

Abstract
We recently identified autocrine interferon (IFN)β as a novel mechanism mediating tumor necrosis factor (TNF)α-induced expression of inflammatory genes in airway smooth muscle (ASM) cells, including CD38, known to regulate calcium signaling. Here, we investigated the putative involvement of IFNβ in regulating TNFα-induced airway hyper-responsiveness (AHR), a defining feature of asthma. Using our pharmacodynamic model to assess ex vivo AHR isolated murine tracheal rings, we found that TNFα-induced enhanced contractile responses to carbachol and bradykinin was abrogated by neutralizing anti-IFNβ antibody or in tracheal rings deficient in CD38. In cultured human ASM cells, where CD38 has been involved in TNFα-induced enhanced calcium signals to carbachol and bradykinin, we found that neutralizing anti-IFNβ prevented TNFα enhancing action only on carbachol responses but not to that induced by bradykinin. In a well-characterized model of allergic asthma (mice sensitized and challenged with Aspergillus fumigatus (Af)), we found heightened expression of both IFNβ and CD38 in the airways. Furthermore, allergen-associated AHR to methacholine, assessed by lung resistance and dynamic compliance, was completely suppressed in CD38-deficient mice, despite the preservation of airway inflammation. These data provide the first evidence that ASM-derived IFNβ and CD38 may play a significant role in the development of TNFα-associated AHR.
Keywords: Allergic asthma, Cytokine, Airway smooth muscle, Inflammation, Calcium signaling, Hypercontractility
Introduction
Considerable in vitro and in vivo evidence suggests that tumor necrosis factor (TNF)α plays a key role in the development of non-specific bronchial hyper-responsiveness (AHR), a central feature of asthma (Boushey et al., 1980). In both normal and asthmatic subjects, Thomas and colleagues demonstrated that oral administration of TNFα increased airway responsiveness to methacholine (MCh) (Thomas, 2001; Thomas et al., 1995). Similarly, oral or intra-peritoneal administration of TNFα to different animal species induces AHR to different G-protein-coupled receptor (GPCR) agonists (Kips et al., 1992; Wheeler et al., 1992). Using the soluble protein inhibitor Ro-45-2981, Renzetti et al. (1996) provided the evidence for a direct role of TNFα in allergen-associated airway inflammation and AHR in sensitized guinea-pigs and Brown Norway rats. Additional studies performed in receptor knockout mice further confirmed the involvement of both TNFα receptors, TNFR1 and TNFR2, in the development of AHR triggered by allergen challenge in sensitized mice (Kanehiro et al., 2002) or following ozone exposure (Shore et al., 2001). Further, others showed a therapeutic benefit of anti-TNFα antibodies (Etanercept) in patients with severe asthma (Berry et al., 2006; Howarth et al., 2005). The precise mechanisms by which TNFα promotes AHR remain unknown, although studies from our laboratory and others showed that modulation of calcium metabolism in airway smooth muscle (ASM), the main effector tissue regulating bronchoconstriction, may represent a molecular mechanism linking TNFα to AHR (Amrani, 2006).
Using isolated airway preparations from different species, TNFα has been shown to enhance ASM responsiveness to agonists by either augmenting ASM reactivity (upward shift of the dose–response curve) and/or ASM sensitivity (leftward shift of the curve), suggesting the involvement of multiple molecular mechanisms in TNFα effects on ASM (Adner et al., 2002; Amrani and Panettieri, 2002; Anticevich et al., 1995; Sakai et al., 2004a; Sukkar et al., 2001). Additionally, we showed that in cultured human ASM cells TNFα potentiated, also in a non-specific manner, calcium signals in response to different GPCR agonists (Amrani, 2006). Parallel studies further supported the potential role of TNFα–ASM interaction in AHR by showing that TNFα acts by inducing Rho-dependent calcium sensitization (Hunter et al., 2003; Hunter and Nixon, 2006; MacEwan, 2002; McFarlane et al., 2001; Parris et al., 1999; Sakai et al., 2004b). Together, these investigators demonstrate that TNFα promotes AHR by modulating ASM contractility, an effect that could result from an altered GPCR-associated calcium signaling.
In our recent studies, we demonstrated that autocrine interferon (IFN)β is a novel signaling molecule that mediates some TNFα effects in ASM cells, including expression of inflammatory proteins such as IL-6 and RANTES (Tliba et al., 2003a, b, 2004). The observation that autocrine IFNβ also partially participates in mediating TNFα-induced expression of CD38 (Tliba et al., 2004), a critical ectoenzyme that was shown to regulate ASM reactivity to contractile agonists in mice (Deshpande et al., 2004, 2003, 2005) prompted us to investigate the potential role of IFNβ–CD38 pathways in TNFα induction of in vitro and ex vivo AHR.
Materials and methods
Animals
In all, 8–12-week-old female C57BL/6 wild-type (WT) or CD38-deficient (CD38−/−) mice (Jackson Laboratories, Bar Harbor, ME), were housed under pathogen-free conditions. This study was approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. Sensitization and challenge to Aspergillus fumigatus (Af) was described previously (Haczku et al., 2000, 2001, 2002). Briefly, 27 days after intra-peritoneal sensitization with Af, sensitized and control mice were anesthetized by isoflurane inhalation, and 25 μl of Af extract or vehicle was applied to the left nares, respectively. All mice were sacrificed 24 h after their intranasal treatment, unless specified otherwise, when the peaks of eosinophil infiltration and airway responses were assumed to occur. Naïve mice that received intranasal glycerol treatment alone showed no difference compared to non-sensitized, normal C57BL/6 mice in any of the study’s parameters investigated, including lung histology, BAL cellular content, immunoglobulin, cytokine profile and airway responses to acetylcholine (data not shown).
In vivo measurement of airway responsiveness to acetylcholine (ACh)
Airway function measurements were carried out as previously described (Haczku et al., 2000, 2001, 2002). Lung function tests were assessed 24 h after the Af or saline challenge. Bronchial reactivity to aerosolized MCh was measured using the FlexiVent® system (Sireq, Montreal, Canada). Lung mechanics were studied in tracheostomized mice under anesthesia by intra-peritoneal injection of ketamine and xylazine. Mice were ventilated with a tidal volume of 8 ml/kg at a rate of 450 breaths/min and a positive end-expiratory pressure of 2 cm H2O by a computerized FlexiVent System. After mechanical ventilation for 2 min, a sinusoidal 1-Hz oscillation was applied to the tracheal tube. The single-compartment model was fitted to these data by multiple linear regression to calculate dynamic resistance and compliance of the airway.
BAL analysis for differential cell count, cytokine content
Lungs were lavaged using 2 ml of sterile saline. The amount of liquid retained after BAL was an average of 1.75 ml. Cytokine levels were determined from cell free supernatant of the BAL by ELISA using antibodies and recombinant cytokines from PharMingen (San Diego, CA). Total and differential cell counts and ELISA analysis were performed as described previously (Haczku et al., 2000, 2001, 2002).
Immunohistochemistry
After BAL, lungs obtained from naïve and sensitized and challenged mice were inflated with 10% formalin in PBS, excised and immunohistochemistry was performed as described previously (Haczku et al., 2006). FITC-conjugated anti-smooth muscle α-actin was purchased from Sigma Aldrich (St. Louis, MO) and isotype control antibodies were purchased from BD Pharmingen (San Diego, CA). Anti-murine IFNβ antibody (R&D Systems, MN) was detected using an anti-rabbit Alexa 594 secondary antibody (Molecular Probes, Eugene, OR). After washing, the glass coverslips were mounted onto glass slides, examined under epifluorescence microscopy (Nikon, Tokyo, Japan), and photographed.
Measurement of isometric force generation
Isometric force generation by murine-cultured tracheal rings was performed as described previously (Chen et al., 2003; Kim et al., 2005; Tliba et al., 2003a, b). Briefly, mouse tracheae obtained from 7- to 12-week-old female Balb/c mice were sectioned into 3–4 mm long rings. The tracheal rings were then cultured overnight in Ham’s F-12 medium supplemented with 100 mM HEPES, 1.0 M NaOH, 10% fetal bovine serum (Hy-Clone, Logan, UT), 0.2 M glutamine, 1.0 M CaCl2, 100 U/ml penicillin, and 100 μg/ml streptomycin. The organ cultures were treated with 50 ng/ml TNFα or diluent alone for 24 h and then washed with Krebs solution containing (in mM) 118 NaCl, 4.7 KCl, 1.2 KH2PO4, 11.1 dextrose, 1.2 MgSO4, 2.8 CaCl2 and 25 NaHCO3. In some experiments, tracheal rings were pre-incubated with 1000 U/ml neutralizing anti-mouse IFNβ antibody before treatment with TNFα or diluent. Contractile response curves to carbachol (Ch) were obtained by using a range of concentrations from 10−9 to 10−5 M, respectively. Tracheae were mounted with a stainless-steel pin, supported by a Plexiglas rod into the base of a double-jacketed, glass organ bath, maintained at 37 °C and bubbled with 95% O2, 5% CO2, pH of 7.4. Changes in tension of the rings were measured using an FT03 isometric transducer (Astro-Med, Inc., West Warwick, RI) and synchronously recorded with an MP 100WS system (BIOPAC Systems, Inc., Santa Barbara, CA). All initial tensions of tracheal rings were set at approximately 0.5 g. After a 20-min equilibration period, cumulative concentration–isometric force curves were then generated.
Human airway smooth muscle cell culture
Human tracheae were obtained from lung transplant donors in accordance with procedures approved by the University of Pennsylvania Committee on Studies Involving Human Beings at the University of Pennsylvania. ASM culture was performed as described previously (Panettieri et al., 1989).
Measurement of intracellular calcium concentration
Fura-2/AM, a cell-permeant, ratiometric calcium indicator dye was used to determine the intracellular calcium concentration in HASM cells. Calcium measurements were determined as described previously (Amrani et al., 1995). Briefly, human ASM cells grown on 100 mm dishes were washed with HBSS containing 10 mM N-2-hydroxyethylpiperazine-N′-ethane sulfonic acid, 11 mM glucose, 2.5 mM CaCl2, and 1.2 mM MgCl2 (pH 7.4) and incubated with 5 μM fura-2/AM for 30 min at 37 °C and 5% CO2. Fura-2-loaded cells were trypsinized, and fluorescence was estimated in cell suspensions by excitation at 340 and 380 nm wavelength. Fluorescence emissions were collected separately for each wavelength at 510 nm using a spectrofluorimeter (Photon Technology International). The ratio of fluorescence intensities at 340 and 380 nm wavelength was determined and converted to the calcium concentrations using the standard equation. The net calcium responses to contractile agonists were calculated by subtracting the basal from that of the peak intracellular calcium concentration. All experiments were performed at 37 °C.
RT-PCR analysis
Total RNA was extracted from tracheal rings cultured with or without TNFα (50 ng/ml) for 24 h and from lung homogenates of naïve and Af-sensitized mice using Trizol reagent (Invitrogen) according to the manufacturer’s instructions. Reverse transcriptase PCR analysis of IFNβ was then performed as reported previously (Tliba et al., 2003a, b, 2004). The primer sequences used for amplification are: IFNβ forward primer 5′-CAC GAC AGC TCT TTC CAT GA-3′; and reverse 5′-AGC CAG TGC TCG ATG AAT CT-3′, mu-CD 38 forward primer 5′-GCA ACA TCA CAA GAG AAG ACT ACG C-3′ and reverse 5′-ACA CAC TGA AGA AAC CTG GCA GGC C-3′, mu-β-actin forward 5′-CCC CAT TGA ACA TGG CAT TG-3′ and reverse primer 5′-ACG ACC AGA GGC ATA CAG G-3′. Additionally, another set of mu-IFNβ primers was used in some experiments with murine lung. The sequences of this set are forward primer 5′-CCA CCA CAG CCC TCT CCA TCA ACT AT-3′ and the reverse primer 5′-CAA GTG GAG AGC AGT TGA GGA CAT C-3′. PCR products were separated on 1% agarose gels and visualized by staining with ethidium bromide.
Data analysis
Tension was calculated as milligram tensions per milligram tracheal smooth muscle weight (mg/mg) and expressed as percentage (%) of 10−5 M Ch-evoked force of the cultured tracheal rings in the absence of TNFα for contraction studies. The concentrations of agonists required to produce half-maximal contraction were determined, and the EC50 of bradykinin (BK) were then converted to log values (EC50). Concentration of agonists required to produce a half-maximal contraction (pD2) was determined with −log values of the EC50. All values were expressed as means±SEM. Comparisons among groups with or without TNFα were performed by a one-way ANOVA. Student’s unpaired t-test was used to compare the effect of drug treatment. A p-value <0.05 was considered significant. For lung function analyses and calcium experiments, statistical analysis was performed with Prism4 software (GraphPad Inc., San Diego, CA). Student t-test was used for pair wise comparisons and two-way-ANOVA for analysis of time courses. Data are expressed as means±SEM; p<0.05 was considered statistically significant.
Reagents
Tissue culture reagents and primers used for PCR were obtained from Invitrogen (Carlsbad, CA). Human recombinant TNFα was provided by Roche Diagnostics (Indianapolis, IN). Neutralizing anti-IFNβ (sheep polyclonal Ab) against both murine and human protein was from PBL Biomedical Laboratories (Piscataway, NJ), isotype-matched goat or mouse IgG were purchased from R&D Systems. Ch, BK, and Fura2/AM were purchased from Sigma (St. Louis, MO, USA). Af was bought from Bayer Pharmaceuticals (Elkhart, IN). Aluminum hydroxide was from Pierce (Imject Alum; Rockford, IL).
Results
IFNβ and CD38 are critical for TNFα-induced AHR in murine tracheal rings
We first assessed whether endogenous IFNβ mediated TNFα modulation of ASM responsiveness to contractile agonists using our ex vivo model of airway hyper-responsiveness (AHR) (Chen et al., 2003; Kim et al., 2005; Tliba et al., 2003a, b). As shown in Fig. 1A, compared to untreated murine tracheae, levels of IFNβ mRNA were significantly up-regulated following 24 h stimulation with 50 ng/ml TNFα. Fig. 1B shows that Ch generates force in murine tracheal rings (pD2 values of 6.5±0.18, n = 7) in a concentration-dependent manner while BK had a very modest contractile response. In TNFα-treated murine tracheal rings, the contractile responses induced by Ch and BK were significantly increased when compared with responses of rings pre-exposed to diluent alone (Fig. 1B and C). TNFα treatment had little effect on pD2 values to Ch (6.4±0.28, n = 6) but significantly enhanced murine ASM sensitivity to all BK concentrations (pD2 = 6.7±0.5, n = 6). These data suggest that enhanced contraction to agonists observed in TNFα-treated rings is associated (BK) or not associated (Ch) with changes in receptor affinity. Interestingly, TNFα-induced enhancement of both Ch- or BK-evoked contractile responses were prevented by neutralizing anti-IFNβ antibody (1000 U/ml, Fig. 1B and C) but not the isotype-matched antibody (data not shown). Because CD38 pathways has been associated with cytokine-induced AHR in cultured ASM cells (Deshpande et al., 2003), we next investigated the involvement of CD38 in this ex vivo model of AHR. Interestingly, we found that TNFα failed to increase Ch-induced contractile responses in CD38-deficient murine tracheal rings (Fig. 2B) when compared to those derived from WT animals (Fig. 2A). Collectively, these data show that TNFα enhances ASM responsiveness to both Ch and BK in an IFNβ- and CD38-dependent manner.
Fig. 1.
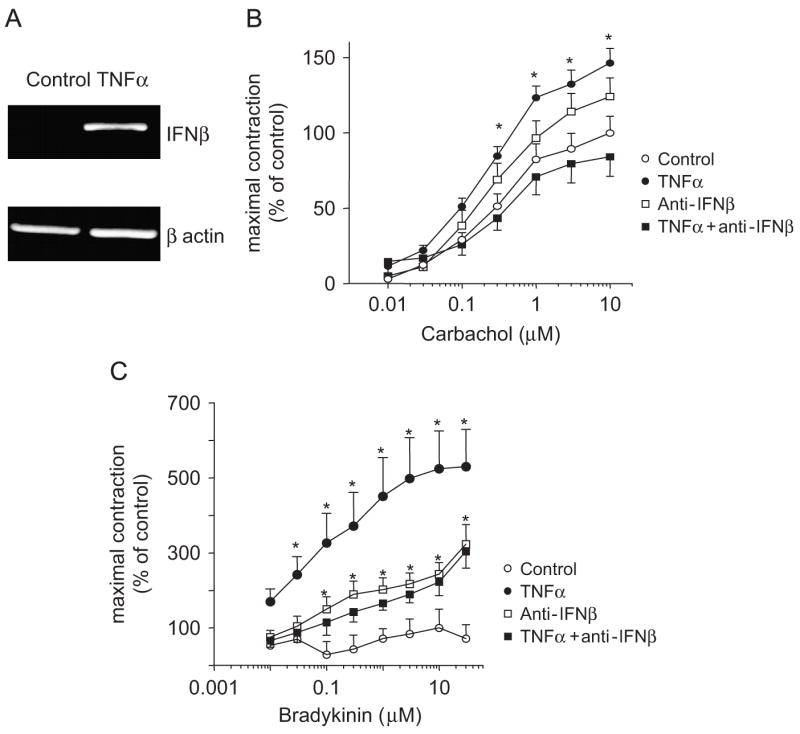
Role of IFNβ in mediating TNFα-induced augmentation of agonist-induced contraction in cultured tracheal rings. (A) Tracheae were dissected from Balb/c mice and stimulated with TNFα for 24 h as described in Methods. Total mRNA was reverse transcribed and amplified with IFNβ and β-actin-specific primers. PCR products were separated on a 1% agarose gel. Data are representative of mRNA obtained from three different experiments. Cumulative concentration–response curves to carbachol (B) or to bradykinin (C) were completed in cultured control (n = 4–5) or in the presence of TNFα (50 ng/ml) (n = 7). In some experiments (n = 7–8), tracheal rings were treated with anti-IFNβ (1000 U/ml) alone or for 30 min prior to incubation with TNFα. Isometric measurements of tracheal reactivity were calculated as changes in milligram tensions per milligram weight (mg/mg) and expressed as percentage of 10−5 mol/l carbachol-induced tensions in control rings. All tension measurements from groups were expressed as means±SEM, *p<0.05 compared with rings treated with diluent alone. Contractile studies in response to BK were done in the presence of 10−5 M D-thiorphan.
Fig. 2.
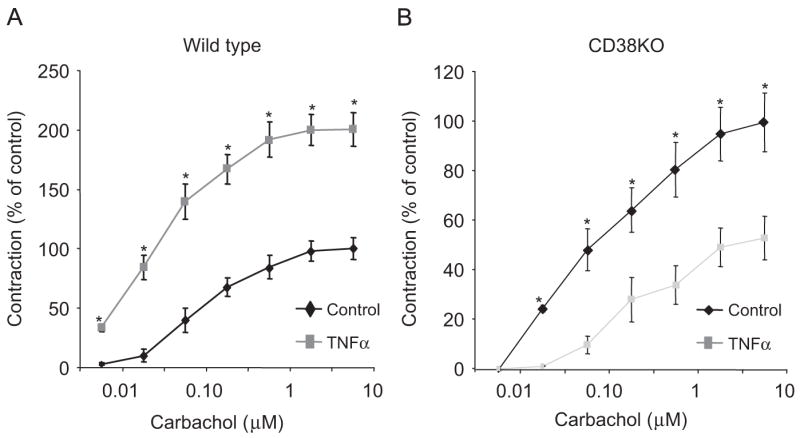
TNFα-induced enhancement of contractile agonist-evoked contractile force is abrogated in CD38 knockout mice. Cumulative concentration–response curves to carbachol in WT (A) and CD38−/− mice (B) in the presence or absence of TNFα (100 ng/ml) treatment for 24 h. All tension measurements from groups are expressed as means±SEM, n = 6–7, *p<0.05 compared with untreated rings.
Differential role of autocrine IFNβ in TNFα-induced airway hyper-responsiveness in human ASM cells
We previously made the unique finding that autocrine IFNβ is essential in mediating TNFα-induced expression of inflammatory genes in human ASM cells (Tliba et al., 2003a, b, 2004). In the present study, we found that autocrine IFNβ also participates in TNFα-induced AHR in isolated murine tracheal rings. The next logical question was to assess whether autocrine IFNβ also mediates TNFα-induced in vitro AHR by using our cellular model of cytokine-induced enhancement of calcium signaling in human ASM cells (Amrani and Panettieri, 2002). In agreement with our previous findings (reviewed in Amrani, 2006), we found that TNFα (24 h, 10 ng/ml) significantly enhanced calcium responses to Ch from 248.5±18.47 nM (n = 10) to 532.8±93.26 nM (n = 9; p<0.001, Fig. 3). Interestingly, pretreatment of ASM cells with neutralizing anti-IFNβ completely abrogated TNFα-induced potentiation of calcium responses to Ch (Fig. 3) but did not prevent TNFα enhancing effects on BK responses (data not shown). Anti-IFNβ antibody alone had no effect on calcium transients in response to Ch (Fig. 3A) or BK (data not shown). These data strongly suggest that autocrine IFNβ also plays a role in mediating cytokine-induced in vitro ASM AHR.
Fig. 3.
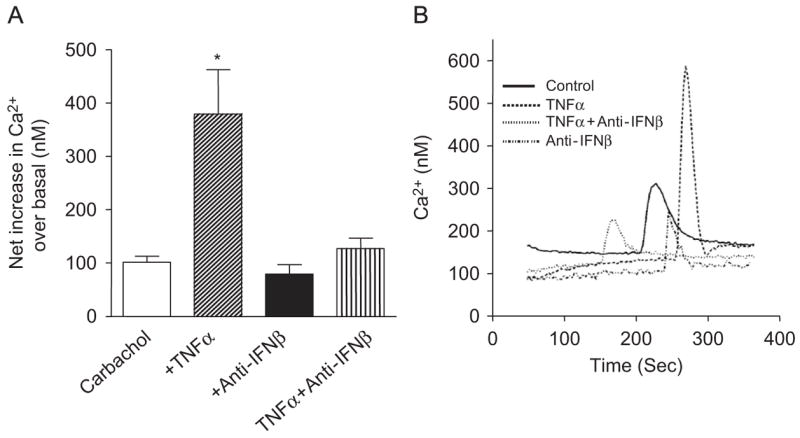
TNFα potentiation of carbachol-mediated calcium transients is abrogated by anti-IFNβ. Human ASM were loaded with Fura-2/AM as described in Materials and methods. (A) TNFα activates carbachol-mediated intracellular Ca2+ increases in cultured HASM which can be abrogated be pre-treating the cells with anti-IFNβ. Values are means±SEM of three independent experiments (n = 9–10 for control and TNFα and three for TNFα±anti-IFNβ). *p<0.001 compared with control. (B) Each tracing represents a cytosolic calcium transient from a single sample.
Af sensitization and challenge is associated with increased expression of IFNβ in the airway tissue
We next used our well-established murine model of allergic AHR (Af-exposed mice, (Haczku, 2006)) to assess whether both IFNβ and CD38 were increased following allergen challenge. Twenty-four hours after Af challenge of sensitized mice, there was a significant AHR to MCh as assessed by lung resistance and dynamic compliance (Fig. 4A). In addition, these mice had increased levels of pro-inflammatory cytokines including IL-4, IL-5 as well as TNFα and had an influx of eosinophils and neutrophils in the airways (Fig. 4B and C). Immunohistochemical studies in lung tissues of mice sensitized and challenged with Af showed increased expression of IFNβ in the ASM and in the epithelium (Fig. 4D) as well as higher levels of IFNβ and CD38 mRNA in lungs by RT-PCR (Fig. 4E). Allergen-induced CD38 expression started at 6 h while IFNβ levels were increased only at 12 h post-challenge. Both cytokines were significantly increased at 48 h post-allergen challenge. These data suggest that (i) allergen challenge is a potent inducer of both CD38 and IFNβ in the lungs and (ii) that ASM and epithelium represent novel sources of IFNβ in the airways.
Fig. 4.

Af-induced allergic airway inflammation and hyper-responsiveness is associated with release of TNFα and an increased expression of IFNβ and CD38 in the airways and lung. Sensitized mice were challenged with Af as described and studied 24 h later. (A) Lung function measurements were performed using the FlexiVent system. (B) BAL cell profile was determined by performing total and differential cell counts on cytospin preparations. (C) Cytokine profile was determined by ELISA (means±SEM of n = 10–12). (D) Immunohistochemistry was performed using a direct FITC-conjugated anti-smooth muscle α-actin monoclonal antibody or anti-IFNβ primary antibody followed by secondary Alexa 594 antibody. Data are representative of immunostaining performed in three different experiments. (E) Total mRNA was extracted at various time points after sensitization and challenge with Af and the level of IFNβ and CD38 mRNA was determined using RT-PCR as described in Methods. Data are representative of three different experiments.
CD38 is essential for Af challenge-induced airway hyper-responsiveness
To assess the role of CD38 in allergen (Af)-induced AHR, we sensitized and challenged WT and CD38−/− mice as described in the “Materials and methods” section. Af-induced airway inflammation of these mice was confirmed by increased total BAL cell counts and higher number of eosinophils and neutrophils in both the groups (Fig. 6). WT mice demonstrated a significant AHR to MCh 24 h after Af challenge in terms of both lung resistance and dynamic compliance (Fig. 5A and B) while these lung responses to MCh were abrogated in similarly challenged CD38−/− mice. Surprisingly, allergen-induced influx of inflammatory cells (macrophages, neutrophils, eosinophils and lymphocytes) in the airways was not affected in CD38-deficient mice (Fig. 6A and B). These data suggest that CD38 pathway is important in mediating allergen-induced AHR but not in the development of airway inflammation.
Fig. 6.
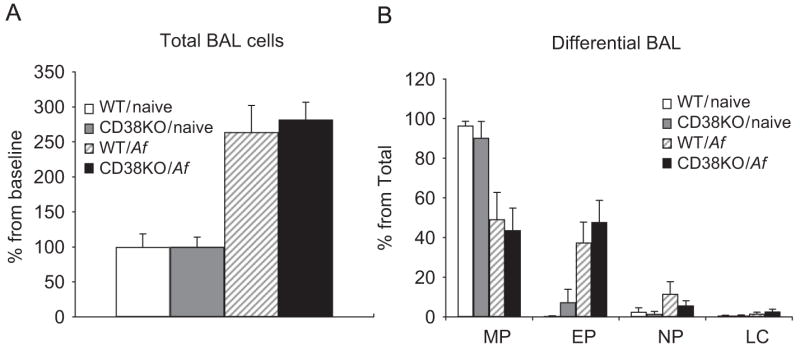
Af-induced inflammatory cell influx in the airways is not affected in CD38−/− mice. The total number of BAL cells (A) was derived from the differential counts in Giemsa-stained cytospin preparations and the total cell counts (B) in each BAL sample as described in Materials and methods. Data are expressed as means±SEM of n = 8–12 mice for each condition.
Fig. 5.
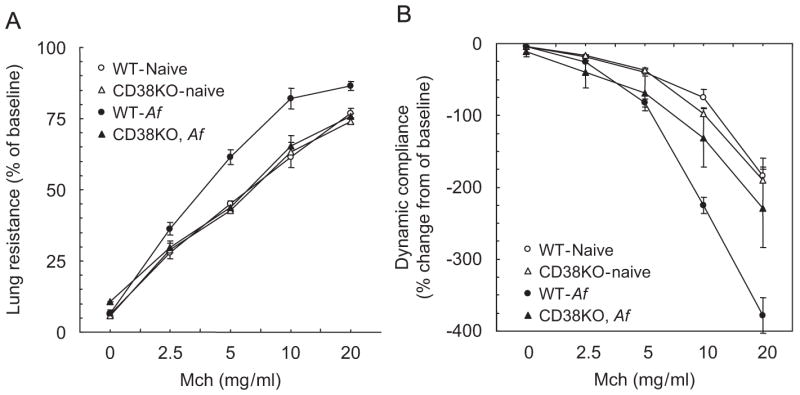
Af-induced AHR is abrogated in CD38−/− mice. Sensitized mice were challenged with Af as described and studied 24 h later using the FlexiVent system. Lung resistance (A) and dynamic compliance (B) was measured after inhalation of saline (0) or different concentrations of methacholine (MCh). While allergen-challenged wild-type animals showed significantly increased lung resistance to increasing doses of MCh compared to naive animals (*p<0.0001), allergen challenge of CD38−/− mice did not significantly increase their lung resistance. In fact lung resistance in Af-challenged CD38−/− mice was significantly lower (p = 0.0135, two-way ANOVA) when compared to Af-challenged WT animals. Similarly, dynamic compliance (Cdyn) did not decrease significantly in Af-challenged CD38−/− mice compared to naive controls. However, Cdyn in Af-challenged wild-type mice was significantly reduced (p<0.0001, two-way ANOVA) compared to naive animals. Values are expressed as % change from baseline. Means±SEM of n = 5–6.
Discussion
In previous reports, we made the interesting finding that TNFα-treated ASM cells secrete active IFNβ, which in turn modulates both cell mitogenesis as well as expression of different inflammatory genes (Tliba et al., 2003a, b, 2004). In this study, we show for the first time that IFNβ, which is increased in the airways following allergen exposure, is essential for promoting ASM hyper-responsiveness induced by TNFα. We also provide the first preclinical evidence of a role of CD38, an IFNβ-inducible protein, in the development of AHR in a murine model of allergic asthma.
Accumulating evidence from both clinical (Berry et al., 2006; Howarth et al., 2005) and animal studies (Choi et al., 2005; Kim et al., 2006) strongly support a critical role of TNFα in the pathogenesis of asthma, including its clinical feature, AHR. A direct action of TNFα on ASM function as a plausible mechanism for the enhanced airway responsiveness is evidenced by previous reports showing (i) enhanced ex vivo contractility to different contractile agonists (Adner et al., 2002; Sakai et al., 2004a, b; Chen et al., 2003) or (ii) impaired responses to β2-adrenoceptor agonists (Chen et al., 2003; Bachar et al., 2005; Wills-Karp et al., 1993) following TNFα treatment. The mechanisms by which TNFα promotes these “pro-asthmatic” responses are still unknown. In previous reports (Tliba et al., 2003a, b, 2004), we made the unique finding that ASM-derived IFNβ differentially regulates TNFα-induced expression of inflammatory genes in cultured ASM cells including RANTES, IL-6 as well as CD38, an ectoenzyme recently involved in AHR in asthma. Interestingly, we found that TNFα also increases IFNβ expression in isolated murine tracheal rings, an ex vivo model often used to study the cellular and molecular mechanisms associated with AHR. The use of neutralizing antibodies allowed us to demonstrate for the first time that IFNβ, in an autocrine manner, plays a key role in mediating TNFα-induced ex vivo ASM hyper-responsiveness to two different contractile agonists Ch (Fig. 1B) and BK (Fig. 1C). Different mechanisms in the ASM have been proposed to explain the action of TNFα in the development of ex vivo AHR. Recent evidence shows that induction of CD38, an ADP-ribosyl cyclase that regulates Ca2+ homeostasis (Malavasi et al., 1992; Mehta et al., 1996; Musso et al., 2001), is playing an key role in mediating TNFα-induced ASM hyper-responsiveness (Deshpande et al., 2004; Kang et al., 2006). The use of CD38-deficient mice allowed us to provide the first experimental evidence showing the implication of CD38 in TNFα-mediating ASM hyper-responsiveness. Our data show that in CD38-deficient murine tracheal rings, not only did TNFα fail to enhance Ch-evoked contractile responsiveness but also that Ch responses were dramatically suppressed in TNFα-treated rings (Fig. 2). It is possible that CD38 signaling pathways are somewhat serving as potent regulators of TNFα signaling. In this regard, we have shown that interfering with CD38 pathways differentially regulates cellular responses induced by TNFα in ASM cells (Tliba et al., 2003a, b). It is therefore conceivable that in isolated airway preparations, a similar phenomenon could occur with TNFα having a dual action on muscle reactivity, i.e. hyper-responsiveness or hypo-responsiveness, depending on the presence or absence of CD38 pathways.
Induction of CD38 by TNFα in ASM cells required the autocrine action of IFNβ (Tliba et al., 2004), we therefore investigated whether IFNβ was playing any role in our cellular model of hyper-responsiveness, i.e. cytokine-induced alterations in calcium handling in ASM cells (Amrani, 2006). Synthesis of de novo protein was required to mediate TNFα effects (Amrani et al., 1997), although the proteins involved have not been identified. A recent study demonstrated that induction of transient receptor potential C3 mediates TNFα-induced increases in agonist-evoked calcium responses (White et al., 2006). We now found that autocrine IFNβ is a novel mediator involved in the potentiating effect of TNFα on calcium signals induced by Ch. These in vitro data contrast with the ex vivo data found in murine tracheal rings where neutralizing IFNβ prevented TNFα-induced ex vivo AHR to both Ch and BK (Amrani, unpublished observation). This discrepancy could be explained by two possibilities: (i) the species differences of our in vitro (human) and in vivo (mice) models or (ii) the cellular complexity of the ex vivo model where TNFα could be acting on different cell types (epithelium and nerves) to promote AHR. Assuming that ASM is the main target of TNFα in the isolated murine airways, then these in vitro observations argue against the fact that changes in calcium metabolism is the only mechanism involved in TNFα-induced AHR (here to BK). In agreement with this hypothesis are the recent studies showing that AHR to BK induced by TNFα strongly correlates with changes in receptor levels in the ASM (Zhang et al., 2004). Future studies will delineate the IFNβ-associated calcium-dependent (transient signals) and -independent (receptor changes) pathways involved in TNFα-induced AHR.
Other novel findings described in this study were made in a well-established mouse model of allergic AHR (i.e., Af-sensitized and challenged mice, Fig. 4A) (Haczku et al., 2000, 2001, 2006). We found a time-dependent (seen at 6 h) and sustained (up to 48 h) increase in IFNβ as well as CD38 expression in the lungs (including in the ASM) of hyper-responsive mice following allergen challenge (Fig. 4E). The pathogenic role of CD38 in the allergic response was demonstrated using knockout mice where allergen challenge-associated AHR to MCh seen in WT animals was completely abrogated in CD38−/− mice; surprisingly, the inflammatory component of the allergic responses (influx of inflammatory cells) was not affected (Figs. 5 and 6). This observation supports the recent finding by Guedes et al. (2006) describing a central role of CD38 in AHR induced by intranasal administration of exogenous IL-13. The authors also found that IL-13-induced AHR could be abrogated in CD38 knockout mice despite no effect on the airway inflammatory response. This suggests that inflammation independent but CD38-dependent pathways, likely involving changes in ASM function, are playing an important role in the development of allergic AHR. The role of IFNβ in the allergic responses has not been investigated in the present report but our ex vivo studies strongly suggest a potential role of IFNβ in AHR. Additional studies are clearly needed to investigate this novel hypothesis.
Together, we found that ASM-derived IFNβ acts as an important factor mediating TNFα-induced ASM hyper-responsiveness to contractile agonists, possibly through its ability to regulate CD38 pathways. These ex vivo data combined to the novel observation that CD38 is critical for mediating allergen-induced AHR, strongly suggest that IFNβ–CD38 axis is a novel area of investigation for studying the cellular/molecular mechanisms underlying AHR in asthma.
Acknowledgments
We thank Mary McNichol for her assistance in the preparation of the manuscript. This study was supported by NIH R01 064063 (YA). We also thank Hasna Baidouri and William B. Jester for excellent technical assistance.
Abbreviation
- Af
Aspergillus fumigatus
- AHR
airway hyper-responsiveness
- ASM
airway smooth muscle
- BK
bradykinin
- (Ca2+)I
intracellular calcium
- Ch
carbachol
- IFN
interferon
- MCh
methacholine
- TNF
tumor necrosis factor
References
- Adner M, Rose AC, Zhang Y, Sward K, Benson M, Uddman R, Shankley NP, Cardell LO. An assay to evaluate the long-term effects of inflammatory mediators on murine airway smooth muscle: evidence that TNFalpha up-regulates 5-HT(2A)-mediated contraction. Br J Pharmacol. 2002;137:971–982. doi: 10.1038/sj.bjp.0704928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrani Y. Airway smooth muscle modulation and airway hyper-responsiveness in asthma: new cellular and molecular paradigms. Exp Rev Clin Immunol. 2006;2:353–364. doi: 10.1586/1744666X.2.3.353. [DOI] [PubMed] [Google Scholar]
- Amrani Y, Panettieri RA., Jr Modulation of calcium homeostasis as a mechanism for altering smooth muscle responsiveness in asthma. Curr Opin Allergy Clin Immunol. 2002;2:39–45. doi: 10.1097/00130832-200202000-00007. [DOI] [PubMed] [Google Scholar]
- Amrani Y, Magnier C, Enouf J, Wuytack F, Bronner C. Ca2+ increase and Ca(2+)-influx in human tracheal smooth muscle cells: role of Ca2+ pools controlled by sarco-endoplasmic reticulum Ca(2+)-ATPase 2 isoform. Br J Pharmacol. 1995;115:1204–1210. doi: 10.1111/j.1476-5381.1995.tb15026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrani Y, Krymskaya V, Maki C, Panettieri RA., Jr Mechanisms underlying TNF-alpha effects on agonist-mediated calcium homeostasis in human airway smooth muscle cells. Am J Physiol. 1997;273:L1020–L1028. doi: 10.1152/ajplung.1997.273.5.L1020. [DOI] [PubMed] [Google Scholar]
- Anticevich SZ, Hughes JM, Black JL, Armour CL. Induction of human airway hyperresponsiveness by tumour necrosis factor-alpha. Eur J Pharmacol. 1995;284:221–225. doi: 10.1016/0014-2999(95)00463-u. [DOI] [PubMed] [Google Scholar]
- Bachar O, Rose AC, Adner M, Wang X, Prendergast CE, Kempf A, Shankley NP, Cardell LO. TNF {alpha} reduces tachykinin, PGE2-dependent, relaxation of the cultured mouse trachea by increasing the activity of COX-2. Br J Pharmacol. 2005;144:220–230. doi: 10.1038/sj.bjp.0706067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry MA, Hargadon B, Shelley M, Parker D, Shaw DE, Green RH, Bradding P, Brightling CE, Wardlaw AJ, Pavord ID. Evidence of a role of tumor necrosis factor alpha in refractory asthma. N Engl J Med. 2006;354:697–708. doi: 10.1056/NEJMoa050580. [DOI] [PubMed] [Google Scholar]
- Boushey HA, Holtzman MJ, Sheller JR, Nadel JA. Bronchial hyperreactivity. Am Rev Respir Dis. 1980;121:389–413. doi: 10.1164/arrd.1980.121.2.389. [DOI] [PubMed] [Google Scholar]
- Chen H, Tliba O, Van Besien CR, Panettieri RA, Jr, Amrani Y. Selected contribution: TNF-{alpha} modulates murine tracheal rings responsiveness to G-protein-coupled receptor agonists and KCl. J Appl Physiol. 2003;95:864–872. doi: 10.1152/japplphysiol.00140.2003. [DOI] [PubMed] [Google Scholar]
- Choi IW, Sun K, Kim YS, Ko HM, Im SY, Kim JH, You HJ, Lee YC, Lee JH, Park YM, Lee HK. TNF-alpha induces the late-phase airway hyperresponsiveness and airway inflammation through cytosolic phospholipase A(2) activation. J Allergy Clin Immunol. 2005;116:537–543. doi: 10.1016/j.jaci.2005.05.034. [DOI] [PubMed] [Google Scholar]
- Deshpande DA, Walseth TF, Panettieri RA, Kannan MS. CD38-cyclic ADP-ribose-mediated Ca2+ signaling contributes to airway smooth muscle hyperresponsiveness. FASEB J Express Article. 2003 doi: 10.1096/fj.1002-0450fje. [DOI] [PubMed] [Google Scholar]
- Deshpande DA, Dogan S, Walseth TF, Miller SM, Amrani Y, Panettieri J, Reynold A, Kannan MS. Modulation of calcium signaling by IL-13 in human airway smooth muscle: role of CD38/cADPR pathway. Am J Respir Cell Mol Biol. 2004:2003–0313OC. doi: 10.1165/rcmb.2003-0313OC. [DOI] [PubMed] [Google Scholar]
- Deshpande DA, White TA, Guedes AG, Milla C, Walseth TF, Lund FE, Kannan MS. Altered airway responsiveness in CD38-deficient mice. Am J Respir Cell Mol Biol. 2005;32:149–156. doi: 10.1165/rcmb.2004-0243OC. [DOI] [PubMed] [Google Scholar]
- Guedes AG, Paulin J, Rivero-Nava L, Kita H, Lund FE, Kannan MS. CD38-deficient mice have reduced airway hyperresponsiveness following IL-13 challenge. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1286–L1293. doi: 10.1152/ajplung.00187.2006. [DOI] [PubMed] [Google Scholar]
- Haczku A. Role and regulation of lung collectins in allergic airway sensitization. Pharmacol Ther. 2006;110:14–34. doi: 10.1016/j.pharmthera.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Haczku A, Takeda K, Hamelmann E, Loader J, Joetham A, Redai I, Irvin CG, Lee JJ, Kikutani H, Conrad D, Gelfand EW. CD23 exhibits negative regulatory effects on allergic sensitization and airway hyperresponsiveness. Am J Respir Crit Care Med. 2000;161:952–960. doi: 10.1164/ajrccm.161.3.9905046. [DOI] [PubMed] [Google Scholar]
- Haczku A, Atochina EN, Tomer Y, Chen H, Scanlon ST, Russo S, Xu J, Panettieri RA, Jr, Beers MF. Aspergillus fumigatus-induced allergic airway inflammation alters surfactant homeostasis and lung function in BALB/c mice. Am J Respir Cell Mol Biol. 2001;25:45–50. doi: 10.1165/ajrcmb.25.1.4391. [DOI] [PubMed] [Google Scholar]
- Haczku A, Atochina EN, Tomer Y, Cao Y, Campbell C, Scanlon ST, Russo SJ, Enhorning G, Beers MF. The late asthmatic response is linked with increased surface tension and reduced surfactant protein B in mice. Am J Physiol Lung Cell Mol Physiol. 2002;283:L755–L765. doi: 10.1152/ajplung.00062.2002. [DOI] [PubMed] [Google Scholar]
- Haczku A, Cao Y, Vass G, Kierstein S, Nath P, Atochina-Vasserman EN, Scanlon ST, Li L, Griswold DE, Chung KF, Poulain FR, Hawgood S, Beers MF, Crouch EC. IL-4 and IL-13 form a negative feedback circuit with surfactant protein-D in the allergic airway response. J Immunol. 2006;176:3557–3565. doi: 10.4049/jimmunol.176.6.3557. [DOI] [PubMed] [Google Scholar]
- Howarth PH, Babu KS, Arshad HS, Lau L, Buckley M, McConnell W, Beckett P, Al Ali M, Chauhan A, Wilson SJ, Reynolds A, Davies DE, Holgate ST. Tumour necrosis factor (TNFalpha) as a novel therapeutic target in symptomatic corticosteroid dependent asthma. Thorax. 2005;60:1012–1018. doi: 10.1136/thx.2005.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter I, Nixon GF. Spatial compartmentalization of tumor necrosis factor (TNF) receptor 1-dependent signaling pathways in human airway smooth muscle cells. Lipid rafts are essential for TNF-alpha-mediated activation of RhoA but dispensable for the activation of the NF-kappaB and MAPK pathways. J Biol Chem. 2006;281:34705–34715. doi: 10.1074/jbc.M605738200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter I, Cobban HJ, Vandenabeele P, MacEwan DJ, Nixon GF. Tumor necrosis factor-alpha-induced activation of RhoA in airway smooth muscle cells: role in the Ca(2+) sensitization of myosin light chain(20) phosphorylation. Mol Pharmacol. 2003;63:714–721. doi: 10.1124/mol.63.3.714. [DOI] [PubMed] [Google Scholar]
- Kanehiro A, Lahn M, Makela MJ, Dakhama A, Joetham A, Rha YH, Born W, Gelfand EW. Requirement for the p75 TNF-alpha receptor 2 in the regulation of airway hyperresponsiveness by gamma delta T cells. J Immunol. 2002;169:4190–4197. doi: 10.4049/jimmunol.169.8.4190. [DOI] [PubMed] [Google Scholar]
- Kang BN, Tirumurugaan KG, Deshpande DA, Amrani Y, Panettieri RA, Walseth TF, Kannan MS. Transcriptional regulation of CD38 expression by tumor necrosis factor-alpha in human airway smooth muscle cells: role of NF-kappaB and sensitivity to glucocorticoids. FASEB J. 2006;20:1000–1002. doi: 10.1096/fj.05-4585fje. [DOI] [PubMed] [Google Scholar]
- Kim JH, Jain D, Tliba O, Yang B, Jester WF, Jr, Panettieri RA, Jr, Amrani Y, Pure E. TGF-beta potentiates airway smooth muscle responsiveness to bradykinin. Am J Physiol Lung Cell Mol Physiol. 2005;289:L511–L520. doi: 10.1152/ajplung.00027.2005. [DOI] [PubMed] [Google Scholar]
- Kim J, McKinley L, Natarajan S, Bolgos GL, Siddiqui J, Copeland S, Remick DG. Anti-tumor necrosis factor-alpha antibody treatment reduces pulmonary inflammation and methacholine hyper-responsiveness in a murine asthma model induced by house dust. Clin Exp Allergy. 2006;36:122–132. doi: 10.1111/j.1365-2222.2005.02407.x. [DOI] [PubMed] [Google Scholar]
- Kips JC, Tavernier J, Pauwels RA. Tumor necrosis factor causes bronchial hyperresponsiveness in rats. Am Rev Respir Dis. 1992;145:332–336. doi: 10.1164/ajrccm/145.2_Pt_1.332. [DOI] [PubMed] [Google Scholar]
- MacEwan DJ. TNF ligands and receptors – a matter of life and death. Br J Pharmacol. 2002;135:855–875. doi: 10.1038/sj.bjp.0704549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malavasi F, Funaro A, Alessio M, DeMonte LB, Ausiello CM, Dianzani U, Lanza F, Magrini E, Momo M, Roggero S. CD38: a multi-lineage cell activation molecule with a split personality. Int J Clin Lab Res. 1992;22:73–80. doi: 10.1007/BF02591400. [DOI] [PubMed] [Google Scholar]
- McFarlane S, Jupp O, Cobban H, Hunter I, Anderson H, Vandenabeele P, Nixon G, MacEwan D. Stimulation of stress-activated but not mitogen-activated protein kinases by tumour necrosis factor receptor subtypes in airway smooth muscle. Biochem Pharmacol. 2001;61:749–759. doi: 10.1016/s0006-2952(01)00530-5. [DOI] [PubMed] [Google Scholar]
- Mehta K, Shahid U, Malavasi F. Human CD38, a cell-surface protein with multiple functions. FASEB J. 1996;10:1408–1417. doi: 10.1096/fasebj.10.12.8903511. [DOI] [PubMed] [Google Scholar]
- Musso T, Deaglio S, Franco L, Calosso L, Badolato R, Garbarino G, Dianzani U, Malavasi F. CD38 expression and functional activities are up-regulated by IFN-gamma on human monocytes and monocytic cell lines. J Leukoc Biol. 2001;69:605–612. [PubMed] [Google Scholar]
- Panettieri RA, Murray RK, DePalo LR, Yadvish PA, Kotlikoff MI. A human airway smooth muscle cell line that retains physiological responsiveness. Am J Physiol. 1989;256:C329–C335. doi: 10.1152/ajpcell.1989.256.2.C329. [DOI] [PubMed] [Google Scholar]
- Parris JR, Cobban HJ, Littlejohn AF, MacEwan DJ, Nixon GF. Tumour necrosis factor-alpha activates a calcium sensitization pathway in guinea-pig bronchial smooth muscle. J Physiol. 1999;518:561–569. doi: 10.1111/j.1469-7793.1999.0561p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renzetti LM, Paciorek PM, Tannu SA, Rinaldi NC, Tocker JE, Wasserman MA, Gater PR. Pharmacological evidence for tumor necrosis factor as a mediator of allergic inflammation in the airways. J Pharmacol Exp Ther. 1996;278:847–853. [PubMed] [Google Scholar]
- Sakai H, Otogoto S, Chiba Y, Abe K, Misawa M. Involvement of p42/44 MAPK and RhoA protein in augmentation of ACh-induced bronchial smooth muscle contraction by TNF-alpha in rats. J Appl Physiol. 2004a;97:2154–2159. doi: 10.1152/japplphysiol.00752.2003. [DOI] [PubMed] [Google Scholar]
- Sakai H, Otogoto S, Chiba Y, Abe K, Misawa M. TNF-alpha augments the expression of RhoA in the rat bronchus. J Smooth Muscle Res. 2004b;40:25–34. doi: 10.1540/jsmr.40.25. [DOI] [PubMed] [Google Scholar]
- Shore SA, Schwartzman IN, Le Blanc B, Murthy GG, Doerschuk CM. Tumor necrosis factor receptor 2 contributes to ozone-induced airway hyperresponsiveness in mice. Am J Respir Crit Care Med. 2001;164:602–607. doi: 10.1164/ajrccm.164.4.2001016. [DOI] [PubMed] [Google Scholar]
- Sukkar MB, Hughes JM, Armour CL, Johnson PR. Tumour necrosis factor-alpha potentiates contraction of human bronchus in vitro. Respirology. 2001;6:199–203. doi: 10.1046/j.1440-1843.2001.00334.x. [DOI] [PubMed] [Google Scholar]
- Thomas PS. Tumour necrosis factor-alpha: the role of this multifunctional cytokine in asthma. Immunol Cell Biol. 2001;79:132–140. doi: 10.1046/j.1440-1711.2001.00980.x. [DOI] [PubMed] [Google Scholar]
- Thomas PS, Yates DH, Barnes PJ. Tumor necrosis factor-alpha increases airway responsiveness and sputum neutrophilia in normal human subjects. Am J Respir Crit Care Med. 1995;152:76–80. doi: 10.1164/ajrccm.152.1.7599866. [DOI] [PubMed] [Google Scholar]
- Tliba O, Deshpande D, Van Besien C, Chen H, Kannan M, Panettieri RA, Amrani Y. IL-13 enhances agonist-evoked calcium signals and contractile responses in airway smooth muscle. Br J Pharmacol. 2003a;140:1159–1162. doi: 10.1038/sj.bjp.0705558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tliba O, Tliba S, Da Huang C, Hoffman RK, DeLong P, Panettieri RA, Jr, Amrani Y. Tumor necrosis factor {alpha} modulates airway smooth muscle function via the autocrine action of interferon {beta} J Biol Chem. 2003b;278:50615–50623. doi: 10.1074/jbc.M303680200. [DOI] [PubMed] [Google Scholar]
- Tliba O, Panettieri RA, Jr, Tliba S, Walseth TF, Amrani Y. Tumor necrosis factor-alpha differentially regulates the expression of proinflammatory genes in human airway smooth muscle cells by activation of interferon-beta-dependent CD38 pathway. Mol Pharmacol. 2004;66:322–329. doi: 10.1124/mol.104.001040. [DOI] [PubMed] [Google Scholar]
- Wheeler AP, Hardie WD, Bernard GR. The role of cyclooxygenase products in lung injury induced by tumor necrosis factor in sheep. Am Rev Respir Dis. 1992;145:632–639. doi: 10.1164/ajrccm/145.3.632. [DOI] [PubMed] [Google Scholar]
- White TA, Xue A, Chini EN, Thompson M, Sieck GC, Wylam ME. Role of transient receptor potential C3 in TNF-alpha-enhanced calcium influx in human airway myocytes. Am J Respir Cell Mol Biol. 2006;35:243–251. doi: 10.1165/rcmb.2006-0003OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills-Karp M, Uchida Y, Lee JY, Jinot J, Hirata A, Hirata F. Organ culture with proinflammatory cytokines reproduces impairment of the beta-adrenoceptor-mediated relaxation in tracheas of a guinea pig antigen model. Am J Respir Cell Mol Biol. 1993;8:153–159. doi: 10.1165/ajrcmb/8.2.153. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Adner M, Cardell LO. Up-regulation of bradykinin receptors in a murine in-vitro model of chronic airway inflammation. Eur J Pharmacol. 2004;489:117–126. doi: 10.1016/j.ejphar.2004.02.033. [DOI] [PubMed] [Google Scholar]