

Abstract
Dietary energy restriction (DER) inhibits mammary carcinogenesis yet mechanisms accounting for its protective activity have not been fully elucidated. In this study we tested the hypothesis that DER exerts effects on intracellular energy sensing pathways resulting in alterations of phosphorylated proteins that play a key role in the regulation of cancer. Experiments were conducted using the 1-methyl-1-nitrosourea-induced mammary cancer model in which rats were 0, 20, or 40% energy restricted during the post initiation stage of carcinogenesis. Parallel experiments were done in non carcinogen treated rats in which effects of DER at 0, 5, 10, 20, or 40% in liver were investigated. In a DER dose dependent manner, levels of Thr-172 phosphorylated AMP-activated protein kinase (AMPK) increased in mammary carcinomas with a concomitant increase in phosphorylated acetyl-CoA-carboxylase, a direct target of AMPK, the phosphorylation of which is regarded as an indicator of AMPK activity. Levels of phosphorylated mTOR decreased with increasing DER and down regulation of mTOR activity was verified by a decrease in the phosphorylation state of two mTOR’s targets, 70-kDa ribosomal protein S6 kinase (p70S6K) and 4E-binding protein 1 (4E-BP1). Coincident with changes in mTOR phosphorylation, levels of activated protein kinase B (Akt) were also reduced. Similar patterns were observed in mammary gland and in liver of non carcinogen treated rats. This work identifies components of intracellular energy sensing pathways, specifically mTOR, its principal up-stream regulators, AMPK and Akt, and its downstream targets, p70S6K and 4E-BP1 as candidate molecules on which to center mechanistic studies of DER.
Keywords: dietary energy restriction, AMP-activated protein kinase, mammalian target of rapamycin, protein kinase B (Akt), mammary carcinogenesis
Introduction
Dietary energy restriction (DER) reduces carcinogenesis at multiple organ sites including breast and liver (1–5). This well-established phenomenon occurs in a dose-dependent manner and is remarkably reproducible across animal models. Much of the effort in this field to elucidate the mechanisms that account for DER’s inhibitory activity has focused either on the identification of the cellular processes affected, i.e. proliferation, apoptosis, and angiogenesis, or on the systemic factors that may mediate protective activity, primarily insulin like growth factor 1 (IGF-1), adrenal cortical steroids, and adipokines such as leptin (6–10). Inhibition of carcinogenesis by DER involves decreased cell proliferation, increased sensitivity to cell death via apoptosis and decreased density in the amount of blood vessels supplying a tumor (4). For a number of years, it was judged that the key to elucidating DER’s powerful effects on these cellular processes might lie in understanding alterations of systemic factors like glucocorticoids and IGF-1, and certainly, those studies have given the field important insights (3;4). But experiments designed to determine whether DER-mediated changes in plasma concentrations of glucocorticoids or IGF-1 are obligatory for protection against mammary cancer indicated that knowledge of effects on these systemic factors was not sufficient to identify all the mechanisms that account for DER mediated protection (7;10).
Despite well recognized changes in energy metabolism known to occur during the development of cancer (11;12), little attention has been given to the possibility that a key to identifying the critical pathways involved in DER-mediated inhibition of carcinogenesis lies within the cell and is triggered in part by the effects of DER on intracellular energy sensing mechanisms. We hypothesized that DER would activate an ancient intracellular fuel sensor, AMP-activated protein kinase (AMPK) and that in concert with DER’s effects on IGF-1 signaling that are mediated via protein kinase B (Akt), would work to down regulate the activity of the mammalian target of rapamycin (mTOR). Measurement of AMPK activation served as a primary endpoint for this work since a growing body of evidence indicates that AMPK functions as a fuel sensor in many tissues and organs where it inhibits anabolic pathways when cellular ATP levels are reduced and when AMP rises in response to limited energy availability (13). Moreover, since activation of AMPK by phosphorylation has been reported to inhibit the activity of mTOR, the phosphorylation mediated activation of 70-kDa ribosomal protein S6 kinase (p70S6K), and phosphorylation dependent inactivation of 4E-binding protein 1 (4E-BP1) (14–16), events linked to the inhibition of cell proliferation and angiogenesis and induction of a pro-apoptotic environment, we hypothesized that mTOR activity would be downregulated by DER; these molecules served as additional endpoints in our analyses. Because down-regulation of the signaling pathway of which insulin like growth factor 1 receptor (IGF1R), phosphatidylinositol 3-kinase (PI3K), and Akt are components has been shown to be associated with DER, and this pathway is linked to the regulation of mTOR (17–20), these molecules were also measured. The linkage of signaling through the IGF1R pathway to mTOR is specifically attributed to one of its activated mediators, phosphorylated Akt, which has been reported to oppose the effects of phosphorylated AMPK on the activity of mTOR and its downstream targets (21), thus providing an additional rationale for the pathways investigated. Our initial analyses were performed on mammary carcinomas and then selectively extended to lysates of mammary gland excised from the same rats in which no evidence of pre-malignant or malignant mammary pathologies was apparent (referred to as pathology-free). Having obtained evidence consistent with an effect of DER on these energy sensing mechanisms, a second experiment was performed in non carcinogen treated rats. This was done to extend our observations to another organ site in which carcinogenesis is inhibited by DER via similar cellular mechanisms (5;22) and to more rigorously test our hypothesis in a tissue that is relatively homogenous in terms of cell types present. This approach permitted more detailed analyses than were possible using the limited amount of pathology-free mammary tissue available from our first experiment and it allowed us to extend the work to less stringent levels (5 and 10%) of DER. Thus, the goal of the work reported herein was to establish whether DER affects the activity of the energy sensing network of which AMPK, Akt and mTOR are components.
Materials and Methods
Chemicals
Primary antibodies used in this study were anti-phospho-AMPK (Thr172), anti-AMPK, anti-phospho-mTOR (Ser2448), anti-mTOR, anti-phospho-p70S6K (Thr389), anti-p70S6K, anti-phospho-4E-BP1 (Thr37/46), anti-4E-BP1, anti-phospho-Akt (Ser473), anti-Akt, , anti-phospho-ACC (Ser79), anti-ACC, anti-PI3Kp85, anti-PI3Kp110α, anti-phospho-TSC2 (Thr1462), anti-TSC2, and anti-rabbit immunoglobulin-horseradish peroxidase-conjugated secondary antibody, as well as LumiGLO reagent with peroxide, all from Cell Signaling Technology (Beverly, MA). Anti-IGF1R antibody and anti-mouse immunoglobulin-horseradish peroxidase-conjugated secondary antibody were from Santa Cruz (Santa Cruz, CA). Anti-LKB1 antibody was from Millipore (Billerica, MA). Mouse anti-β-actin primary antibody was obtained from Sigma Chemical Co. (St. Louis, MO). A mTOR Kinase Activity Assay kit was purchased from (EMD, San Diego, CA). Tris-glycine gels were purchased from Invitrogen (Carlsbad, CA).
Animals, housing and diets
Female Sprague Dawley rats were obtained from Taconic Farms, Germantown NY at 20 days of age (DOA) and were housed individually in stainless steel metabolic cages with wire mesh bottoms from 23 DOA to the end of an experiment. The cages were equipped with adjustable width external tunnel feeders that permitted accurate quantification of food intake. All rats were fed a modified AIN-93G diet formulation as previously described (23). The animal facility in which the rats were housed is AAALAC accredited. The animal room was maintained at 22 ± 1°C with 50% relative humidity and a 12-h light/12-h dark cycle. The work reported was reviewed and approved by Colorado State University Animal Care and Use Committee and conducted according to that committee’s guidelines.
Experiment 1
Experimental Design
At 21 DOA, rats were injected with 50 mg of 1-methyl-1-nitrosourea/kg body weight (i.p.) as previously described (24). Five days following carcinogen injection, all rats were randomized into three groups (n=30/gp) and meal fed either AIN-93G control diet ad libitum (control), a modified AIN-93G diet in an amount that was 80% of ad libitum intake of the control (20%DER) or a modified AIN-93G diet in an amount that was 60% of ad libitum intake of the control (40%DER) (23). All of the rats were meal fed twice daily (8:00–11:00 AM and 2:00–5:00 PM), 7 days per week in order to reduce possible confounding due to inter-group variation in meal timing, meal number, and duration of fasting between meals. Rats in the control group were allowed access to an unlimited amount of diet each meal while rats in DER groups were given a restricted amount that was equally divided to two meals and provided in each meal twice per day. The diet fed to 20% or 40% DER rats was formulated to insure an intake of all nutrients equivalent to the control group while limiting total dietary energy by reducing carbohydrate. All rats were palpated three times per week for detection of mammary tumors beginning at 19 days post carcinogen until study termination at 52 days post carcinogen (73 DOA).
Necropsy and Sample Collection
Following overnight fast and inhalation of gaseous carbon dioxide and cervical dislocation, rats were then skinned and the skin area with the mammary grand chain was examined under translucent light to locate mammary tumors. The tumors were excised and snap frozen in liquid nitrogen. The contralateral abdominal inguinal mammary gland chain was excised, prepared as a whole mount on transparency film, and snap frozen in liquid nitrogen. All samples were stored at −80°C. Only mammary adenocarcinomas and pathology-free mammary gland number 4 from the same tumor bearing rat were evaluated (7 to 9 rats/group).
Experiment 2
Experimenal Design
Rats were randomized by body weight at 23 DOA and divided into each of five groups, 8 rats per group. Rats in all groups were meal fed the same diet formulations as described in Experiment 1, 3 hours per meal, 2 meals per day, 7 days per week. Rats in the control (DER 0%) group were allowed access to an unlimited amount of diet each meal while rats in DER groups were given a restricted amount that was equally divided to two meals and provided in each meal twice per day. The four groups of restricted-fed rats were given 95 (DER 5%), 90 (DER 10%), 80 (DER 20%) or 60% (DER 40%) of the ad libitum intake of the unrestricted rats. Since the objective of this experiment was to study the effects of DER on energy sensing pathways in the liver, DER was maintained for 4 weeks at which time the rats were euthanized.
Necropsy and Sample Collection
All rats were euthanized over a 3-hour time interval each day. Following the routine overnight fast to which rats were accustomed, rats were euthanized via inhalation of gaseous carbon dioxide. The sequence in which rats were euthanized was stratified across groups so as to minimize the likelihood that order effects would masquerade as treatment associated effects. After the rats lost consciousness, the livers were immediately excised and frozen between clamps that were pre-cooled in liquid nitrogen. The livers were put in plastic bags and the bags were sealed, and then placed in liquid nitrogen. After transport from the animal facility, the frozen livers were stored at −80Cº until they were analyzed.
Western Blotting
Mammary carcinomas, mammary gland, or liver were homogenized in lysis buffer [40 mM Tris-HCl (pH 7.5), 1% Triton X-100, 0.25 M sucrose, 3 mM EGTA, 3 mM EDTA, 50 μM β-mercaptoethanol, 1 mM phenyl-methylsulfony fluoride and complete protease inhibitor cocktail (Calbiochem, San Diego, CA)]. The lysates were centrifuged at 7,500 × g for 10 min in a tabletop centrifuge at 4ºC and clear supernatant fractions were collected and stored at −80ºC. The protein concentration in the supernatants was determined by the Bio-Rad protein assay (Bio-Rad, Hercules, CA).
Western blotting was performed as described previously (6). Briefly, 40–60 μg of protein lysate per sample was subjected to 8–16% sodium dodecyl sulfate-polyacrylamide gradient gel electrophoresis (SDS-PAGE) after being denatured by boiling with SDS sample buffer [63 mM Tris-HCl (pH 6.8), 2% SDS, 10% glycerol, 50 mM DTT, and 0.01% bromophenol blue] for 5 minutes and the proteins were transferred to a nitrocellulose membrane. The levels of LKB1, phospho-AMPK (Thr172), AMPK, phospho-mTOR (Ser2448), mTOR, phospho-p70S6K (Thr389), p70S6K, phospho-4E-BP1(Thr37/46), 4E-BP1, phospho-ACC (Ser79), ACC, phospho-Akt (Ser473), Akt, IGF1R, PI3Kp85, PI3Kp110α, and β-actin were determined using specific primary antibodies, followed by treatment with the appropriate peroxidase-conjugated secondary antibodies and visualized by LumiGLO reagent Western blotting detection system. The chemiluminescence signal was captured using a ChemiDoc densitometer (Bio-Rad, Hercules, CA) run under the control of Quantity One software (Bio-Rad, Hercules, CA). The actin-normalized scanning density data were reported.
ELISA-based mTOR Kinase Activity Assay, K-LISA (EMD, San Diego, CA)
Two steps were preceded as following.
Immunoprecipitation and kinase activity
Clamp-frozen liver was homogenized in a specified lysis buffer [50mM Tris HCL, 100 mM NaCl, 50 mM Beta-glycerophosphate, 10% glycerol (W/V), 1% Tween-20 detergent (W/V), 1mM EDTA, 20 nM microcystin-LR, 25 mM NaF, and a cocktail of protease inhibitors (EMD, San Diego, CA, Cat. No.539134)]. Homogenates were centrifuged at 16,000 × g for 10 min at 4ºC and the supernatant was transferred to a clean tube. The supernatant (0.5 ml, protein concentration at 7 mg/ml) was pre-cleared by adding 15 μl Protein G-Plus agarose beads, incubating for 15 min at 4ºC, and centrifuged at 4,000 × g for 5 min at 4ºC to pellet the agarose beads. 10 μl of mTOR antibody (EMD, San Diego, CA) was added to the pre-cleared lysates in fresh tubes and rotated for 1h at 4ºC. Then, 50 μl of protein G-Plus agarose beads was added in the tube and rotated for 90 min at 4ºC. The agarose beads were washed with the lysis buffer for 3 times and with 1 × Kinase Assay Buffer (EMD, San Diego, CA) once and centrifuged at 4,000 × g for 5 min at 4ºC. The beads were gently mixed with 2x Kinase Assay Buffer working solution (2x kinase assay buffer, 20mM ATP, and 100mM DTT) (EMD, San Diego, CA) and recombinant p70S6K-GST fusion protein (EMD, San Diego, CA) to be evenly suspend, and incubated at 30ºC for 30 min. The reaction was stopped by adding Stop Solution (EMD, San Diego, CA) and centrifuge at 4,000 × g for 5 min at 4ºC and the supernatant was stored at −80ºC until the assay can be completed.
ELISA
100 μl of supernatant obtained from the kinase activity was added to each well of Glutathione-Coated 96-Well Plates (EMD, San Diego, CA) and incubated for 60 min at 30ºC, followed by three times wash with Plate Wash Buffer (EMD, San Diego, CA). Then, 100 μl of anti-p70S6K-T389 Working Solution (EMD, San Diego, CA) was added to each well and incubated for 1h at room temperature. After three washes with the Plate Wash Buffer, 100 μl of HRP Antibody-Conjugate (EMD, San Diego, CA) was added to each well and incubated for 1h at room temperature. After another three washes with the Plate Wash Buffer, 100 μl of TMB Substrate (EMD, San Diego, CA) was added and incubated for 20 min at room temperature followed by adding 100 μl of ELISA Stop Solution (EMD, San Diego, CA) to each well. The plate was read at 450 nm – 595 nm. A serially diluted standards (enriched rat brain fraction, EMD, San Diego, CA), which were 50 μl, 16.67 μl, 5.56 μl, 1.85 μl, 0.62 μl, and 0.21 μl, was performed at the same time. Since the activity unit of the mTOR standard was not provided by the company EMD, the data were expressed as arbitrary unit, i.e. the microliter of standard. It has been suggested that the data can be expressed as the percent of the control.
Statistical Analyses
Differences among groups in the incidence and multiplicity of mammary adenocarcinomas were evaluated, respectively, by chi-square analysis (25) or ANOVA after square root transformation of tumor count as recommended in (26). Differences among groups in the body weight were analyzed by ANOVA and post hoc comparisons were done by method of Bonferoni (27). For Western blots, representative western bands were shown in the figures. The data displayed in the bar graphs of the figures were either the actin-normalized scanning data or the ratio of the actual scanning units derived from the densitometric analysis of each Western blot for the phospho-protein divided by its non-phosphorylated counterpart for proteins involved in energy sensing pathways. For statistical analyses, the actin-normalized scanning density data obtained from the ChemiDoc scanner (Bio-Rad, Hercules, CA) were first rank transformed. This approach is particularly suitable for semi-quantitative measurements that are collected as continuously distributed data as is the case with Western blots The ranked data were then subjected to analysis of variance (Experiment 1) or regression analysis (Experiment 2) (28). Ratio data were computed from the scanning units derived from the densitometric analysis, i.e. the arbitrary units of optical density for variables stated and then the ratios were rank transformed and evaluated via analysis of variance or regression analysis (28). All analyses were performed used Systat statistical analysis software, Version 12.
Results
Experiment 1
Effect of DER on the carcinogenic response and body weight
Cancer incidence and multiplicity were reduced by DER in a dose dependent manner as shown in Table 1. Cancer incidence was 96, 60, and 23% (p<0.001) and cancer multiplicity was 2.4, 1.1, and 0.3 carcinomas per rat (p<0.001) in control, 20% DER, and 40% DER treatment groups, respectively. DER resulted in lower final body weights that were 180, 150, or 123 grams in the control, 20% or 40% DER, respectively, p<0.001 (Table 1).
Table 1.
The overall effects of different levels of dietary energy restriction (DER) on body weight and mammary carcinogenesis1
| DER (%) | Experiment 1 | Experiment 2 | |||
|---|---|---|---|---|---|
| Final body weight (g) | Cancer incidence (%) | Cancer multiplicity (No./rat) | Final body weight (g) | Total body weight gain (g) | |
| 0 | 180 ± 3 a | 96a | 2.4 ± 0.3 a | 143 ± 3 a | 95 ± 2 a |
| 5 | - | - | - | 113 ± 1 b | 68 ± 1 b |
| 10 | - | - | - | 111 ± 1 b | 66 ± 1 b |
| 20 | 150 ± 1 b | 60b | 1.1 ± 0.2 b | 106 ± 1 bc | 61 ± 1 b |
| 40 | 123 ± 1 c | 23c | 0.3 ± 0.083 c | 94 ± 1 c | 48 ± 1 c |
Experiment 1 was a mammary carcinogenesis study as described in the Materials and Methods section. The rats were fed either an unlimited amount of diet (DER 0%) or were restricted to 80 or 60% of ad libitum intake. In Experiment 2, rats were not injected with carcinogen. Rats were fed either an unlimited amount of diet or were restricted to 95, 90, 80, and 60% of the ad libitum intake (DER 5%, 10%, 20% and 40%) as described in the Materials and Methods. Values are means ± SE for body weight and cancer multiplicity (n=30 in Experiment 1, n=8 in Experiment 2). Different superscripts (a–c) within the same column are statistically significant among the levels of energy intake (p<0.05).
Mammary carcinomas
Since we have previously shown DER to dramatically decreased tumor mass by inhibiting cell proliferation and tumor vascularization and by promoting a proapoptotic environment (6;8;9;29), the initial focus of our investigation was on mammary carcinomas.
AMPK
As shown in Figure 1, panel A by Western blot analysis on mammary carcinomas, DER induced a 1.5-fold increase in the amount of LKB1, the kinase that phosphorylates AMPK on Thr172 (p<0.001). While DER caused a numerical decrease in the amount of AMPK present in mammary carcinomas (p=0.32), DER induced an increase in phosphorylated AMPK (Thr172), p<0.001; the amount of phosphorylated AMPK to AMPK was 1.6 and 2.8-fold higher at 20% and 40% DER versus control (0% DER), respectively. Parallel to the increase in AMPK activation, an increase in the ratio of phosphorylated ACC (Ser79)/ACC was observed in mammary carcinomas at both 20% (1.3 fold) and 40% DER (1.9 fold) (p=0.029).
Figure 1.

Effects of dietary energy restriction (DER) at 0% (control, n=9), 20% (n=9) or 40% (n=7) on LKB1, the phosphorylation of AMP-activated protein kinase (AMPK, Thr172) and acetyl-CoA-carboxylase (ACC, Ser79) in mammary carcinoma of rats (panel A); on the phosphorylation of mammalian target of rapamycin (pmTOR, Ser2448), 70-kDa ribosomal protein S6 kinase (pP70S6K, Thr389), and eukaryote initiation factor 4E binding protein 1 (p4E-BP1, Thr37/46) in mammary carcinoma of rats (panel B); on the expression of the following proteins: insulin like growth factor 1 receptor alpha (IGF1Rα), phosphatidylinositol 3-kinase p110 alpha (PI3Kp110α), phosphorylated Akt (pAkt, Ser473) in mammary carcinoma of rats (panel C); and on phosphorylation of acetyl-CoA-carboxylase (ACC, Ser79) and mammalian target of rapamycin (pmTOR, Ser2448) in mammary gland of rats (panel D). Representative western blot images and bar graphs for the levels of proteins (normalized to beta actin) or the ratio of phosphorylated to unphosphorylated form were shown in each panel. The images shown are those directly acquired from the ChemiDoc work station that is equipped with a CCD camera having a resolution of 1300 × 1030. The normalized intensity data from the ChemiDoc were evaluated; statistical analyses were done on the ranks of the absorbance data via ANOVA and/or regression analysis. Bars, SE. #, p< 0.05 versus 0% DER; †, p < 0.05 versus 20%DER.
mTOR, p70S6K, and 4E-BP1
As shown in Figure 1, panel B, DER significantly decreased the ratio of phosphorylated mTOR (Ser2448)/mTOR, (p<0.001); pP70S6K(Thr389)/P70S6K (p<0.05) and phosphorylated 4E-BP1(Thr37/46)/4E-BP1 (p<0.001). Compared to control (0% DER), the reduction was 11% or 58% for phosphorylated mTOR; 27% or 45% for phosphorylated P70S6K; and 52% or 77% for phosphorylated 4E-BP1 at 20% or 40% DER, respectively.
IGF1R, PI3K and Akt
As shown in Figure 1, panel C, DER was associated with a reduction in levels of IGF1Rα (p<0.001), PI3Kp110α (p=0.033), and the ratio of phosphorylated Akt (Ser473)/Akt (p=0.033). The reduction was 19% or 34% for IGF1Rα, 8% or 32% for PI3Kp110α, and 26% or 37% for phosphorylated Akt (Ser473) for 20% or 40% DER versus control (0% DER).
Mammary gland
AMPK activity was measured by the ratio of phosphorylated to total ACC. As shown in Figure 1, panel D, Western blot analysis indicated that the ratio of pACC (Ser79)/ACC was increased 1.2 or 1.7 fold by 20% or 40% DER, p=0.034. Moreover, the ratio of phosphorylated mTOR (Ser2448)/mTOR was reduced by 15% or 67% for 20% or 40% DER, p=0.004.
Experiment 2
As described in the Materials and Methods section, a range of restricted energy intake was investigated that encompassed but expanded on those used in Experiment 1. Western blot analyses were performed on livers from all rats (n=8/group) in this experiment so that we could robustly test our hypothesis about DER on AMPK, Akt, and mTOR using regression analysis. The overall effects of different levels of energy intake on body weight gain over the duration of this experiment are shown in Table 1. All rats were in positive energy balance, i.e. they gained weight.
Liver
AMPK
Representative Western blots showing the effects of DER on hepatic levels of AMPK and phosphorylated AMPK protein are shown in Figure 2, panel A. The amount of phosphorylated AMPK (Thr172) and their ratio increased (r= 0.82, p<0.001, and r=0. 93, p<0.001, respectively) with increasing DER.
Figure 2.
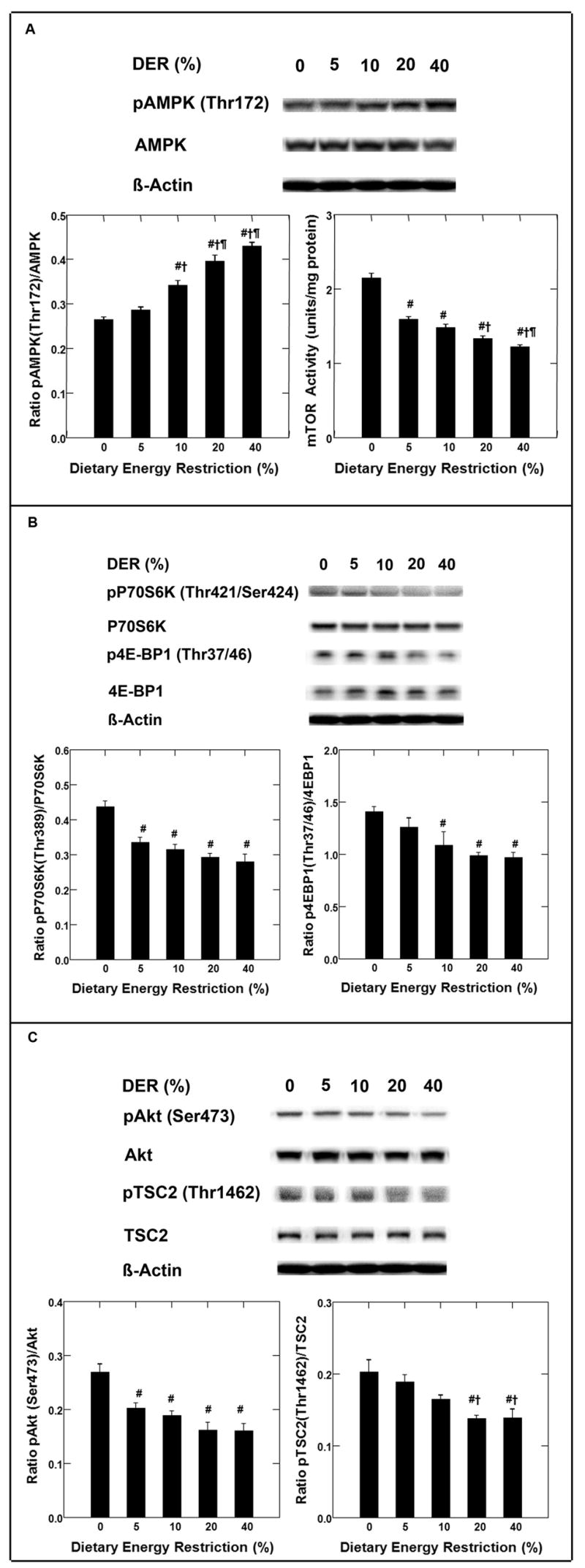
Effects of dietary energy restriction (DER) at 0%, 5%, 10%, 20% or 40% on the phosphorylation of AMP-activated protein kinase (AMPK, Thr172) and the activity of mammalian target of rapamycin (mTOR) in liver of rats (panel A); on the phosphorylation of 70-kDa ribosomal protein S6 kinase (pP70S6K, Thr389) and eukaryote initiation factor 4E binding protein 1 (p4E-BP1, Thr37/46) (panel B); on the phosphorylation of Akt (pAkt, Ser473) and tuberous sclerosis complex 2 (TSC2, Thr1462) (panel C) in liver of rats (n=8). Representative western blot images and bar graphs for the levels of proteins or activity or the ratio of phosphorylated to unphosphorylated form were shown in each panel. The images shown are those directly acquired from the ChemiDoc work station that is equipped with a CCD camera having a resolution of 1300 × 1030. The normalized intensity data from the ChemiDoc were evaluated; statistical analyses were done on the ranks of the absorbance data via regression analysis. The mTOR activity was determined by ELISA-based mTOR Kinase Activity Assay and the data was analyzed by regression analysis. Bars, SE. #, p < 0.05 versus 0% DER; †, p < 0.05 versus 5%DER; ¶, p < 0.05 versus 10%DER.
mTOR activity
Hepatic mTOR activity was assessed directly using an ELISA-based assay as described in the Materials and Methods section. As shown in Figure 2, panel A, mTOR activity was inversely proportional to DER (r= −0.89, p<0.001). The correlation between mTOR activity and phosphorylated mTOR (Ser2448) was also measured; the two measurements were highly correlated (r=0.97, p<0.0001, data not shown).
p70S6K, and 4E-BP1
Levels of p70S6K and phosphorylated p70S6K protein both decreased with increasing DER (p<0.01; representative Western blots shown in Figure 2, Panel B) and therefore we computed their ratio. As shown in Figure 2, Panel B, there was a decrease in that ratio with increasing DER (r= −0.81, p<0.001).
The effects of DER on amounts of 4E-BP1 and phosphorylated 4E-BP1 (Thr37/46) are also shown in Figure 2, panel B. Levels of phosphorylated 4E-BP1 were marginally affected by DER (p=0.001); whereas, there was a negative association (r= −0.65, p=0.001) between DER and phosphorylated 4E-BP1 (Thr37/46). As shown in the panel B, there was a decrease in the ratio of phosphorylated 4E-BP1 to 4E-BP1 with increasing DER (r= −0.66, p<0.001).
Akt
A representative Western blot for Akt and phosphorylated Akt (Ser473) is shown in Figure 2, panel C. The level of Akt in the liver was not affected by DER, p=0.51; whereas, there was a strong inverse association between the level of phosphorylated Akt (Ser473) (r=−0.71, p<0.001) and the ratio of phosphorylated Akt to Akt (r=−0.76, p<0.001) with increasing DER. Based on the effects of DER on levels of phosphorylated Akt, we predicted that there would be corresponding changes in levels of phosphorylated tuberous sclerosis complex (TSC2) at Thr1462 which is a substrate for phosphorylated Akt. Figure 2, panel C shows representative Western blots for TSC2 and phosphorylated TSC2. Regression analysis of these Western data on DER indicated that the ratio of phosphorylated TSC2 to TSC2 decreased with increasing DER (r= −0.68, r<0.001). A logical parallel to these analyses would have been the Western blot analysis of phosphorylated TSC2 (Ser 1341/1337), the sites phosphorylated by AMPK; however, to date none of the commercially available anti-phospho-protein antisera have been of sufficient specificity for use in Western blot analyses of rat tissue extracts.
Discussion
Although a number of the candidate mechanisms by which DER inhibits carcinogenesis have been proposed (3), the hypothesis that DER acts by modulating the activity of intracellular energy sensing pathways has not previously been tested. As shown in Figure 1, panel A, this study reports the first evidence that DER induces the activation of a highly sensitive intracellular energy sensor, i.e the phosphorylation of AMPK in mammary carcinomas, and that activation of AMPK in response to DER, also occurs in mammary gland (Figure 1, panel D) and in the liver of non carcinogen treated rats (Figure 2, panel A). Furthermore, the effect of DER on this intracellular energy sensor appears to be coupled, in part, with its effects on the extracellular environment that are mediated via insulin-related growth factor signaling in a manner that affects the activity of mTOR. Key steps in the pathways affected by DER are diagrammed in Figure 3 and are discussed in the following paragraphs.
Figure 3.
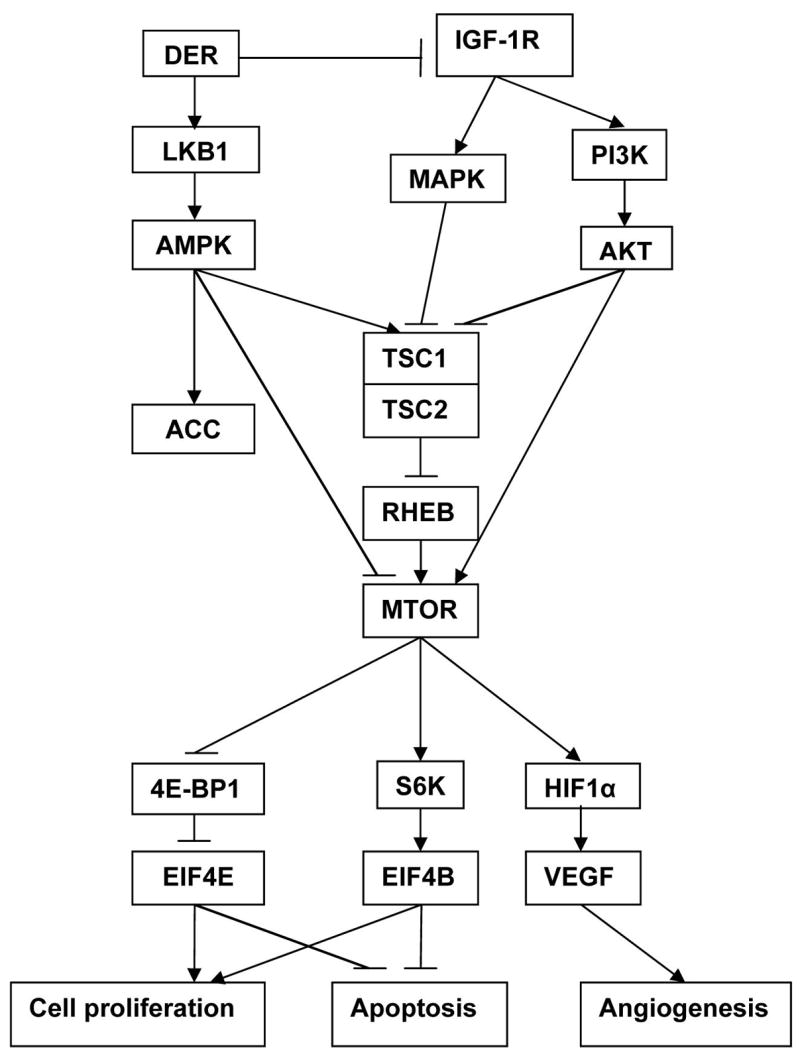
Energy sensing pathways affected by dietary energy restriction (DER).
AMPK is an exquisitely sensitive detector of small changes in the cellular availability of ATP, and some investigators have even proposed that AMPK plays a central role in homeostatic regulation of whole body energy metabolism (30). AMPK is activated via phosphorylation of Thr172, located on the α catalytic subunit of this hetero-trimeric protein. AMPK is phosphorylated on Thr172 by constitutively active LKB1, a reaction that occurs due to conformational changes induced in the γ regulatory subunit of AMPK by its binding of AMP, an event that occurs with greater prevalence when the ratio of AMP/ATP increases within the cell (13). We studied the effects of DER on the levels of AMPK and phosphorylated AMPK (Thr172) and on LKB1. It was observed via Western blot analyses that the amount of phosphorylated AMPK and the ratio of phospho-AMPK/AMPK increased with increasing DER (p<0.001), indicative of strong activation response of AMPK in response to DER. To confirm that DER actually increased AMPK activity, the site specific phosphorylation of ACC, a known substrate for AMPK, was measured. The phosphorylation of ACC (Ser79) increased in response to DER. Given that ACC is not directly related to the energy sensing pathway that we were investigating, such independent confirmation of increased AMPK activity strengthens the observation that DER activates AMPK in mammary carcinomas. Moreover, given that inhibition of fatty acid synthesis has also been associated with anti-cancer activity, and that phosphorylation of Ser79-ACC by AMPK inhibits ACC’s activity, this observation suggests yet another mechanism by which DER may inhibit carcinogenesis (31;32).
LKB1 is generally reported to be constitutively active and is not usually considered a factor in determining whether AMPK responds to changes in intracellular energy availability signaled by the AMP/ATP ratio (33). However, Western blot analysis (Figure 1, panel A) indicated that the amount of LKB1 protein in mammary carcinomas was significantly increased in response to DER. The significance of this finding is unclear since the kinase activity of LKB1 was not measured, but available evidence does indicate that loss of LKB1 kinase activity underlies Peutz-Jeghers syndrome and this has led to the observation that LKB1 is a tumor suppressor that exerts its effect at least in part via its kinase mediated activation of AMPK (reviewed in (32)). Hence, the possibility that DER induces the tumor suppressor activity of the protein complex in which LKB1 participates (STE-related adaptor (STRAD), mouse protein 25 (MO25), LKB1) merits further investigation.
We extended our investigation of DER and AMPK to mammary gland but focused on AMPK activity as indicated by phospho-ACC (Ser79) because of the limited amount of mammary gland that was available from Experiment 1. As shown in Figure 1, panel D, the amount of phospho-ACC and the ratio pACC/ACC increased with DER suggesting that the effect of DER extends to cells that are not directly involved in carcinogenesis. We further tested this idea by assessing activation of AMPK in liver of non carcinogen treated rats with the benefit of the relative homogeneity of cell types within liver versus either mammary carcinoma or mammary gland. A highly significant positive relationship was observed between activated AMPK and magnitude of DER (Figure 2, Panel A); in fact DER accounted for 61% of the variation in activated AMPK. Collectively, these observations were consistent with emerging evidence that AMPK functions as a cellular energy sensor (34) and that DER activates AMPK in both cancer cells and in cells of tissues not involved in the carcinogenic process. AMPK activity was highest when energy intake was lowest and activity was down regulated as energy intake increased.
mTOR
Activated AMPK has a number of targets that are likely to be directly relevant to carcinogenesis (32) but in this study we focused on mTOR, an evolutionarily conserved serine-threonine kinase that is a key regulator of protein translation and synthesis. This kinase is centrally involved in cell growth, i.e. increase in cell size and cell mass and these processes are tightly coupled to cell division (reviewed in (32)). The mTOR pathway integrates nutrient, energy, and mitogen signals to regulate cell growth and cell division (35–37). Activated AMPK has been reported to suppress mTOR activity through the maintenance of the GTPase activity of RHEB protein via site specific phosphorylation of TSC2 at Ser 1337 and 1341. As shown in Figure 1, panel B, DER reduced levels of phospho mTOR which we showed to be highly correlated with mTOR activity in liver. We next determined the effect of DER on phospho-mTOR in the mammary gland and as shown in Figure 1, panel D, DER significantly reduced its phosphorylation. As shown in Figure 2, panel A, the down regulation of mTOR activity in liver was shown by a direct assessment of its activity.
p70S6 kinase and 4E-BP1
Since mTOR affects a complex array of signaling events, we decided to further investigate effects of DER on the phosphorylation of two proteins, p70S6K and 4E-BP1 that are substrates of mTOR (38–40). Activated mTOR phosphorylates p70S6K and this leads to increased ribosomal biogenesis (41;42). 4E-BP1 is a repressor of translation initiation (40;43). Activated mTOR phosphorylates 4E-BP1 which inactivates the protein. When it is hypophosphorylated, 4E-BP1 binds to and inhibits the rate-limiting translation initiation factor eIF4E (eukaryotic translation initiation factor 4E). Upon phosphorylation, eIF4E is released from 4E-BP1, allowing eIF4E to assemble with other translation initiation factors to initiate cap-dependent translation (44). These analyses were only performed in mammary carcinomas and liver. As shown in Figures 1 panel B and figure 2 panel B, phosphorylation of p70S6 kinase and 4E-BP1 were significantly reduced in mammary carcinomas and liver by DER. These findings are consistent with the down regulation of mTOR activity by DER and indicate that DER is likely to inhibit cell growth and cell division via mTOR mediated effects on protein synthesis. Relative to a chronic disease process such as carcinogenesis, this suggests that the propensity for altered patterns of gene expression that occur during clonal selection and expansion of transformed cell populations would be suppressed by DER, a finding consistent with our previous observations (23;29;45).
Akt
DER has been reported by us to decrease circulating levels of IGF-1 (10) and reduced levels of circulating IGF-1 would be expected to down-regulate signaling via the pathway of which IGF1R, PI3K, and Akt are components. Of these proteins, activated Akt, a serine/threonine kinase, is the critical affector molecule. Phospho-Akt serves important roles in cell proliferation, cell survival and new blood vessel formation that are associated with tumor development (3). Therefore, our investigation was extended to these molecules because emerging evidence indicates that this pathway’s effects are also, in part, mediated via mTOR (17–20). The linkage of this pathway to mTOR is specifically attributed to phosphorylated Akt, which has been reported to oppose the effects of phosphorylated AMPK on the activity of mTOR and its upstream targets via phosphorylation of Ser1345 and Thr1227 on TSC2 (21). As shown in Figure 1 panel C and Figure 2 panel C, effects on activated Akt were marked and DER was observed to reduce levels of phospho-Akt (total amount or the ratio of pAkt/Akt) in both mammary carcinomas and liver. While it is clear that reduced levels of activated Akt are likely to affect proliferation, apoptosis and angiogenesis by mechanisms independent of mTOR (reviewed in (46)), the finding that DER induces dual events, AMPK activation and down regulation of growth factor signaling via Akt, that are integrated via TSC2 to decrease mTOR activity is a currently unappreciated dimension in elucidating the mechanisms that account for DER’s profound ability to inhibit the carcinogenic process.
Summary and Implications
These findings provide important insights about DER’s mechanisms of action and are likely to have significant translational implications. While evidence continues to grow indicating that limiting energy availability to an organism has beneficial effects against a number of chronic diseases and also prolongs survival (i.e. retards aging), most efforts to identify common causal links have focused on signaling via insulin and insulin-like growth factors and processes such as reduced cellular oxidation. Our finding that DER induces the activation of AMPK provides a new lens through which to search for common causal mechanisms that account for the health benefits of DER. For many years, the control of type-2 diabetes and hyperlipidemia syndromes has been a goal of drug development efforts that directly or indirectly target the activation of AMPK. One of the most widely used of the drugs thus far developed is metformin (47), and emerging evidence is suggestive of reduced cancer rates in individuals who have taken metformin for the control of type-2 diabetes (48). We have also recently reported that 2-deoxy glucose induces the activation of AMPK in vivo and we suspect that this accounts, at least part, for its ability to inhibit experimentally induced breast cancer (49). Moreover, to the extent that activation of AMPK and down regulation of Akt by DER mediate its effects on carcinogenesis via mTOR, the area of DER now intersects with three signaling pathways of current interest as new targets for development of cancer therapeutics. Those efforts may now be seen as opportunities to co-develop energy restriction mimetic agents as defined in (49). In summary, this work identifies components of intracellular energy sensing pathways, specifically mTOR, its principal up-stream regulators, AMPK and Akt, and its downstream targets, p70S6K and 4E-BP1 as candidate molecules on which to center mechanistic studies of DER.
Footnotes
This work was supported by United States Public Health Services Grant CA52626 from the National Cancer Institute.
The abbreviations used are: AMPK, AMP-activated protein kinase; DER, dietary energy restriction; DOA, days of age; MNU, 1-methyl-1-nitrosourea; ACC, acetyl-CoA-carboxylase; 4E-BP1, eukaryote initiation factor 4E binding protein 1; IGF-1, insulin like growth factor 1; IGF1R, insulin like growth factor 1 receptor; mTOR, mammalian target of rapamycin; PI3K, phosphatidylinositol 3-kinase; P70S6K, 70-kDa ribosomal protein S6 kinase; TSC2, tuberous sclerosis complex 2; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
Reference List
- 1.Birt DF, Kris ES, Choe M, Pelling JC. Dietary energy and fat effects on tumor promotion. Cancer Res. 1992;52:2035s–9s. [PubMed] [Google Scholar]
- 2.Fu PP, Dooley KL, Von Tungeln LS, Bucci T, Hart RW, Kadlubar FF. Caloric restriction profoundly inhibits liver tumor formation after initiation by 6-nitrochrysene in male mice. Carcinogenesis. 1994;15:159–61. doi: 10.1093/carcin/15.2.159. [DOI] [PubMed] [Google Scholar]
- 3.Hursting SD, Lavigne JA, Berrigan D, Perkins SN, Barrett JC. Calorie restriction, aging, and cancer prevention: mechanisms of action and applicability to humans. Annu Rev Med. 2003;54:131–52. doi: 10.1146/annurev.med.54.101601.152156. [DOI] [PubMed] [Google Scholar]
- 4.Thompson HJ, Zhu Z, Jiang W. Dietary energy restriction in breast cancer prevention. J Mammary Gland Biol Neoplasia. 2003;8:133–42. doi: 10.1023/a:1025743607445. [DOI] [PubMed] [Google Scholar]
- 5.Yoshida K, Inoue T, Hirabayashi Y, Nojima K, Sado T. Calorie restriction and spontaneous hepatic tumors in C3H/He mice. J Nutr Health Aging. 1999;3:121–6. [PubMed] [Google Scholar]
- 6.Jiang W, Zhu Z, Thompson HJ. Effect of energy restriction on cell cycle machinery in 1-methyl-1-nitrosourea-induced mammary carcinomas in rats. Cancer Res. 2003;63:1228–34. [PubMed] [Google Scholar]
- 7.Jiang W, Zhu Z, McGinley JN, Thompson HJ. Adrenalectomy does not block the inhibition of mammary carcinogenesis by dietary energy restriction in rats. J Nutr. 2004;134:1152–6. doi: 10.1093/jn/134.5.1152. [DOI] [PubMed] [Google Scholar]
- 8.Thompson HJ, Zhu Z, Jiang W. Identification of the apoptosis activation cascade induced in mammary carcinomas by energy restriction. Cancer Res. 2004;64:1541–5. doi: 10.1158/0008-5472.can-03-3108. [DOI] [PubMed] [Google Scholar]
- 9.Thompson HJ, McGinley JN, Spoelstra NS, Jiang W, Zhu Z, Wolfe P. Effect of dietary energy restriction on vascular density during mammary carcinogenesis. Cancer Res. 2004;64:5643–50. doi: 10.1158/0008-5472.CAN-04-0787. [DOI] [PubMed] [Google Scholar]
- 10.Zhu Z, Jiang W, McGinley J, Wolfe P, Thompson HJ. Effects of dietary energy repletion and IGF-1 infusion on the inhibition of mammary carcinogenesis by dietary energy restriction. Mol Carcinog. 2005;42:170–6. doi: 10.1002/mc.20071. [DOI] [PubMed] [Google Scholar]
- 11.Pan JG, Mak TW. Metabolic targeting as an anticancer strategy: dawn of a new era? Sci STKE. 2007;2007:pe14. doi: 10.1126/stke.3812007pe14. [DOI] [PubMed] [Google Scholar]
- 12.Shaw RJ. Glucose metabolism and cancer. Curr Opin Cell Biol. 2006;18:598–608. doi: 10.1016/j.ceb.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 13.Hardie DG, Carling D. The AMP-activated protein kinase--fuel gauge of the mammalian cell? Eur J Biochem. 1997;246:259–73. doi: 10.1111/j.1432-1033.1997.00259.x. [DOI] [PubMed] [Google Scholar]
- 14.Bolster DR, Crozier SJ, Kimball SR, Jefferson LS. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem. 2002;277:23977–80. doi: 10.1074/jbc.C200171200. [DOI] [PubMed] [Google Scholar]
- 15.Kimura N, Tokunaga C, Dalal S, Richardson C, Yoshino K, Hara K, et al. A possible linkage between AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) signalling pathway. Genes Cells. 2003;8:65–79. doi: 10.1046/j.1365-2443.2003.00615.x. [DOI] [PubMed] [Google Scholar]
- 16.Krause U, Bertrand L, Hue L. Control of p70 ribosomal protein S6 kinase and acetyl-CoA carboxylase by AMP-activated protein kinase and protein phosphatases in isolated hepatocytes. Eur J Biochem. 2002;269:3751–9. doi: 10.1046/j.1432-1033.2002.03074.x. [DOI] [PubMed] [Google Scholar]
- 17.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294:1942–5. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 18.Gerasimovskaya EV, Tucker DA, Weiser-Evans M, Wenzlau JM, Klemm DJ, Banks M, et al. Extracellular ATP-induced proliferation of adventitial fibroblasts requires phosphoinositide 3-kinase, Akt, mammalian target of rapamycin, and p70 S6 kinase signaling pathways. J Biol Chem. 2005;280:1838–48. doi: 10.1074/jbc.M409466200. [DOI] [PubMed] [Google Scholar]
- 19.Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J. 1999;344:427–31. [PMC free article] [PubMed] [Google Scholar]
- 20.Peterson RT, Beal PA, Comb MJ, Schreiber SL. FKBP12-rapamycin-associated protein (FRAP) autophosphorylates at serine 2481 under translationally repressive conditions. J Biol Chem. 2000;275:7416–23. doi: 10.1074/jbc.275.10.7416. [DOI] [PubMed] [Google Scholar]
- 21.Kwiatkowski DJ, Manning BD. Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet. 2005;14:R251–R258. doi: 10.1093/hmg/ddi260. [DOI] [PubMed] [Google Scholar]
- 22.James SJ, Muskhelishvili L. Rates of apoptosis and proliferation vary with caloric intake and may influence incidence of spontaneous hepatoma in C57BL/6 x C3H F1 mice. Cancer Res. 1994;54:5508–10. [PubMed] [Google Scholar]
- 23.Zhu Z, Haegele AD, Thompson HJ. Effect of caloric restriction on pre-malignant and malignant stages of mammary carcinogenesis. Carcinogenesis. 1997;18:1007–12. doi: 10.1093/carcin/18.5.1007. [DOI] [PubMed] [Google Scholar]
- 24.Thompson HJ. Methods for the Induction of Mammary Carcinogenesis in the Rat Using either 7,12-Dimethylbenz(a)antracene or 1-Methyl-1-Nitrosurea. In: Ip MM, Asch BB, editors. Methods in Mammary Gland biology and Breast Cancer Research. New York: Kluwer Academic/Plenum Publishers; 2000. pp. 19–29. [Google Scholar]
- 25.Snedecor GW, Cochran WG. Statistical Methods. 8. Ames, IA: Iowa State University Press; 1989. [Google Scholar]
- 26.Sokal RR, Rohlf FJ. Biometry the principles and practice of statistics in biological research. 3. New York: W.H. Freeman; 1995. [Google Scholar]
- 27.Hochberg Y, Tamhane AC. Multiple Comparison Procedures. New York, NY: John Wiley & Sons, Inc; 1987. [Google Scholar]
- 28.Morrison DF. Multivariate Statistical Methods. 3. New York: McGraw-Hill Publishing Co; 1990. [Google Scholar]
- 29.Zhu Z, Jiang W, Thompson HJ. Effect of energy restriction on tissue size regulation during chemically induced mammary carcinogenesis. Carcinogenesis. 1999;20:1721–6. doi: 10.1093/carcin/20.9.1721. [DOI] [PubMed] [Google Scholar]
- 30.Hardie DG. The AMP-activated protein kinase pathway--new players upstream and downstream. J Cell Sci. 2004;117:5479–87. doi: 10.1242/jcs.01540. [DOI] [PubMed] [Google Scholar]
- 31.Brunet J, Vazquez-Martin A, Colomer R, Grana-Suarez B, Martin-Castillo B, Menendez JA. BRCA1 and acetyl-CoA carboxylase: The metabolic syndrome of breast cancer. Mol Carcinog. 2008;47:157–63. doi: 10.1002/mc.20364. [DOI] [PubMed] [Google Scholar]
- 32.Motoshima H, Goldstein BJ, Igata M, Araki E. AMPK and cell proliferation--AMPK as a therapeutic target for atherosclerosis and cancer. J Physiol. 2006;574:63–71. doi: 10.1113/jphysiol.2006.108324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hardie DG. New roles for the LKB1-->AMPK pathway. Curr Opin Cell Biol. 2005;17:167–73. doi: 10.1016/j.ceb.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 34.Hardie DG, Hawley SA, Scott JW. AMP-activated protein kinase - development of the energy sensor concept. J Physiol. 2006;574:7–15. doi: 10.1113/jphysiol.2006.108944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–45. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 36.Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 37.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–84. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 38.Gingras AC, Kennedy SG, O’Leary MA, Sonenberg N, Hay N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev. 1998;12:502–13. doi: 10.1101/gad.12.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 40.Martin KA, Blenis J. Coordinate regulation of translation by the PI 3-kinase and mTOR pathways. Adv Cancer Res. 2002;86:1–39. doi: 10.1016/s0065-230x(02)86001-8. [DOI] [PubMed] [Google Scholar]
- 41.Lee CC, Huang CC, Wu MY, Hsu KS. Insulin stimulates postsynaptic density-95 protein translation via the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway. J Biol Chem. 2005;280:18543–50. doi: 10.1074/jbc.M414112200. [DOI] [PubMed] [Google Scholar]
- 42.Martin KA, Rzucidlo EM, Merenick BL, Fingar DC, Brown DJ, Wagner RJ, et al. The mTOR/p70 S6K1 pathway regulates vascular smooth muscle cell differentiation. Am J Physiol Cell Physiol. 2004;286:C507–C517. doi: 10.1152/ajpcell.00201.2003. [DOI] [PubMed] [Google Scholar]
- 43.Gingras AC, Raught B, Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001;15:807–26. doi: 10.1101/gad.887201. [DOI] [PubMed] [Google Scholar]
- 44.Sonenberg N, Gingras AC. The mRNA 5′ cap-binding protein eIF4E and control of cell growth. Curr Opin Cell Biol. 1998;10:268–75. doi: 10.1016/s0955-0674(98)80150-6. [DOI] [PubMed] [Google Scholar]
- 45.Zhu Z, Jiang W, Thompson HJ. Effect of energy restriction on the expression of cyclin D1 and p27 during premalignant and malignant stages of chemically induced mammary carcinogenesis. Mol Carcinog. 1999;24:241–5. doi: 10.1002/(sici)1098-2744(199904)24:4<241::aid-mc1>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 46.Younes H, Leleu X, Hatjiharissi E, Moreau AS, Hideshima T, Richardson P, et al. Targeting the phosphatidylinositol 3-kinase pathway in multiple myeloma. Clin Cancer Res. 2007 Jul 1;13(13):3771–5. doi: 10.1158/1078-0432.CCR-06-2921. [DOI] [PubMed] [Google Scholar]
- 47.Hardie DG. AMP-activated protein kinase as a drug target. Annu Rev Pharmacol Toxicol. 2007;47:185–210. doi: 10.1146/annurev.pharmtox.47.120505.105304. [DOI] [PubMed] [Google Scholar]
- 48.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–5. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu Z, Jiang W, McGinley JN, Thompson HJ. 2-Deoxyglucose as an energy restriction mimetic agent: effects on mammary carcinogenesis and on mammary tumor cell growth in vitro. Cancer Res. 2005;65:7023–30. doi: 10.1158/0008-5472.CAN-05-0453. [DOI] [PubMed] [Google Scholar]