

Abstract
Obesity and insulin resistance, cardinal features of metabolic syndrome, are closely associated with a state of low-grade inflammation1,2. In adipose tissue chronic overnutrition leads to macrophage infiltration, resulting in local inflammation that potentiates insulin resistance3,4. For instance, transgenic expression of Mcp1 in adipose tissue increases macrophage infiltration, inflammation, and insulin resistance5,6. Conversely, disruption of Mcp1 or its receptor, Ccr2, impairs migration of macrophages into adipose tissue, thereby lowering adipose tissue inflammation and improving insulin sensitivity5,7. These findings together suggest a correlation between adipose tissue macrophage content (ATM) and insulin resistance. However, resident macrophages in tissues display tremendous heterogeneity in their activities and functions, primarily reflecting their local metabolic and immune microenvironment8. While Mcp-1 directs recruitment of pro-inflammatory classically activated macrophages to sites of tissue damage5,8, resident macrophages, such as those present in adipose tissue of lean mice, display the alternatively activated phenotype9. Despite their higher reparative capacity10, the precise role of alternatively activated macrophages in obesity-induced insulin resistance remains unknown. Using mice with macrophage-specific deletion of peroxisome proliferator activated receptor-γ (PPARγ), we show here that PPARγ is required for maturation of alternatively activated macrophages. Disruption of PPARγ in myeloid cells impairs alternative macrophage activation, thereby predisposing these animals to development of diet-induced obesity, insulin resistance, and glucose intolerance. Furthermore, gene expression profiling revealed that downregulation of oxidative phosphorylation gene expression in skeletal muscle and liver leads to decreased insulin sensitivity in these tissues. Together, our findings demonstrate that resident alternatively activated macrophages have a beneficial role in regulating nutrient homeostasis and suggest that macrophage polarization towards the alternative state might be a useful strategy for treating type 2 diabetes.
To distinguish between the pathogenic and reparative functions of macrophages in metabolic disease, we sought to identify transcriptional regulators that control alternative macrophage activation. Notably, PPARγ, a genetic sensor of fatty acids11, is markedly induced in macrophages stimulated with interleukin-4 (IL-4)12,13, prompting us to examine its role in alternative macrophage activation. Since Balb/c, but not C57Bl/6 mice, can fully support maturation of alternatively activated macrophages14, we generated macrophage-specific PPARγ knockout (Mac-PPARγ KO) mice on the Th2-permissive Balb/c strain (see methods). Quantitative PCR analysis of genomic DNA from peritoneal elicited cells and bone marrow-derived macrophages (PECs and BMDMs, respectively) showed high excision efficiency (~85-90%) in Mac-PPARγ KO animals (Supplementary Fig. S1a). Immunoblot analysis further confirmed the absence of PPARγ protein in BMDMs from Mac-PPARγ KO mice (Supplementary Fig. S1b). Verifying a critical role for PPARγ in alternative activation, arginase I mRNA and activity, hallmarks of alternatively activated macrophages10, were reduced by 40% and 50%, respectively, in IL-4 stimulated PPARγ null BMDMs (Supplementary Fig. S1c and Fig. 1a). To determine whether the arginase I gene is a direct transcriptional target of PPAR/RXR heterodimers, we analyzed its promoter region for PPAR response elements (PPREs). A putative PPRE was identified in the distal enhancer of the arginase I gene, a region known to be essential for its expression in alternatively activated macrophages15. Electromobility gel shift assays confirmed that PPAR/RXR heterodimers bound to the identified site in sequence specific manner (Supplementary Fig. S1d). To verify that PPARγ/RXR heterodimers can activate the arginase I promoter in vivo, RAW264.7 cells were transiently transfected with luciferase reporter construct driven by ~4 kb of mouse arginase I promoter/enhancer. Treatment of transfected macrophages with IL-4 led to ~9-fold increase in luciferase activity (Fig. 1b). Moreover, the addition of PPARγ and its ligand, rosiglitazone, enhanced the ability of IL-4 to activate the arginase I promoter (~2-fold over IL-4 stimulated level), demonstrating that PPARγ directly regulates this important facet of macrophage activation. Because alternatively activated macrophages can counteract excessive secretion of pro-inflammatory cytokines13, we examined whether IL-4 could appropriately attenuate lipopolysaccharide (LPS)-induced TNFα and IL-6 secretion. Although LPS-stimulated release of TNFα and IL-6 was not significantly different between the two genotypes (Fig. 1c and Supplementary Fig. S1e), IL-4 failed to suppress the secretion of IL-6 in macrophages deficient in PPARγ (Fig. 1c), indicating that a subset of IL-4 dependent anti-inflammatory responses are regulated by PPARγ.
Figure 1.
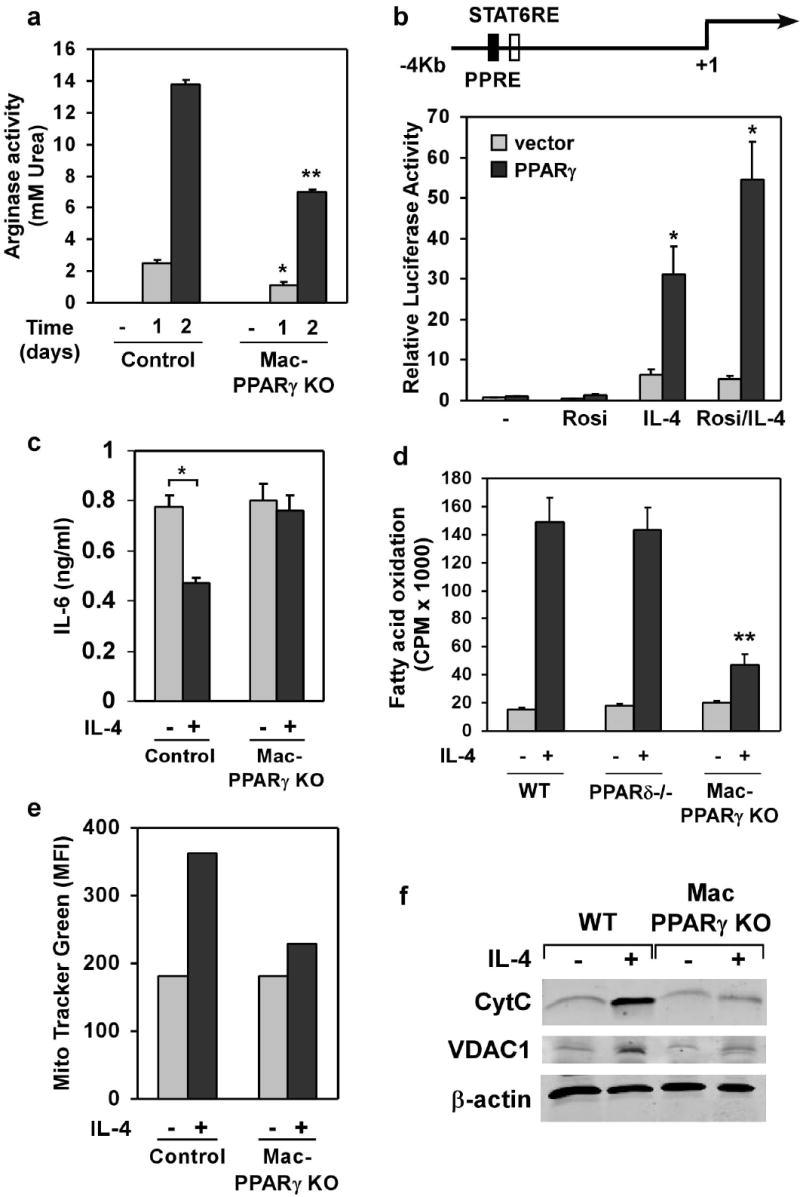
PPARγ regulates alternative macrophage activation. a, Decreased induction of arginase activity by IL-4 in PPARγ null macrophages. BMDM from control and Mac-PPARγ KO mice were stimulated with IL-4 (10ng/ml) for 24 or 48 hours prior to quantification of cell-associated arginase activity. b, Activation of arginase I promoter by PPARγ/RXR heterodimers. c, PPARγ is required for suppression of IL-6 production in alternatively activated macrophages. Macrophages pre-treated with IL-4 (10 ng/ml) for 24 hours were subsequently stimulated with LPS (5 ng/ml) for 6 hours (TNFα) or 24 hours (IL-6). d, PPARγ is required for macrophage oxidative metabolism. Fatty acid oxidation rates were quantified in control, PPARδ null and PPARγ null BMDMs 96 hours after stimulation with IL-4. e-f, IL-4 fails to induce mitochondrial biogenesis in PPARγ deficient macrophages, as measured by (e) Mito Tracker Green and (f) CytC and VDAC1 protein levels. Equivalent loading was confirmed by immunoblotting for β-actin. Rosiglitazone (Rosi).
We recently reported that a switch to oxidative metabolism is an integral component of alternative macrophage activation13. Because PPARs regulate fatty acid homeostasis in many cell types16, we investigated the requirement for PPARγ or δ, the two major PPAR subtypes expressed in murine macrophages, in controlling this oxidative switch. Surprisingly, PPARγ, rather than PPARδ, was required for IL-4 induced increase in β-oxidation of fatty acids, as evidenced by the ~70% reduction in the rate of fatty acid oxidation in IL-4 stimulated PPARγ null macrophages (Fig. 1d). In agreement with this genetic data, ligand activation of PPARγ led to ~2 fold increase in the rate of β-oxidation (Supplementary Fig. S1f). Moreover, quantitative RT-PCR analyses revealed that expression of genes controlling lipolysis (Lpl), fatty acid uptake (CD36), and oxidation (Acadm, Acadl) was reduced in IL-4 stimulated PPARγ null macrophages (Supplementary Fig. S1g). In contrast, fatty acid uptake rates were similar in control and Mac-PPARγ KO macrophages (Supplementary Fig. S1h). Since an increase in cellular mitochondrial content accompanies this metabolic switch, we tested the requirement for PPARγ in mitochondrial biogenesis. Cellular staining with MitoTracker Green revealed an absolute requirement for PPARγ in mediating the biogenic effects of IL-4 (Fig. 1e). Fluorescence microscopy with MitoTracker Red further verified that reduction in respiring mitochondria largely accounted for the observed decrease in total mitochondria in these cells (Supplementary Fig. S1i). Consistent with these findings, IL-4 failed to induce the mitochondrial proteins cytochrome C and ATP synthase α in PPARγ deficient macrophages (Fig. 1f). Lastly, the observed impairment in alternative activation was independent of the upstream IL-4 signaling pathway, because PPARγ-deficient macrophages expressed similar levels of IL-4Rα protein on their cell surface and stimulation with IL-4 resulted in equivalent levels of STAT6 phosphorylation (Supplementary Fig. S2a, b).
To provide an independent test for whether PPARγ can regulate the macrophage program of alternative activation in vivo, we employed an immunological model for studying macrophage Th2-type responses. Cutaneous infection with Leishmania major in Th2-biased Balb/c mice leads to non-healing lesions, whereas the Th1-prone C57Bl/6 mice are resistant to acute infection17. Importantly, since impairment in alternative macrophage activation can delay disease progression in Balb/c mice18, we investigated whether Mac-PPARγ KO mice were also resistant to cutaneous Leishmaniasis. Indeed, Mac-PPARγ KO mice had significantly less footpad swelling 5-7 weeks after injection of L. major promastigotes (Fig. 2a). While lesions in Mac-PPARγ KO started to stabilize at 7 weeks, footpads of control mice continued to enlarge and rapidly underwent necrosis (Fig. 2b). Consistent with local extension of disease, the draining popliteal lymph nodes were hypercellular and enlarged in control mice (Supplementary Fig. S3). Because these results mimic the phenotype of mice lacking alternatively activated macrophages, our data strongly suggest that PPARγ is required for acquisition and maintenance of the alternatively activated phenotype.
Figure 2.
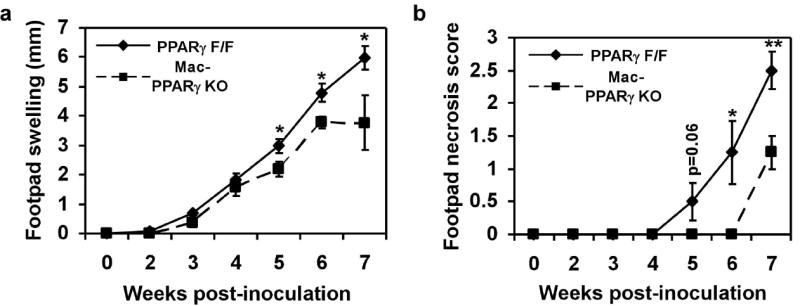
Mac-PPARγ KO mice are less susceptible to infection by Leishmania major. a, Footpad swelling in control and Mac-PPARγ KO mice after infection with L. major (n=5/genotype). b, Decreased necrosis in footpads of Mac-PPARγ KO mice.
On the basis of these results, we tested whether genetic deletion of PPARγ in macrophages would exacerbate the development of metabolic syndrome. Cohorts of control and Mac-PPARγ KO mice were challenged with a high fat diet (HFD) for 18 weeks to promote maximal infiltration of macrophages into adipose tissue4. Surprisingly, Mac-PPARγ KO mice gained more weight than control mice on the HFD. After 17 weeks on a HFD, the body weight of Mac-PPARγ KO mice (46.7 ± 2.3 g) exceeded that of control mice (40.3 ± 1.2 g) by ~15% (Fig. 3a). Dual energy x-ray absorptiometry (DEXA) showed a 20% increase in total fat mass and a 12% increase in adiposity in Mac-PPARγ KO animals (Fig. 3b). Consistent with an increase in adiposity, epididymal fat pads were larger (~18% by mass) and serum leptin levels were higher (~57%) in Mac-PPARγ KO mice (Supplementary Figs. S4a, b)19. However, adipocyte cell size was not significantly different (Supplementary Fig. S4c, d). To better define the alterations in adipose tissue, we quantified transcript levels of genes important in adipocyte differentiation and fatty acid metabolism. As shown in Fig. 3c, mRNA levels of a large number of genes involved in nutrient uptake, fatty acid synthesis, and β-oxidation were reduced by ~50-80% in white adipose tissue (WAT) of Mac-PPARγ KO mice, indicating global suppression of adipocyte function. Direct co-culture of PPARγ-deficient macrophages with adipocytes led to a marked reduction of insulin-stimulated glucose uptake in adipocytes (Fig. 3d), suggesting a causal relationship between PPARγ-deficient ATMs and dysregulation of adipocyte metabolism. Remarkably, this was confirmed by Q-PCR analysis of co-cultured adipocytes which showed suppression of adipocyte gene expression by PPARγ-deficient macrophages (Supplementary Fig. S4e).
Figure 3.

Alterations in adipose tissue mass and function in Mac-PPARγ KO mice. a, Weight gain of control and Mac-PPARγ KO mice on a HFD. Arrow denotes period when mice were fasted for glucose and insulin tolerance tests. b, Body composition as determined by DEXA (n=5/genotype). c, Q-PCR analyses of gonadal adipose tissue gene expression. Relative transcript levels of genes involved in adipocyte differentiation and function. Lpl, lipoprotein lipase; Cd36, fatty acid translocase; Slc27a1, fatty acid transporter 1; Slc2a4, glucose transporter 4; Fabp4, fatty acid binding protein 4; Lipe, hormone sensitive lipase; Fasn, fatty acid synthase; Acaca, acetyl-Coenzyme A carboxylase a; Acox1, acyl-Coenzyme A oxidase 1; Cpt1a, carnitine palmitoyltransferase 1a; Acadm and Acadl, medium- and long-chain acyl-CoA dehydrogenase; Adipoq, adiponectin; Srebf1, sterol regulatory element binding factor 1c. d, Co-culture of macrophages with adipocytes decreases insulin-stimulated glucose uptake. BMDMs from control or Mac-PPARγ KO mice were co-cultured with differentiated 3T3-L1 adipocytes for 48 hours. 2-deoxyglucose uptake was assessed 30 minutes after stimulation with insulin. e, Q-PCR analyses of macrophage gene expression in WAT from control and Mac-PPARγ KO mice. Emr1, F4/80; Cd68, macrosialin; Arg1, arginase I; Mrc1, mannose receptor; Clec7a, dectin-1; Retnla, resistin like alpha; Nos2, inducible nitric oxide synthase; IL-6, interleukin-6. f-g, Macrophage content of epididymal adipose tissue. Representative sections of epididymal fad pads stained with F4/80 (Emr1) antibody (f). Fraction of ATMs is equal to F4/80-stained cells/total cells counted in the fields (g), and statistically analyzed using the paired t-test.
To determine whether macrophage infiltration or aberrant activation contributed to adiposity, we analyzed macrophage-specific gene expression in WAT of control and Mac-PPARγ KO mice. Despite being more obese, transcript levels of macrophage-specific markers, Emr1 and CD68, were reduced by ~70% in WAT of Mac-PPARγ KO mice (Fig. 3e). Consistent with the Q-PCR data, histological analysis showed marked reduction in the macrophage content of WAT of Mac-PPARγ KO mice (Fig. 3f,g). Importantly, expression of genes preferentially expressed in alternatively activated macrophages10, such as ArgI, Mrc1, and Clec7a, was also decreased by ~70-80% in WAT of Mac-PPARγ KO mice (Fig. 3e), suggesting that macrophage-specific deletion of PPARγ leads to specific reduction in alternatively activated macrophages. The absence of alternatively activated ATMs increased local inflammation in WAT, as evidenced by higher expression of IL-6 and Nos2 (Fig. 3e). Lastly, although ATM content is much lower in lean mice3,9, Q-PCR analysis of WAT revealed that chow fed Mac-PPARγ KO mice expressed much lower levels of genes associated with the alternative state (Supplementary Fig. S5a).
While the Th2 cytokines IL-4 and IL-13 are required for maturation of alternatively activated macrophages during parasitic infections10,14, the importance of this signaling pathway in ATM biology has not been formally tested. To determine whether IL-4/IL-13 signaling is required for acquisition of the alternative phenotype by ATMs, we analyzed the molecular signature of ATMs in mice defective in IL-4/IL-13 signaling, in particular, the STAT6 null and Mac-IL-4Rα KO mice14,20. Consistent with a critical role for Th2 cytokines in alternative activation, molecular signature of alternatively activated ATMs was dramatically reduced in lean STAT6 null and Mac-IL-4Rα KO mice (Supplementary Fig. S5b, c). Moreover, the expression profile of ATMs in obese STAT6 null mice was identical to the one observed in obese Mac-PPARγ KO mice (Supplementary Fig. S5d and Fig. 3e). Since the molecular signatures of ATMs in three distinct genetic models (Mac-PPARγ KO, STAT6 null and Mac-IL-4Rα KO) were similar, our data show that an intact IL-4/IL-13/STAT6/PPARγ axis is required for maturation of alternatively activated ATMs. Furthermore, to investigate whether stimulation of ATMs by IL-4 is sufficient to polarize them towards the alternative state, obese control and Mac-PPARγ KO mice were injected with recombinant IL-4, and ATM gene expression was monitored by Q-PCR. Supplementary Figure S5e shows that IL-4 induced the expression of alternatively activated markers (Arg1, Chi3l3, Mrc1 and Jag1) in a PPARγ-dependent manner.
To explore the role of alternatively activated macrophages in obesity-induced insulin resistance, we performed glucose and insulin tolerance tests in control and Mac-PPARγ KO mice. Oral glucose tolerance tests revealed that Mac-PPARγ KO mice were significantly more glucose intolerant after an 18 week HFD challenge (Fig. 4a). As would be expected with a decrease in insulin sensitivity, Mac-PPARγ KO mice were more resistant to the glucose lowering effects of exogenous insulin (Fig. 4b). Moreover, after a 4-hour fast, paired blood glucose and serum insulin levels were much higher in Mac-PPARγ KO mice, with blood glucose levels at 170 ± 9.2 mg/dl vs. 125 ± 2.3 mg/dl (p < 0.0007) and serum insulin levels at (3.00 ± 0.77 ng/ml vs. 0.24 ± 0.02 ng/ml (p < 0.002), (Fig. 4c). The calculated homeostasis model assessment (HOMA) measure of insulin resistance was significantly higher in Mac-PPARγ KO mice (Fig. 4d). To investigate the potential sites of insulin resistance, control and Mac-PPARγ KO mice were injected with saline or insulin (5mU/g) through their inferior vena cava, and liver and skeletal muscle were quickly harvested for biochemical analysis. Strikingly, insulin-stimulated phosphorylation of AKT was markedly decreased in liver and skeletal muscle of Mac-PPARγ KO mice (Fig. 4e, f), findings consistent with the presence of insulin resistance in these tissues.
Figure 4.

Impaired glucose homeostasis in high fat fed male Mac-PPARγ KO mice. a, Oral glucose tolerance tests (1 g/kg) in male mice after 19 weeks of feeding the HFD (n=5/genotype). b, Insulin tolerance test. Obese mice were fasted for 4 hours prior to intraperitoneal injection of insulin (0.65 u/kg). c, Fasting serum insulin levels in control and Mac-PPARγ KO mice after 4 hr fast. d, Homa-IR index of insulin sensitivity (insulin [ng/ml] x glucose [mM]). e-f, Decreased insulin signaling in obese Mac-PPARγ KO mice. Control and Mac-PPARγ KO mice were injected with insulin (5 mU/g), and cellular lysates were immunoblotted for total and serine phosphorylated (S473) Akt; liver (e) and quadriceps (f). g-h, Relative transcript levels of genes involved in β-oxidation and oxidative phosphorylation, and of transcriptional regulators controlling these pathways in quadriceps (g) and liver (h). i, Circulating levels of adiponectin in control and Mac-PPARγ KO mice.
Mitochondrial dysfunction in muscle is associated with the onset of insulin resistance and type 2 diabetes21-24, prompting us to examine mitochondrial gene expression in skeletal muscle of obese mice. Q-PCR analyses showed that mRNAs encoding key enzymes in fatty acid oxidation (Cpt1b, Acox1) and oxidative phosphorylation (Ndufs1, Sdh, Atp5j and Atp5b) were reduced by 30-70% in quadriceps muscles of Mac-PPARγ KO mice (Fig. 4g). Moreover, expression level of transcription factors and coactivator proteins controlling mitochondrial biogenesis25,26, including Tfam, Nrf-1, Pgc-1α and Pgc-1β, was also reduced by 35-75% in the quadriceps of Mac-PPARγ KO mice (Fig. 4g). Similarly, expression of genes in the electron transport chain and their transcriptional regulators was reduced by 30-60% in livers of Mac-PPARγ KO mice (Fig. 4h). Nonetheless, livers of Mac-PPARγ KO mice did not display any overt signs of steatosis (Supplementary Fig. S6c, d), potentially reflecting their intact capacity to undergo β-oxidation (Supplementary Fig. S6e). These findings suggest that direct or indirect effects of alternatively activated macrophages are important in maintaining oxidative capacity in skeletal muscle and liver. Because we did not observe significant differences in macrophage content or activation in liver and skeletal muscle of control and Mac-PPARγ KO mice (Supplementary Fig. S6a, b and data not shown), we focused on factors secreted by adipocytes that modulate oxidative metabolism in peripheral tissues19,27. Notably, recent studies have shown that adiponectin, a hormone specifically secreted by adipocytes, can induce PGC-1α and mitochondrial biogenesis in skeletal muscle28. Consistent with the global reduction in adipocyte function (Fig. 3c) and skeletal muscle oxidative capacity (Fig. 4g), circulating level of adiponectin was reduced by ~18% in Mac-PPARγ KO mice (Fig. 4i). In contrast, serum levels of resistin, total cholesterol and triglycerides were similar in both strains of mice (Supplementary Table S1).
In summary, the requirement for PPARγ in expression of the alternatively activated phenotype, the absence of alternatively activated ATMs in Mac-PPARγ KO mice, and the observation that Mac-PPARγ KO mice are more susceptible to obesity and insulin resistance suggest that this program of macrophage activation protects against the metabolic sequelae of obesity. In this setting of excess caloric intake, homeostatic functions performed by alternatively activated ATMs might allow animals to more efficiently store and oxidize incoming lipids, thereby maintaining insulin sensitivity and glucose tolerance. However, our findings raise additional issues that will require further investigation. First, to validate whether alternatively activated macrophages can be therapeutically exploited to treat type 2 diabetes, genetic and pharmacologic approaches that activate or inhibit IL-4 signaling in macrophages need to be tested. Second, although the presence of alternatively activated ATMs attenuates inflammation, we can not exclude the possibility that remodelling activities of these cells might also lead to improvement in adipose tissue function. Lastly, in addition to their paracrine effects, it is plausible that peptides or lipids secreted by alternatively activated ATMs act in an endocrine fashion to modulate peripheral insulin sensitivity.
Methods summary
Detailed protocols utilized in this manuscript are provided in the online Methods section. For generation of Mac-PPARγ KO mice, we backcrossed the PPARγfl/+ mice, generated by the Gonzalez laboratory29, for 10 generations onto the Th2-permissive Balb/c background. To generate mice in which the PPARγ genes were disrupted in macrophages, PPARγfl/fl Balb/c mice were mated with LysMCre mice, also on the Balb/c background14. Cohorts of PPARγfl/fl (control) and PPARγfl/fl; LysMCre (Mac-PPARγ KO) mice were produced by intercrossing PPARγfl/fl and PPARγfl/fl;LysMCre animals. Mice were genotyped for the presence of floxed and deleted alleles as described previously29, and littermates were used to assemble the cohorts utilized in these studies.
Methods
Functional analysis for alternative macrophage activation
Bone marrow-derived macrophages (BMDM) from control, Mac-PPARγ KO and PPARδ -/- (on the 129Sv background) mice were cultured as described in previously13. To promote alternative macrophage activation, BMDM were stimulated with IL-4 (10 ng/ml) for 24-96 hours. Arginase activity was monitored via a colorimetric assay that detects the production of urea15. Fatty acid oxidation rates in macrophages were quantified using the sodium hydroxide trap in a modified tissue culture flask13. Cellular mitochondrial content was quantified using MitoTracker dyes (Molecular Probes), as per manufacturer’s instructions. All assays were performed in duplicate or triplicate and repeated at least three independent times, and total cell number or protein content was used for normalization of the data.
Gel mobility shift assays and transient transfections
In vitro translated proteins (TNT kit, Promega) and 32P end-labeled oligonucleotides were used to carry out the gel mobility shift assays. Competition assays were performed with excess unlabeled oligonucleotides. The acyl-CoA oxidase (AOx)-PPRE served as a positive control for these studies. The sequence of arginase I PPRE is AAGTCA G AGAGCA. Transient transfection experiments were performed in RAW 264.7 cells. Briefly, cells were electroporated via the GenePulser II (BioRad) at 300V, 1000 μF, and allowed to recover overnight. Luciferase assays were performed 16-20 hours after stimulation with IL-4 (10 ng/ml) or rosiglitazone (1 μM) with the Dual-Luciferase reporter assay system (Promega). phRL-null was used as an internal control to monitor transfection efficiency. All transfections were done in triplicate and repeated at least three times.
Immunoblots, ELISAs and immunohistochemistry
Total cellular proteins were immunoblotted for cytochrome C (1:1000, BD Pharmingen), VDAC1 (1:2000, Molecular Probes), and β-actin (1:5000, Sigma). IR™Dye 800 conjugated secondary antibodies (1:30000, Rockland) were used for protein detection. For ELISAs, macrophages were stimulated with cytokines or LPS for 6-24 hours, and cytokine secretion was quantified by ELISA, per manufacturer’s protocols (BD Pharmingen). All assays were done in triplicate and repeated at least three independent times. Macrophage in the gonadal fat pads were visualized by F4/80 immunostaining and quantified as described previously7.
Gene expression analysis
Trizol reagent (Invitrogen) was used to prepare total RNA from macrophages or tissues. Total RNA (2 μg) was treated with DNase (1U/ml) and reverse transcribed using a first strand cDNA synthesis kit (Marligen). Q-PCR assays were carried out on the DNA Engine Opticon 2 real time PCR detection system. Data was normalized to L32 levels using the ΔΔ C(t) method and is presented as relative transcript levels.
Leishmania infection
Leishmania major (WHOM/IR/-/173) promastigotes were cultured at 27° C in M199 containing 15% FBS, 100U/ml penicillin, 2mM glutamine, 100μM adenine, 5μg/ml hemin, and 40mM HEPES. Stationary phase metacyclic promastigotes (2 × 106) were injected into the left hind footpad18. Footpad thickness was measured weekly using a caliper (Mitutoyo), and reported as the difference in thickness between the infected and uninfected footpads. Footpad necrosis was graded using the following scale: 0- no visible abscesses; 1- one or more visible subcutaneous abscesses; 2- point surface necrosis; 3- surface necrosis < 50% of foot pad; 4- surface necrosis > 50% of foot pad; and 5- death of the foot.
Metabolic studies
Mice were fed the HFD (Bio-Serv F3282) to promote obesity. Oral glucose tolerance tests (1 g/kg) were performed after an overnight fast. For the insulin tolerance test, mice were fasted for 4 hours prior to injection of human regular insulin (0.65 units/kg). Tail blood glucose levels were monitored at 0, 15, 30, 60, and 120 min time points with the Bayer Elite glucometer. For biochemical analysis of insulin signaling, obese mice of both genotypes were injected with insulin (5mU/g) through their inferior vena cava after a 4 hour fast. Liver and quadriceps were isolated 2 and 5 minutes, respectively, after insulin injection. Homogenized proteins were immunoblotted for total AKT and phopho-AKT (S473, Cell Signaling). Serum levels of adipokines (leptin, adiponectin, and resistin), cytokines (TNFα, IL-6, and IL-1β), lipids (total cholesterol, and triglycerides), and insulin in fasted mice was quantified using commercially available kits.
Macrophage-adipocyte co-culture
Macrophage/adipocyte direct co-culture experiments were performed as described previously30. Briefly, 100,000 BMDMs (day 10) were co-incubated with differentiated 3T3-L1 adipocytes (day 8) for 48 hours. Cultures were stimulated with insulin (100 nM), and glucose uptake was monitored using 0.2 mM 2-[3H] deoxyglucose (1μCi/ml) over 5 minutes. Incorporated radioactivity was normalized for protein content, and expressed as fold increase over untreated samples.
Statistical analysis
Data is presented as mean ± SEM, and Student’s t-test (two-tailed distribution, two-sample unequal variance) was used to calculate the P value. Statistical significance is displayed as P < 0.05 (one asterisk) or P < 0.01 (two asterisks).
Supplementary Material
Supplementary Information accompanies the paper on Nature’s website (http://www.nature.com).
Acknowledgments
We thank members of the Chawla lab for valuable comments, and A. Loh and C.H. Lee for critique on the manuscript. We also thank Peter Murray, Jim McKerrow and Owen McGuinness for providing key reagents and technical guidance. This work was supported by grants made available to AC: NIH (DK062386 and HL076746), Astellas Foundation, Takeda Pharmaceuticals North America, Rockefeller Brothers Fund and by Goldman Philanthropic Partnerships. AC is a Charles E. Culpeper Medical Scholar. Support was provided by Stanford MSTP (JIO and ARE), AHA (JIO), HHMI Gilliam fellowship (ARE), NRSA AI066402 (RRRG), and NIH Training Grant AI07290 (LM). All animal care was in accordance with Stanford University’s A-PLAC committee guidelines.
Footnotes
Competing interests statement The authors declare that they have no competing financial interests.
References
- 1.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hotamisligil GS. Inflammation and metabolic disorders. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 3.Weisberg SP, et al. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu H, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–30. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kanda H, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494–505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamei N, et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J Biol Chem. 2006;281:26602–14. doi: 10.1074/jbc.M601284200. [DOI] [PubMed] [Google Scholar]
- 7.Weisberg SP, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. 2005:JCI24335. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–64. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 9.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–84. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 11.Lehrke M, Lazar MA. The many faces of PPARgamma. Cell. 2005;123:993–9. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- 12.Huang JT, et al. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature. 1999;400:378–82. doi: 10.1038/22572. [DOI] [PubMed] [Google Scholar]
- 13.Vats D, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006;4:13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herbert DR, et al. Alternative macrophage activation is essential for survival during schistosomiasis and downmodulates T helper 1 responses and immunopathology. Immunity. 2004;20:623–35. doi: 10.1016/s1074-7613(04)00107-4. [DOI] [PubMed] [Google Scholar]
- 15.Pauleau AL, et al. Enhancer-mediated control of macrophage-specific arginase I expression. J Immunol. 2004;172:7565–73. doi: 10.4049/jimmunol.172.12.7565. [DOI] [PubMed] [Google Scholar]
- 16.Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10:355–361. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 17.Alexander J, Satoskar AR, Russell DG. Leishmania species: models of intracellular parasitism. J Cell Sci. 1999;112(Pt 18):2993–3002. doi: 10.1242/jcs.112.18.2993. [DOI] [PubMed] [Google Scholar]
- 18.Holscher C, Arendse B, Schwegmann A, Myburgh E, Brombacher F. Impairment of alternative macrophage activation delays cutaneous leishmaniasis in nonhealing BALB/c mice. J Immunol. 2006;176:1115–21. doi: 10.4049/jimmunol.176.2.1115. [DOI] [PubMed] [Google Scholar]
- 19.Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature. 2006;444:847–853. doi: 10.1038/nature05483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rutschman R, et al. Cutting edge: Stat6-dependent substrate depletion regulates nitric oxide production. J Immunol. 2001;166:2173–7. doi: 10.4049/jimmunol.166.4.2173. [DOI] [PubMed] [Google Scholar]
- 21.Mootha VK, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–73. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 22.Patti ME, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A. 2003;100:8466–71. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petersen KF, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–2. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vianna CR, et al. Hypomorphic mutation of PGC-1beta causes mitochondrial dysfunction and liver insulin resistance. Cell Metab. 2006;4:453–64. doi: 10.1016/j.cmet.2006.11.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357–68. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- 26.Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metabolism. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 27.Lazar MA. How obesity causes diabetes: not a tall tale. Science. 2005;307:373–5. doi: 10.1126/science.1104342. [DOI] [PubMed] [Google Scholar]
- 28.Civitarese AE, et al. Role of adiponectin in human skeletal muscle bioenergetics. Cell Metab. 2006;4:75–87. doi: 10.1016/j.cmet.2006.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akiyama TE, et al. Conditional disruption of the peroxisome proliferator-activated receptor gamma gene in mice results in lowered expression of ABCA1, ABCG1, and apoE in macrophages and reduced cholesterol efflux. Mol Cell Biol. 2002;22:2607–19. doi: 10.1128/MCB.22.8.2607-2619.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lumeng CN, Deyoung SM, Saltiel AR. Macrophages block insulin action in adipocytes by altering expression of signaling and glucose transport proteins. Am J Physiol Endocrinol Metab. 2007;292:E166–74. doi: 10.1152/ajpendo.00284.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information accompanies the paper on Nature’s website (http://www.nature.com).