

Abstract
We previously demonstrated that bone marrow (BM) cells migrate to Ewing’s tumors and differentiate into endothelial cells within the tumor vasculature. Recent evidence suggests that the roles of BM cells in tumors are more diverse. We investigated whether non-endothelial cell types critical for tumor vessel development are also derived from migrated BM cells.
We utilized BM transplantation with GFP+ transgenic mice as BM donors and nude mice as recipients to track the fate of migrated BM cells. After engraftment, we injected recipient mice either subcutaneously or intramuscularly with Ewing’s sarcoma cells. We labeled functional tumor vessels using intravascular perfusion staining with tomato lectin. We assessed BM cell recruitment/differentiation within the tumor microenvironment using immunohistochemistry. Ewing’s tumors contained BM-derived cells that had differentiated into endothelial cells lining perfused tumor vessels. A substantial fraction of recruited BM cells also resided in the vessel vicinity and expressed desmin and PDGFR-β, indicating smooth muscle cell differentiation. To further characterize the role of stem/progenitor cells in Ewing’s sarcoma, we sorted Tie2− BM cells from Tie2-GFP transgenic mice and then injected them intravenously into Ewing’s tumor-bearing mice. Tie2− BM progenitors migrated to Ewing’s tumors and differentiated into Tie2+ cells occupying a perivascular residence and expressing α-smooth muscle actin, desmin and PDGFR-β, as well as VEGFR-2. We did not observe differentiation of Tie2− cells into Tie2+ perivascular cells in VEGF165-inhibited TC/siVEGF7-1 tumors. The differentiation of Tie2− BM cells into Tie2+ cells in parental but not VEGF165-inhibited tumors indicates that the tumor microenvironment may influence the differentiation pathway.
Keywords: bone marrow cells, Ewing’s sarcoma, pericytes, vascular smooth muscle cells, vasculogenesis
Introduction
Both angiogenesis and vasculogenesis play important roles in the establishment and growth of blood vessels during normal physiologic responses including wound healing and myocardial repair as well as pathologic situations such as proliferative retinopathy and tumor growth. We previously demonstrated that bone marrow (BM) cells participate in the formation of blood vessels that support the growth of Ewing’s sarcoma tumors[1, 2]. We further demonstrated that BM cells that migrate into Ewing’s sarcoma tumors differentiate into endothelial cells that co-localize with these blood vessels and that this process is controlled by vascular endothelial growth factor 165 (VEGF165) [1–3]. Others have also shown direct incorporation of BM-derived endothelial progenitor cells into the expanding vascular networks that are essential for tumor growth [4–6]. Furthermore, intravascular staining using lectin perfusion demonstrated that these BM-derived endothelial cells line the lumens of functional tumor vessels [6].
The contribution of recruited BM cells to tumor growth may extend beyond differentiation into endothelial cells. Studies have shown that BM-derived cells differentiate into pericytes (also known as mural cells and vascular smooth muscle cells) in tumor blood vessels [7–9]. Rajantie et al. demonstrated that periendothelial mural and hematopoietic cell progenitors from BM rather than endothelial cell progenitors contributed to neoangiogenesis [8]. Also, De Palma et al. reported that Tie2-expressing monocytes homed to tumors and promoted angiogenesis in a paracrine fashion; specifically eliminating these cells led to suppression of tumor angiogenesis and growth [10]. Finally, Gr+CD11b+ myeloid immune suppressor cells were found to not only home to tumors and incorporate into tumor endothelium, but also to promote tumor angiogenesis and growth indirectly via release of matrix metalloproteinase 9 [11].
In the present study, we performed transplantation of BM obtained from green fluorescent protein (GFP) and Tie2-GFP transgenic mice into nude BALB/cAnN mice to track the migration and fate of BM cells. We used intravascular lectin perfusion staining to identify the functional blood vessels in order to better understand how stem/progenitor cells contribute to tumor vessel development. In addition to differentiating into endothelial cells and directly incorporating into perfused, functional tumor blood vessels, we demonstrated that BM cells recruited to Ewing’s tumors differentiated into vascular smooth muscle cells/pericyte-like cells expressing desmin, platelet-derived growth factor receptor-β (PDGFR-β) and α-smooth muscle actin (α-SMA). We further demonstrated that differentiation of these perivascular supporting cells was induced by VEGF165.
Material and methods
Ewing’s sarcoma cells and culture conditions
TC71 and VEGF165 small interfering RNA (siRNA)-inhibited clone (TC/siVEGF7-1) human Ewing’s sarcoma cells were cultured in Eagle’s modified essential medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 1 mM nonessential amino acids, 1 mM penicillin-streptomycin, and a twofold vitamin solution (Gibco BRL, Grand Island, NY). The t(11; 22) translocation in Ewing’s sarcoma cells was detected using reverse transcriptase-polymerase chain reaction. TC/siVEGF7-1 cells were created by transfection of TC71 cells with a vector-based, VEGF165-targeted small interfering RNA (siRNA) expression system [5]. VEGF165 expression in TC/siVEGF7-1 cells is decreased by 80% with no alteration in VEGF189 expression. TC/siVEGF7-1 cells produced 98% less VEGF165 protein compared to TC71 cells.
Mice and murine BM cells
All animal experiments were approved by the Institutional Animal Care and Use Committee of The University of Texas M. D. Anderson Cancer Center. Female nude BALB/cAnN mice that were 6 – 8 weeks old were purchased from the National Cancer Institute (Bethesda, MD). GFP transgenic mice (Jackson Laboratories strain 003115) were obtained from the M. D. Anderson Cancer Center Genetic Engineering Mouse Facility. Tie2-GFP transgenic mice (Jackson Laboratories strain 003658) were provided by Bing Su (Yale University School of Medicine, New Haven, CT). All of the mice were housed in a specific-pathogen-free facility. GFP and Tie2-GFP transgenic mice were used as BM donors, and nude BALB/cAnN mice were used as BM recipients. BM cells were isolated from donor mice by flushing their hind femurs with phosphate-buffered saline (PBS). Freshly isolated BM cells were resuspended in PBS and kept on ice until they were injected into the tail veins of the recipient mice.
BM transplantation using GFP transgenic donors
Recipient BALB/cAnN mice were irradiated using an external cesium source (137Cs Mark 1 irradiator; J.L. Sheperd & Associates, Glendale, CA). Specifically, the mice underwent whole-body irradiation at 8 Gy in one dose. BM cells (3 × 106 cells/0.2 ml PBS) isolated from GFP transgenic mice were injected via the lateral tail vein into recipient mice within 1 hr after irradiation. Engraftment of the BM cells in the recipient mice was assessed 6 weeks after BM transplantation. Whole BM was obtained from recipient mice by flushing their hind femurs with PBS and analyzed for GFP expression using a flow cytometer (Beckman Coulter, Fullerton, CA).
Ewing’s sarcoma mouse model
Nude mice were injected either subcutaneously or intramuscularly with TC71 or TC/siVEGF7-1 Ewing’s sarcoma cells in a growth factor-depleted Matrigel basement membrane matrix (2.5 × 106 cells/0.3 ml BD Matrigel Basement Membrane Matrix; BD Biosciences, San Jose, CA). For the intramuscular tumor cell injections, the animals were first anesthetized via intraperitoneal administration of pentobarbital sodium (Nembutal), after which TC71 cells in Matrigel were injected into their left calf muscles. All injections were performed with a 30-gauge needle and a 1-cc disposable syringe. Two weeks after tumor cell injections, the animals were euthanized and their Ewing’s sarcoma tumors were resected, placed in Optimal Cutting Temperature (OCT) compound (Sakura Finetek USA, Torrance, CA), snap-frozen in liquid nitrogen, and stored at −80°C.
Lectin perfusion
Animals received an intravenous injection of fluorescein-conjugated Lycopersicon Esculentum (tomato) lectin (0.4 mg; Vector Laboratories, Burlingame, CA). After 2 min, the animals were euthanized. Their Ewing’s sarcoma tumors were immediately resected, placed in OCT compound, snap-frozen in liquid nitrogen, and stored at −80°C.
Isolation of human and murine stem/progenitor cell subpopulations
Human CD34+ cells were isolated from umbilical cord blood obtained from the St. Louis Cord Blood Bank (St. Louis, MO) as previously described [2]. Alternatively, frozen, bone marrow-derived CD34+ cells were purchased from the National Disease Research Interchange (NDRI, Philadelphia, PA). CD34+ cells were cultured at 37°C in Serum-free Expansion Medium (Stemcell Technologies, Vancouver, British Columbia) supplemented with Flt-3 Ligand, Stem Cell Factor, IL-3 and IL-6 (StemSpan CC100 cytokine cocktail, Stemcell Technologies). Culture-expanded CD34+ cells were analyzed using phycoerythrin (PE)-conjugated mouse anti-human CD34 (BD Biosciences, San Jose, CA) and fluorescein isothiocyanate (FITC)-conjugated mouse anti-human CD45 (BD Biosciences). Isotypic mouse IgG1κ antibodies conjugated to either PE or FITC were used as negative controls (BD Biosciences). Fluorescence-activated cell sorting (BD FACSAria) was utilized to isolate CD34+/CD45+ cells. In order to permit tracking of these human progenitor cells in vivo, CD34+/CD45+ cells were cultured for 24 hours with the adenovirus vector Ad5/F35-GFP. Ad5/F35 binds cells in a CAR-independent manner, allowing effective gene delivery to human stem/progenitor cells [12].
Whole bone marrow from GFP transgenic mice was used as the source of murine Sca1+/Gr1+ and VEGFR2+ progenitor cells and analyzed using the following antibodies: PE-conjugated anti-mouse Flk1 (VEGF-R2, Ly-73) (BD Biosciences), PE-conjugated anti-mouse Sca-1 (Ly-6A/E) (BD Biosciences) and allophycocyanin (APC)-conjugated anti-mouse Gr-1 (Ly-6G) (BD Biosciences). Isotypic mouse IgG1κ antibodies conjugated to either PE (BD Biosciences) or APC (R & D Systems) were used as negative controls. Fluorescence-activated cell sorting (BD FACSAria) of freshly-isolated whole mouse BM was utilized to isolate Flk1+ and Sca1+/Gr1+ cells.
Assessment of in vivo migration of progenitor subpopulations to Ewing’s tumors
Nude mice were injected subcutaneously with TC71 Ewing’s sarcoma cells resuspended in growth factor-depleted matrigel basement membrane matrix (BD Matrigel Basement Membrane Matrix, BD Biosciences) (2 × 106 cells/0.3 mL Matrigel). One week following tumor cell inoculation, each of the three progenitor subpopulations of interest (murine Sca1+/Gr1+, murine VEGFR2+ or human Ad5/F35-infected CD34+/CD45+ cells) was separately injected intravenously into a Ewing’s tumor-bearing mouse. Mice were euthanized one week later, tumors resected, placed in Optimal Cutting Temperature (OCT) compound, snap-frozen in liquid nitrogen and stored at −80°C.
Tie2-GFP transplant model
Whole BM cells were obtained from Tie2-GFP transgenic mice by flushing their hind femurs with PBS and sorted using a fluorescence-activated cell sorter (BD FACSAria; BD Biosciences) to obtain the GFP− population. Sorted GFP− BM cells were resuspended in 0.2 ml of PBS and kept on ice until use. These cells were injected into the tail veins of nude mice (~10 × 106 cells/mouse) that had been inoculated with either TC71 or TC/siVEGF7-1 cells (in Matrigel) 1 week earlier. One week after the BM cell injection, the mice were euthanized, and their tumors were resected and analyzed.
Immunohistochemical analysis and microscopy
For immunofluorescence staining, frozen tumor tissue sections were fixed in acetone and blocked using 4% fish gelatin in PBS. For detection of GFP-expressing cells, cryostat sections were incubated with rabbit anti-GFP (Santa Cruz Biotechnology, Santa Cruz, CA) followed by goat anti-rabbit Alexa Fluor 594 (Molecular Probes, Eugene, OR). Fluorescein-lectin-marked tumor vessel endothelium was imaged directly via fluorescence microscopy. Immunostaining for desmin was performed by incubating with rabbit anti-desmin (Abcam, Cambridge, MA) followed by goat anti-rabbit Alexa Fluor 488 (Molecular Probes). For detection of PDGFRβ-expressing cells, sections were incubated with rabbit anti-PDGFRβ (P-20; Santa Cruz Biotechnology) followed by goat anti-rabbit Alexa Fluor 488 (Molecular Probes). For detection of α-SMA-expressing cells, sections were incubated with mouse anti-SMA (Abcam) followed by goat anti-mouse Alexa Fluor 488 (Molecular Probes). Immunostaining for CD31 was performed by incubating with rat anti-mouse CD31 (BD Biosciences) followed by goat anti-rat Alexa Fluor 488 (Molecular Probes). Immunohistochemical detection of Tie2 was performed using rabbit anti-Tie2 (C-20; Santa Cruz Biotechnology) followed by goat anti-rabbit Alexa Fluor 488 (Molecular Probes). Immunohistochemical detection of VEGFR2 was performed using rabbit anti-mouse VEGFR2 (Sigma, St. Louis, MO) followed by goat anti-rabbit Alexa Fluor 488 (Molecular Probes). When using mouse monoclonal antibodies, slides were preblocked overnight with F(ab) fragment (Jackson ImmunoResearch, West Grove, PA) prior to incubation with the primary antibody. Hoescht 33342 dye (Molecular Probes) was used to stain the cell nuclei. Fluorescent images were captured at 10× (eyepiece) and 10× or 20× (objective) with Optimas imaging software (San Diego, CA).
For confocal microscopy, the following secondary antibodies were utilized: goat anti-mouse Cy-3 (Cy-3 –conjugated AffiniPure F(ab′)2 Fragment Goat Anti-Mouse IgG + IgM, Jackson Immunoresearch Laboratories Inc, West Grove, PA), goat anti-mouse Cy-5 (Jackson Immunoresearch), goat anti-rabbit Cy-3 (Jackson Immunoresearch), and goat anti-rabbit Cy-5 (Jackson Immunoresearch). Sytox Green (Invitrogen, Carlsbad, California) was used to stain cell nuclei. Images were collected using a Zeiss Laser Confocal Microscope (Carl Zeiss MicroImaging, Inc. Thornwood, New York).
Results
Migration of BM cells into perfused functional Ewing’s tumor vessels
We employed a BM transplantation model using GFP transgenic mice as BM donors and nude mice as recipients to track the migration of BM cells to Ewing’s sarcoma tumors. To demonstrate successful engraftment of donor stem/progenitor cells, we collected recipient BM samples 6 weeks after transplantation and analyzed them for GFP expression using flow cytometry. These BM samples had substantially higher GFP expression (>69% GFP+ cells) when compared with BM samples obtained from control (untransplanted) nude mice (<1% GFP+ cells) confirming engraftment. Transplanted mice were then injected with TC71 cells. Mice were sacrificed 2 weeks later and their tumors removed. Prior to sacrifice, we performed intravascular perfusion staining with fluorescein-conjugated lectin to identify the functional vessels and determine whether BM cells were recruited to these perfused vessels. The intensity of spontaneous green fluorescence emitted by GFP-expressing bone marrow cells recruited to tumors in vivo was too low to detect these cells in cryostat sections. For this reason, we utilized an anti-GFP antibody to identify the migrated GFP+ cells. GFP-expressing cells within tumors were detected in cryostat sections by incubating with rabbit anti-GFP, followed by a secondary antibody conjugated to the Alexa594 fluorophore. GFP+ cells were therefore red. This also helped distinguish the lectin+ green vessels from the GFP+ cells. Following i.v. infusion of tumor-bearing mice with fluorescein-conjugated lectin, perfused (functional) tumor vessel endothelium displayed very strong spontaneous green fluorescence (Fig. 1A and 1B). Superimposition of the GFP and lectin images revealed numerous areas throughout the TC71 Ewing’s tumor in which BM-derived GFP+ cells (red-stained) resided in the vicinity of perfused (green-stained) blood vessels (Fig. 1A and 1B). Within these vascular areas, GFP and lectin co-localized at specific sites, resulting in a yellow fluorescence in the merged image (Fig. 1A, arrow). Only perfused vessel endothelium was lectin-marked, which suggested that BM-derived cells can differentiate into endothelial cells lining the lumens of functional vessels in TC71 tumors. A much more substantial fraction of recruited BM cells were distinct from the lectin+ vascular cells. These BM-derived cells resided in close proximity to the perfused tumor vessels, did not stain positively for lectin, and formed clusters that were outside but closely associated with the vascular endothelial lining (Fig. 1B).
Fig. 1.

Bone marrow cells differentiate into both endothelial and vascular smooth muscle cells within perfused, functional Ewing’s tumor neovessels. (A) TC71 Ewing’s tumors were implanted in mice previously transplanted with GFP+ BM. Donor-derived cells were detected by staining tumor sections with anti-GFP, followed by a secondary antibody conjugated to the Alexa 594 fluorophore (red). Functional tumor vessel endothelium was labeled green by intravascular perfusion with fluorescein-conjugated lectin. The arrow in the merged image indicates BM cells that differentiated into endothelial cells within perfused tumor vessels (yellow). Images were analyzed using a 20× objective. (B) Migrated BM cells (red) reside in close association with numerous lectin-marked functional TC71 tumor neovessels (green). (C, D) TC71 tumor sections were co-stained for GFP+ BM cells and either PDGFRβ (C) or desmin (D). Extensive networks of PDGFRβ+ (1C) and desmin+ (1D) cells are seen throughout the tumor area. The merged images indicate that BM-derived cells express PDGFRβ and desmin (denoted by yellow fluorescence), suggesting vascular smooth muscle cell differentiation. Hoescht 33342 staining confirmed that marker expression was associated with cells. The tumor areas shown were analyzed with a 20× objective. Arrows (Fig. 1D) indicate that the vascular smooth muscle cell network is a mosaic of locally-derived and BM-derived cells.
Differentiation of recruited BM cells into vascular smooth muscle cells
We have demonstrated that recruited BM cells differentiate into endothelial cells that co-localized with the tumor vessels. To determine whether recruited stem/progenitor cells also differentiate into vascular smooth muscle (pericyte-like) cells within the Ewing’s sarcoma tumor vasculature, we co-stained tumors obtained from GFP+ BM-transplanted mice for GFP and either desmin or PDGFRβ. As described above, the intensity of spontaneous green fluorescence emitted by GFP-expressing bone marrow cells recruited to tumors in vivo was too low to detect these cells in cryostat sections without utilizing an anti-GFP antibody. Therefore co-immunostaining with anti-GFP (Alexa594) to detect BM-derived cells and anti-desmin/anti-PDGFRβ (Alexa488) to detect pericytes was used. Tumor sections displayed multiple cells co-expressing GFP and PDGFRβ (Fig. 1C) or GFP and desmin (Fig. 1D). We observed desmin+ and PDGFRβ+ cells in large clusters that extended from the vessel lumens deeper into the tumor microenvironment, suggesting the presence of an extensive smooth muscle cell/pericyte framework supporting vascular endothelium. Superimposition of the GFP and desmin/PDGFRβ images revealed distinct sites of yellow fluorescence (Fig. 1D, Merge, white arrows - BM-derived cells) immediately adjacent to sites of green fluorescence (Fig. 1D, Merge, red arrows –non-BM-derived cells), indicating that the pericyte network was a mosaic of both locally derived and BM-derived cells. To demonstrate that the co-localization observed was not merely the result of artifact, adjacent serial tumor sections were single stained for either anti-GFP or anti-desmin/anti-PDGFRβ alone. These single immunostained sections (data not shown) confirmed that both GFP+ and desmin+/PDGFRβ+ cells were present, as observed in the co-stained sections.
Characterization of specific BM-derived progenitor subpopulations that migrate to Ewing’s sarcoma tumors and differentiate into vascular smooth muscle cells
After demonstrating the participation of whole, unfractionated BM in Ewing’s sarcoma neovasculature expansion, we next determined which specific BM subpopulations migrate to Ewing’s tumors and contribute to the pericyte/vascular smooth muscle cell network. Specifically, we investigated the ability of CD34+/CD45+, Sca1+Gr1+, and VEGFR2+ cells to differentiate into vascular smooth muscle cells within Ewing’s tumors. Human CD34+ cells derived from umbilical cord blood or BM were GFP-labeled with Ad5/F35-GFP. Murine Sca1+/Gr1+ and VEGFR2+ cells were isolated from fresh GFP+ mouse BM. Each progenitor cell type was separately i.v.-injected into nude mice bearing TC71 Ewing’s tumors. One week later, mice were sacrificed, tumors excised and sectioned. Migrated progenitor cells were detected within tumors by immunostaining with anti-GFP. Vascular smooth muscle cell differentiation of migrated BM cell subpopulations was assessed by co-staining of tumor sections for GFP and the pericyte markers α-smooth muscle actin (SMA) and desmin. Yellow fluorescence in the merged image (Fig. 2A, arrows) demonstrated that some of the migrated Sca1+/Gr1+ cells expressed SMA, indicating they had differentiated into vascular smooth muscle cells within the Ewing’s tumor vasculature. Using confocal microscopy, we demonstrated that incorporation of Sca1+/Gr1+-derived SMA+ cells (Fig. 2A, arrows) resulted in a mosaic pattern of BM progenitor-derived (yellow) and locally-derived (green) pericytes adjacent to each other within the Ewing’s tumor vascular smooth muscle cell network (Fig. 2A). VEGFR2+ cells were similarly found to migrate to Ewing’s tumors and differentiate into vascular smooth muscle cells expressing desmin (Fig. 2B, arrows) and SMA (data not shown). In addition, migrated, human CD34+- derived cells were also detected within the Ewing’s tumor neovasculature. Confocal analysis demonstrated that some of the CD34+ cells that had homed to the Ewing’s tumor vasculature expressed alpha-SMA (Fig. 2C, arrows) and desmin (data not shown), indicating that a fraction of this migrated progenitor subpopulation had differentiated into pericytes/vascular smooth muscle cells.
Fig. 2.
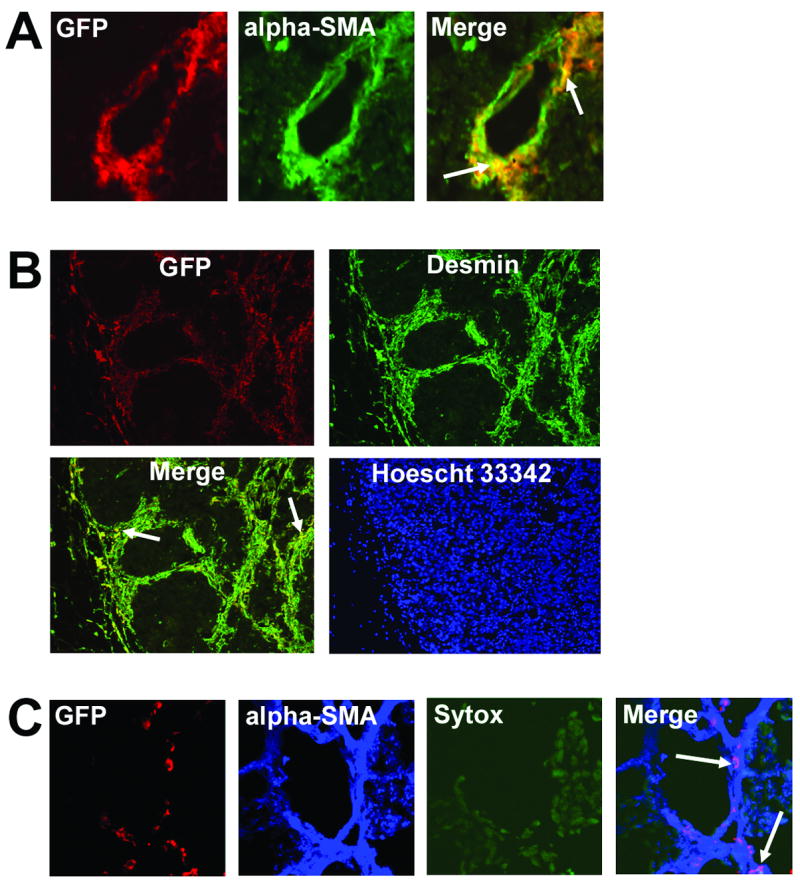
CD34+/CD45+, Sca1+/Gr1+, and VEGFR2+ cells migrate to Ewing’s tumors and differentiate into vascular smooth muscle cells. GFP-labeled Sca1+/Gr1+, VEGFR2+ and CD34+/CD45+ cells were i.v.-injected into nude mice with TC71 tumors. (A) Confocal microscopy (40×); Sca1+/Gr1+ cells differentiated into α-SMA+ pericytes (arrows), which resided adjacent to locally-derived cells (green) within the vascular smooth muscle cell network. (B) Migrated VEGFR2+ cells (red) were detected throughout the Ewing’s tumors area. Many of these GFP+ VEGFR2+-derived cells also expressed desmin (arrows), indicating pericyte differentiation (10× objective). (C) Confocal microscopy (20×); CD34+/CD45+ cells also differentiated into α-SMA+ pericytes (arrows) within the tumor vascular smooth muscle cell network.
Tie2− BM precursors migrate to Ewing’s tumors and differentiate into Tie2+ vascular smooth muscle cells in a VEGF165-dependent manner
To further explore the differentiation of BM-derived cells in Ewing’s tumors and the role of the tumor microenvironment, we utilized BM from Tie2-GFP transgenic mice, in which GFP is expressed under the direction of the Tek (formerly Tie2) promoter. Freshly isolated Tie2-GFP mouse BM was >99% GFP-negative by flow cytometric analysis, indicating that these cells were Tie2 negative (Fig. 3A). To determine whether the microenvironment plays a role in the differentiation of BM-derived cells that are recruited to Ewing’s tumors, the GFP-negative BM fraction (indicated by P8, Fig. 3A) was isolated from whole Tie2-GFP mouse BM using FACS and subsequently i.v.-injected into nude mice bearing subcutaneous TC71 Ewing’s sarcoma tumors. Tumors were resected 1 week later. Immunostaining revealed clusters of GFP+ cells throughout the tumors, indicating that the i.v.-injected Tie2− cells had migrated to the tumors and differentiated into Tie2+ cells (Fig. 3B). Co-expression of Tie2 and GFP in cells within the tumors was confirmed using immunohistochemical analysis (data not shown). These migrated GFP+Tie2+ cells were found clustered in the vicinity of CD31+ vessels (Fig. 3B). However, we were not able to demonstrate co-localization of GFP and CD31, suggesting that these BM-derived Tie2+ cells had not differentiated into endothelial cells. These cells resided adjacent to and surrounding CD31+ tumor microvessels and expressed the pericyte/smooth muscle cell differentiation marker desmin (Fig. 3C), as well as PDGFRβ and α-smooth muscle actin (data not shown). We also found that VEGF receptor-2 (VEGFR2) was expressed by these Tie2+ perivascular cells (Fig. 3D).
Fig. 3.
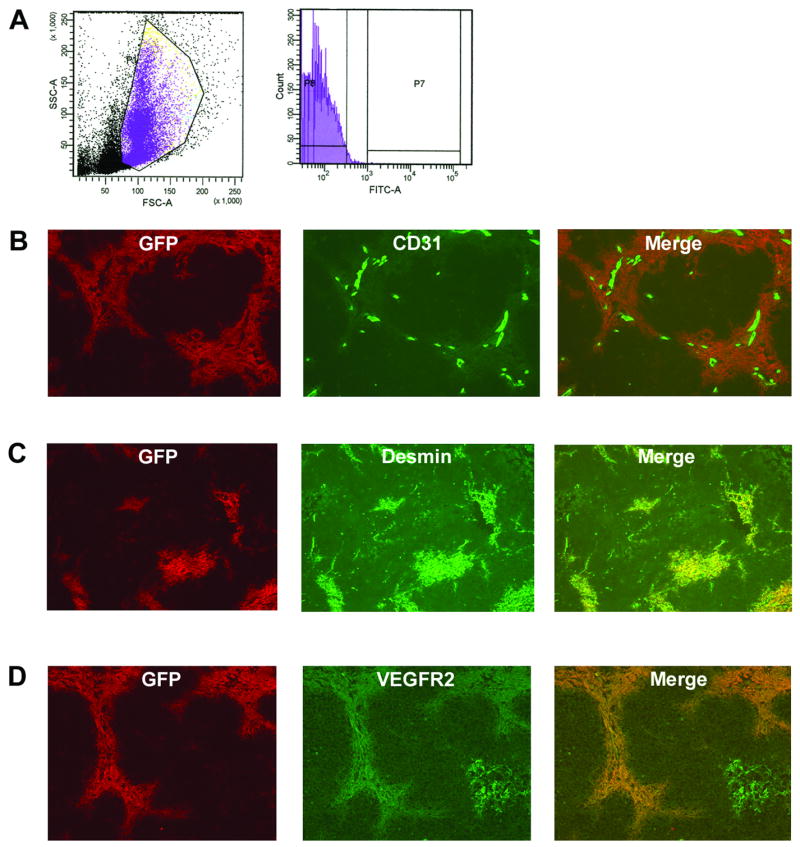
Tie2− BM precursors migrate to Ewing’s tumors and differentiate into Tie2+ periendothelial vascular smooth muscle cells. (A) Flow cytometric analysis of whole mouse BM from Tie2-GFP animals demonstrated >99% GFP− (Tie2−) cells. (B) GFP-negative BM cells were i.v.-injected into nude mice bearing s.c. TC71 tumors. Tumors were excised 1 week later. GFP+ (Tie2+) cells (red) were detected throughout the tumor. Migrated Tie2+ cells (red) were clustered near CD31+ vessels (green). Colocalization of these GFP+Tie2+ cells with CD31 was not observed (10× objective). (C) BM-derived Tie2+ cells expressed the pericyte/vascular smooth muscle cell differentiation marker desmin (10× objective). (D) Tie2+GFP+ cells within TC71 tumors express VEGFR-2. Colocalization of GFP (red) and VEGFR-2 (green) is seen.
To determine whether the migration of Tie2− BM cells into Ewing’s tumors and their subsequent differentiation into Tie2+ vascular smooth muscle cells was specifically mediated by VEGF165 within the tumor microenvironment, we repeated all of the above experiments using a VEGF165 siRNA-inhibited TC71 clone (TC/siVEGF7-1) [3]. Using the GFP-transplant and i.v. lectin infusion schema described above, we first established that TC/siVEGF7-1 tumors contained small, discontinuous lectin-marked vessels, which was consistent with our previous findings [2, 3]. Despite this disruption in vessel size/morphology, multiple areas contained clusters of BM-derived cells surrounding perfused tumor vessels (Fig. 4A). While the number of migrated cells was smaller than that seen in the parental tumors (Fig. 1B), this indicated that BM cells continued to migrate into the tumor area despite the low levels of VEGF165. Having demonstrated that BM cells can migrate into the TC/siVEGF7-1 tumors, we next injected GFP-negative cells from Tie2-GFP BM into mice with TC/siVEGF7-1 tumors. We resected the tumors 1 week later. Immunostaining of the resected TC/siVEGF7-1 tumor tissue revealed few Tie2+GFP+ cells (Fig. 4B, arrows). This data indicates that VEGF165 may influence not only BM cell migration to the tumor area, but also the fate of the migrated cells.
Fig. 4.
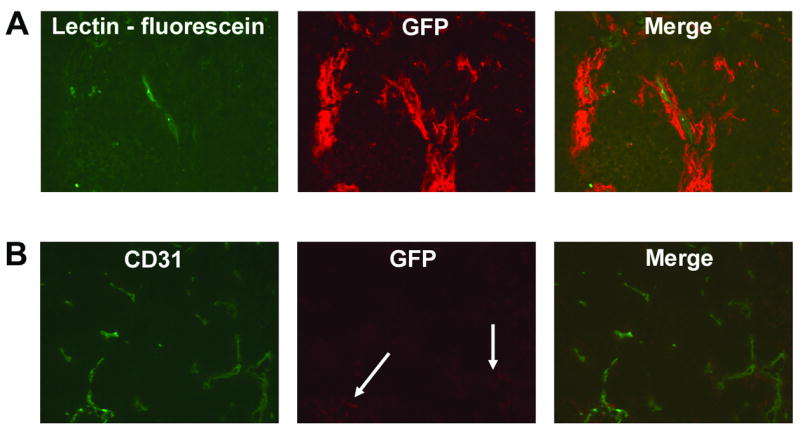
Differentiation of Tie2− BM-derived progenitor cells into Tie2+ vascular smooth muscle cells is inhibited in VEGF165-inhibited Ewing’s tumors, despite migration of BM cells into the tumor area. (A) VEGF165-inhibited TCsiVEGF7-1 Ewing’s tumors were implanted in mice previously transplanted with GFP+ whole BM. Migrated BM cells (red) were found in close association with lectin-marked functional TCsiVEGF7-1 tumor neovessels (green) (20× objective). (B) Sorted GFP-negative BM cells from Tie2-GFP transgenic mice were i.v.-injected into nude mice bearing s.c. TCsiVEGF7-1 tumors. Compared to parental TC71 tumors, VEGF165-inhibited TCsiVEGF7-1 tumors demonstrated a dramatic reduction in the number of Tie2+GFP+ cells (in red; arrows) within the tumor area one week after i.v. injection (10× objective).
Discussion
Previous work in our laboratory has demonstrated that BM cells migrate to Ewing’s sarcoma tumors and differentiate into endothelial cells [1, 2]. In the present study, we demonstrated that a substantial fraction of BM-derived cells reside in the vicinity of perfused functional tumor vessels. These cells have differentiated into vascular smooth muscle/pericyte-like cells in addition to endothelial cells. Pericytes are known to envelop the inner endothelial lining of blood vessels, providing support and regulating blood vessel formation/maturation via communication with endothelial cells [9]. Studies have shown that paracrine signaling, specifically via PDGF-BB, is important for proper interaction between pericytes and endothelial cells [13, 14]. Also, inhibition of pericyte function and detachment of pericytes from vascular endothelium result in blood vessel disruption and tumor growth inhibition [15]. Our finding that BM cells differentiate into vascular smooth muscle cells in addition to endothelial cells within Ewing’s tumors adds further support to the idea that stem/progenitor cells and vasculogenesis contribute to solid tumor neovascularization and growth. These BM-derived cells expressing pericyte markers were not only in direct contact with the vascular endothelium, as has been traditionally reported, but also were shown to form large, multi-layered networks that extended outwards from the vessel lumen into the tumor tissue. Interfering with the homing of BM-derived endothelial/pericyte progenitors to Ewing’s tumors may provide a dual insult to the tumor vasculature by interfering with the supply of endothelial cells and pericytes that are needed to form functional tumor vessels. By disrupting vasculature expansion, this strategy could potentially allow better inhibition of tumor growth.
In addition to demonstrating the recruitment of native host BM cells to the tumor site and their differentiation into vascular smooth muscle cells within the Ewing’s tumor, we further identified three specific BM subsets that home to Ewing’s tumors and participate in this process. The contribution of each individual BM subpopulation was characterized by sorting the specific GFP+ cell populations and then separately injecting each of them i.v. into a different individual mouse bearing a Ewing’s tumor. CD34+/CD45+, VEGFR2+ and Sca1+/Gr1+ cells all established residence within the expanding tumor vascular network and differentiated into cells expressing markers characteristic of pericyte differentiation. Our findings suggest that a portion of the vascular smooth muscle cell/pericyte precursors that contribute to the formation of the tumor vasculature are derived from these subsets of pluripotent hematopoietic stem cells. Because these cells are also capable of differentiating into myeloid cells, it is likely that the tumor microenvironment contributes to the specific fate of migrated, BM-derived cells.
We demonstrated that Tie2− BM cells migrated into Ewing’s sarcoma tumors and differentiated into Tie2+ cells, which expressed pericyte/smooth muscle cell markers and resided close to the tumor microvessels. This is consistent with the findings of De Palma et al., who recently reported that Tie2 is expressed by BM-derived mesenchymal pericyte precursors [16]. Our novel demonstration of the differentiation of Tie2− BM cells into Tie2+ cells in Ewing’s tumors suggests that this tumor may be capable of influencing the differentiation of the migrated cells to a phenotype that will enhance and support the tumor blood supply thus providing a growth advantage. It could be argued that our finding of Tie2+GFP+ cells within the tumor tissue after i.v.-injection of Tie2−GFP− cells was secondary to a small contaminating population of GFP+ cells present in the original i.v. inoculum. However, this explanation is highly unlikely for several reasons. First, as shown in Fig. 3A, the GFP-negative population isolated using FACS was strongly-negative; weakly-GFP-negative cells were not selected for i.v. injection into tumor-bearing mice. Furthermore, analysis of tumor sections demonstrated massive infiltration of GFP+ cells throughout the tumor tissue, with greater than 50% of low-power fields containing evidence of these BM-derived cells, following two separate in vivo experiments. It is unlikely that such a small, contaminating population of GFP+ cells which escaped the stringent selection requirements imposed during FACS was responsible for the abundance of GFP+ BM-derived cells within the tumor. Indeed, previous work in our laboratory has demonstrated that the detection of migrated, GFP+ BM cells in tumors by immunohistochemical analysis is very difficult when only a small number of such cells is recruited to the tumor site. Thus, one would also have to propose that this small number of GFP+ contaminating cells migrated and expanded to achieve the number of GFP+ cells seen in the tumor sections. Even considering a 24 h doubling time it is highly unlikely that this small number of GFP+ cells could expand and account for all of the GFP+ cells seen. Based on these considerations, the presence of Tie2+GFP+ cells within Ewing’s tumors as demonstrated here is most likely the result of differentiation of migrated, Tie2−GFP− BM precursors to Tie2+GFP+ cells.
Given its overexpression in Ewing’s tumors, we investigated whether VEGF165 was responsible for the migration and differentiation of this population of BM-derived pericyte precursors. We have previously shown that VEGF165 siRNA-inhibited (TC/siVEGF7-1) Ewing’s tumors demonstrate delayed in vivo growth and decreased vessel density [2, 3]. In the present study, we found that whole GFP+ BM-derived cells can still migrate to TC/siVEGF7-1 tumors, suggesting that other chemotactic factors in addition to VEGF165 are involved in recruiting progenitor cells to Ewing’s tumors. However, TC/siVEGF7-1 tumors displayed a dramatically lower number of Tie2+GFP+ cells present following the i.v. injection of GFP−Tie2− BM cells when compared with TC71 parental tumors. The transplant model we employed in the present study did not permit simultaneous evaluation of Tie2− BM cell migration and differentiation of these cells into Tie2+ cells within the same tumor-bearing animal, as we could not identify the migrated Tie2− cells. Simultaneous detection of GFP+Tie2+ cells and undifferentiated, carbocyanine dye (CM-Dil)-labeled Tie2− cells using immunohistochemical methods was unsuccessful. However, taken together, our findings suggest that VEGF165 in the tumor microenvironment plays a key role in influencing the differentiation of infiltrating stem/progenitor cells.
Understanding the functions of recruited BM cells within tumors and how they contribute to tumor vascular expansion, as well as the molecular cross-talk mediating their interactions with tumor cells, may allow us to identify novel therapeutic targets that can be exploited to inhibit tumor growth. Such novel therapies are needed for cancers like Ewing’s sarcoma, the prognosis for which has remained unchanged over the past 15 years.
Acknowledgments
NIH, Grant number: R01 CA103986 (ESK), Core Grant CA 16672
Abbreviations
- BM
bone marrow
- GFP
green fluorescent protein
- IHC
immunohistochemistry
- PDGFR-β
Platelet-derived growth factor receptor-β
- TC/siVEGF7-1
VEGF165 small interfering RNA (siRNA)-inhibited clone
- VEGFR-2
Vascular endothelial growth factor receptor-2
References
- 1.Bolontrade MF, Zhou RR, Kleinerman ES. Vasculogenesis Plays a Role in the Growth of Ewing’s Sarcoma In Vivo. Clin Cancer Res. 2002;8:3622–3627. [PubMed] [Google Scholar]
- 2.Lee TH, Bolontrade MF, Worth LL, Guan H, Ellis LM, Kleinerman ES. Production of VEGF165 by Ewing’s sarcoma cells induces vasculogenesis and the incorporation of CD34+ stem cells into the expanding tumor vasculature. Int J Cancer. 2006;119:839–846. doi: 10.1002/ijc.21916. [DOI] [PubMed] [Google Scholar]
- 3.Guan H, Zhou Z, Wang H, Jia S-F, Liu W, Kleinerman ES. A small interfering RNA targeting vascular endothelial growth factor inhibits Ewing’s sarcoma growth in a xenograft mouse model. Clin Cancer Res. 2005;11:2662–2669. doi: 10.1158/1078-0432.CCR-04-1206. [DOI] [PubMed] [Google Scholar]
- 4.Lyden D, Hattori K, Dias S, Costa C, Blaikie P, Butros L, Chadburn A, Heissig B, Marks W, Witte L, Wu Y, Hicklin D. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194–1201. doi: 10.1038/nm1101-1194. [DOI] [PubMed] [Google Scholar]
- 5.Dwenger A, Rosenthal F, Machein M, Waller C, Spyridonidis A. Transplanted bone marrow cells preferentially home to the vessels of in situ generated murine tumors rather than of normal organs. Stem Cells. 2004;22:86–92. doi: 10.1634/stemcells.22-1-86. [DOI] [PubMed] [Google Scholar]
- 6.Duda DG, Cohen KS, Kozin SV, Perentes JY, Fukumura D, Scadden DT, Jain RK. Evidence for incorporation of bone marrow-derived endothelial cells into perfused blood vessels in tumors. Blood. 2006;107:2774–2776. doi: 10.1182/blood-2005-08-3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song S, Ewald AJ, Stallcup W, Werb Z, Bergers G. PDGFRβ+ perivascular progenitor cells in tumors regulate pericyte differentiation and vascular survival. Nat Cell Biol. 2005;7:870–879. doi: 10.1038/ncb1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rajantie I, Ilmonen M, Alminaite A, Ozerdem U, Alitalo K, Salven P. Adult bone marrow-derived cells recruited during angiogenesis comprise precursors for periendothelial vascular mural cells. Blood. 2004;104:2084–2086. doi: 10.1182/blood-2004-01-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lamagna C, Bergers G. The bone marrow constitutes a reservoir of pericyte progenitors. J Leukoc Biol. 2006;80:677–681. doi: 10.1189/jlb.0506309. [DOI] [PubMed] [Google Scholar]
- 10.De Palma M, Venneri MA, Roca C, Naldini L. Targeting exogenous genes to tumor angiogenesis by transplantation of genetically modified hematopoietic stem cells. Nat Med. 2003;9:789–795. doi: 10.1038/nm871. [DOI] [PubMed] [Google Scholar]
- 11.Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, Matrisian LM, Carbone DP, Lin PC. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6:409–421. doi: 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 12.Yotnda P, Onishi H, Heslop HE, Shayakhmetov D, Lieber A, Brenner M, Davis A. Efficient infection of primitive hematopoietic stem cells by modified adenovirus. Gene Ther. 2001;8:930–7. doi: 10.1038/sj.gt.3301488. [DOI] [PubMed] [Google Scholar]
- 13.Hellstrom M, Kalen M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047–3055. doi: 10.1242/dev.126.14.3047. [DOI] [PubMed] [Google Scholar]
- 14.Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–5. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- 15.Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111:1287–1295. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, Naldini L. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8:211–226. doi: 10.1016/j.ccr.2005.08.002. [DOI] [PubMed] [Google Scholar]