

Abstract
Optically active anti-β-substituted γ,δ-unsaturated amino acids are important synthetic building blocks in organic synthesis and for peptidomimetics. A novel asymmetric Eschenmoser–Claisen rearrangement with use of a C2-symmetric chiral auxiliary was developed to generate this type of amino acid. Excellent diastereoselectivities and high enantioselectivities (87–93% ee) were obtained after the chiral auxiliary was removed via iodolactonization/zinc reduction.
γ,δ-Unsaturated amino acids are important naturally occurring nonproteinogenic amino acids found in plants1 and microorganism.2 They are also very important building blocks in organic synthesis for the potential conversion of their terminal double bonds to many other functionalities, and for their applications in peptidomimetic drug discovery.3 A well-known strategy to synthesize this type of amino acid is via a Claisen rearrangement.4 The related methodologies to synthesize optically active syn-β-substituted γ,δ-unsaturated amino acids have also been developed by using chiral ligands,5 or chirality transfer from available chiral sources.6 However, no satisfactory Claisen rearrangement has been reported for optically active anti-β-substituted γ,δ-unsaturated amino acids. In general, the chirality transfer method can also be expanded to the anti-products; however, it suffers from limited chiral starting materials and epimerization to the syn epimer.6 In Welch’s report of an asymmetric Eschenmoser–Claisen rearrangement,7 the remote chiral center provided low diastereofacial selectivity as a consequence of the C–N bond rotation in the N,O-ketene acetal intermediate.
Most recently, we have reported that racemic anti-β-substituted γ,δ-unsaturated amino acids can be generated with good diastereoselectivities via the Eschenmoser–Claisen rearrangement.8 We envisioned that an asymmetric Eschenmoser–Claisen rearrangement also could be performed using a C2-symmetric chiral auxiliary. The use of a C2-symmetric chiral auxiliary was expected to preclude the rotamer problem mentioned above and therefore provide an improved asymmetric induction. We report here a novel synthesis of the optically active anti-β-substituted γ,δ-unsaturated amino acids using a C2-symmetric chiral auxiliary.
The C2-symmetric chiral auxiliary (2R,5R)-dimethylpyrrolidine 1 was synthesized in excellent ee according to a literature reported method.9 It was then coupled to a Cbz-protected glycine using DIC/HOAT to afford amide 2 in excellent yield despite the steric hindrance of the secondary amine (Scheme 1). The Meerwein salt formation and rearrangement was conducted in similar conditions as reported previously for the synthesis of the racemic anti-β-substituted γ,δ-unsaturated amino acids. Although, in general, high temperature would provide low diastereoselectivities, a couple of these reactions had to be heated to 60 °C (entries 2 and 3) to make the rearrangement happen. Prolonged heating dramatically decreased the reaction yield and cleanness.10
Scheme 1.
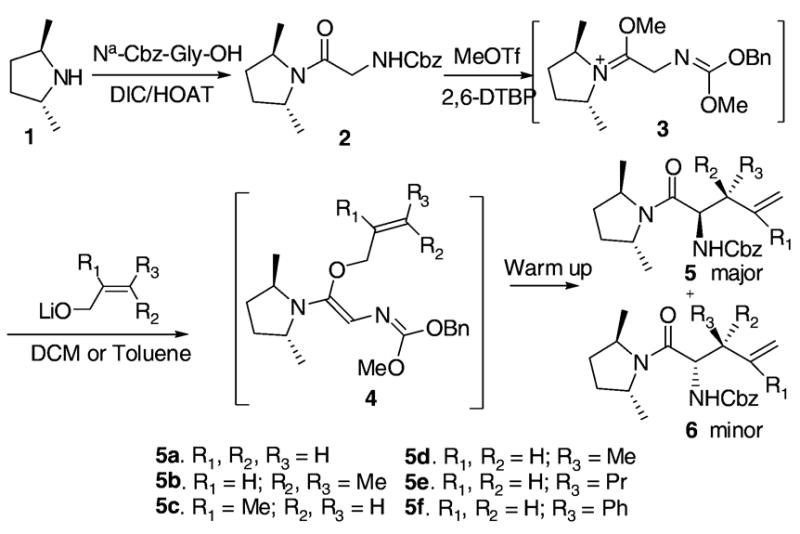
The Meerwein Salt Formation and Asymmetric Eschenmoser–Claisen Rearrangement
The rearranged products are mixtures of diastereomers and their 1H NMR peaks are often too close together to distinguish even with a purified sample. In such cases, HPLC was used to separate the components and determine their anti/syn ratios and de values. For comparison, racemic amino acids 10e and 10f that also contain some syn isomers (Scheme 2) obtained from our previous work were coupled to the chiral auxiliary 1 to provide authentic samples. In this way, we were able to determine the anti/syn ratios and de values in entries 4 and 7 (Table 1). The anti/syn ratios and de values of other products were determined by analogy. The details of the chromatographic identification are reported in the Supporting Information. The absolute configurations were not confirmed until the removal of the chiral auxiliary.
Scheme 2.
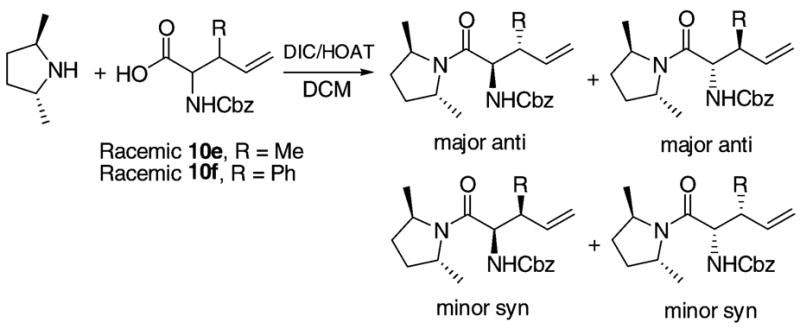
Synthesis of the Authentic Samples
Table 1.
The Results of the Asymmetric Eschenmoser–Claisen Rearrangement
| entry | allylation agent | anti:syn | de (%, 5/6) | yield (%)a |
|---|---|---|---|---|
| 1 |
|
NA | 88b | 75.2 |
| 2 |
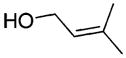
|
NA | 49c | 80.6 |
| 3 |
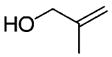
|
NA | 91b | 81.6 |
| 4 |
|
>98:2b | 87b | 84.6 |
| 5 |
|
>96:4b | 86b | 83.1 |
| 6 |
|
>87:13b | 54b | 65.0 |
| 7 |
|
>98:2c | 93c | 70.0 |
Isolated yield of total isomers.
Determined by chiral HPLC.
Determined by 1H NMR.
The rearrangement results from seven primary allylic alcohols are summarized in Table 1. The excellent diastereoselectivities from the trans allylic alcohols were attributed to the (Z)-N,O-ketene acetal formation and pseudochairlike conformations of the rearrangement intermediates discussed in our previous work. Compared with the bulkier non-C2-symmetric chiral auxiliaries that gave an average relative asymmetric induction of 4.7 to 1 in the work of Welch et al., the C2-symmetric chiral auxiliary provided much improved relative asymmetric induction despite its small size. Presumably, this is due to the elimination of the rotamer problem.11 The two substituents R1 and R3 are pointed away from the C2-symmetric chiral auxiliary in the transition state and their size is not expected to greatly affect the diastereoselectivities. This is supported by the results from entries 3–5 and 7. However, a cis configured substituent of the starting allylic alcohol can destabilize the chairlike transition state and lead to low de as in entries 2 and 6.
Many high-quality trans allylic primary alcohols are commercially available or can be easily synthesized from allylic oxidation of olefins, and reduction of conjugated aldehydes or esters.12 Thus, the developed method is ready to introduce many pharmacologically interesting functionalities at the β-position. Many desired anti-β-substituted γ,δ-unsaturated amino acid derivatives can be generated with excellent diastereoselectivities from inexpensive glycine via this efficient strategy.
The stereochemistry outcome is consistent with Welch’s work. However, it cannot be simply explained by using the model Rawal et al. suggested for the thio-Claisen rearrangement.11 Conformational energy differences, other than the straightforward steric factor, must also be considered. The much lower diastereoselectivities from entries 2 and 6 suggest that the transition state is different from what Rawal et al. suggested. A computer molecular modeling study of the transition state is underway.
A two-step strategy was introduced here to remove the chiral auxiliary.13 The rearrangement products were subject to iodolactonization in different solvent combinations. It turned out that the reaction did not take place in DME/H2O, probably due to the poor solubility of the substrates. However, we were pleased to find that the reaction went very well in THF/H2O and a mixture of iodolactones 8 and 9 was formed in a ratio of about 9 to 1 by 1H NMR (Scheme 3, only major products are shown). The chiral auxiliary in the aqueous phase can be recovered by extraction with ethyl ether and distillation.9 The iodolactones 8 and 9 were then easily converted to the same amino acids 10 via zinc reduction as expected.14 This iodolactonization/zinc reduction pathway worked well for 5a–e with no observable syn products from the 1H NMR spectra of 10d and 10e. However, a large amount of unidentified byproducts were formed from the iodolactonization of 5f. Subsequent zinc reduction of the iodolactones gave 10f (R1, R2 = H; R3 = Ph), along with 20% of the syn isomer (from 1H NMR). It is likely that the electron-withdrawing phenyl group disfavors the formation of the desired iodolactones or the iodolactones are not stable under the reaction conditions. Therefore, 10f was obtained via a reduction/oxidation method we reported previously.8
Scheme 3.
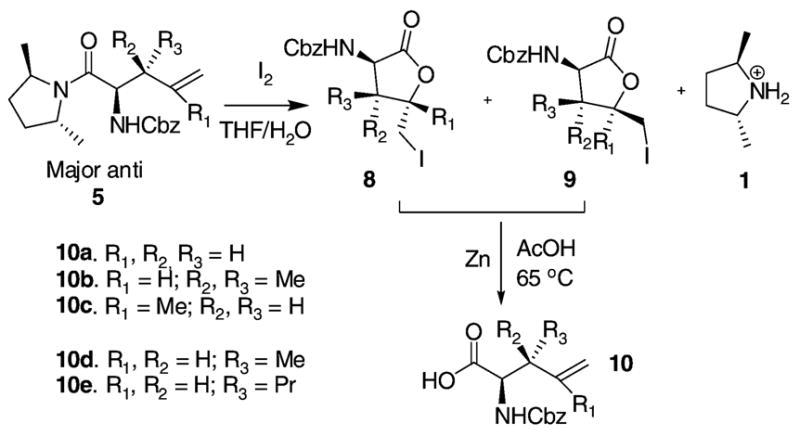
The Iodolactonization and Zinc Reduction of 5
The results of the iodolactonization/zinc reduction for 5a–e and the reduction/oxidation for 5f are shown in Table 2. The anti/syn ratios and ee values of those acids are consistent with the anti/syn ratios and de values of the starting amides, indicating that both methods worked well without noticeable epimerization. Occasionally an enriched ee was observed due to the loss of the minor anti diastereomer during chromatography purification of the iodolactones. The advantages of the iodolactonization/zinc reduction strategy are mild reaction conditions, easy recovery of the chiral auxiliary, and clean final products. It should be noted that this iodolactonization provides a window to enrich the optical purity of the major products. In fact many iodolactone derivatives solidified after workup and flash column chromatography purification. A potential recrystallization of the iodolactones will be studied in the future scale up of the reaction. On the other hand, this strategy takes a longer time compared with the reduction/oxidation pathway and does not work for the amide analogues with a β-phenyl substituent.
Table 2.
The Results of the Chiral Auxiliary Cleavage
| compd 10 | anti:syna | eeb (%) | yield (%) (two-step) |
|---|---|---|---|
| a | NA | 82 | 70.7 |
| b | NA | 40 | 65.4 |
| c | NA | 89 | 74.4 |
| d | >98:2 | 87 | 65.1 |
| e | >98:2 | 89 | 69.1 |
| f | >98:2 | 93 | 66.4 |
Determined by 1H NMR.
Determined by chiral HPLC by comparison with the racemic compounds synthesized from the previously reported racemic version of the Eschenmoser–Claisen rearrangement.
To validate the absolute stereochemistry assignments of products 10 as well as compounds 5 that were suggested by Yamazaki et al.,7 we compared the optical rotation and NMR spectra of product 10a with the published data.15 It was confirmed that the assignment was correct. In addition, product 10d was subjected to hydrogenation and the resulting product was determined unambiguously to be the enantiomer of L-isoleucine (2S,3S) by its opposite sign of optical rotation and identical NMR spectra. This further confirms the stereochemistry assignments.
To summarize, we have developed a four-step general method to synthesize optically active anti-β-substituted γ,δ-unsaturated amino acids via the asymmetric Eschenmoser–Claisen rearrangement. We have demonstrated that the C2-symmetric chiral auxiliary (2R,5R)-dimethylpyrrolidine provided excellent diastereoselectivities for both aliphatic and aromatic β-substituted amino acid analogues. These optically active amino acids are ready to be scaled up and their applications in peptidomimetics will be presented in the future.
Acknowledgments
This work was supported by U.S. Public Health Service grants DK 17420 and the National Institute of Drug Abuse DA 06284 and DA 13449. We also thank Prof. Richard Glass (the University of Arizona) for useful discussions.
Footnotes
Supporting Information Available: Experimental procedures and spectroscopic characterization (1H NMR, 13C NMR, HRMS, IR) of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Gramer U, Rehfeldt AG, Spener F. Biochemistry. 1980;19:3074. doi: 10.1021/bi00554a037. [DOI] [PubMed] [Google Scholar]
- 2.(a) Katagiri K, Tori K, Kimura Y, Nagasaki T, Minato H. J Med Chem. 1967;10:1149. doi: 10.1021/jm00318a035. [DOI] [PubMed] [Google Scholar]; (b) Tsubotani S, Funabashi Y, Takamoto M, Hakoda S, Harada S. Tetrahedron. 1991;47:8079. [Google Scholar]
- 3.(a) Morimoto Y, Takaishi M, Kinoshita T, Sakaguchi K, Shibata K. Chem Commun. 2001;18:1820. doi: 10.1039/b108752e. [DOI] [PubMed] [Google Scholar]; (b) Gu X, Ying J, Min B, Cain J, Davis P, Willey P, Edita N, Yamamura H, Porreca F, Hruby VJ. Biopolymers. 2005;80:151. doi: 10.1002/bip.20208. [DOI] [PubMed] [Google Scholar]; (c) Ruties TPJT, Wolf L, Schoemaker HE. J Chem Soc, Perkin Trans 1. 2000;24:4197. [Google Scholar]
- 4.(a) Bartlett PA, Barstow JF. J Org Chem. 1982;47:3933. [Google Scholar]; (b) Kazmaier U. Angew Chem, Int Ed Engl. 1994;33:998. [Google Scholar]; (c) Kazmaier U. Synlett. 1995;11:1138. [Google Scholar]; (d) Kazmaier U, Maier S. Chem Commun. 1995;19:1991. [Google Scholar]; (e) Kazmaier U. J Org Chem. 1996;61:3694. doi: 10.1021/jo960014g. [DOI] [PubMed] [Google Scholar]; (f) Kubel B, Hofle G, Steglich W. Angew Chem, Int Ed Engl. 1975;14:58. [Google Scholar]
- 5.(a) Kazmaier U, Krebs A. Angew Chem, Int Ed Engl. 1995;34:2012. [Google Scholar]; (b) Mues H, Kazmaier U. Synthesis. 2001;3:487. [Google Scholar]; (c) Kazmaier U, Mues H, Krebs A. Chem Eur J. 2002;8:1850. doi: 10.1002/1521-3765(20020415)8:8<1850::AID-CHEM1850>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 6.(a) Sakaguchi K, Suzuki H, Ohfune Y. Chirality. 2001;13:357. doi: 10.1002/chir.1045. [DOI] [PubMed] [Google Scholar]; (b) Sakaguchi K, Yamamoto M, Kawamoto T, Ymada T, Shinada T, Shimamoto K, Ohfune Y. Tetrahedron Lett. 2004;45:5869. [Google Scholar]
- 7.(a) Welch JT, Eswarakrishnan S. J Am Chem Soc. 1976;109:6716. [Google Scholar]; (b) Yamazaki T, Welch JT, Plummer JS, Gimi RH. Tetrahedron Lett. 1991;32:4267. [Google Scholar]
- 8.Qu H, Gu X, Min BJ, Liu Z, Hruby VJ. Org Lett. 2006;8:4215. doi: 10.1021/ol061414l. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Short RP, Kennedy RM, Masamune S. J Org Chem. 1989;54:1755. [Google Scholar]; (b) The chiral auxiliary can be synthesized in one step from a commercially available precursor (Toronto Research Chemicals Inc.)
- 10.A major byproduct (usually <5% yield) in the rearrangement was isolated by flash column chromatography. The compound was identified as 7 (see the Supporting Information) by NMR and mass spectrometry.
- 11.He S, Kozmin SA, Rawal VH. J Am Chem Soc. 2000;122:190. [Google Scholar]
- 12.Heydari A, Khaksar S, Akbari J, Esfandyari M, Pourayoubi M, Tajbakhsh M. Tetrahedron Lett. 2007;48:1135. [Google Scholar]
- 13.Tamaru Y, Mizutani M, Furukawa Y, Kawamura S, Yoshida Z, Yanagi K, Minobe M. J Am Chem Soc. 1984;106:1079. [Google Scholar]
- 14.Metz P, Hungerhoff B. J Org Chem. 1997;62:4442. doi: 10.1021/jo970123a. [DOI] [PubMed] [Google Scholar]
- 15.Harding CI, Dixon DJ, Ley SV. Tetrahedron. 2004;60:7679. [Google Scholar]