bülow G-U, Bram RJ. NF-AT activation induced by a CAML-interacting member of the tumor necrosis factor receptor superfamily. Science. 1997;278:138–141. doi: 10.1126/science.278.5335.138. [DOI] [PubMed] [Google Scholar]
- 12.Thompson JS, Bixler SA, Qian F, Vora K, Scott ML, Cachero TG, Hession C, Schneider P, Sizing ID, Mullen C, Strauch K, Zafari M, Benjamin CD, Tschopp J, Browning JL, Ambrose C. BAFF-R, a novel TNF receptor that specifically interacts with BAFF. Science. 2001;293:2108–2111. doi: 10.1126/science.1061965. [DOI] [PubMed] [Google Scholar]
- 13.Yan M, Brady JR, Chan B, Lee WP, Hsu B, Harless S, Cancro M, Grewal IS, Dixit VM. Identification of a novel receptor for B lymphocyte stimulator that is mutated in a mouse strain with severe B cell deficiency. Curr Biol. 2001;11:1547–1552. doi: 10.1016/s0960-9822(01)00481-x. [DOI] [PubMed] [Google Scholar]
- 14.Thompson JS, Schneider P, Kalled SL, Wang L, Lefevre EA, Cachero TG, MacKay F, Bixler SA, Zafari M, Liu Z-Y, Woodcock SA, Qian F, Batten M, Madry C, Richard Y, Benjamin CD, Browning JL, Tsapis A, Tschopp J, Ambrose C. BAFF binds to the tumor necrosis factor receptor-like molecule B cell maturation antigen and is important for maintaining the peripheral B cell population. J Exp Med. 2000;192:129–135. doi: 10.1084/jem.192.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Do RKG, Hatada E, Lee H, Tourigny MR, Hilbert D, Chen-Kiang S. Attenuation of apoptosis underlies B lymphocyte stimulator enhancement of humoral immune response. J Exp Med. 2000;192:953–964. doi: 10.1084/jem.192.7.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Batten M, Groom J, Cachero TG, Qian F, Schneider P, Tschopp J, Browning JL, Mackay F. BAFF mediates survival of peripheral immature B lymphocytes. J Exp Med. 2000;192:1453–1465. doi: 10.1084/jem.192.10.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harless SM, Lentz VM, Sah AP, Hsu BL, Clise-Dwyer K, Hilbert DM, Hayes CE, Cancro MP. Competition for BLyS-mediated signaling through Bcmd/BR3 regulates peripheral B lymphocyte numbers. Curr Biol. 2001;11:1986–1989. doi: 10.1016/s0960-9822(01)00598-x. [DOI] [PubMed] [Google Scholar]
- 18.Hsu BL, Harless SM, Lindsley RC, Hilbert DM, Cancro MP. Cutting edge: BLyS enables survival of transitional and mature B cells through distinct mediators. J Immunol. 2002;168:5993–5996. doi: 10.4049/jimmunol.168.12.5993. [DOI] [PubMed] [Google Scholar]
- 19.Avery DT, Kalled SL, Ellyard JI, Ambrose C, Bixler SA, Thien M, Brink R, Mackay F, Hodgkin PD, Tangye SG. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J Clin Invest. 2003;112:286–297. doi: 10.1172/JCI18025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hatada EN, Do RKG, Orlofsky A, Liou H-C, Prystowsky M, MacLennan ICM, Caamano J, Chen-Kiang S. NF-κB1 p50 is required for BLyS attentuation of apoptosis but dispensible for processing of NF-κB2 p100 to p52 in quiescent mature B cells. J Immunol. 2003;171:761–768. doi: 10.4049/jimmunol.171.2.761. [DOI] [PubMed] [Google Scholar]
- 21.Rolink AG, Tschopp J, Schneider P, Melchers F. BAFF is a survival and maturation factor for mouse B cells. Eur J Immunol. 2002;32:2004–2010. doi: 10.1002/1521-4141(200207)32:7<2004::AID-IMMU2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 22.Tardivel A, Tinel A, Lens S, Steiner Q-G, Sauberli E, Wilson A, Mackay F, Rolink AG, Beermann F, Tschopp J, Schneider P. The anti-apoptotic factor Bcl-2 can functionally substitute for the B cell survival but not for the marginal zone B cell differentiation activity of BAFF. Eur J Immunol. 2004;34:509–518. doi: 10.1002/eji.200324692. [DOI] [PubMed] [Google Scholar]
- 23.Litinskiy MB, Nardelli B, Hilbert DM, He B, Schaffer A, Casali P, Cerutti A. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol. 2002;3:822–829. doi: 10.1038/ni829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamada T, Zhang K, Yamada A, Zhu D, Saxon A. B lymphocyte stimulator activates p38 mitogen-activated protein kinase in human Ig class switch recombination. Am J Respir Cell Mol Biol. 2005;32:388–394. doi: 10.1165/rcmb.2004-0317OC. [DOI] [PubMed] [Google Scholar]
- 25.Castigli E, Wilson SA, Scott S, Dedeoglu F, Xu S, Lam K-P, Bram RJ, Jabara H, Geha RS. TACI and BAFF-R mediate isotype switching in B cells. J Exp Med. 2005;201:35–39. doi: 10.1084/jem.20032000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, Tschopp J, Browning JL. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med. 1999;190:1697–1710. doi: 10.1084/jem.190.11.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khare SD, Sarosi I, Xia X-Z, McCabe S, Miner K, Solovyev I, Hawkins N, Kelley M, Chang D, Van G, Ross L, Delaney J, Wang L, Lacey D, Boyle WJ, Hsu H. Severe B cell hyperplasia and autoimmune disease in TALL-1 transgenic mice. Proc Natl Acad Sci USA. 2000;97:3370–3375. doi: 10.1073/pnas.050580697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Groom JR, Fletcher CA, Walters SN, Grey ST, Watt SV, Sweet MJ, Smyth MJ, Mackay CR, Mackay F. BAFF and MyD88 signals promote a lupuslike disease independent of T cells. J Exp Med. 2007;204:1959–1971. doi: 10.1084/jem.20062567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gross JA, Dillon SR, Mudri S, Johnston J, Littau A, Roque R, Rixon M, Schou O, Foley KP, Haugen H, McMillen S, Waggie K, Schreckhise RW, Shoemaker K, Vu T, Moore M, Grossman A, Clegg CH. TACI-Ig neutralizes molecules critical for B cell development and autoimmune disease: impaired B cell maturation in mice lacking BLyS. Immunity. 2001;15:289–302. doi: 10.1016/s1074-7613(01)00183-2. [DOI] [PubMed] [Google Scholar]
- 30.Schiemann B, Gommerman JL, Vora K, Cachero TG, Shulga-Morskaya S, Dobles M, Frew E, Scott ML. An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science. 2001;293:2111–2114. doi: 10.1126/science.1061964. [DOI] [PubMed] [Google Scholar]
- 31.Jacob CO, Pricop L, Putterman C, Koss MN, Liu Y, Kollaros M, Bixler SA, Ambrose CM, Scott ML, Stohl W. Paucity of clinical disease despite serological autoimmunity and kidney pathology in lupus-prone New Zealand Mixed 2328 mice deficient in BAFF. J Immunol. 2006;177:2671–2680. doi: 10.4049/jimmunol.177.4.2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Read S, Malmström V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25+CD4+ regulatory cells that control intestinal inflammation. J Exp Med. 2000;192:295–302. doi: 10.1084/jem.192.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, Mak TW, Sakaguchi S. Immunologic self-tolerance maintained by CD25+CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303–309. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perez VL, van Parijs L, Biuckians A, Zheng XX, Strom TB, Abbas AK. Induction of peripheral T cell tolerance in vivo requires CTLA-4 engagement. Immunity. 1997;6:411–417. doi: 10.1016/s1074-7613(00)80284-8. [DOI] [PubMed] [Google Scholar]
- 35.Eager TN, Karandikar NJ, Bluestone JA, Miller SD. The role of CTLA-4 in induction and maintenance of peripheral T cell tolerance. Eur J Immunol. 2002;32:972–981. doi: 10.1002/1521-4141(200204)32:4<972::AID-IMMU972>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 36.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 37.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 38.Chambers CA, Sullivan TJ, Allison JP. Lymphoproliferation in CTLA-4-deficient mice is mediated by costimulation-dependent activation of CD4+ T cells. Immunity. 1997;7:885–895. doi: 10.1016/s1074-7613(00)80406-9. [DOI] [PubMed] [Google Scholar]
- 39.Chambers CA, Cado D, Truong T, Allison JP. Thymocyte development is normal in CTLA-4-deficient mice. Proc Natl Acad Sci USA. 1997;94:9296–9301. doi: 10.1073/pnas.94.17.9296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waterhouse P, Bachmann MF, Penninger JM, Ohashi PS, Mak TW. Normal thymic selection, normal viability and decreased lymphoproliferation in T cell receptor-transgenic CTLA-4-deficient mice. Eur J Immunol. 1997;27:1887–1892. doi: 10.1002/eji.1830270811. [DOI] [PubMed] [Google Scholar]
- 41.Chambers CA, Sullivan TJ, Truong T, Allison JP. Secondary but not primary T cell responses are enhanced in CTLA-4-deficient CD8+ T cells. Eur J Immunol. 1998;28:3137–3143. doi: 10.1002/(SICI)1521-4141(199810)28:10<3137::AID-IMMU3137>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 42.Gajewski TF, Fallarino F, Fields PE, Rivas F, Alegre M-L. Absence of CTLA-4 lowers the activation threshold of primed CD8+ TCR-transgenic T cells: lack of correlation with Scr homology domain 2-containing protein tyrosine phosphatase. J Immunol. 2001;166:3900–3907. doi: 10.4049/jimmunol.166.6.3900. [DOI] [PubMed] [Google Scholar]
- 43.Greenwald RJ, Boussiotis VA, Lorsbach RB, Abbas AK, Sharpe AH. CTLA-4 regulates induction of anergy in vivo. Immunity. 2001;14:145–155. doi: 10.1016/s1074-7613(01)00097-8. [DOI] [PubMed] [Google Scholar]
- 44.Gozalo-Sanmillan S, McNally JM, Lin MY, Chambers CA, Berg LJ. Cutting edge: two distinct mechanisms lead to impaired T cell homeostasis in Janus kinase 3- and CTLA-4-deficient mice. J Immunol. 2001;166:727–730. doi: 10.4049/jimmunol.166.2.727. [DOI] [PubMed] [Google Scholar]
- 45.Stohl W, Xu D, Kim KS, David CS, Allison JP. MHC class II-independent and -dependent T cell expansion and B cell hyperactivity in vivo in mice deficient in CD152 (CTLA-4) Int Immunol. 2004;16:895–904. doi: 10.1093/intimm/dxh091. [DOI] [PubMed] [Google Scholar]
- 46.Stohl W, Xu D, Kim KS, Koss MN, Jorgensen TN, Deocharan B, Metzger TE, Bixler SA, Hong YS, Ambrose CM, Mackay F, Morel L, Putterman C, Kotzin BL, Kalled SL. BAFF overexpression and accelerated glomerular disease in mice with an incomplete genetic predisposition to systemic lupus erythematosus. Arthritis Rheum. 2005;52:2080–2091. doi: 10.1002/art.21138. [DOI] [PubMed] [Google Scholar]
- 47.Deocharan B, Marambio P, Edelman M, Putterman C. Differential effects of interleukin-4 in peptide induced autoimmunity. Clin Immunol. 2003;108:80–88. doi: 10.1016/s1521-6616(03)00096-2. [DOI] [PubMed] [Google Scholar]
- 48.Zhao Z, Burkly LC, Campbell S, Schwartz N, Molano A, Choudhury A, Eisenberg RA, Michaelson JS, Putterman C. TWEAK/Fn14 interactions are instrumental in the pathogenesis of nephritis in the chronic graft-versus-host model of systemic lupus erythematosus. J Immunol. 2007;179:7949–7958. doi: 10.4049/jimmunol.179.11.7949. [DOI] [PubMed] [Google Scholar]
- 49.Kallies A, Hasbold J, Tarlinton DM, Dietrich W, Corcoran LM, Hodgkin PD, Nutt SL. Plasma cell ontogeny defined by quantitative changs in Blimp-1 expression. J Exp Med. 2004;200:967–977. doi: 10.1084/jem.20040973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quandt D, Hoff H, Rudolph M, Fillatreau S, Brunner-Weinzierl MC. A new role of CTLA-4 on B cells in thymus-dependent immune responses in vivo. J Immunol. 2007;179:7316–7324. doi: 10.4049/jimmunol.179.11.7316. [DOI] [PubMed] [Google Scholar]
- 51.Kouki T, Sawai Y, Gardine CA, Fisfalen M-E, Alegre M-L, DeGroot LJ. CTLA-4 gene polymorphism at position 49 in exon 1 reduces the inhibitory function of CTLA-4 and contributes to the pathogenesis of Graves’ disease. J Immunol. 2000;165:6606–6611. doi: 10.4049/jimmunol.165.11.6606. [DOI] [PubMed] [Google Scholar]
- 52.Ueda H, Howson JMM, Esposito L, Heward J, Snook H, Chamberlain G, Rainbow DB, Hunter KMD, Smith AN, Di Genova G, Herr MH, Dahlman I, Payne F, Smyth D, Lowe C, Twells RCJ, Howlett S, Healy B, Hutland S, Rance HE, Everett V, Smink LJ, Lam AC, Cordell HJ, Walker HM, Bordin C, Hulme J, Motzo C, Cucca F, Hess JF, Metzker ML, Rogers J, Gregory S, Allahabadia A, Nithiyananthan R, Tuomilehto-Wolf E, Tuomilehto J, Bingley P, Gillespie KM, Undlien DE, Ronningen KS, Guja C, Ionescu-Tirgoviste C, Savage DA, Maxwell AP, Carson DJ, Patterson CC, Franklyn JA, Clayton DG, Peterson LB, Wicker LS, Todd JA, Gough SCL. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506–511. doi: 10.1038/nature01621. [DOI] [PubMed] [Google Scholar]
- 53.Lee YH, Harley JB, Nath SK. CTLA-4 polymorphisms and systemic lupus erythematosus (SLE): a meta-analysis. Hum Genet. 2005;116:361–367. doi: 10.1007/s00439-004-1244-1. [DOI] [PubMed] [Google Scholar]
- 54.Matsui T, Kurokawa M, Kobata T, Oki S, Azuma M, Tohma S, Inoue T, Yamamoto K, Nishioka K, Kato T. Autoantibodies to T cell costimulatory molecules in systemic autoimmune diseases. J Immunol. 1999;162:4328–4335. [PubMed] [Google Scholar]
- 55.Sutherland APR, Ng LG, Fletcher CA, Shum B, Newton RA, Grey ST, Rolph MS, Mackay F, Mackay CR. BAFF augments certain Th1-associated inflammatory responses. J Immunol. 2005;174:5537–5544. doi: 10.4049/jimmunol.174.9.5537. [DOI] [PubMed] [Google Scholar]
- 56.Vora KA, Wang LC, Rao SP, Liu Z-Y, Majeau GR, Cutler AH, Hochman PS, Scott ML, Kalled SL. Cutting edge: geminal centers formed in the absence of B cell-activating factor belonging to the TNF family exhibit impaired maturation and function. J Immunol. 2003;171:547–551. doi: 10.4049/jimmunol.171.2.547. [DOI] [PubMed] [Google Scholar]
- 57.Rahman ZSM, Rao SP, Kalled SL, Manser T. Normal induction but attenuated progression of germinal center responses in BAFF and BAFF-R signaling-deficient mice. J Exp Med. 2003;198:1157–1169. doi: 10.1084/jem.20030495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kayagaki N, Yan M, Seshasayee D, Wang H, Lee W, French DM, Grewal IS, Cochran AG, Gordon NC, Yin J, Starovasnik MA, Dixit VM. BAFF/BLyS receptor 3 binds the B cell survival factor BAFF ligand through a discrete surface loop and promotes processing of NF-κB2. Immunity. 2002;17:515–524. doi: 10.1016/s1074-7613(02)00425-9. [DOI] [PubMed] [Google Scholar]
- 59.Ramanujam M, Wang X, Huang W, Liu Z, Schiffer L, Tao H, Frank D, Rice J, Diamond B, Yu KOA, Porcelli S, Davidson A. Similarities and differences between selective and nonselective BAFF blockade in murine SLE. J Clin Invest. 2006;116:724–734. doi: 10.1172/JCI26385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ju ZL, Shi GY, Zuo JX, Zhang JW, Sun J. Unexpected development of autoimmunity in BAFF-R-mutant MRL-lpr mice. Immunology. 2007;120:281–289. doi: 10.1111/j.1365-2567.2006.02500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Furie R, Stohl W, Ginzler E, Becker M, Mishra N, Chatham W, Merrill JT, Weinstein A, McCune WJ, Zhong J, Freimuth W Lymphostat-B Study Group. Safety, pharmacokinetic and pharmacodynamic results of a phase 1 single and double dose-escalation study of Lymphostat-B (human monoclonal antibody to BLyS) in SLE patients. Arthritis Rheum. 2003;48:S377. [Google Scholar]
- 62.Wallace DJ, Lisse J, Stohl W, McKay J, Boling E, Merrill JT, Furie R, Petri M, Ginzler E, Chatham W, Fernandez V, Zhong J, Chevrier M, Freimuth W LBSL02 Study Grp. Belimumab (BmAb) reduces SLE disease activity and demonstrates durable bioactivity at 76 weeks. Arthritis Rheum. 2006;54:S790. [Google Scholar]