

Abstract
The DNA base excision-repair pathway is responsible for the repair of DNA damage caused by oxidation/alkylation and protects cells against the effects of endogenous and exogenous agents. Removal of the damaged base creates a baseless (AP) site. AP endonuclease1 (Ape1) acts on this site to continue the BER-pathway repair. Failure to repair baseless sites leads to DNA strand breaks and cytotoxicity. In addition to the repair role of Ape1, it also functions as a major redox-signaling factor to reduce and activate transcription factors such as AP1, p53, HIF-1α, and others that control the expression of genes important for cell survival and cancer promotion and progression. Thus, the Ape1 protein interacts with proteins involved in DNA repair, growth-signaling pathways, and pathways involved in tumor promotion and progression. Although knockdown studies with siRNA have been informative in studying the role of Ape1 in both normal and cancer cells, knocking down Ape1 does not reveal the individual role of the redox or repair functions of Ape1. The identification of small-molecule inhibitors of specific Ape1 functions is critical for mechanistic studies and translational applications. Here we discuss small-molecule inhibition of Ape1 redox and its effect on both cancer and endothelial cells. Antioxid. Redox Signal. 10, 1853–1867.
Introduction
The DNA base excision-repair (BER) pathway is responsible for the repair of DNA caused by oxidative, alkylation, and ionizing radiation and thus protects cells against the toxic effects of endogenous and exogenous agents. Removal of the incorrect or damaged base by a DNA glycosylase, either a simple glycosylase such as the methylpurine DNA glycosylase (MPG or AAG) or a complex glycosylase (Ogg1, Nth1, NEIL1, etc.) composes the first step of the BER pathway (Fig. 1). MPG repairs the alkylated DNA major cytotoxic lesion, 3-methyladenine (3-me A) and also functions to cleave a major product of lipid oxidation; 1, N(6)-ethenoadenine. Ogg1, Nth1, NEIL1, and NEIL2 recognize and remove both purine and pyrimidine oxidative DNA-damaged adducts. All of these glycosylases require the further processing of the initiating step in the BER pathway by Ape1 (Fig. 1). Ape1 (apurinic/apyrimidinic endonuclease 1) is the major repair protein for abasic sites, accounting for 95% of all AP endonuclease activity (77). It cleaves 5′ of the abasic site generating a normal 3′-hydroxyl group and an abasic deoxyribose-5-phosphate, which is processed by subsequent enzymes of the BER pathway. The removal of a damaged base results in an AP site that, if not repaired, may result in a block to DNA replication and genetic instability (77). DNA polymerase β is recruited to the abasic site by Ape1 and can use the 3′OH as a substrate once it has removed the 5′ deoxyribose phosphate terminus. DNA ligase I then ligates the remaining break in the phosphodiester backbone, completing the repair. Other enzymatic repair activities associated with Ape1 include 3′-repair diesterase or phosphatase activity (62), which is important for repair of DNA damaged by IR, and a 3′–5′ exonuclease activity, reported to play a role in the excision of deoxyribonucleoside analogues (7–10).
FIG. 1.
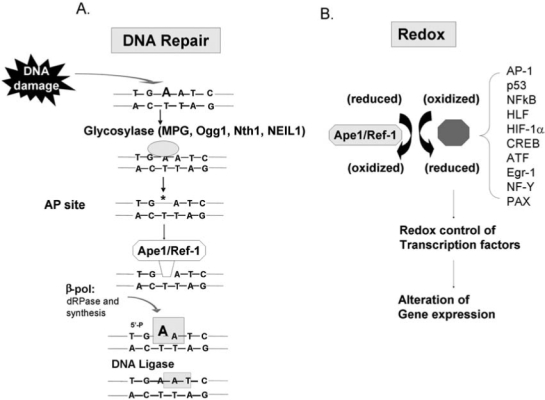
Oxidative and alkylation DNA damage is effectively repaired by the DNA BER pathway. This is a very simplified drawing of the DNA BER pathway. For a more detailed discussion, see Fishel et al. (21). (A) Damaged bases (in this case an N3-adenine) are removed by glycosylases that result in baseless or apurinic (AP) sites. Ape1 acts on AP sites, produces a nick that allows É-polymerase to insert the correct base and DNA ligase to connect the DNA backbone. (B) In addition to its repair function, Ape1 also serves as a redox factor maintaining transcription factors in an active reduced state. Some example targets are shown. Therefore, the protection of Ape1 from DNA damage may be twofold: directly through its DNA-repair function and indirectly through its redox activity by enabling transcription factors to bind to DNA and respond to the DNA damage. Lack of Ape1 activity may lead to cell death.
However, Ape1 also has another major cellular function: Ape1 functions as a reduction–oxidation (redox) factor and stimulates the DNA-binding activity of numerous transcription factors that are involved in cancer promotion and progression, such as AP-1 (Fos/Jun), NF-κB, PAX, HIF-1α, HLF, p53, and others (Fig. 1) (16, 21, 71). The redox function of Ape1 is only found in mammals and not in other vertebrates, as demonstrated by the lack of redox function of the zebrafish Ape1 (zApe1) (Fig. 2). The acquisition of the redox function in Ape1 proteins is discussed in a recent publication (25). Each of the molecularly distinct functional domains of Ape1, redox and repair, are completely independent in their function [i.e., mutations of the cysteine at position 65 Cys65 (C65) removes the redox function but does not affect the repair function (Fig. 3), whereas mutation of a variety of amino acids required for DNA repair activity, such as His309 (H309) and others (51) (Fig. 3) does not affect the redox function]. Whereas the DNA-repair active site of Ape1 has been clearly delineated (26), the redox domain or region is less obvious. It appears that the only required Cys for full redox function is Cys65, which is buried within the Ape1 protein (Fig. 3). This was recently confirmed by Georgiadis and her co-workers (25) when they mutated a Thr (T) in zebrafish Ape1 located at the same position of the Cys in mammalian Ape1s to a Cys, and the subsequent protein gained redox function (25).
FIG. 2.

Comparison of selected Ape1 family members from bacteria to mammals. The percentage displayed represents the amino acid similarity between the respective protein and human Ape1. Only mammals have acquired the redox function.
FIG. 3.
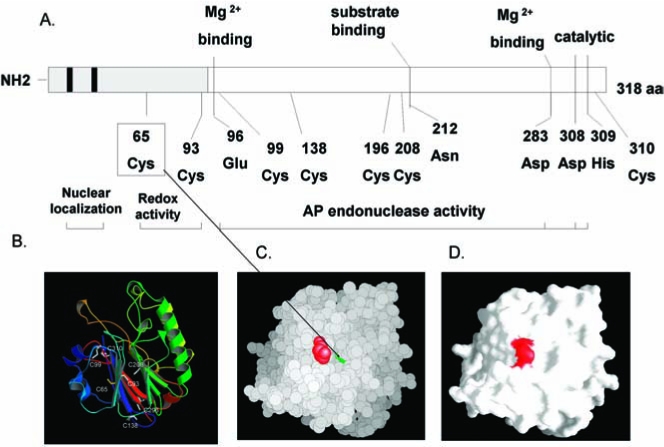
Cartoon of major amino acids involved in the DNA repair or redox function of Ape1. (A) Seven cysteines exist in Ape1, but only Cys65 appears to be involved in redox activity. Of the Cys residues, Cys65 is unique to mammals (25). The lower left panel (B) shows the relative positions of the seven Cys residues on a ribbon rendering of Ape1. The lower right two panels demonstrate that Cys65 is a buried residue. All atoms of Ape1 are shown in a van der Waals surface rendering in (C), with C65 in green and P112 in red. (D) A molecular surface rendering colored in the same way and in the same orientation as in (C). Cys 65 is not visible because it is not surface accessible (i.e., it is buried). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
Interestingly, the zebrafish crystal structure is quite similar to human Ape1, but only five of the seven Cys found in mammalian Ape1s are structurally conserved. The two that are not conserved are Cys65 and Cys138 (25). An evolutionary analysis of the Cys residues within vertebrates suggests that the presence of Cys65 distinguishes mammalian from nonmammalian vertebrate sequences (25).
The importance of Ape1 to the cell is demonstrated by the fact that it has not been possible to generate an animal knockout model. Ape1 mouse knockouts are embryonic lethal on days E5 to E9, and no viable cell lines have been established that are completely deficient for Ape1 (79). Previously, a small number of studies using DNA antisense methods have implicated Ape1 in cellular resistance to a variety of agents (6, 16, 59, 74). By using antisense Ape1, hypersensitivity of human HeLa, rat glioma, or human lung carcinoma cells to alkylating and oxidative agents as well as ionizing radiation but not UV radiation was indicated (16). More recently, a similar enhanced sensitivity for osteogenic sarcoma cells to DNA-damaging agents has been reported (75). Cells lacking Ape1 activity are hypersensitive to alkylating agents that induce the formation of AP sites (13, 24). Targeted reduction of Ape1 protein by specific anti-sense oligonucleotides renders mammalian cells hypersensitive to methylmethane sulfonate (MMS), H2O2, bleomycin, temozolomide (TMZ), and gemcitabine (2, 32, 43, 59, 74). Additionally, blocking the repair function of Ape1 has led to enhanced effectiveness of DNA-damaging agents such as temozolomide (TMZ), and other alkylating and oxidative DNA-damaging agents (19–21, 30, 44–47, 51, 70, 75, 81). Finally, blocking the activity of Ape1 after IR has lead to an increase in tumor cell killing (37, 68, 73).
However, the use of antisense methods in cancer cells removes not only the repair function of Ape1, but also its redox function and all protein–protein interactions mediated by Ape1. Ape1 is thought to interact with a large number of other repair-related proteins including Ogg1 (66), DNA glycosylases, DNA polymerase β (61), x-ray cross-complementing-1 (XRCC1) (4, 53, 61), proliferating cell nuclear antigen (PCNA) (14), Cockayne syndrome B (CSB) (78), and flap endonuclease 1 (FEN1) (14, 61, 64).
Additional functions that have been ascribed to Ape1 also complicate its analysis with siRNA. For example, it has been implicated in nucleotide incision repair (NIR) (76), involved in NK cell–mediated killing through granzyme A (GzmA) (17, 50), prevents oxidative stress by negatively regulating Rac1/GTPase activity(60), regulates endothelial NO production and vascular tone (38), and in a recent finding that is important for our studies, Ape1 appears to suppress the activation of PARP1 [poly (ADP-ribose) polymerase 1] during the repair process of oxidative DNA damage, which may allow cells to avoid cell death (63). Other findings of a relation between Ape1 and Bcl2 levels (39) and acting as a negative regulator of the parathyroid hormone gene have been shown (1, 11, 42, 58). Finally, recent data link Ape1 with PTEN (18), a major tumor suppressor that has been implicated as playing a crucial role in a variety of cancers, including brain tumors (5).
Thus, by using antisense RNA or similar technology, it will not be possible to determine precisely the role of the endonuclease or redox function of Ape1 in cancer or normal cells without specific inhibitors of each function independently. Therefore, with the long-term goal of developing cancer therapeutic agents, as well as small-molecule inhibitors of each the major functions of Ape1, we present data further characterizing the Ape1 redox mechanism of action and demonstrating the role of the redox function of Ape1 in cancer and retinal endothelial cells (RECs) in survival and growth by using a specific small-molecule inhibitor of just the redox function of Ape1. Inhibition of the redox function blocks endothelial cell growth and blocks the growth of tumor cell lines as a single agent. This effect is independent of the Ape1 DNA-repair function.
Materials and Methods
Synthesis of E3330, 3-[5-(2,3-dimethoxy-6-methyl-1,4-benzoquinoyl)]-2-nonyl-2-propionic acid
Reagents were used as received from commercial sources. THF and CH2Cl2 were distilled before use, and acetone was dried over activated 4-Å molecular sieves for 2 h under argon. Flash chromatographic separations were performed by using 32- to 63-μm silica gel; thin-layer chromatography was performed with Analtech 250-μm GHLF plates with 254-nm fluorescent indicator. All 1H-NMR spectra were obtained on a Bruker 300-MHz NMR equipped with a multinuclear (1H, 13C, 19F, and 31P) 5-mm probe. 1H spectra were calibrated to CHCl3 at 7.24 ppm. 13C NMR was calibrated to CHCl3 at 77.0 ppm. Elemental analyses were performed at the Purdue University Microanalysis Laboratory by Dr. Daniel Lee.
1,2,3,4-Tetramethoxybenzene (2)
By following a modified procedure by Tremblay et al. (72), 2,3,4-trimethoxybenzaldehyde 1 (6.08 g, 31.0 mmol) was dissolved in MeOH (60 ml), and then H2SO4 (0.6 ml) and 35% aq. H2O2 (4.0 ml, ~40 mmol) was added at room temperature. The reaction was then heated at reflux for 2 h. The solution was cooled and extracted with ethyl acetate. The layers were separated, and the organic layer was washed with brine, dried over MgSO4, and filtered. Flash chromatography (1:4 EtOAc:hexanes) gave 2,3,4-trimethoxyphenol (5.56 g, 98%) as a light-red oil. Rf = 0.29 (1:4 EtOAc:hexanes); 1H NMR (CDCl3): δ 3.79 (s, 3H); 3.87 (s, 3H); 3.94 (s, 3H); 5.34 (bs, 1H, OH); 6.57 (q, 2H).
The intermediate phenol (3.85 g, 20.9 mmol) was dissolved in dry acetone (90.0 ml), and K2CO3 (9.36 g, 67.7 mmol) was added, followed by MeI (9.0 ml, 140 mmol). The reaction mixture was heated under reflux for 26 h, cooled to room temperature, and filtered. The solvent was removed under reduced pressure, and the residue was taken up in CH2Cl2 (50 ml). The suspension was filtered, dried over MgSO4, filtered again, and condensed. The crude product was purified by either flash chromatography (1:4 EtOAc:hexanes) or re-crystallization from Et2O/hexanes to provide 2 (3.41 g, 82%) as white needles, mp = 86–87°C (Lit. 87–87.5°C) (3). 1H NMR (CDCl3): δ 3.80 (s, 6H); 3.88 (s, 6 H); 6.56 (s, 2H).
6-Methyl-2,3,4,5-tetramethoxybenzaldehyde (3)
By following a modified procedure by Hansen et al. (28), tetramethoxybenzene 2 (2.40 g, 12.1 mmol) was added to a 100-ml flame-dried round-bottom flask to which THF (50 ml) was added. The solution was cooled to 0°C, and n-BuLi (2.5 M in hexanes, 6.0 ml, 15 mmol) was added slowly at 0°C. The solution was stirred for 1 h at 0°C; then MeI (1.2 ml, 19.3 mmol) was added slowly, the reaction was stirred at 0°C for 2 h, and the reaction mixture was warmed to room temperature and stirred an additional hour. The reaction was acidified with dilute HCl, washed twice with brine, dried over MgSO4, filtered, and condensed. After flash chromatography (1:9 EtOAc/hexanes), the tetramethoxytoluene product was collected as a yellow oil (2.27 g, 88%). Rf = 0.57 (7:13 EtOAc:hexanes); 1H NMR (CDCl3): δ 2.43 (s, 3H); 3.74 (s, 3H); 3.89 (s, 3H); 3.92 (s, 3H); 4.00 (s, 3H); 10.41 (s, 1H).
According to the procedure by Ohkawa et al. (57), tetramethoxytoluene (0.690 g, 3.25 mmol) was dissolved in CH2Cl2 (2.0 ml) in a flame-dried 25-ml round-bottom flask and cooled to 0°C. α,α-Dichloromethyl methyl ether (0.85 ml, 9.6 mmol) was then added at 0°C, followed by TiCl4 (1 M in CH2Cl2, 9.0 ml, 9.0 mmol) at 0°C. The reaction was warmed slowly to room temperature and stirred for 6 h under argon. The reaction was then poured into chilled water and stirred for 10 min. The reaction was diluted with EtOAc, washed with brine, dried over MgSO4, filtered, and condensed. The crude oil was then purified by flash chromatography (3:17 EtOAc:hexanes) to provide 3 (0.696 g, 89%) as a yellow oil. Rf = 0.26 (3:17 EtOAc:hexanes); 1H NMR (CDCl3): δ 2.44 (s, 3H); 3.74 (s, 3H); 3.89 (s, 3H); 3.92 (s, 3H); 4.00 (s, 3H); 10.24 (s, 1H).
Ethyl (E)-3-(6-methyl-2,3,4,5-tetramethoxyphenyl)-2-nonylpropenoate (4)
By following modification to the procedure by Shinkawa et al. (55), NaH (0.24 g, 10.0 mmol) was added to a flame-dried 50-ml three-neck round-bottom flask connected to a water-jacketed reflux condenser. The flask was purged with argon, and a drying tube was attached to the top. THF (15 ml) was added to the flask, followed by triethyl 2-phosphonoundecanoate (1.90 g, 5.44 mmol) dissolved in THF (5 ml) at room temperature. The reaction was heated under reflux for 30 min, and then aldehyde 3 (1.04 g, 4.32 mmol) was dissolved in THF (8 ml) and added slowly under reflux. The reaction was stirred for another 3 h under reflux and was then cooled, acidified with dilute HCl, diluted with ethyl acetate, and washed with brine. The organic layer was dried (MgSO4), filtered, and condensed. The resulting oil was purified via flash-column chromatography (3:17 EtOAc:hexanes) to provide 4 (1.28 g, 68%) as a colorless oil. Rf = 0.40 (3:17 EtOAc:hexanes); 1H NMR (CDCl3): δ 0.84 (t, 3H); 1.08 – 1.22 (m, 11H); 1.24 – 1.35 (m, 3H); 1.33 (t, 3H); 2.03 (s, 3H); 2.12 (m, 2H); 3.69 (s, 3H); 3.78 (s, 3H); 3.88 (s, 3H); 3.93 (s, 3H); 4.25 (q, 2H); 7.37 (s, 1H).
(E)-3-(4,5-dimethoxy-2-methyl-3,6-dioxocyclohexa-1,4-dienyl)-2-nonylpropenoic acid (5)
Ester 4 (1.57 g, 3.60 mmol) was dissolved in EtOH (12 ml), and then KOH (0.41 g, 7.3 mmol) was added. The solution was heated to reflux and stirred for 30 min. The reaction was then cooled, acidified with HCl, and extracted with ethyl acetate. The organic layer was washed with brine, dried over MgSO4, filtered, and condensed. The resulting acid was then used in the following reaction without purification. Alternatively, the product can be purified via flash chromatography (2:3 Et2O:hexanes 0.5% AcOH) to provide the free acid (1.43 g, 97%) as a colorless oil: Rf = 0.36 (2:3 Et2O:hexanes 0.5% AcOH).1H NMR (CDCl3): δ 0.84 (t, 3H, J = 6.9 Hz); 1.13 (m, 12H); 1.36 (m, 2H); 2.04 (s, 3H); 2.14 (m, 2H); 3.70 (s, 3H); 3.79 (s, 3H); 3.89 (s, 3H); 3.93 (s, 3H); 7.54 (s, 1H).
By following modifications to the procedures by Shinkawa et al. and Flader et al. (23, 56), the crude acid obtained earlier (1.13 g, 2.77 mmol) was dissolved in ethyl acetate (15.0 ml) at room temperature; then HNO3 (1.5 ml) and AcOH (6 drops) were added, and the reaction was stirred for 4 h. The mixture was then diluted with EtOAc (20 ml) washed with brine, dried over MgSO4, filtered, and condensed. The red oil was then purified by either flash column chromatography (2:3 Et2O/hexanes, 0.5% AcOH) or recrystallization from Et2O/hexanes to afford 5 (0.443 g, 42%) as a red solid; mp = 56–57°C (Lit. 68°C) (56). Elemental analysis: calc C, 66.65%, H, 7.99%; found C, 66.32%, H, 7.89%. 1H NMR (CDCl3): δ 0.84 (t, 3H), 1.18 (bs, 14H); 1.39 (bs, 2H); 1.94 (d, 3H); 2.09 (t, 3H); 3.99 (s, 3H); 4.02 (s, 3H); 7.26 (d, 1H).
Expression and purification of proteins
Proteins were purified as previously described (25, 29, 36, 40, 80).
Ape1 DNA repair assays
Oligonucleotide gel-based Ape1 endonuclease activity assays were performed as previously described (20, 41). The base excision repair (BER) pathway analysis was performed as described (67).
Redox assays
Electrophoretic mobility shift assay (EMSA)
EMSAs were performed as described (15) with the following modifications. The 10-μg/μl purified Ape1 protein was reduced with 1.0 mM DTT at 37°C for 10 min and diluted in PBS buffer to yield final concentrations of 2 μg/μl protein and 0.2 mM DTT. Two μl of reduced Ape1 protein was added to EMSA reaction buffer [10 mM Tris (pH 7.5), 50 mM NaCl, 1 mM MgCl2, 1 mM EDTA, 5% (vol/vol) glycerol] with 6 μg of nuclear extract (treated with 0.01mM diamide for 10 min) from Hey-C2 cells as the source of AP1 protein in a total volume of 18 μl and incubated for 30 min. at room temperature. One μl poly(dI-dC) · poly(dI-dC) (1μg/ul) (Amersham Biosciences, Piscataway, NJ) was added for 5 min followed by 1 μl of the 5′ hexachloro-fluorescein phosphoramidite (HEX)-labeled double-stranded oligonucleotide DNA (The Midland Certified Reagent Company, Midland, TX) containing the AP-1 consensus sequence (5′CGCTTGATGACTCAGCCGGAA-3′) (0.1 pmol), and the mixture was further incubated for 30 min at room temperature. The final concentration of DTT in the redox reactions was 0.02 mM. Samples were loaded on a 5% nondenaturing polyacrylamide gel and subjected to electrophoresis in 0.5X TBE buffer (200 V for 1 h at 4°C) and detected by using the Hitachi FMBio II Fluorescence Imaging System (Hitachi Genetic Systems, South San Francisco, CA). The HEX fluorophore is excited by a solid-state laser at 532 nm (Perkin-Elmer) and emits a fluorescent light signal at 560 nm, which is then measured using a 585-nm filter.
Transactivation assay
E3330 assays
SKOV-3X and Hey-C2 ovarian cancer cell lines were co-transfected with the NF-κB-Luc or AP-1-Luc genes (luciferase gene with the NF-κB or AP-1 responsive promoter) by using plasmid pNF-κB-Luc or pAP-1-Luc (Stratagene, La Jolla, CA) and lipofectamine TM 2000 (Invitrogen Life Technologies, Carlsbad, CA). The cells were cotransfected with a control pcDNA empty vector plasmid, pcDNA-wtApe1 or the redox mutant, pcDNA-C65A, and a Renilla luciferase control reporter vector pRL-CMV (Promega Corp., Madison, WI) in a 10:1 ratio by using lipofectamine TM 2000. After 24-h transfection, cells were lysed, and the Firefly and Renilla luciferase activities were assayed by using the Dual Luciferase Reporter Assay System (Promega Corp.) with Renilla luciferase activity for normalization in a 96-well microtiter plate luminometer (Thermolabs Systems, Franklin, MA). All of the transfection experiments were performed in triplicate and repeated at least 3 times in independent experiments.
Ape1 transactivation mutation assays
The stable Skov-3X ovarian cancer cell line with NF-κB-Luc gene (luciferase gene with the NF-κB–responsive promoter) was established by transfecting Skov-3X cells with plasmid pNF-κB-Luc (Stratagene) by using lipofectamine TM 2000 (Invitrogen Life Technologies) and screening the luciferase-positive colonies by using a Luciferase Assay Kit (Promega Corp.). The stable Skov-3X cells with the NF-κB-Luc gene were cotransfected with plasmid pcDNA-wtApe1 or the redox mutant pcDNA-C65A and a Renilla luciferase control reporter vector pRL-CMV (Promega Corp.) in a 1:10 ratio by using lipofectamine TM 2000. The analysis was conducted as described earlier for the E3330 studies.
Cell survival/killing assays
3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymeyhoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTT) dye assay for cell growth was performed as previously published (20, 75).
Murine retinal endothelial cell isolation and culture
Mouse retinal vascular endothelial cells (RVECs) were isolated as described before with modifications (69). In brief, retinal tissues from young adult wild-type mice carrying a SV40 transgene from the Charles River Laboratories (ImmortoMouse) were digested with 0.2 mg/ml collagenase type I (Worthington, Lakewood, NJ) in DMEM at 37°C for 3 h. After titration and filtration through a 40-μm nylon membrane (Fisher Scientific, Hanover Park, IL), the dissociated cells were incubated with sheep anti-rat magnetic beads (Dynal Biotech, Lake Success, NY) precoated with a rat anti-mouse PECAM-1 monoclonal antibody (BD Biosciences, San Jose, CA) for affinity binding at 4°C for 45 min. Bead-bound cells were separated by magnetic force, plated in a 24-well plate precoated with 2 μg/ml of human fibronectin (BD Biosciences, San Jose, CA) and grown in an endothelial growth media (EBM-2MV, Cambrex). Cells were allowed to grow out of the beads reaching confluence for ~10 days before using for assays.
Cell-proliferation assay
Equal numbers of RVECs were seeded at 2,000 cell/well in a 96-well plate. Cells were cultured with or without 10 ng/ml recombinant mouse basic fibroblast growth factor (bFGF; R&D Systems, Inc.) supplement in various concentrations of E3330 (10, 25, 50, 100 μM). Each group consisted at least five wells (n = 5) including a vehicle control group. Two days after seeding, the total number of cells was assayed by using the CellTiter 96 AQueous One Solution (Promega Corporation). The proliferation rate was calculated according to manufacturer's instructions. Proliferations of RVECs from different groups were compared with the vehicle control group for statistical significance.
Tube-formation assay
Matrigel (BD Biosciences, Bedford, MA) was used to coat each well (50 μl) of a precooled 96-well plate. RVECs at 5,000 cells per well were seeded and incubated in EBM with 1% FBS and 10-ng/ml bFGF at 37°C for 24 h. The formation of capillary-like structures by RVECs on the Matrigel was quantified by counting the number of closed tube units in each well. The percentage of the tube formation to the vehicle control group was calculated for each E3330 (1, 5, 20 μM) treated group (n = 3).
Results
A number of studies have implicated Cys65 in the redox function of Ape1. However, some disagreement still exists whether this Cys is the only Cys involved, or just the major Cys involved in redox activity. Recent data using a zebrafish Ape1 (zApe1) model demonstrates that the acquisition of the Cys65 in human Ape1 converts a redox-inactive zApe1 to a redox-active zApe1 (25). To confirm this, we mutated the Cys65 in human Ape1 to an Ala (A) and performed cellular transactivation reporter analysis by using AP-1 and NF-κB as the downstream targets of Ape1 (Fig. 4). When Cys65 is mutated to Ala, Ape1 loses all of its redox activity in this cell-based reporter system, whereas addition of wild-type Ape1 results in enhanced redox activity (Fig. 4), regardless of the downstream target.
FIG. 4.
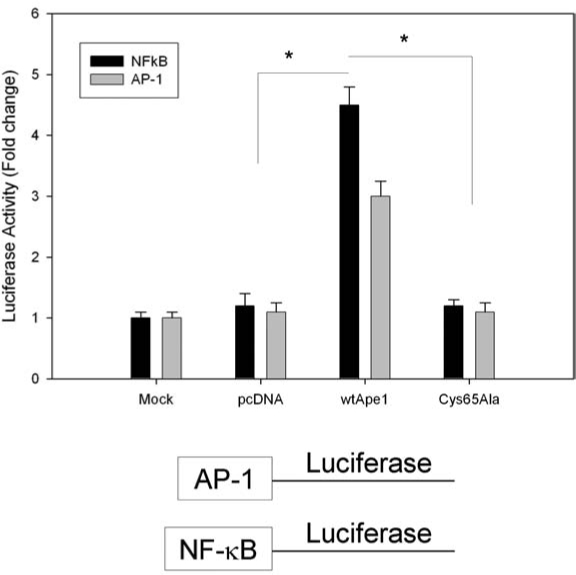
Effect of human Ape1 and Cys65 redox Ape1 mutant on NF-κB and AP-1 transactivation in a cell-based reporter assay system. SKOV-3X cells were transfected with AP-1 or NF-κB–Luc construct containing an NF-κB or AP-1–response promoter and driving the expression of a luciferase gene. The cells were cotransfected with plasmid pcDNA–wild-type hApe1 or the redox mutant (pcDNA-C65A) and a Renilla luciferase control reporter vector pRL-CMV. After a 24-h transfection period, cells were lysed, and Firefly and Renilla luciferase activities were assayed by using Renilla luciferase activity for normalization. All of the transfection experiments were performed in triplicate and repeated at least 3 times in independent experiments. Data are expressed as mean ± standard error from a representative experiment, and Student's t tests were performed. *Significant difference at the p < 0.05 level comparing the Cys65 mutant and wt-Ape1 and wt-Ape1 and mock or pcDNA.
Our next series of experiments involved the use of a small molecule redox inhibitor of Ape1, 3-[5-(2,3-dimethoxy-6-methyl-1,4-benzoquinoyl)]-2-nonyl-2-propionic acid (E3330) (82). This molecule has previously been shown to block Ape1 function in studies with NF-κB (35) and in hematopoietic cells (82) and is very specific for Ape1 with a high binding affinity and specificity (34, 65). To continue these studies, E3330, which is not commercially available, had to be synthesized by using a new approach, which we developed and described in the Methods section. This route was developed after encountering difficulties with the previously reported synthetic route (22, 23, 55). The result of this synthesis is shown in Fig. 5.
FIG. 5.

Synthesis of 3-[5-(2,3-dimethoxy-6-methyl-1,4-benzoquinoyl)]-2-nonyl-2-propionic acid (E3330) by using a novel approach.
By using E3330 in a series of EMSA experiments, we demonstrate that E3330 blocks the redox function of Ape1 with AP-1 as the downstream target in vitro (Fig. 6A) as well as after the treatment of ovarian cancer cells with E3330 (Fig. 6B). Additionally, we demonstrate that E3330 blocks Ape1 redox activity with HIF-1α demonstrating that the redox inhibition is not specific for the downstream target (Fig. 6C). Similar results with NF-κB in EMSA studies have also been observed (data not shown).
FIG. 6.
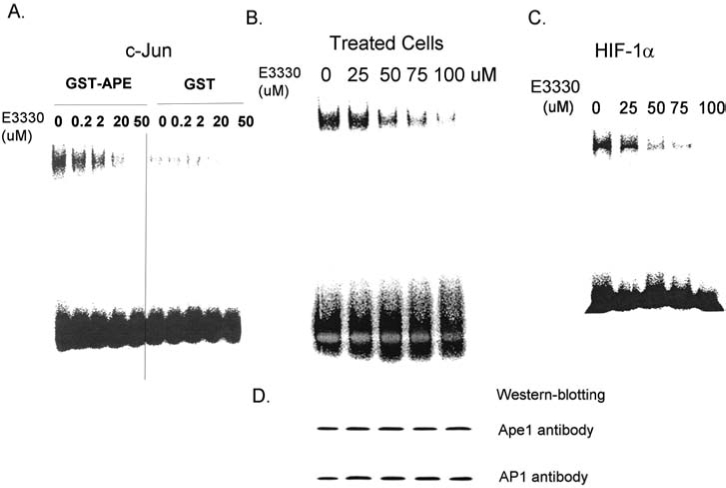
EMSA studies of AP-1 or HIF-1α binding after E3330 treatments. Increasing levels of E3330 decrease purified c-Jun binding in vitro (A) and in SKOV-3X ovarian cancer cells treated with E3330 (B). HIF-1α is also a downstream target of the redox function of Ape1, and inhibition of Ape1 redox by E3330 blocks HIF-1α binding (C). (D) A Western blot analysis of the samples in (B) by using Ape1 and AP-1 antibodies, demonstrating that the level of Ape1 and AP-1 in the cell extracts was identical, and the results observed are not due to a decrease in the respective proteins.
Although E3330 blocked the redox function of Ape1, it had no effect on Ape1 repair endonuclease activity (Fig. 7A), nor did it affect the ability of proteins involved in the BER pathway to perform the repair of AP sites (Fig. 7B) by using the established BER-pathway assay (67). These studies demonstrate the specificity of E3330 for the redox, but not the repair function of Ape1.
FIG. 7.
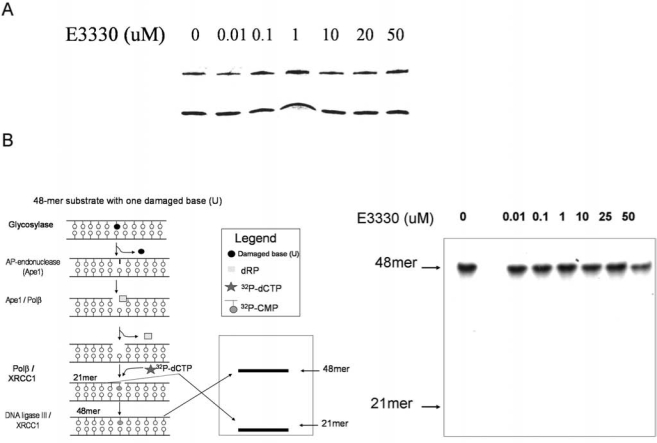
Oligonucleotide gel-based endonuclease and BER assays. Analysis was performed as described in Methods (20, 41). (A) One nanogram of purified Ape1 protein was used with an increasing amount of E3330, as indicated above the gel lanes. Ape1 protein extracts were added to a reaction mixture containing 0.2 pmol of HEX-labeled double-stranded tetrahydrofuran oligonucleotide, 50 mM HEPES, 50 mM KCl, 10 mM MgCl2, 2 mM DTT, 1% BSA, and 0.05% Triton X-100 (pH 7.5). Reactions with or without E3330 were incubated for 15 min at 37°C. Reactions were halted by adding 10 μl of formamide. Ape1 assay products were separated on a 20% polyacrylamide gel containing 7 M urea. The upper band (26-mer) represents uncleaved AP oligonucleotides, whereas the lower band (14-mer) is the reacted oligonucleotide. (B) BER pathway in vitro analysis by using the gap-filling assay, as described by Singhal et al. (67). The gap-filling assay is initiated from a uracil in an oligonucleotide substrate, which is then removed by using uracil-glycosylase. If the BER pathway is fully functional, a 48-mer band will be observed on the gel, whereas a 21-mer band indicates incomplete BER-pathway activity. E3330 was added to the reactions, as indicated, and had no effect on the pathway, indicating no effect on Ape1-repair function.
As an additional approach to demonstrate the specificity of E3330 to Ape1, we used the mutant zebrafish (T58C), which had been converted from a redox-inactive Ape1 protein to a redox-active protein (25), and reacted it with E3330. We then performed the redox EMSA by using AP-1 as the target protein. As shown in Fig. 8, E3330 blocked the ability of the T58C zebrafish Ape1, which had acquired the redox function, in a similar fashion to that with which it blocks human Ape1 redox activity. The concentration of E3330 required to inhibit 50% of the redox activity was 6.5 μM for wild-type hApe1 and 5 μM for T58C zApe. Although not conclusive, these data demonstrate that E3330 is not affecting the other cysteines in the Ape1 proteins and that the Cys65 in human Ape1 is the primary target of E3330 and the main cysteine involved in the redox function of Ape1. This was further confirmed in studies in which we mutated each cysteine in human Ape1 individually, and only the Cys65 had any major effect on the redox function of Ape1 (data not shown). Experiments are in progress to demonstrate unequivocally the interaction of E3330 and Ape1 protein and the mechanism involved (Georgiadis and Kelley, personal communication, 2008).
FIG. 8.
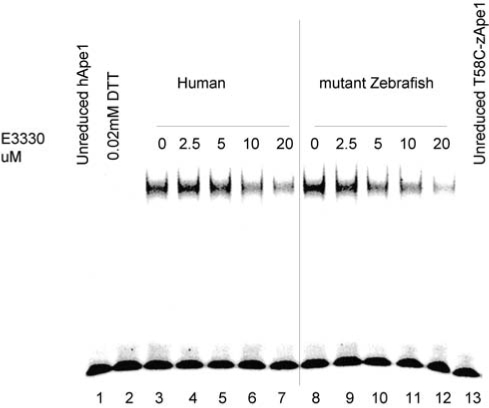
Redox EMSA showing E3330 inhibition of mutant zebrafish Ape1. An increasing amount of E3330 was incubated with 2 μl purified hApe1(lanes 1–7) or mutant zebrafish Ape1 (T58C-zApe1) (lanes 8–13; reduced with 1.0 mM DTT and then diluted to 2 μg/μl with 0.2 mM DTT in PBS) in EMSA reaction buffer [10 mM Tris (pH 7.5), 50 mM NaCl, 1 mM MgCl2, 1 mM EDTA, 5% (vol/vol) glycerol] with a total volume of 10 μl for 30 min, and then the EMSA assay was performed. E3330 inhibited the AP1-DNA binding stimulated by hApe1 and T58C-zApe1 to the same extent. Lane 1 is unreduced hApe1 control; lane 2 is control for DTT carryover, and lane 13 is unreduced T58C-zApe1 control. Lanes 3–7 are the human Ape1 reacted with increasing amounts of E3330. Lanes 8–12 show T58C zebrafish Ape1 reacted with increasing amounts of E3330.
Further to confirm the activity of E3330 as an Ape1 redox inhibitor in cell-based assays, we co-transfected two ovarian cancer cell lines, Hey-C2 and SKOV-3X, with either the pNF-κB-Luc or pAP-1-Luc constructs and the Renilla luciferase reporter vector pRL-CMV as a control for transfection. After transfection, we treated the cells with increasing amounts of E3330. As observed in Fig. 9, increasing amounts of E3330 causes an analogous decrease in the amount of AP-1 or NF-κB activation, as evidenced by a decrease in the amount of AP-1 or NF-κB activation of the luciferase reporter gene. These results indicate decreased binding of these proteins due to the blockage of their conversion from an oxidized to a reduced state by Ape1 because of E3330 blocking Ape1 redox function.
FIG. 9.

Effect of E3330 on Ape1 redox in transactivation assay. Effect of E3330 on endogenous cellular Ape1 redox activity by using a functional transactivation assay as described in Methods and Fig. 4. Increasing amounts of E3330 were added to either Hey-C2 or SKOV-3X ovarian cancer cell lines. At 24 h after transfection, the ratio of firefly luciferase activity to Renilla luciferase activity was determined to measure AP-1 or NF-κB activity. Data are expressed as the mean ± SEM of three independent experiments performed in triplicate and are presented as percentage transactivation compared with the normalized control of no E3330.
Because E3330 does not affect the repair functions of Ape1, as shown in Fig. 7, we tested the effect of blocking Ape1 redox function on two ovarian cancer cell lines. As shown in Fig. 10, E3330 causes a decrease in the amount of cell proliferation with increasing amounts of drug on both cell lines with an IC50 of 33 and 37 μM, respectively. We observed similar results with other cancer cell lines (data not shown). These results indicate that the redox activity functions of Ape1 in the growth or survival response of the cancer cells is independent of the DNA repair activity of Ape1. These findings confirm similar findings we observed in a hematopoietic model of embryonic stem cells (82). Further mechanistic studies as to how this is occurring are currently under way, although preliminary studies indicate that the redox-inhibitory effect is not necessarily due to cell killing, but to a block in cell proliferation or cytostatic effect similar to that observed by using Ape1 siRNA in vivo (19, 20).
FIG. 10.

Cell-growth assay by using E3330 on two ovarian cancer cell lines. The MTT growth/survival assay was performed as described in Methods. Increasing amounts of E3330 resulted in decreasing cell growth and decreased cell numbers. Data are expressed as the mean ± SEM of three independent experiments performed in triplicate.
E3330 inhibits RVEC proliferation and angiogenesis in vitro
Proliferation of endothelial cells (ECs) is an important index for the angiogenic ability of EC in vitro. The MTS assay is a simple and highly reproducible method of quantifying cell proliferation and was used to determine the effect of E3330 on the proliferation rate of RVECs. As shown in Fig. 11A, addition of E3330 inhibited RVEC growth in a dose-dependent manner. Supplement of bFGF in the basal media significantly boosted the proliferation rate. However, a similar inhibition effect was still evident with various doses of E3330 treatments. The difference between each E3330-treated group and its corresponding vehicle control of the same medium was statistically significant (p < 0.01).
FIG. 11.
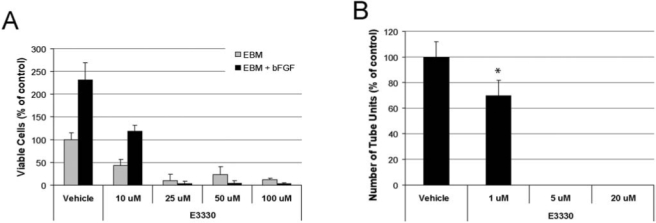
Inhibition of redox activity of Ape1 reduces retinal vascular endothelial cell angiogenesis in vitro. (A) In the MTS proliferation assay, RVECs were treated with or without bFGF and with various doses of E3330 (10–100 μM) in the culture for 2 days. A dose-dependent inhibition of RVEC growth was evident in both basal media (EBM) and bFGF-supplemented (10 ng/ml) media (EBM + bFGF). The number of viable cells in all E3330-treated groups was significantly different (p < 0.01) from its corresponding vehicle control cells in the same medium. (B) In the tube-formation assay, RVECs cultured in bFGF medium on Matrigel were treated with various doses of E3330 (1–20 μM) for 1 day. RVEC formation of capillary-like structure was significantly inhibited at a much lower dose range. The 30% reduction of tube units in 1 μM E3330 was statistically different from the vehicle control (p < 0.05).
An in vitro angiogenesis assay also was used to determine the effect of E3330 on RVEC formation of capillary-like structures on Matrigel. The inhibition effect of E3330 was similar to the proliferation assay but in a much lower dose range (Fig. 11B). E3330 at 1 μM was sufficient to elicit a 30% reduction of tube formation. Quantitative measurement of the number of tube units revealed that the difference was statistically significant (p < 0.05). The tube formation was completely abolished at 5 μM or higher concentration.
These results demonstrate that inhibition of redox activity significantly attenuates RVEC proliferation and capillary formation in vitro. The capillary formation of RVECs appears much more sensitive to redox inhibition.
Discussion
It has long been recognized that DNA-damaging agents are useful in killing cancer cells and are still the mainstay of cancer treatments (31, 49). However, a significant problem with the success of DNA-damaging agents is the upregulation or activation of a number of different pathways that confer resistance of the cancer cells to the treatment with DNA-damaging agents. This includes pathways such at those involved in signaling, multidrug resistance, cell-cycle checkpoints, antiangiogenesis, and others as potential approaches to treat and kill cancer. Much less work has focused on blocking the ability of a cancer cell to recognize and repair the damaged DNA that results from front-line cancer treatments (chemotherapy and radiation). One of these systems includes DNA-repair enzymes, such as Ape1, leading to resistance and failure of the agent to kill the cancer cells (16, 21). These findings suggest that one possible strategy for preventing resistance and improving efficacy of the DNA-targeting agents would be to inhibit the DNA-repair enzyme responsible for resistance. This potential has been recognized, as evidenced by the identification of small-molecule inhibitors of several DNA-repair enzymes, including MGMT, polyADP-ribose polymerase (PARP1), ataxia–telangiectasia mutated kinase (ATM kinase), Ape1, and DNA PKcs in varying degrees of success and development (12, 21, 48).
Toward the goal of developing inhibitors of Ape1 as a potential cancer therapeutic, as well as developing specific, independent inhibitors of each of the major functions of Ape1 (redox and repair), we focused on the blocking of the Ape1 redox function. Although overwhelming evidence supports the observation that Cys65 is the main cysteine involved in the redox role of human Ape1, we further confirm this premise through site-directed mutagenesis of the Cys65 site, leading to the lack of NF-κB or AP-1 transactivation in a cell-model system (Fig. 4) and inhibition of Ape1 redox by using a small molecule–specific inhibitor, E3330 (Figs. 6 and 9). Furthermore, we demonstrate that the humanized zebrafish Ape1, which we previously demonstrated gained redox activity (25), is now inhibited by E3330 (Fig. 8). In addition, the redox inhibition of Ape1 has no effect on the repair function of Ape1, demonstrated by the lack of inhibition in two separate DNA-repair assays (Fig. 7). E3330 was also able to block the redox function of Ape1 in two ovarian cancer cell lines by using a transactivation reporter assay in a dose-dependent manner (Fig. 9). These data support the cellular role of Ape1 in redox activation of downstream targets and demonstrate E3330 as a small molecule to block transcription-factor activation; particularly transcription factors that are involved in cancer cell growth and proliferation, such as NF-κB, AP-1, and HIF-1α. These data are in line with our previous findings demonstrating that blocking the redox, but not repair function of Ape1 led to blocking cell proliferation (82). Although previous studies demonstrated that altering Ape1 levels leads to blockage of cell growth and increased cancer cell sensitivity (2, 6, 16, 19, 20, 30, 33, 37, 43–47, 51, 59, 68, 70, 73–75, 81), these studies used overexpression of Ape1, Ape1 antisense oligonucleotides, or Ape1 siRNA: The quandary with this approach, although valid, is that each of these approaches changes the total cellular content level of Ape1 and removes all of the Ape1 functions, not just the repair or redox activities. Because Ape has multiple functions as well as protein–protein interactions with other DNA repair and signaling proteins, the increase or decrease of Ape1 protein may result in prejudiced or inexact results. Use of specific small-molecule inhibitors such as E3330 will be important to delineate the true role of Ape1 in various cancer, disease, and normal cellular functions. Ultimately, using Ape1 redox inhibitors with Ape1-specific endonuclease repair inhibitors will give a clearer picture of the multiple activites of Ape1.
Toward the goal of developing specific Ape1 redox inhibition to study this role of Ape1 in normal and cancer cells, we performed single-agent dose–response studies by using E3330 in ovarian cancer cell lines and determined that E3330 does have single-agent cancer cell killing abilities (Fig. 10). This finding is not restricted to just ovarian cancer cell lines and has been observed with cell lines representing colon, lung, breast, brain, pancreatic, prostate, and multiple myeloma cancers (data not shown). In contrast, we do not see significant growth inhibition in our studies with normal cells, such as hematopoietic embryonic cells (82), or in RVEC (Fig. 11) E3330 inhibits RVEC cell growth (MTS assay in the results). Additionally, we do not see cell killing in human CD34+ progenitor cells (unpublished data). These data are novel in that they implicate the redox role of Ape in cancer, but not in “normal” cell survival. This implicates the redox role as independent of the Ape1 DNA-repair function and independent of the addition of DNA-damaging agents. This is supported by our recently published data in ovarian cancer studies in xenografts, demonstrating that the knockdown of Ape1 results in the blocking of cell growth and proliferation, but not cell death (19). This is the first time cancer cell killing has been reported by using a small-molecule inhibitor of Ape1 redox function.
One of the most exciting results presented here details the potential role of Ape1 in angiogenesis (Fig. 11). As demonstrated in our RVEC studies, inhibition of Ape1 redox function by using E3330 blocks RVEC cell growth, even with bFGF stimulation (Fig. 11). Additional studies using RVEC formation of capillary-like structures on Matrigel resulted in the decreased angiogenesis, as measured by this assay, but at a much lower dose range than in the MTS assays (Fig. 11). These results demonstrate that inhibition of redox activity significantly attenuates RVEC proliferation and capillary formation in vitro, but does not cause cell death. Furthermore, the capillary formation of RVECs appears much more sensitive to redox inhibition of Ape1 than the proliferation. This is the first time this role of Ape1 has been clearly demonstrated, and it necessitated the use of a specific small-molecule Ape1 redox inhibitor to identify this novel activity.
As discussed, Ape1 was originally identified as the primary target of E3330. E3330 was immobilized on beads, and Ape1 was identified from a nuclear extract of Jurkat cells as a protein that specifically binds to E3330. By using surface plasmon resonance, a kinetic constant (KD) of 1.6 nM was obtained for the binding of E3330 to Ape1, suggesting a specific interaction. It was further shown that E3330 blocked the ability of Ape1 to reduce NF-κB, thus interfering with the redox activity of Ape1 (27, 35, 52, 65). Through a series of N- and C-terminal truncation mutants, a peptide including residues 72-88 of Ape1 was proposed to bind to E3330, suggesting that E3330 interacts with a specific site on Ape1 (34, 54, 65). However, we believe that this approach was imperfect, in that the truncated Ape1 molecules are unlikely to fold correctly, and thus the identification of the binding site is suspect. In addition, the proposed binding site is somewhat puzzling in light of the structure of Ape1, as an obvious binding site is not noted (data not shown). These residues (aa 72–80) form a ridge on the surface of the molecule with no obvious cavities or binding pockets that are sufficiently large to bind E3330. Currently, we are pursuing studies to delineate the binding site or region of E3330 on Ape1.
In conclusion, Ape1 is a multifunctional protein with both important DNA-repair and redox capabilities. However, to delineate the various functions of Ape1, small-molecule inhibitors of each function will be necessary ultimately to conclude which function is required in normal and cancer cell function. Additionally, we show data that demonstrate a new role of Ape1 with its involvement in angiogenesis, and subsequent inhibition of Ape1s redox function abrogates this role. The use of E3330 and new analogues currently under development will be critical reagents in the future studies of the cellular functions of Ape1 and have potential therapeutic applications.
Acknowledgments
Financial support for this work was provided by the National Institutes of Health, National Cancer Institute CA94025, CA106298, CA114571, and CA121168 to M.R.K., CA 114571 to M.M.G, IU Simon Cancer Center Translational initiative pilot funding (ITRAC) to M.R.K. and M.M.G., and the Riley Children's Foundation (M.R.K.). R.L.N. was supported on training grant 5T32CA009634.
Abbreviations
AP, apurinic/apyrimidinic; Ape1, AP endonuclease1; ATM, ataxia–telangiectasia mutated; BER, base excision repair; CSB, Cockayne syndrome B; EC, endothelial cell; EMSA, electrophoretic mobility shift assay; FEN1, flap endonuclease 1; GZMA, granzyme A; HEX, 5′ hexachloro-fluorescein phosphoramidite; IR, incision repair; ITRAC, IU Simon Cancer Center Translational Research Acceleration Collaboration; MMS, methylmethane sulfonate; NIR, nucleotide incision repair; NK, natural killer; NMR, nuclear magnetic resonance; NO, nitric oxide; PARP1, polyADP-ribose polymerase 1; PCNA, proliferating cell nuclear antigen; PTEN, phosphatase and tensin homologue; REC, retinal endothelial cell; redox, reduction oxygenation; RVEC, retinal vascular endothelial cell; THF, tetrahydrofuran; TMZ, temozolomide; XRCC1, x-ray cross complementing 1.
References
- 1.Bhakat KK. Izumi T. Yang SH. Hazra TK. Mitra S. Role of acetylated human AP-endonuclease (APE1/Ref-1) in regulation of the parathyroid hormone gene. EMBO J. 2003;22:6299–6309. doi: 10.1093/emboj/cdg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bobola MS. Finn LS. Ellenbogen RG. Geyer JR. Berger MS. Braga JM. Meade EH. Gross ME. Silber JR. Apurinic/ apyrimidinic endonuclease activity is associated with response to radiation and chemotherapy in medulloblastoma and primitive neuroectodermal tumors. Clin Cancer Res. 2005;11:7405–7414. doi: 10.1158/1078-0432.CCR-05-1068. [DOI] [PubMed] [Google Scholar]
- 3.Brimble MA. McEwan JF. Turner P. Asymmetric Diels-Alder addition of cyclopentadiene to chiral naphthoquinones tetrahedron. Asymmetry. 1998;9:1239–1255. [Google Scholar]
- 4.Campalans A. Marsin S. Nakabeppu Y. O'Connor TR. Boiteux S. Radicella JP. XRCC1 interactions with multiple DNA glycosylases: a model for its recruitment to base excision repair. DNA Repair (Amst) 2005;4:826–835. doi: 10.1016/j.dnarep.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 5.Chakravarti A. Delaney MA. Noll E. Black PM. Loeffler JS. Muzikansky A. Dyson NJ. Prognostic and pathologic significance of quantitative protein expression profiling in human gliomas. Clin Cancer Res. 2001;7:2387–2395. [PubMed] [Google Scholar]
- 6.Chen DS. Olkowski ZL. Biological responses of human apurinic endonuclease to radiation-induced DNA damage. Ann NY Acad Sci. 1994;726:306–308. doi: 10.1111/j.1749-6632.1994.tb52834.x. [DOI] [PubMed] [Google Scholar]
- 7.Chou KM. Cheng YC. The exonuclease activity of human apurinic/apyrimidinic endonuclease (APE1): biochemical properties and inhibition by the natural dinucleotide Gp4G. J Biol Chem. 2003;278:18289–18296. doi: 10.1074/jbc.M212143200. [DOI] [PubMed] [Google Scholar]
- 8.Chou KM. Cheng YC. An exonucleolytic activity of human apurinic/apyrimidinic endonuclease on 3′ mispaired DNA. Nature. 2002;415:655–659. doi: 10.1038/415655a. [DOI] [PubMed] [Google Scholar]
- 9.Chou KM. Kukhanova M. Cheng YC. A novel action of human apurinic/apyrimidinic endonuclease: excision of L-configuration deoxyribonucleoside analogs from the 3′ termini of DNA. J Biol Chem. 2000;275:31009–31015. doi: 10.1074/jbc.M004082200. [DOI] [PubMed] [Google Scholar]
- 10.Chou KM. Lam W. Cheng YC. A novel DNA exonuclease activity of human apurinic/apyrimidinic endonuclease (APE1): implication in DNA mismatch repair. Proc Am Assoc Ca Res. 2001;42:2988. [Google Scholar]
- 11.Chung U. Igarashi T. Nishishita T. Iwanari H. Iwamatsu A. Suwa A. Mimori T. Hata K. Ebisu S. Ogata E. Fujita T. Okazaki T. The interaction between Ku antigen and REF1 protein mediates negative gene regulation by extracellular calcium. J Biol Chem. 1996;271:8593–8598. doi: 10.1074/jbc.271.15.8593. [DOI] [PubMed] [Google Scholar]
- 12.Damia G. D'Incalci M. Targeting DNA repair as a promising approach in cancer therapy. Eur J Cancer. 2007;43:1791–1801. doi: 10.1016/j.ejca.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 13.Demple B. Harrison L. Repair of oxidative damage to DNA: enzymology and biology. Annu Rev Biochem. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- 14.Dianova II. Bohr VA. Dianov GL. Interaction of human AP endonuclease 1 with flap endonuclease 1 and proliferating cell nuclear antigen involved in long-patch base excision repair. Biochemistry. 2001;40:12639–12644. doi: 10.1021/bi011117i. [DOI] [PubMed] [Google Scholar]
- 15.DiGiuseppe JA. Hunting DJ. Dresler SL. Aphidicolin-sensitive DNA repair synthesis in human fibroblasts damaged with bleomycin is distinct from UV-induced repair. Carcinogen Carcinogenesis. 1990;11:1021–1026. doi: 10.1093/carcin/11.6.1021. [DOI] [PubMed] [Google Scholar]
- 16.Evans AR. Limp-Foster M. Kelley MR. Going APE over ref-1. Mutat Res. 2000;461:83–108. doi: 10.1016/s0921-8777(00)00046-x. [DOI] [PubMed] [Google Scholar]
- 17.Fan Z. Beresford PJ. Zhang D. Xu Z. Novina CD. Yoshida A. Pommier Y. Lieberman J. Cleaving the oxidative repair protein Ape1 enhances cell death mediated by granzyme A. Nat Immunol. 2003;4:145–153. doi: 10.1038/ni885. [DOI] [PubMed] [Google Scholar]
- 18.Fantini D. Vacotto C. Deganuto M. Bivi N. Gustincich S. Marcon G. Quadrioglio FDG. Bhakat K. Mitra S. APE1/Ref-1 regulates PTEN expression mediated by Egr-1. Free Radic Res. 2008;42:20–29. doi: 10.1080/10715760701765616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fishel ML. He Y. Reed AM. Chin-Sinex H. Hutchins GD. Mendonca MS. Kelley MR. Knockdown of the DNA repair and redox signaling protein Ape1/Ref-1 blocks ovarian cancer cell and tumor growth. DNA Repair (Amst) 2008;7:177–186. doi: 10.1016/j.dnarep.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fishel ML. He Y. Smith ML. Kelley MR. Manipulation of base excision repair to sensitize ovarian cancer cells to alkylating agent temozolomide. Clin Cancer Res. 2007;13:260–267. doi: 10.1158/1078-0432.CCR-06-1920. [DOI] [PubMed] [Google Scholar]
- 21.Fishel ML. Kelley MR. The DNA base excision repair protein Ape1/Ref-1 as a therapeutic and chemopreventive target. Mol Aspects Med. 2007;28:375–395. doi: 10.1016/j.mam.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 22.Flader C. Liu J. Borch RF. Development of novel quinone phosphorodiamidate prodrugs targeted to DT-diaphorase. J Med Chem. 2000;43:3157–3167. doi: 10.1021/jm000179o. [DOI] [PubMed] [Google Scholar]
- 23.Flader C. Liu J. Borch RF. Development of novel quinone phosphorodiamidate prodrugs targeted to DT-diaphorase. J Med Chem. 2000;43:3157–3167. doi: 10.1021/jm000179o. [DOI] [PubMed] [Google Scholar]
- 24.Fung H. Demple B. A vital role for Ape1/Ref1 protein in repairing spontaneous DNA damage in human cells. Mol Cell. 2005;17:463–470. doi: 10.1016/j.molcel.2004.12.029. [DOI] [PubMed] [Google Scholar]
- 25.Georgiadis M. Luo M. Gaur R. Delaplane S. Li X. Kelley M. Evolution of the redox function in mammalian apurinic/apyrimidinic. Mutat Res. 2008 doi: 10.1016/j.mrfmmm.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gorman MA. Morera S. Rothwell DG. de la Fortelle E. Mol CD. Tainer JA. Hickson ID. Freemont PS. The crystal structure of the human DNA repair endonuclease HAP1 suggests the recognition of extra-helical deoxyribose at DNA abasic sites. EMBO J. 1997;16:6548–6558. doi: 10.1093/emboj/16.21.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goto M. Yamada K. Katayama K. Tanaka I. Inhibitory effect of E3330, a novel quinone derivative able to suppress tumor necrosis factor-alpha generation, on activation of nuclear factor-kappa B. Mol Pharmacol. 1996;49:860–873. [PubMed] [Google Scholar]
- 28.Hansen CA. Dean AB. Draths KM. Frost JW. Synthesis of 1,2,3,4-tetrahydroxybenzene from D-glucose: exploiting myo-inositol as a precursor to aromatic chemicals. J Am Chem Soc. 1999;121:3799–3800. [Google Scholar]
- 29.Hansen WK. Deutsch WA. Yacoub A. Xu Y. Williams DA. Kelley MR. Creation of a fully functional human chimeric DNA repair protein: combining O6-methylguanine DNA methyltransferase (MGMT) and AP endonuclease [APE/redox effector factor 1 (Ref 1)] DNA repair proteins. J Biol Chem. 1998;273:756–762. doi: 10.1074/jbc.273.2.756. [DOI] [PubMed] [Google Scholar]
- 30.Harrison JF. Rinne ML. Kelley MR. Druzhyna NM. Wilson GL. LeDoux SP. Altering DNA base excision repair: use of nuclear and mitochondrial-targeted N-methylpurine DNA glycosylase to sensitize astroglia to chemotherapeutic agents. Glia. 2007;55:1416–1425. doi: 10.1002/glia.20556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Havelka AM. Berndtsson M. Olofsson MH. Shoshan MC. Linder S. Mechanisms of action of DNA-damaging anticancer drugs in treatment of carcinomas: is acute apoptosis an “off-target” effect? Mini Rev Med Chem. 2007;7:1035–1039. doi: 10.2174/138955707782110196. [DOI] [PubMed] [Google Scholar]
- 32.Herring CJ. Deans B. Elder RH. Rafferty JA. MacKinnon J. Barzilay G. Hickson ID. Hendry JH. Margison GP. Expression levels of the DNA repair enzyme HAP1 do not correlate with the radiosensitivities of human or HAP1-transfected rat cell lines. Br J Cancer. 1999;80:940–945. doi: 10.1038/sj.bjc.6690447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herring CJ. West CM. Wilks DP. Davidson SE. Hunter RD. Berry P. Forster G. MacKinnon J. Rafferty JA. Elder RH. Hendry JH. Margison GP. Levels of the DNA repair enzyme human apurinic/apyrimidinic endonuclease (APE1, APEX, Ref-1) are associated with the intrinsic radiosensitivity of cervical cancers. Br J Cancer. 1998;78:1128–1133. doi: 10.1038/bjc.1998.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hiramoto M. Shimizu N. Nishi T. Shima D. Aizawa S. Tanaka H. Hatakeyama M. Kawaguchi H. Handa H. High-performance affinity beads for identifying anti-NF-kappa B drug receptors. Methods Enzymol. 2002;353:81–88. doi: 10.1016/s0076-6879(02)53038-x. [DOI] [PubMed] [Google Scholar]
- 35.Hiramoto M. Shimizu N. Sugimoto K. Tang J. Kawakami Y. Ito M. Aizawa S. Tanaka H. Makino I. Handa H. Nuclear targeted suppression of NF-kappa B activity by the novel quinone derivative E3330. J Immunol. 1998;160:810–819. [PubMed] [Google Scholar]
- 36.Hsieh MM. Hegde V. Kelley MR. Deutsch WA. Activation of APE/Ref-1 redox activity is mediated by reactive oxygen species and PKC phosphorylation. Nucleic Acids Res. 2001;29:3116–3122. doi: 10.1093/nar/29.14.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ide H. Kotera M. Human DNA glycosylases involved in the repair of oxidatively damaged DNA. Biol Pharm Bull. 2004;27:480–485. doi: 10.1248/bpb.27.480. [DOI] [PubMed] [Google Scholar]
- 38.Jeon BH. Gupta G. Park YC. Qi B. Haile A. Khanday FA. Liu YX. Kim JM. Ozaki M. White AR. Berkowitz DE. Irani K. Apurinic/apyrmidinic endonuclease 1 regulates endothelial NO production and vascular tone. Circ Res. 2004;95:902–910. doi: 10.1161/01.RES.0000146947.84294.4c. [DOI] [PubMed] [Google Scholar]
- 39.Jin Z. May WS. Gao F. Flagg T. Deng X. Bcl2 suppresses DNA repair by enhancing c-Myc transcriptional activity. J Biol Chem. 2006;281:14446–14456. doi: 10.1074/jbc.M511914200. [DOI] [PubMed] [Google Scholar]
- 40.Kelley MR. Hansen W. Xu Y. Yacoub A. Williams D. Deutsch WA. Combining O6-methylguanine DNA methyltransferase (MGMT) and AP endonuclease (APE) DNA repair activities in chimeric proteins: use in retroviral gene therapy and chemotherapeutic dose intensification. Proc Am Assoc Cancer Res. 1997;38:383. [Google Scholar]
- 41.Kreklau EL. Limp-Foster M. Liu N. Xu Y. Kelley MR. Erickson LC. A novel fluorometric oligonucleotide assay to measure O(6)-methylguanine DNA methyltransferase, methylpurine DNA glycosylase, 8-oxoguanine DNA glycosylase and abasic endonuclease activities: DNA repair status in human breast carcinoma cells overexpressing methylpurine DNA glycosylase. Nucleic Acids Res. 2001;29:2558–2566. doi: 10.1093/nar/29.12.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuninger DT. Izumi T. Papaconstantinou J. Mitra S. Human AP-endonuclease 1 and hnRNP-L interact with a nCaRE-like repressor element in the AP-endonuclease 1 promoter. Nucleic Acids Res. 2002;30:823–829. doi: 10.1093/nar/30.3.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lau JP. Weatherdon KL. Skalski V. Hedley DW. Effects of gemcitabine on APE/ref-1 endonuclease activity in pancreatic cancer cells, and the therapeutic potential of antisense oligonucleotides. Br J Cancer. 2004;91:1166–1173. doi: 10.1038/sj.bjc.6602080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu L. Gerson SL. Therapeutic impact of methoxyamine: blocking repair of abasic sites in the base excision repair pathway. Curr Opin Invest Drugs. 2004;5:623–627. [PubMed] [Google Scholar]
- 45.Liu L. Nakatsuru Y. Gerson SL. Base excision repair as a therapeutic target in colon cancer. Clin Cancer Res. 2002;8:2985–2991. [PubMed] [Google Scholar]
- 46.Liu L. Yan L. Donze JR. Gerson SL. Blockage of abasic site repair enhances antitumor efficacy of 1,3-bis-(2-chloroethyl)-1-nitrosourea in colon tumor xenografts. Mol Cancer Ther. 2003;2:1061–1066. [PubMed] [Google Scholar]
- 47.Liu L. Yan L. Mahajan V. Donze JR. Gerson SL. Inhibition of AP site repair coupled with action of topoisomerase II poison: a potential strategy to enhance efficacy of chemotherapeutic alkylating agents. Proc Am Assoc Ca Res. 2003;44 [Google Scholar]
- 48.Madhusudan S. Hickson I. DNA repair inhibition: a selective tumour targeting strategy. Trends Mol Med. 2005;11:503–511. doi: 10.1016/j.molmed.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 49.Martin F. Solary E. Tumor cell resistance to DNA-damaging agents: from apoptosis to neiosis. Curr Med Chem Anticancer Agents. 2004;4:461–463. doi: 10.2174/1568011043352768. [DOI] [PubMed] [Google Scholar]
- 50.Martinvalet D. Zhu P. Lieberman J. Granzyme a induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity. 2005;22:355–370. doi: 10.1016/j.immuni.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 51.McNeill DR. Wilson DM., 3rd A dominant-negative form of the major human abasic endonuclease enhances cellular sensitivity to laboratory and clinical DNA-damaging agents. Mol Cancer Res. 2007;5:61–70. doi: 10.1158/1541-7786.MCR-06-0329. [DOI] [PubMed] [Google Scholar]
- 52.Miyamoto K. Nagakawa J. Hishinuma I. Hirota K. Yasuda M. Yamanaka T. Katayama K. Yamatsu I. Suppressive effects of E3330, a novel quinone derivative, on tumor necrosis factor-alpha generation from monocytes and macrophages. Agents Actions. 1992;37:297–304. doi: 10.1007/BF02028123. [DOI] [PubMed] [Google Scholar]
- 53.Nazarkina ZK. Khodyreva SN. Marsin S. Lavrik OI. Radicella JP. XRCC1 interactions with base excision repair DNA intermediates. DNA Repair (Amst) 2007;6:254–264. doi: 10.1016/j.dnarep.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 54.Nishi T. Shimizu N. Hiramoto M. Sato I. Yamaguchi Y. Hasegawa M. Aizawa S. Tanaka H. Kataoka K. Watanabe H. Handa H. Spatial redox regulation of a critical cysteine residue of NF-kappa B in vivo. J Biol Chem. 2002;277:44548–44556. doi: 10.1074/jbc.M202970200. [DOI] [PubMed] [Google Scholar]
- 55.Nobuo S. Tomio C. Tomio T. Hirofumi K. Eezai Kagaku KK. Japan: Eisai Co Ltd; 1991. Production of quinone derivative (C07C 66/00, B01J 27/10, C07C 46/02, C07C 51/373, C07D213/46, C07B 61/00 ed.) [Google Scholar]
- 56.Nobuo S. Tomio I. Masahiko T. Tomio T. Hirofumi K. Eezai Kagaku KK. Japan: Eisai Co Ltd; 1991. Production OF P-Dimethoxybenzene derivative (C07C 43/205, C07C 41/01, C07C 41/16, C07C 43/23 ed.) [Google Scholar]
- 57.Ohkawa S. Terao S. Terashita Z. Shibouta Y. Nishikawa K. Dual inhibitors of thromboxane A2 synthase and 5-lipoxygenase with scavenging activity of active oxygen species: synthesis of a novel series of (3-pyridylmethyl)benzoquinone derivatives. J Med Chem. 1991;34:267–276. doi: 10.1021/jm00105a042. [DOI] [PubMed] [Google Scholar]
- 58.Okazaki T. Chung U. Nishishita T. Ebisu S. Usuda S. Mishiro S. Xanthoudakis S. Igarashi T. Ogata E. A redox factor protein, ref1, is involved in negative gene regulation by extracellular calcium. J Biol Chem. 1994;269:27855–27862. [PubMed] [Google Scholar]
- 59.Ono Y. Furuta T. Ohmoto T. Akiyama K. Seki S. Stable expression in rat glioma cells of sense and antisense nucleic acids to a human multifunctional DNA repair enzyme, APEX nuclease. Mutat Res. 1994;315:55–63. doi: 10.1016/0921-8777(94)90028-0. [DOI] [PubMed] [Google Scholar]
- 60.Ozaki M. Suzuki S. Irani K. Redox factor-1/APE suppresses oxidative stress by inhibiting the rac1 GTPase. FASEB J. 2002;16:889–890. doi: 10.1096/fj.01-0664fje. [DOI] [PubMed] [Google Scholar]
- 61.Parlanti E. Giada L. Giovanni M. Eugenia D. Human base excision repair complex is physically associated to DNA replication and cell cycle regulatory proteins. Nucleic Acids Res. 2007;35:1569–1577. doi: 10.1093/nar/gkl1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parsons JL. Dianova II. Dianov GL. APE1 is the major 3′-phosphoglycolate activity in human cell extracts. Nucleic Acids Res. 2004;32:3531–3536. doi: 10.1093/nar/gkh676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Peddi SR. Chattopadhyay R. Naidu CV. Izumi T. The human apurinic/apyrimidinic endonuclease-1 suppresses activation of poly(adp-ribose) polymerase-1 induced by DNA single strand breaks. Toxicology. 2006;224:44–55. doi: 10.1016/j.tox.2006.04.025. [DOI] [PubMed] [Google Scholar]
- 64.Ranalli TA. Tom S. Bambara RA. AP endonuclease 1 coordinates flap endonuclease 1 and DNA ligase I activity in long patch base excision repair. J Biol Chem. 2002;277:41715–41724. doi: 10.1074/jbc.M207207200. [DOI] [PubMed] [Google Scholar]
- 65.Shimizu N. Sugimoto K. Tang J. Nishi T. Sato I. Hiramoto M. Aizawa S. Hatakeyama M. Ohba R. Hatori H. Yoshikawa T. Suzuki F. Oomori A. Tanaka H. Kawaguchi H. Watanabe H. Handa H. High-performance affinity beads for identifying drug receptors. Nat Biotechnol. 2000;18:877–881. doi: 10.1038/78496. [DOI] [PubMed] [Google Scholar]
- 66.Sidorenko VS. Nevinsky GA. Zharkov DO. Mechanism of interaction between human 8-oxoguanine-DNA glycosylase and AP endonuclease. DNA Repair. 2007;6:317–328. doi: 10.1016/j.dnarep.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 67.Singhal RK. Prasad R. Wilson SH. DNA polymerase beta conducts the gap-filling step in uracil-initiated base excision repair in a bovine testis nuclear extract. J Biol Chem. 1995;270:949–957. doi: 10.1074/jbc.270.2.949. [DOI] [PubMed] [Google Scholar]
- 68.Spitz DR. Azzam EI. Li JJ. Gius D. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: a unifying concept in stress response biology. Cancer Metastasis Rev. 2004;23:311–322. doi: 10.1023/B:CANC.0000031769.14728.bc. [DOI] [PubMed] [Google Scholar]
- 69.Su X. Sorenson CM. Sheibani N. Isolation and characterization of murine retinal endothelial cells. Mol Vis. 2003;9:171–178. [PubMed] [Google Scholar]
- 70.Taverna P. Liu L. Hwang HS. Hanson AJ. Kinsella TJ. Gerson SL. Methoxyamine potentiates DNA single strand breaks and double strand breaks induced by temozolomide in colon cancer cells. Mutat Res. 2001;485:269–281. doi: 10.1016/s0921-8777(01)00076-3. [DOI] [PubMed] [Google Scholar]
- 71.Tell G. Damante G. Caldwell D. Kelley MR. The intracellular localization of APE1/Ref-1: more than a passive phenomenon? Antioxid Redox Signal. 2005;7:367–384. doi: 10.1089/ars.2005.7.367. [DOI] [PubMed] [Google Scholar]
- 72.Tremblay MS. Sames D. A new fluorogenic transformation: development of an optical probe for coenzyme Q. Org Lett. 2005;7:2417–2420. doi: 10.1021/ol0507569. [DOI] [PubMed] [Google Scholar]
- 73.Trzeciak AR. Nyaga SG. Jaruga P. Lohani A. Dizdaroglu M. Evans MK. Cellular repair of oxidatively induced DNA base lesions is defective in prostate cancer cell lines, PC-3 and DU-145. Carcinogenesis. 2004;25:1359–1370. doi: 10.1093/carcin/bgh144. [DOI] [PubMed] [Google Scholar]
- 74.Walker LJ. Craig RB. Harris AL. Hickson ID. A role for the human DNA repair enzyme HAP1 in cellular protection against DNA damaging agents and hypoxic stress. Nucl Acids Res. 1994;22:4884–4889. doi: 10.1093/nar/22.23.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang D. Luo M. Kelley MR. Human apurinic endonuclease 1 (APE1) expression and prognostic significance in osteosarcoma: enhanced sensitivity of osteosarcoma to DNA damaging agents using silencing RNA APE1 expression inhibition. Mol Cancer Ther. 2004;3:679–686. [PubMed] [Google Scholar]
- 76.Wiederhold L. Leppard JB. Kedar P. Karimi-Busheri F. Rasouli-Nia A. Weinfeld M. Tomkinson AE. Izumi T. Prasad R. Wilson SH. Mitra S. Hazra TK. AP endonuclease-independent DNA base excision repair in human cells. Mol Cell. 2004;15:209–220. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 77.Wilson DM., 3rd Barsky D. The major human abasic endonuclease: formation, consequences and repair of abasic lesions in DNA. Mutat Res. 2001;485:283–307. doi: 10.1016/s0921-8777(01)00063-5. [DOI] [PubMed] [Google Scholar]
- 78.Wong H-K. Muftuoglu M. Beck G. Imam SZ. Bohr VA. Wilson DM., III Cockayne syndrome B protein stimulates apurinic endonuclease 1 activity and protects against agents that introduce base excision repair intermediates. Nucleic Acids Res. 2007;35:4103–4113. doi: 10.1093/nar/gkm404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xanthoudakis S. Smeyne RJ. Wallace JD. Curran T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc Natl Acad Sci U S A. 1996;93:8919–8923. doi: 10.1073/pnas.93.17.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yacoub A. Kelley MR. Deutsch WA. The DNA repair activity of human redox/repair protein APE/ref-1 is inactivated by phosphorylation. Cancer Res. 1997;57:5457–5459. [PubMed] [Google Scholar]
- 81.Yan L. Bulgar A. Miao Y. Mahajan V. Donze JR. Gerson SL. Liu L. Combined treatment with temozolomide and methoxyamine: blocking apurininc/pyrimidinic site repair coupled with targeting topoisomerase II{alpha} Clin Cancer Res. 2007;13:1532–1539. doi: 10.1158/1078-0432.CCR-06-1595. [DOI] [PubMed] [Google Scholar]
- 82.Zou GM. Luo MH. Reed A. Kelley MR. Yoder MC. Ape1 regulates hematopoietic differentiation of embryonic stem cells through its redox functional domain. Blood. 2007;109:1917–1922. doi: 10.1182/blood-2006-08-044172. [DOI] [PubMed] [Google Scholar]