

Abstract
MMP2200 is a novel glycopeptide opioid agonist with similar affinities for mu and delta receptors. Glycosylation promoted brain penetration and production of centrally mediated behavioral effects in mice; however, it is unknown if the magnitude of enhanced brain penetration is sufficient to permit central mediation of drug effects and production of synergistic mu/delta antinociceptive interactions after systemic administration in primates. To address this issue, the present study compared the effects of MMP2200 and the mu agonist morphine in four behavioral procedures in rhesus monkeys. In an assay of thermal nociception, morphine (1.0–5.6 mg/kg) produced dose-dependent antinociception, whereas MMP2200 (10–56 mg/kg) was ineffective. In an assay of capsaicin-induced thermal allodynia, both morphine (0.01–1.0 mg/kg) and MMP2200 (0.032–3.2 mg/kg) produced dose-dependent antiallodynic effects. MMP2200-induced antiallodynia was blocked by the moderately mu-selective antagonist naltrexone (0.01 mg/kg), the delta-selective antagonist naltrindole (1.0 mg/kg), and the peripherally selective opioid antagonist quaternary naltrexone (0.32 mg/kg). In an assay of schedule-controlled behavior, both morphine (0.01–1.0mg/kg) and MMP2200 (10–56 mg/kg) decreased response rates. Morphine effects were antagonized by naltrexone (0.001–0.01 mg/kg); however, the effects of MMP2200 were not antagonized by either naltrexone (0.01 mg/kg) or naltrindole (1.0 mg/kg). In an assay of drug self-administration, morphine (0.0032–0.32 mg/kg/inj) produced reinforcing effects, whereas MMP2200 (0.032–0.32 mg/kg/inj) did not. These results suggest that systemically administered MMP2200 acted as a peripheral, mu/delta opioid agonist with limited distribution to the CNS in rhesus monkeys. These results also suggest the existence of species differences in the pharmacokinetics and brain penetration of glycopeptides.
INTRODUCTION
Opioids act at three main types of opioid receptor--the mu, delta and kappa receptors--and most opioid analgesics function primarily as agonists at mu receptors (Gutstein and Akil, 2005). One strategy to improve the therapeutic utility of opioids has been to combine mu receptor activation with pharmacological manipulation of other targets to produce a net increase in desirable versus undesirable effects. For example, simultaneous activation of mu and delta opioid receptors may produce enhanced antinociception with fewer undesirable effects than selective activation of either mu or delta receptors alone in both rodents (Vaught and Takemori, 1979; Heyman et al., 1989; Adams et al., 1993) and non-human primates (Dykstra et al., 2002; Stevenson et al., 2003).
The simultaneous activation of mu and delta receptors can be achieved either with a mixture of selective mu and delta opioids or with a single compound that has mixed activity at both mu and delta receptors. Endogenous opioid neurotransmitters such as met- and leu-enkephalin represent one class of opioid receptor ligands that produce relatively non-selective agonist effects at both mu and delta receptors, and many synthetic peptides have also been developed with both selective and non-selective mu/delta activity (Hruby and Mosberg, 2004). Although these peptides have played an important role in delineating the pharmacological consequences of combined mu/delta receptor activation, opioid peptides have historically been poor drug candidates in part because of their limited ability to cross the blood-brain barrier and access central sites of action after systemic administration. However, recent structure-activity studies suggest that glycosylation may promote distribution of peptides across the blood-brain barrier and enhance the potency of systemically administered (IV, IP, SC) peptides to produce centrally mediated behavioral effects (Bilsky et al., 2000; Egleton et al., 2001; Lowery et al., 2007). For example, MMP2200 is a glycosylated derivative of the leu-enkephalin analog Tyr-D-Thr-Gly-Phe-Leu-Ser-CONH2 (Figure 1). Like its unglycosylated parent peptide, MMP2200 displays similar nanomolar affinities for both mu and delta opioid receptors in rat brain homogenates and functions as an agonist in both guinea pig ileum and mouse vas deferens smooth muscle assays, which are thought to be sensitive to mu and delta receptor agonists, respectively (Elmagbari et al., 2004). More recent studies have confirmed that MMP2200 also binds with similar affinities to human mu and delta opioid receptors expressed in Chinese hamster ovary cells, is selective for mu and delta vs. kappa receptors, and functions as a high efficacy mu and delta agonist in assays of agonist-stimulated GTPγS binding (Jean Bidlack, unpublished data). However, MMP2200 was approximately 10-fold more potent than the parent compound in producing antinociception in mice after systemic (IV) administration (compare compounds 1 and 9 in (Elmagbari et al., 2004). These results were interpreted to suggest that glycosylation increased stability and/or penetration of the blood-brain barrier, and subsequent kinetic studies provided evidence to support both possibilities (Egleton et al., 2005). Taken together, these in vitro studies and behavioral studies suggest that MMP2200 may be a promising lead compound in the development of glycosylated mu/delta opioid peptides as analgesic drugs.
Figure 1.
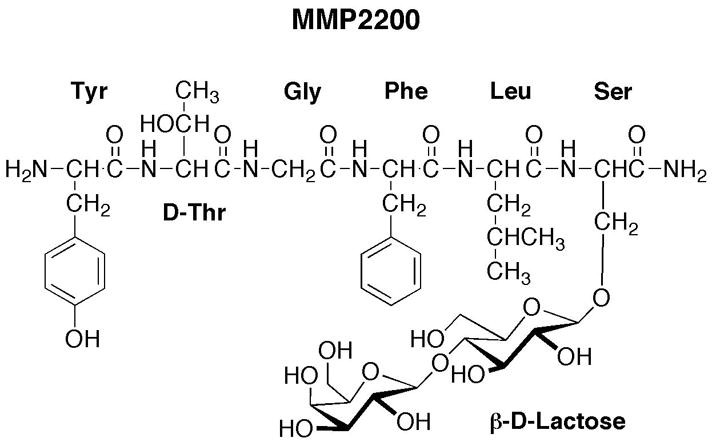
The chemical structure of MMP2200: H2N-Tyr-D-Thr-Gly-Phe-Leu-Ser-(O-β-D-lactose)- CONH2.
The purpose of the present study was to evaluate the behavioral pharmacology of MMP2200 in comparison to morphine in rhesus monkeys. The pharmacology of MMP2200 in non-human primates has not been described previously. However, there are significant species differences in the anatomy and neural functions of opioid receptors (Mansour et al., 1988; Mennicken et al., 2003; Berger et al., 2006), and in the pharmacokinetics and blood-brain barrier penetration of many opioids and other drugs (Ward and Smith, 2004; Ward et al., 2005; Ohtsuki and Terasaki, 2007). Consequently, non-human primates may provide an important complement to rodents for translational research designed to predict opioid pharmacology in humans. The primary goal of the study was to compare the effects of MMP2200 and morphine in four different behavioral assays. First, drug effects were evaluated in an assay of thermal antinociception, which measures withdrawal responses from an acute, noxious thermal stimulus (50°C water). Opioid effects in this procedure are generally considered to be centrally mediated (Negus et al., 1993b; Ko et al., 1998b). Second, drug effects were examined in an assay of capsaicin-induced thermal allodynia. Allodynia is defined as a pain-like response to a normally innocuous stimulus, and it is a common component of pain associated with inflammation. In this assay, drugs are evaluated for their ability to normalize hypersensitive withdrawal responses, and drug effects may be mediated by either peripheral or central receptors (Negus et al., 1993b; Ko et al., 1998b; Butelman et al., 2004). Third, drug effects were examined in an assay of schedule-controlled responding for food reinforcement. This assay provides a measure of the degree to which opioids produce a general disruption of motivated behavior independent of nociception, and rate-decreasing effects of opioid agonists in this procedure are considered to be mediated by central opioid receptors (Young and Woods, 1982). Lastly, drug effects were evaluated in an assay of drug self-administration. Existing opioid analgesics such as morphine produce reinforcing effects in assays of drug self-administration and display high abuse liability, and the reinforcing effects of mu agonists are also considered to be centrally mediated (Koob et al., 1984).
METHODS
Subjects
Seven adult male and female rhesus monkeys (Macaca mulatta) were used in studies of thermal nociception and thermal allodynia, five adult male rhesus monkeys were used in studies of schedule-controlled responding, and four adult male rhesus monkeys were used in studies of drug self-administration. Subjects weighed between 6.9 and 12.4 kg during the course of these studies. All monkeys had prior exposure to drugs (primarily dopaminergic and opioid compounds) and to the behavioral procedures in which they were tested. The subjects were individually housed, and water was freely available. Their diet consisted of PMI Feeds Jumbo monkey diet (2–6 biscuits/day; PMI Feeds, Inc., St. Louis, MO) supplemented with fresh fruit twice daily. In addition, monkeys in the assay of schedule-controlled behavior could earn additional food pellets during experimental sessions. A 12 hr light/12 hr dark cycle was in effect (lights on from 7AM–7PM).
Animal maintenance and research were conducted in accordance with the guidelines provided by the NIH Committee on Laboratory Animal Resources. The facility was licensed by the United States Department of Agriculture, and all animal protocols were approved by the Institutional Animal Care and Use Committee. The health of the monkeys was monitored daily by the staff veterinarian. Monkeys had visual, auditory and olfactory contact with other monkeys throughout the study. Monkeys also had access to puzzle feeders, mirrors and chew toys to provide environmental enrichment. Nature videotapes or music were played daily in all housing rooms.
Behavioral and Pharmacological Procedures
Behavioral studies were conducted using four procedures: (1) an assay of thermal nociception using warm water as the thermal stimulus, (2) an assay of capsaicin-induced thermal allodynia using warm water as the thermal stimulus, (3) an assay of schedule-controlled responding for food presentation, and (4) an assay of drug self-administration. These procedures have been used extensively by our laboratory to examine the effects of other opioids and of delta/mu interactions in rhesus monkeys (Negus et al., 1998; Brandt et al., 2001; Stevenson et al., 2003; Stevenson et al., 2005). In each assay, the effects of MMP2200 were compared to the effects of the prototype mu opioid analgesic morphine.
Assay of Thermal Nociception
Monkeys (N=3) were seated in acrylic restraint chairs so that their tails hung down freely. The bottom 10 cm of each monkey’s shaved tail was immersed in a thermal container of warm water. If the subject did not withdraw its tail within 20 sec, the tail was removed from the water by the experimenter, and a latency of 20 sec was assigned to that measurement. During each set of measurements, tail-withdrawal latencies were measured from water heated to 38 and 50°C. The order in which the temperatures were presented varied from one set of measurements to the next. A stopwatch was used to measure and record time intervals.
Each test session consisted of multiple cycles of tail-withdrawal latency measurements. Before the first cycle, baseline latencies to tail-withdrawal from 38 and 50°C water were determined. Testing continued only if tail withdrawal from 38°C water did not occur before the 20 sec cutoff, and if tail withdrawal occurred in ≤3 sec from 50°C water. This criterion was met during every session with every monkey in this study. Subsequently, either saline or a single dose of morphine (1.0–5.6 mg/kg) or MMP2200 (10–56 mg/kg) was administered IM, and tail witdrawal latencies from 38 and 50°C water were redetermined after 10, 30 and 100 min. After the 100 min measurement, subjects were returned to their home cages. Test sessions were conducted no more than twice a week, with at least 3 days between tests.
MMP2200 at doses up to 56 mg/kg IM failed to produce significant antinociception (see Results). Because previous studies in mice had reported antinociception in mice after IV administration of MMP2200 (Elmagbari et al., 2004), the highest dose of 56 mg/kg was also tested after IV administration in two subjects. MMP2200 was administered into the sapphenous vein.
Raw tail-withdrawal latency data from 50°C water were converted to Percent Maximum Possible Effect (%MPE) using the equation %MPE = [(Test Latency-Control Latency)/(20-Control Latency)*100], where test latency was the tail withdrawal latency from 50°C water obtained after drug administration, and control latency was the latency obtained at the beginning of the session prior to drug administration.
Assay of Capsaicin-Induced Thermal Allodynia
Monkeys (N=4) were seated in acrylic restraint chairs as described above. The base of each monkey’s tail was shaved, and baseline tail withdrawal latencies were measured from water heated to 38, 42, 46 and 50°C. The temperatures were presented in random order. At each temperature determination, if the subject did not withdraw its tail within 20 sec, the tail was removed from the water by the experimenter, and a latency of 20 sec was assigned to that measurement. By this procedure, temperature-effect curves were determined in each monkey at the beginning of each session, and the highest temperature that failed to elicit tail withdrawal was determined (i.e. the highest temperature to produce a tail withdrawal latency of 20 sec). Water heated to this temperature then served as the thermal stimulus for subsequent studies of allodynia during that session. For one monkey, the thermal stimulus was 42°C throughout the study, and for the other three monkeys, the thermal stimulus was 46°C throughout the study.
Allodynia was elicited by topical application of capsaicin as described previously (Butelman et al., 2003). Following baseline tail withdrawal latency determinations, each subject’s tail was degreased with an alcohol pad. A topical capsaicin patch was prepared as described below (see “Drugs”), and the patch was applied to a region approximately 5 cm from the bottom of the tail for 15 min. After 15 min, the patch was removed and tail withdrawal latencies were redetermined using the thermal stimulus identified from the baseline temperature-effect curve.
Preliminary experiments compared the allodynic effects of patches prepared with active capsaicin and with capsaicin vehicle, and the time course of capsaicin-induced allodynia was measured from 15–120 min after removal of the capsaicin patch. The time of peak capsaicin effect was determined to be 15 min after patch removal. For all subsequent studies, tail withdrawal latencies at this time of peak capsaicin effect were used for data analysis. For dose-effect studies, morphine (0.032–1.0 mg/kg), MMP2200 (0.032–3.2 mg/kg) or saline was administered IM before application of the capsaicin patch. Pretreatment times were 30 min for morphine and 15 min for MMP2200 and saline. For time course studies, morphine (0.32 mg/kg) or MMP2200 (3.2 mg/kg) was administered IM 15, 60, 120 or 180 min before application of the capsaicin patch. For antagonism studies, the moderately mu-selective antagonist naltrexone (0.01 mg/kg) or the delta-selective antagonist naltrindole (1.0 mg/kg) was administered IM 15 min before morphine or MMP2200, and quaternary naltrexone (0.32 mg/kg) was administered SC 30 min before MMP2200. Antagonist doses, pretreatment times and routes of administration were selected based on previous studies of the opioid antagonist effects of these compounds in rhesus monkeys (Ko et al., 1998a; Negus et al., 1998; Bowen et al., 2002; Butelman et al., 2004). The capsaicin patch was then applied 30 min after morphine or 15 min after MMP2200. Test sessions were conducted no more than twice a week, with at least 3 days between tests, and with the sites of the topical capsaicin administration varied between sessions. In addition, tests of capsaicin alone were repeated at least three times in each monkey during the course of the study to assess the stability of capsaicin-induced allodynia. Each set of test conditions was conducted in a group of three monkeys.
Raw tail withdrawal latencies obtained 15 min after removal of the capsaicin patch were converted to Percent Maximum Possible Effect (%MPE) using the equation %MPE=[(Test Latency-Capsaicin Alone Latency)/(20- Capsaicin Alone Latency)*100)], where test latency was the tail withdrawal latency obtained after saline or drug pretreatment, and capsaicin alone latency was the latency obtained after the most recent test of capsaicin alone.
Assay of Schedule-Controlled Responding
Five monkeys were used for studies of schedule-controlled responding, and experiments were conducted in each monkey’s home cage. The home cages of all monkeys were modified to include an operant response panel (28 × 28 cm) mounted on the front wall. Three square translucent response keys (6.4 × 6.4 cm) were arranged 2.54 cm apart in a horizontal row 3.2 cm from the top of the operant panel. Each key could be transilluminated by red, green or yellow stimulus lights. The operant panel also supported an externally-mounted pellet dispenser (Gerbrands, Model G5210; Arlington, MA) that delivered 1 gm banana-flavored food pellets (Purina Test Diet, Richmond, IN) to a food receptacle mounted on the cage beneath the operant response panel. The panel and pellet dispenser were controlled by a MED-PC interface and an IBM compatible computer programmed in MEDSTATE Notation (MED Associates, Inc., East Fairfield, VT).
Daily sessions consisted of multiple cycles. For training sessions, each of 5 cycles was 15 min long and consisted of two components: a 10-min timeout period followed by a 5-min response period. During the timeout period, no stimulus lights were illuminated and responding had no scheduled consequences. During the response period, the center key was transilluminated yellow, and the subjects could respond for up to 10 food pellets under a fixed-ratio 30 schedule of reinforcement. If all 10 food pellets were earned before 5 min had elapsed, the lights were turned off, and responding had no scheduled consequences for the remainder of that response period. All monkeys were trained until they responded at rates greater than 1.0 response/sec during all five cycles for 10 consecutive days, and we have shown previously that monkeys respond at relatively stable rates across successive response periods in this procedure (Negus et al., 1993a).
Test sessions were conducted using a time course procedure. In this procedure, a single dose of morphine (1.0–5.6 mg/kg) or MMP2200 (10–56 mg/kg) was administered, and 5-min response periods identical to those described above began 10, 30, 100 and 300 min after the drug injection. In some cases, an additional test cycle was initiated 24hr after the drug injection. In followup antagonism studies, naltrexone (0.001–0.1 mg/kg) or naltrindole (1.0 mg/kg) was administered 15 min before time course studies with the highest doses of morphine (5.6 mg/kg) and MMP2200 (56 mg/kg). Training sessions were usually conducted on Mondays, Wednesdays and Thursdays, and test sessions were usually conducted on Tuesdays and Fridays. Test sessions were always separated by at least three days. In addition, test sessions were conducted only after a training session during which the monkeys responded at rates greater than 1.0 response/sec for all five cycles.
Raw response rate data were converted to percent control rates using the equation %Control Rate = [(Test Rate/Control Rate) * 100], where test rate was the response rate measured during a response period after drug administration, and control rate was the mean response rate during the training day immediately preceding that test day.
Assay of Drug Self-Administration
Four monkeys were used for studies of drug self-administration. Aseptic procedures were used to implant double-lumen, silicone IV catheters into a femoral, internal jugular or external jugular vein as described previously (Negus and Mello, 2004). The catheter exited in the mid-scapular region of the back. The external portion of the catheter was protected by a tether system consisting of a custom-fitted nylon vest connected to a flexible stainless steel cable and fluid swivel mounted to the top of each cage (Lomir Biomedical, Malone, NY). This flexible tether system permitted monkeys to move freely in the cage. Catheter patency was periodically evaluated by IV administration of ketamine (5 mg/kg) or the short-acting barbiturate methohexital (3 mg/kg) through the catheter lumen. The catheter was considered to be patent if IV administration of ketamine or methohexital produced a loss of muscle tone within 10 sec.
Experiments were conducted in each monkey’s home cage, and home cages were modified to include an operant response panel as described above. In addition, each cage was also equipped with two syringe pumps (Model BSP-lE, Braintree Scientific, Braintree, MA; or Model PHM-100, Med Associates Inc., St. Albans, VT), one for each lumen of the double lumen catheter. One syringe pump was used to deliver self-administered morphine or MMP2200 injections through one lumen of the double-lumen catheter. Each injection was delivered in a volume of 0.1 ml in 1 sec. To assist in the maintenance of catheter patency, the second syringe pump was used to deliver saline through the second lumen of the catheter. This second pump was programmed to deliver 0.1 ml infusions every 20 min from 10:00 a.m. each day until 9:00 a.m. the next morning. The panel and syringe pumps were controlled by a MED-PC interface and an IBM compatible computer programmed in MEDSTATE Notation (MED Associates, Inc., East Fairfield, VT).
Daily drug self-administration sessions lasted 2hr. Session onset was signaled by illumination of green stimulus lights in the center key of the response panel, and drug injections were available by pressing this key under a fixed-ratio 10/timeout 1 min schedule of reinforcement. During each timeout period, all stimulus lights were extinguished, and responding had no scheduled consequences. All monkeys were initially trained to respond under alternating conditions of cocaine availability (0.01 or 0.032 mg/kg/inj) and saline availability until cocaine reliably maintained >30 injections/session and saline reliably maintained <30 injections/session. Subsequently, testing was initiated using 3-day blocks. Specifically, saline, the maintenance dose of cocaine (0.01 or 0.032 mg/kg/inj), or a single dose of morphine (0.0032–0.032 mg/kg/inj) or MMP2200 (0.032–0.32 mg/kg/inj) was substituted as the solution available for self-administration, and these conditions remained in place for 3 consecutive days. Each 3-day block of test sessions was preceded by a session during which saline was available and the subject self-administered <30 injections. Thus, all tests conditions were introduced from a “saline extinction” baseline. At the conclusion of each 3-day block of test sessions, either saline or the maintenance dose of cocaine was reintroduced as the available solution depending on performance during the test sessions. Specifically, if the test solution maintained >30 injections/session on the last day of its availability, then saline was made available until responding declined to <30 injections/session, at which time another test solution was made available. If the test solution maintained <30 injections/session on the last day, then the maintenance dose of cocaine was reinstated until self-administration exceeded 30 injections/session. Saline was then made available until responding declined to <30 injections/session, at which time another test solution was made available. Drugs and doses were tested in a pseudo-random order between monkeys.
The primary dependent variable was the number of injections/session. For each 3-day block of test sessions, data from the last two days were averaged to yield a mean number of injections/session for the test condition in that subject.
Data Analysis
Where applicable, data were analyzed by one- or two-way ANOVA using Prism 4.0c for Macintosh (Graphpad Software, San Diego, CA). A significant ANOVA was followed by the Bonferroni post hoc test. The criterion for significance was p<0.05. In addition, ED50 values were determined in individual animals as the dose required to produce 50%MPE (assay of thermal nociception, assay of capsaicin-induced thermal allodynia) or 50% Control Rate (assay of schedule-controlled responding). ED50 values were calculated by interpolation when only two data points were available (one below and one above 50% effect) or by linear regression when at least three data points were available on the linear portion of the dose-effect curve. Individual ED50 values were averaged to yield mean ED50s and 95% confidence limits. ED50s were considered to be significantly different if 95% confidence limits did not overlap.
Drugs
MMP2200 [H2N-Tyr-D-Thr-Gly-Phe-Leu-Ser-(O-β-D-lactose)-CONH2] was manufactured in compliance with Good Manufacturing Practices (GMP) regulations by PolyPeptide Labs, Torrence, CA, and by UCB-Bioproducts (now Lonza) in Belgium, and was administered as the acetate salt (• HOAc). Morphine sulfate and naltrexone HCl were provided by the Drug Supply Program of the National Institute on Drug Abuse (Bethesda, MD), and naltrindole HCl was provided by Kenner C. Rice (NIDA and NIAAA, Bethesda, MD). MMP2200, morphine, naltrexone and naltrindole were dissolved in sterile water and administered IM in the thigh in all assays except drug self-administration (see below). In addition, the highest dose of 56 mg/kg MMP2200 was also administered IV in the assay of thermal nociception. Naltrexone methylbromide (Mallinckrodt, Hazelwood, MO) was also dissolved in sterile water, and it was administered SC as described previously (Butelman et al., 2004). For drug self-administration studies, MMP2200 and morphine were filtered (MillexGV 0.22μm filter, Millpore, Billerica, MA) and delivered IV. Capsaicin (Sigma Chemical Co., St. Louis, MO) was dissolved in a vehicle composed of 70% alcohol and 30% sterile water approximately 15 min prior to use, and it was delivered transdermally (topical patch) as described previously (Butelman et al., 2003). Specifically, the patch consisted of a 1-cm2 piece of two-ply gauze (5 cm wide; Henry Schein, IN) affixed to adhesive backing (23 mm wide; Nexcare Active, NM). The patch was then attached to a 12 cm long elastic tape (5 cm wide; Elastikon, Johnson & Johnson). The capsaicin solution (0.3 ml of 1.22 mg/ml) was slowly injected onto the patch. Within 30 s of preparing the capsaicin patch, it was secured onto the monkey’s tail with the elastic tape and left on for 15 min. All doses were determined based on the forms of the drugs listed above.
RESULTS
Assay of Thermal Nociception
Under baseline conditions, monkeys never withdrew their tails from water heated to 38°C, and they rapidly withdrew their tails from water heated to 50°C. Over the course of the study, the mean±SEM tail withdrawal latency from 50°C water was 0.79±0.09 sec.
Figure 2 shows the time course of antinociceptive effects produced by saline, morphine (1–5.6 mg/kg) and MMP2200 (10–56 mg/kg) in the 50°C warm-water tail-withdrawal assay. After saline injection, tail withdrawal latencies were stable at <1sec for 100 min. Morphine produced dose- and time-dependent antinociception, with peak effects >90%MPE occurring at 100 min (3.2 mg/kg) or 30–100 min (5.6 mg/kg) after morphine administration. The ED50 (95% CL) for morphine at the time of peak effect (100 min) was 1.74 mg/kg (1.67–1.80). In contrast, MMP2200 at doses up to 56 mg/kg failed to produce significant antinociception. Although the main effect of MMP2200 dose approached significance (p=0.054), no dose of MMP2200 produced an effect greater than 16%MPE at any time. Higher doses of MMP2200 were not administered because (a) 56 mg/kg decreased activity and muscle tone in monkeys tested in the warm-water tail-withdrawal assay (data not shown), and (b) 56 mg/kg produced robust rate-decreasing effects in the assay of schedule-controlled responding, and these effects were not reversed by opioid antagonists (see below).
Figure 2.

Time course of effects produced by IM morphine and MMP2200 in the 50°C warm-water, tail-withdrawal assay. Abscissae: Time in min after the injection. Ordinates: Percent maximum possible effect (%MPE). All points show mean ± SEM for three monkeys. For morphine, there was a significant main effect of dose [F(3,8)=182.8, p<0.001] and time [F(2,16)=191.7, p<0.001] and a significant dose x time interaction [F(6,16)=89.58, p<0.001]. For MMP2200, the main effect of dose approached significance [F(3,8)=3.91, p=0.054], and there was not a significant main effect of time [F(2,16)=0.60, p=0.56] or a significant dose x time interaction [F(6,16)=0.36, p=0.89). Asterisks indicate points significantly different from saline control as indicated by the Bonferroni post hoc test. *p<0.05, **p<0.01.
The highest dose of 56 mg/kg MMP2200 was also administered IV to two monkeys in the assay of thermal nociception. As with the IM route, IV MMP2200 decreased activity and muscle tone, but it did not produce antinociception. The highest levels of antinociception observed at any time were 7% MPE in one monkey and 4%MPE in the second monkey.
Assay of Capsaicin-Induced Thermal Allodynia
Under baseline conditions, the highest temperature that failed to elicit tail withdrawal was 42°C in one monkey and 46°C in the other three monkeys, and these threshold temperatures for tail withdrawal were stable throughout the study. Capsaicin treatment produced robust allodynic responses to these normally innocuous thermal stimuli, reducing tail withdrawal latencies from 20±0 sec to 3.04±1.01 sec 15 min after patch removal (mean±SEM data averaged across all monkeys for all determinations of capsaicin alone). These allodynic effects then dissipated, and tail withdrawal latencies recovered to baseline values after 120 min (data not shown). Capsaicin-induced allodynia was stable and did not systematically increase or decrease with repeated determinations in individual monkeys. For example, mean±SEM tail withdrawal latencies during the first, middle and last determinations of capsaicin alone were 3.43±1.36 sec, 3.78±1.14 sec and 2.25±1.00 sec, respectively, and these values were not significantly different. In contrast to the reliable effects of treatment with active capsaicin, application of a capsaicin vehicle patch did not produce allodynia (tail withdrawal latencies of 20±0 sec).
Saline pretreatment did not alter the allodynic effects of capsaicin (mean±SEM %MPE = −12.53±6.09). Figure 3a shows the dose effect curves for morphine (0.032–1.0 mg/kg) and MMP2200 (0.1–3.2 mg/kg) on capsaicin-induced allodynia. Both morphine and MMP2200 produced dose-dependent and statistically significant antiallodynic effects. ED50 values (95%CL) were 0.056 mg/kg (0.01–0.32) for morphine and 0.91 mg/kg (0.18–4.46) for MMP2200. Thus, morphine was approximately 16 times more potent than MMP2200. The large 95% confidence limits in ED50 values are indicative of individual variability in sensitivity to morphine and MMP2200, and sensitivities to both drugs were similar between monkeys. Thus, the subject most sensitive to morphine was also most sensitive to MMP2200, and the monkey least sensitive to morphine was also least sensitive to MMP2200.
Figure 3.

Antiallodynic effects of IM morphine and MMP2200 in the assay of capsaicin-induced allodynia. (a) Abscissa: Dose morphine or MMP2200 in mg/kg, log scale. Point above “Sal” shows the effects of saline administered before capsaicin. Ordinate: Percent maximum possible effect (%MPE). There was a significant effect of morphine dose [F(4,9)=4.48, p=0.029] and MMP2200 dose [F(5,12)=3.53, p=0.034). Asterisks indicate significant difference from saline control as indicated by the Bonferroni post hoc test (*p<0.05). (b) Time course of antiallodynic effects of morphine and MMP2200. Abscissa: Time after injection in min, log scale. Ordinate: %MPE. (c) Effects of 0.32 mg/kg morphine alone or after pretreatment with naltrexone (NTX; 0.01 mg/kg) or naltrindole (NTI; 1.0 mg/kg). Abscissa: Treatment. Ordinate: %MPE. There was a significant effect of treatment [F(2,6)=26.08, p=0.001]. Asterisks indicate significant difference from morphine alone as indicated by the Bonferroni post hoc test (**p<0.01). (d) Effects of 3.2 mg/kg MMP2200 alone or after pretreatment with naltrexone, naltrindole or quaternary naltrexone (QNTX; 0.32 mg/kg). There was a significant effect of treatment [F(3,8)=48.19, p<0.001]. Asterisks indicate significant difference from MMP2200 alone as indicated by the Bonferroni post hoc test (**p<0.01). All points show mean ± SEM from three monkeys except 1.0 mg/kg morphine in panel a and 3.2 mg/kg MMP2200 at 180 min in panel b (N=2).
Figure 3b shows the time course of anti-allodynic effects induced by 0.32 mg/kg morphine and 3.2 mg/kg MMP2200. Morphine produced time-dependent antiallodynia, with the peak effect occurring at 15 min, and approximately 50% antiallodynia still present after 180 min. MMP2200 also produced time-dependent antiallodynia that peaked after 15 min. However, the antiallodynic effects of 3.2 mg/kg MMP2200 had dissipated after 120 min.
Figure 3c and 3d shows the effects of naltrindole, naltrexone, and/or quaternary naltrexone pretreatment on the antiallodynic effects of 0.32 mg/kg morphine and 3.2 mg/kg MMP2200. Naltrexone (0.01 mg/kg) antagonized the antiallodynic effects of morphine, but naltrindole (1.0 mg/kg) was ineffective. The effects of morphine after quaternary naltrexone pretreatment were not examined; however, a previous study reported that quaternary naltrexone doses up to 3.2 mg/kg failed to attenuate the antiallodynic effects of the mu agonist fentanyl in a similar assay of capsaicin-induced thermal allodynia in rhesus monkeys (Ko et al., 1998b; Butelman et al., 2004). In contrast, the effects of MMP2200 were significantly reduced by naltrexone, naltrindole and quaternary naltrexone.
Assay of Schedule-Controlled Responding
The average control response rate (±SEM), determined on the days before test days throughout the study, was 2.43 (±0.44) responses/sec. Response rates were similar during each of the five cycles of the multiple cycle sessions (2.39±0.50, 2.44±0.45, 2.42±0.42, 2.48±0.42, 2.44±0.43 responses/sec).
Figures 4a and 4b shows the time courses for different doses of morphine and MMP2200. Morphine produced a dose- and time-dependent decrease in response rates. Doses of 3.2 and 5.6 mg/kg morphine significantly decreased response rates after 30 and 100 min, and for the higher dose, response rates continued to be suppressed at 300 min. The ED50 value for morphine at the time of peak effect (100 min) was 1.87 mg/kg (1.47–2.38). MMP2200 also produced a dose- and time-dependent reduction in response rates, although these effects did not quite reach the level of statistical significance (p=0.061). The highest dose of 56 mg/kg MMP2200 reduced response rates to ≤20% of control in all three monkeys after 10 min, and response rates gradually recovered over the course of 24hr. The ED50 for MMP2200 at the time of peak effect (15 min) was 32.58 mg/kg (17.01–62.41).
Figure 4.

Effects of IM morphine and MMP2200 in the assay of schedule controlled responding. (a and b) Time course of morphine and MMP2200 effects. Abscissae: Time in min or hr after injection of morphine (a) or MMP2200 (b), log scale. Ordinates: Percent control response rate. All points show mean ± SEM for four monkeys (morphine) or three monkeys (MMP2200). For morphine, there were significant main effects of dose [F (3,12)=17.50, p<0.001) and time [F(3,36)=17.27, p<0.001), and a significant dose x time interaction [F(9,36)=3.78, p=0.002)]. For MMP2200, the main effect of dose approached significance [F(3,8)=3.73, p=0.061], and there was not a main effect of time [F(3,24)=0.31, p=0.82] or a significant dose x time interaction [F(9,24)=1.45, p=0.22]. Asterisks indicate points significantly different from control as determined by the Bonferroni post hoc test. *p<0.05, **p<0.01. (c and d) Rate decreasing effects of 5.6 mg/kg morphine (c, 30 min after morphine administration) and 56 mg/kg MMP2200 (d, 10 min after MMP2200 administration) administered alone or after pretreatment with naltrexone (NTX; 0.001–0.01 mg/kg) or naltrindole (NTI; 1.0 mg/kg). Abscissae: Treatment condition. Ordinate: Percent control response rate. All bars show mean±SEM for four monkeys (morphine) or three monkeys (MMP2200). There was a significant effect of treatment for both morphine studies [F(4,12)=10.34, p<0.001] and MMP2200 studies [F(3,6)=8.50, p=0.014]. Asterisks indicate a significant difference from control (*p<0.05), and crosses indicate a significant difference from 5.6 morphine alone or 56 MMP2200 alone († p<0.05) as indicated by the Bonferroni post hoc test.
Figures 4c and 4d show the effects of naltrindole and/or naltrexone pretreatment on the rate-decreasing effects of 5.6 mg/kg morphine and 56 mg/kg MMP2200. Naltrexone produced a dose-dependent antagonism of the rate-decreasing effects of morphine, with doses of 0.0032 and 0.01 mg/kg naltrexone significantly attenuating the morphine effect. By contrast, a dose of 0.01 mg/kg naltrexone did not significantly attenuate the effects of 56 mg/kg MMP2200. The rate-decreasing effects were also not blocked by pretreatment with 1.0 mg/kg naltrindole.
Assay of Drug Self-Administration
Figure 5 shows the effects of morphine and MMP2200 in the assay of drug self-administration. Saline maintained relatively low rates of self-administration (mean±SEM=18.9±2.8 injections/session), whereas the maintenance dose of cocaine maintained significantly higher rates of self-administration (72.9±11.7 injections/session). Morphine maintained self-administration according to an “inverted-U shaped” dose-effect curve. Peak rates of self-administration were maintained by 0.01 mg/kg/inj morphine (60.8±12.1 injections/session), and both 0.01 and 0.032 mg/kg/inj morphine maintained higher rates of self-administration than saline. Conversely, MMP2200 at unit doses of 0.032–0.32 mg/kg/inj failed to maintain rates of self-administration different from those maintained by saline.
Figure 5.

Self-administration maintained by IV saline, cocaine (0.01 or 0.032 mg/kg/inj), morphine (0.0032–0.032 mg/kg/inj) or MMP2200 (0.032–0.32 mg/kg/inj). Abscissa: Dose morphine or MMP2200 available for self-administration in units of mg/kg/inj. Points above “S” and “C” show data for self-administration of saline and the maintenance dose of cocaine, respectively. Ordinate: Number of injections per session. All points show mean data for four monkeys (saline, cocaine, morphine) or three monkeys (MMP2200). There was a significant effect of morphine dose [F(3,9)=5.89, p=0.016] but not of MMP2200 dose [F(3,6)=1.82, p=0.24). Asterisks indicate a significant difference from saline control (p<0.05) as indicated by the Bonferroni post hoc test.
DISCUSSION
The primary finding of this study was that the novel mu/delta glycopeptide MMP2200 produced dose-dependent antiallodynic effects in an assay of capsaicin-induced thermal allodynia in rhesus monkeys. Following IM administration, MMP2200 was 16-fold less potent than morphine and displayed a slightly shorter duration of action. Results of antagonism studies suggest that the antiallodynic effects of MMP2200 were mediated by peripheral mu and delta opioid receptors. Although glycosylation of MMP2200 and related peptides has been reported to promote distribution across the blood-brain barrier in rodents after IV, IP and SC administration (Bilsky et al., 2000; Elmagbari et al., 2004; Egleton et al., 2005), IM and IV administered MMP2200 did not produce several effects thought to be mediated by central opioid receptors in the present study in rhesus monkeys. Overall, these results suggest that systemically administered MMP2200 acted as a peripheral mu/delta opioid agonist with limited distribution to the CNS in rhesus monkeys.
Effects of morphine and MMP2200 in the assay of thermal allodynia
Both morphine and MMP2200 produced dose-dependent antiallodynia in the assay of capsaicin-induced thermal allodynia. Morphine was approximately 30-fold more potent in this assay of thermal allodynia than in the assays of thermal nociception or schedule-controlled responding (see below). This agrees with previous determinations of the relative potencies of morphine in these procedures in rhesus monkeys (Brandt et al., 2001; Stevenson et al., 2003). These results also agree with the more general finding that (a) systemically administered opioids are often more potent in assays of chemically-induced allodynia than in more conventional assays of antinociception, and (b) this enhanced potency results at least in part from actions mediated by peripheral opioid receptors in both non-human primates (Negus et al., 1993b; Ko et al., 1998b; Butelman et al., 2004) and rodents (Stein et al., 1989; Kayser et al., 1991).
In the present study, the effects of morphine were completely antagonized by the moderately mu-selective opioid antagonist naltrexone, but not by the delta-selective antagonist naltrindole, suggesting that morphine effects were mediated by mu opioid receptors. The role of central vs. peripheral opioid receptors was not evaluated for morphine in this study; however, previous studies have provided ample evidence to suggest that peripheral mu receptors contribute to the thermal antiallodynic effects of morphine and other mu opioids in rhesus monkeys (Negus et al., 1993b; Ko et al., 1998b; Butelman et al., 2004).
Like morphine, MMP2200 was approximately 30-fold more potent in the assay of thermal allodynia than in the assay of schedule-controlled responding. The antiallodynic effects of MMP2200 were completely antagonized by naltrexone, naltrindole and quaternary naltrexone. There are three implications of these antagonism studies. First, these results suggest that both mu and delta receptors contribute to the antiallodynic effects of MMP2200. This is consistent with the profile of MMP2200 as both a mu agonist and delta agonist in in vitro binding and functional assays (Elmagbari et al., 2004). Second, the effects of MMP2200 were also blocked by a relatively low dose of quaternary naltrexone, which functions as a peripherally selective antagonist after systemic administration (Valentino et al., 1981; Negus et al., 1993b; Ko et al., 1998b; Butelman et al., 2004). This finding suggests that the effects of MMP2200 were mediated by peripheral opioid receptors.
Finally, these results are also consistent with other data suggesting that co-activation of mu and delta receptors can produce superadditive antinociceptive effects. Previous studies have reported that mixtures of selective mu and delta agonists produce superadditive antinociception in both non-human primates (Dykstra et al., 2002; Stevenson et al., 2003; Stevenson et al., 2005) and rodents (Vaught and Takemori, 1979; Heyman et al., 1989; Adams et al., 1993). One consequence of superadditivity is that enhanced antinociception produced by mixtures of mu and delta agonists requires activity at both mu and delta receptors and is sensitive to either mu or delta antagonists (e.g. (Stevenson et al., 2003). The sensitivity of MMP2200-induced antiallodynia to antagonism by either naltrexone or naltrindole suggests that both mu and delta receptors were required for MMP2200 antiallodynia, and this in turn suggests a positive interaction between the mu and delta receptor-mediated effects of MMP2200. Additional studies would be required to determine the degree to which this interaction of mu and delta receptor-mediated effects might be superadditive.
Effects of morphine and MMP2200 in the assays of thermal nociception, schedule-controlled responding, and drug self-administration
In agreement with previous studies, morphine produced dose-dependent thermal antinociception (Dykstra and Woods, 1986; Stevenson et al., 2003), reductions in schedule-controlled responding (Young and Woods, 1982; Stevenson et al., 2003) and maintenance of drug self-administration (Woods and Schuster, 1971). In contrast, MMP2200 at doses up to 30 times the effective morphine doses did not produce antinociception or maintain drug self-administration. MMP2200 decreased rates of schedule-controlled responding with a potency approximately 17-fold lower than that of morphine, which was similar to the potency difference between MMP2200 and morphine in the assay of thermal allodynia. However, the rate-decreasing effects of MMP2200 were not blocked by mu or delta opioid antagonists, suggesting that these effects were not mediated by opioid receptors. The antinociceptive, reinforcing and rate-decreasing effects of opioid agonists are thought to be mediated primarily by opioid receptors located in the central nervous system (Young and Woods, 1982; Koob et al., 1984; Negus et al., 1993b; Ko et al., 1998b). Consequently, the failure of MMP2200 to produce opioid receptor-mediated effects in any of these procedures suggests that MMP2200 did not cross the blood-brain barrier in sufficient concentrations to occupy a functionally relevant number of central mu or delta opioid receptors.
The failure of MMP2200 to produce thermal antinociception in rhesus monkeys contrasts with the finding that systemically administered MMP2200 potently produced thermal antinociception in mice tested using a very similar warm-water tail-withdrawal procedure (Elmagbari et al., 2004). Indeed, MMP2200 and morphine were roughly equipotent on a mg/kg basis after systemic administration in this assay in mice (cf. morphine and compound 5 in (Egleton et al., 2005). These behavioral findings in mice were consistent with pharmacokinetic studies showing that glycosylated peptides related to MMP2200 crossed the blood-brain barrier and gained access to brain after systemic administration (Egleton et al., 2005).
The ability of systemically administered MMP2200 to produce antinociception in mice but not in rhesus monkeys is probably not related to details of the behavioral procedure. In particular, the present study used a lower intensity thermal stimulus than previous mouse studies (50 vs. 55°C), and lower stimulus intensities are typically associated with greater rather than lower levels of antinociception (Walker et al., 1993). The differences are also probably not attributable to differential distribution associated with different routes of MMP2200 administration. Studies in mice used IV, SC and IP routes of administration for MMP2200 and related glycopeptides (Bilsky et al., 2000; Elmagbari et al., 2004; Egleton et al., 2005), whereas the present study used IV and IM routes. Rather, the contrast in results from mouse and non-human primate studies suggests that there may be species differences in pharmacokinetics or pharmacodynamics of MMP2200. Regarding pharmacokinetics, the present results are consistent with the conclusion that there are species differences in distribution of MMP2200 across the blood-brain barrier, with relatively higher degrees of brain penetration in mice than in non-human primates. These apparent species differences in brain penetration of MMP2200 would be consistent with reports of species differences in the pharmacokinetics and brain penetration of some other drugs (Ohtsuki and Terasaki, 2007). It remains unknown whether mice or non-human primates would serve as better predictors of brain penetration by MMP2200 and related glycopeptides in humans; however, non-human primates have generally been found to be better predictors than rodents or dogs for drug pharmacokinetics in humans (Ward and Smith, 2004; Ward et al., 2005). Regarding pharmacodynamics, species differences in MMP2200 effects may be related to species differences in mu/delta interactions on neural substrates that mediate antinociception. For example, there may be species differences in the degree to which delta agonists modulate the antinociceptive effects of low, intermediate and high efficacy mu agonists (Heyman et al., 1989; Stevenson et al., 2003).
Implications for clinical use of MMP2200 and related mu/delta glycopeptides
A major purpose for development and evaluation of MMP2200 and related glycopeptides has been to promote brain penetration of peptides and thereby increase the potential utility of peptides as therapeutics. MMP2200 was of particular interest in the present study because of its combined agonist activity at mu and delta receptors and its consequent potential for producing synergistic mu and delta receptor-mediated analgesic effects. The results of the present study support the potential value of mixed-action mu/delta agonists as candidate analgesics. MMP2200 was potent and fully efficacious in the assay of thermal allodynia, and the antagonism data provided evidence for a synergistic interaction between the mu and delta receptor-mediated antiallodynic effects of MMP2200. However, MMP2200 did not produce several effects that require agonist activation of central opioid receptors, indicating that glycosylation did not permit a functionally relevant degree of distribution across the blood-brain barrier. There are two general implications of these findings for the potential clinical development of these compounds.
First, glycopeptide mu/delta opioid agonists such as MMP2200 may warrant further investigation as peripherally selective opioids for treatment of inflammatory pain. The peripheral actions of these compounds may be sufficient to produce modest pain relief by reducing inflammation-associated allodynia, and restricted access to the periphery would limit the emergence of centrally mediated undesirable effects such as abuse-related reinforcing effects. However, peripherally selective mu/delta agonists would still be expected to produce peripherally mediated undesirable effects, such as inhibited bladder motility (Sheldon et al., 1989).
Second, to the degree that central actions are desirable, these results suggest that it will be necessary to develop compounds with higher rates of brain penetration than MMP2200. At this point, it is unclear whether this might be achieved with alternate glycosylation strategies, or by the use of existing methods of glycosylation on other peptides. However, regardless of the strategy employed for synthesis, the present results suggest that there may be important species differences in the consequences of peptide glycosylation for transport across the blood-brain barrier. These species differences should be considered in the design of in vivo studies of pharmacokinetics and drug effects.
Acknowledgments
The authors would like to thank Ashley Bear for expert technical assistance.
This work was supported by grants R01-DA11460 from the National Institute on Drug Abuse, R01-NS052727 from the National Institute of Neurological Disorders and Stroke, and 14-02-01-0471 and 14-05-1-0807 from the Office of Naval Research. A portion of this work was also supported by the Intramural Research Programs of the National Institute on Drug Abuse and the National Institute on Alcohol Abuse and Alcoholism.
Abbreviations
- MMP2200
H2N-Tyr-D-Thr-Gly-Phe-Leu-Ser-(O-β-D-lactose)- CONH2
References
- Adams JU, Tallarida RJ, Geller EB, Adler MW. Isobolographic superadditivity between delta and mu opioid agonists in the rat depends on the ratio of compounds, the mu agonist and the analgesic assay used. J Pharmacol Exp Ther. 1993;266:1261–1267. [PubMed] [Google Scholar]
- Berger B, Rothmaier AK, Wedekind F, Zentner J, Feuerstein TJ, Jackisch R. Presynaptic opioid receptors on noradrenergic and serotonergic neurons in the human as compared to the rat neocortex. Br J Pharmacol. 2006;148:795–806. doi: 10.1038/sj.bjp.0706782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilsky EJ, Egleton RD, Mitchell SA, Palian MM, Davis P, Huber JD, Jones H, Yamamura HI, Janders J, Davis TP, Porreca F, Hruby VJ, Polt R. Enkephalin glycopeptide analogues produce analgesia with reduced dependence liability. J Med Chem. 2000;43:2586–2590. doi: 10.1021/jm000077y. [DOI] [PubMed] [Google Scholar]
- Bowen CA, Fischer BD, Mello NK, Negus SS. Antagonism of the antinociceptive and discriminative stimulus effects of heroin and morphine by 3-methoxynaltrexone and naltrexone in rhesus monkeys. J Pharmacol Exp Ther. 2002;302:264–273. doi: 10.1124/jpet.302.1.264. [DOI] [PubMed] [Google Scholar]
- Brandt MR, Furness MS, Mello NK, Rice KC, Negus SS. Antinociceptive effects of delta-opioid agonists in Rhesus monkeys: effects on chemically induced thermal hypersensitivity. J Pharmacol Exp Ther. 2001;296:939–946. [PubMed] [Google Scholar]
- Butelman ER, Ball JW, Harris TJ, Kreek MJ. Topical capsaicin-induced allodynia in unanesthetized primates: pharmacological modulation. J Pharmacol Exp Ther. 2003;306:1106–1114. doi: 10.1124/jpet.103.052381. [DOI] [PubMed] [Google Scholar]
- Butelman ER, Harris TJ, Kreek MJ. Antiallodynic effects of loperamide and fentanyl against topical capsaicin-induced allodynia in unanesthetized primates. J Pharmacol Exp Ther. 2004;311:155–163. doi: 10.1124/jpet.104.068411. [DOI] [PubMed] [Google Scholar]
- Dykstra LA, Granger AL, Allen RM, Zhang X, Rice KC. Antinociceptive effects of the selective delta opioid agonist SNC80 alone and in combination with mu opioids in the squirrel monkey titration procedure. Psychopharmacology (Berl) 2002;163:420–429. doi: 10.1007/s00213-002-1100-8. [DOI] [PubMed] [Google Scholar]
- Dykstra LA, Woods JH. A tail withdrawal procedure for assessing analgesic activity in rhesus monkeys. J Pharmacol Methods. 1986;15:263–269. doi: 10.1016/0160-5402(86)90056-2. [DOI] [PubMed] [Google Scholar]
- Egleton RD, Bilsky EJ, Tollin G, Dhanasekaran M, Lowery JJ, Alves I, Davis P, Porreca F, Yamamura HI, Yeomans L, Keyari CM, Polt R. Biousian glycopeptides penetrate the blood-brain barrier. Tetrahedron: Asymmetry. 2005;16:65–75. [Google Scholar]
- Egleton RD, Mitchell SA, Huber JD, Palian MM, Polt R, Davis TP. Improved blood-brain barrier penetration and enhanced analgesia of an opioid peptide by glycosylation. J Pharmacol Exp Ther. 2001;299:967–972. [PubMed] [Google Scholar]
- Elmagbari NO, Egleton RD, Palian MM, Lowery JJ, Schmid WR, Davis P, Navratilova E, Dhanasekaran M, Keyari CM, Yamamura HI, Porreca F, Hruby VJ, Polt R, Bilsky EJ. Antinociceptive structure-activity studies with enkephalin-based opioid glycopeptides. J Pharmacol Exp Ther. 2004;311:290–297. doi: 10.1124/jpet.104.069393. [DOI] [PubMed] [Google Scholar]
- Gutstein H, Akil H. Opioid analgesics. In: Brunton L, Lazo J, Parker K, editors. The Pharmacological Basis of Therapeutics. McGraw-Hill; New York: 2005. pp. 547–590. [Google Scholar]
- Heyman JS, Vaught JL, Mosberg HI, Haaseth RC, Porreca F. Modulation of mu-mediated antinociception by delta agonists in the mouse: selective potentiation of morphine and normorphine by [D-Pen2, D-Pen5]enkephalin. Eur J Pharmacol. 1989;165:1–10. doi: 10.1016/0014-2999(89)90764-4. [DOI] [PubMed] [Google Scholar]
- Hruby V, Mosberg HI. Endogenous peptides for delta opioid receptors and analogues. In: Chang K-J, Porreca F, Woods JH, editors. The Delta Receptor. Marcel Dekker; New York: 2004. pp. 159–174. [Google Scholar]
- Kayser V, Chen YL, Guilbaud G. Behavioural evidence for a peripheral component in the enhanced antinociceptive effect of a low dose of systemic morphine in carrageenin-induced hyperalgesic rats. Brain Res. 1991;560:237–244. doi: 10.1016/0006-8993(91)91238-v. [DOI] [PubMed] [Google Scholar]
- Ko MC, Butelman ER, Traynor JR, Woods JH. Differentiation of kappa opioid agonist-induced antinociception by naltrexone apparent pA2 analysis in rhesus monkeys. J Pharmacol Exp Ther. 1998a;285:518–526. [PMC free article] [PubMed] [Google Scholar]
- Ko MC, Butelman ER, Woods JH. The role of peripheral mu opioid receptors in the modulation of capsaicin-induced thermal nociception in rhesus monkeys. J Pharmacol Exp Ther. 1998b;286:150–156. [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Pettit HO, Ettenberg A, Bloom FE. Effects of opiate antagonists and their quaternary derivatives on heroin self-administration in the rat. J Pharmacol Exp Ther. 1984;229:481–486. [PubMed] [Google Scholar]
- Lowery JJ, Yeomans L, Keyari CM, Davis P, Porreca F, Knapp BI, Bidlack JM, Bilsky EJ, Polt R. Glycosylation improves the central effects of DAMGO. Chem Biol Drug Des. 2007;69:41–47. doi: 10.1111/j.1747-0285.2007.00462.x. [DOI] [PubMed] [Google Scholar]
- Mansour A, Khachaturian H, Lewis ME, Akil H, Watson SJ. Anatomy of CNS opioid receptors. Trends Neurosci. 1988;11:308–314. doi: 10.1016/0166-2236(88)90093-8. [DOI] [PubMed] [Google Scholar]
- Mennicken F, Zhang J, Hoffert C, Ahmad S, Beaudet A, O’Donnell D. Phylogenetic changes in the expression of delta opioid receptors in spinal cord and dorsal root ganglia. J Comp Neurol. 2003;465:349–360. doi: 10.1002/cne.10839. [DOI] [PubMed] [Google Scholar]
- Negus SS, Burke TF, Medzihradsky F, Woods JH. Effects of opioid agonists selective for mu, kappa and delta opioid receptors on schedule-controlled responding in rhesus monkeys: antagonism by quadazocine. J Pharmacol Exp Ther. 1993a;267:896–903. [PubMed] [Google Scholar]
- Negus SS, Butelman ER, Al Y, Woods JH. Prostaglandin E2-induced thermal hyperalgesia and its reversal by morphine in the warm-water tail-withdrawal procedure in rhesus monkeys. J Pharmacol Exp Ther. 1993b;266:1355–1363. [PubMed] [Google Scholar]
- Negus SS, Gatch MB, Mello NK, Zhang X, Rice K. Behavioral effects of the delta-selective opioid agonist SNC80 and related compounds in rhesus monkeys. J Pharmacol Exp Ther. 1998;286:362–375. [PubMed] [Google Scholar]
- Negus SS, Mello NK. Effects of chronic methadone treatment on cocaine- and food-maintained responding under second-order, progressive-ratio and concurrent-choice schedules in rhesus monkeys. Drug Alcohol Depend. 2004;74:297–309. doi: 10.1016/j.drugalcdep.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Ohtsuki S, Terasaki T. Contribution of carrier-mediated transport systems to the blood-brain barrier as a supporting and protecting interface for the brain; importance for CNS drug discovery and development. Pharm Res. 2007;24:1745–1758. doi: 10.1007/s11095-007-9374-5. [DOI] [PubMed] [Google Scholar]
- Sheldon RJ, Nunan L, Porecca F. Differential modulation of [D-Pen2, D-Pen5]enkephalin and dynorphin A-(1–17) of the inhibitory bladder motility effects of selected mu agonists in vivo. J Pharmacol Exp Ther. 1989;249:462–469. [PubMed] [Google Scholar]
- Stein C, Millan MJ, Shippenberg TS, Peter K, Herz A. Peripheral opioid receptors mediating antinociception in inflammation. Evidence for involvement of mu, delta and kappa receptors. J Pharmacol Exp Ther. 1989;248:1269–1275. [PubMed] [Google Scholar]
- Stevenson GW, Folk JE, Rice KC, Negus SS. Interactions between delta and mu opioid agonists in assays of schedule-controlled responding, thermal nociception, drug self-administration, and drug versus food choice in rhesus monkeys: studies with SNC80 [(+)-4-[({alpha}R)-{alpha}-((2S,5R)-4-Allyl-2,5-dimethyl-1-piperazinyl)-3- methoxybenzyl]-N, N-diethylbenzamide] and heroin. J Pharmacol Exp Ther. 2005;314:221–231. doi: 10.1124/jpet.104.082685. [DOI] [PubMed] [Google Scholar]
- Stevenson GW, Linsenmayer DC, Folk JE, Rice KC, Negus SS. Opioid interactions in rhesus monkeys: effects of delta + mu and delta + kappa agonists on schedule-controlled responding and thermal nociceptionn. J Pharmacol Exp Ther. 2003;307:1054–1064. doi: 10.1124/jpet.103.056515. [DOI] [PubMed] [Google Scholar]
- Valentino RJ, Herling S, Woods JH, Medzihradsky F, Merz H. Quaternary naltrexone: evidence for the central mediation of discriminative stimulus effects of narcotic agonists and antagonists. J Pharmacol Exp Ther. 1981;217:652–659. [PubMed] [Google Scholar]
- Vaught JL, Takemori AE. Differential effects of leucine and methionine enkephalin on morphine-induced analgesia, acute tolerance and dependence. J Pharmacol Exp Ther. 1979;208:86–90. [PubMed] [Google Scholar]
- Walker EA, Butelman ER, Decosta BR, Woods JH. Opioid thermal antinociception in rhesus monkeys: Receptor mechanisms and temperature dependency. J Pharmacol Exp Ther. 1993;267:280–286. [PubMed] [Google Scholar]
- Ward KW, Nagilla R, Jolivette LJ. Comparative evaluation of oral systemic exposure of 56 xenobiotics in rat, dog, monkey and human. Xenobiotica. 2005;35:191–210. doi: 10.1080/00498250400028197. [DOI] [PubMed] [Google Scholar]
- Ward KW, Smith BR. A comprehensive quantitative and qualitative evaluation of extrapolation of intravenous pharmacokinetic parameters from rat, dog, and monkey to humans. II. Volume of distribution and mean residence time. Drug Metab Dispos. 2004;32:612–619. doi: 10.1124/dmd.32.6.612. [DOI] [PubMed] [Google Scholar]
- Woods JH, Schuster CR. Opiates as reinforcing stimuli. In: Thompson T, Pickens R, editors. Stimulus Properties of Drugs. Apple-Century-Crofts; New York: 1971. pp. 163–175. [Google Scholar]
- Young AM, Woods JH. Limitations on the antagonistic actions of opioid. 1982 [PubMed] [Google Scholar]