

Abstract
IFN-γ production by T cells is pivotal for defense against many pathogens, and the proximal promoter of IFN-γ, −73 to −48 bp upstream of the transcription start site, is essential for its expression. However, transcriptional regulation mechanisms through this promoter in primary human cells remain unclear. We studied the effects of CREB/ATF and AP-1 transcription factors on the proximal promoter of IFN-γ in human T cells stimulated with M. tuberculosis. Using EMSA, supershift assays and promoter pulldown assays, we demonstrated that CREB, ATF-2 and c-Jun, but not cyclic AMP response element modulator, ATF-1 or c-Fos, bind to the proximal promoter of IFN-γ upon stimulation, and coimmunoprecipitation indicated the possibility of interaction among these transcription factors. Chromatin immunoprecipitation confirmed the recruitment of these transcription factors to the IFN-γ proximal promoter in live antigen-activated T cells. Inhibition of ATF-2 activity in T-cells with a dominant-negative ATF-2 peptide or with siRNA markedly reduced the expression of IFN-γ and decreased the expression of CREB and c-Jun. These findings suggest that CREB, ATF-2 and c-Jun are recruited to the IFN-γ proximal promoter, and upregulate IFN-γ transcription in response to microbial antigen. In addition, ATF-2 controls expression of CREB and c-Jun during T-cell activation.
Keywords: IFN-γ, transcription factors, promoter, T cells, tuberculosis
Introduction
Interferon (IFN)-γ, produced primarily by T cells and NK cells in response to microbial infection, plays an important role in protection against many pathogens, including intracellular bacteria such as Mycobacterium tuberculosis (1, 2), viruses (3), fungi (4) and protozoa (5) and in immune surveillance to prevent development of cancer (6). IFN-γ has also been used to treat patients with idiopathic pulmonary fibrosis (7). On the other hand, IFN-γ is associated with inflammatory diseases, including sarcoidosis (8) and multiple sclerosis (9). Although expression of IFN-γ is controlled to some extent by posttranscriptional processes (10), the dominant mechanism for regulating IFN-γ gene expression is through coordinated, cell-specific interactions between transcription factors and regulatory elements of the IFN-γ gene (11).
Among the IFN-γ regulatory elements, the proximal promoter of IFN-γ, located −73 to −48 bp upstream of the transcription start site, is necessary and sufficient for IFN-γ gene expression by activated T cells (12). The proximal promoter of IFN-γ is highly conserved in mammals (13), and methylation of the CpG motif at −53 bp is a major epigenetic regulatory mechanism, that is believed to render the promoter region inaccessible to transcription factor binding in Th2 cells that do not produce IFN-γ, whereas this site is unmethylated in Th1 cells that express IFN-γ (14–17). Despite the central importance of the proximal promoter in controlling IFN-γ transcription, limited information is available on the detailed mechanisms by which transcription factors interact with this promoter. The cAMP response element binding protein (CREB) is known to bind to this promoter, and we found that CREB enhances transcription of IFN-γ by primary human T cells that are stimulated with M. tuberculosis (18, 19). Activating transcription factor (ATF) and activator protein (AP)-1 also bind to the proximal promoter, and glucocorticoids inhibit this binding and reduce IFN-γ gene expression in Jurkat T-cells (13). However, both ATF and AP-1 are composed of several transcription factors and their individual effects on IFN-γ transcription remain unclear. Furthermore, most studies of transcription factors that control IFN-γ expression in T-cells have been performed in cell lines and in transgenic mice, and limited information is available on this subject in primary human T cells.
In the current study, we identified the transcription factors of the CREB/ATF/AP-1 family that bind to the proximal promoter of IFN-γ in primary human T cells during a physiologically relevant response to antigens from M. tuberculosis, an intracellular pathogen for which immunologic control requires IFN-γ mediated responses. We found that CREB, ATF-2 and c-Jun are recruited to bind the proximal promoter of IFN-γ and enhance IFN-γ gene expression in response to antigenic stimulation of T cells. In addition, ATF-2 regulates expression of CREB and c-Jun during T-cell activation.
Materials and Methods
Isolation of peripheral blood lymphocytes and cell culture
Heparinized blood samples were collected from 18 tuberculin skin test-positive subjects and 8 tuberculin skin test-negative subjects, under protocols approved by the institutional review boards of The University of Texas Health Center (Tyler, Texas) and the University of North Texas Health Science Center (Fort Worth, Texas). Cells from tuberculin reactors were used in all studies that involved culture of cells with M. tuberculosis antigens, as cells from tuberculin-negative donors would not respond to M. tuberculosis. Cells from tuberculin-negative donors and tuberculin reactors were used for experiments in which cells were cultured with PMA, or anti-CD3 and anti-CD28.
Peripheral blood mononuclear cells (PBMC) were isolated by Ficoll-Paque (Pharmacia Fine Chemicals, Piscataway, NJ) gradient centrifugation. In some cases, CD3+ T cells were isolated from PBMC by negative selection with the Pan T cell Isolation Kit II (Miltenyi Biotec Inc., Auburn, CA), with purity of >95%, as measured by flow cytometry with a FACSCalibur (BD Biosciences, San Jose, CA), using FITC anti-CD3 monoclonal Abs (eBiosciences, San Diego, CA). Monocytes were prepared by culturing 107 PBMC per well in a 12-well plate in 1 ml RPMI-1640 with 10% pooled human AB serum at 37° C, 5% CO2 for 1 h, followed by washing extensively with RPMI-1640 to remove non-adherent cells. Monocyte purity was >90 %, based on flow cytometry using FITC anti-CD14 monoclonal Abs (eBiosciences).
T-cell clone
For some experiments, we utilized B9, a human CD4+ T-cell clone that recognizes a 10-mer peptide from the N-terminus of the 10 kDa M. tuberculosis culture filtrate protein (CFP1076–85) in the context of HLA-DRB1*0401 (20).
Mycobacterial antigen and reagents to activate T cells
Heat-killed M. tuberculosis Erdman, provided by Dr. Patrick Brennan, Colorado State University, was used to stimulate PBMC or CD3+ cells and autologous monocytes. To stimulate the human T cell clone, we cultured Bare Lymphocyte Syndrome cells expressing HLA DRB1*04, the clone and its cognate peptide, CFP1076–85. Monoclonal Abs to CD3 (OKT3, Ortho Biotechnology, Raritan, NJ) and CD28 (BD Biosciences), and phorbol myristate acetate (PMA) and ionomycin (both from Sigma, Saint Louis, MO) were used to stimulate purified CD3+ cells.
Preparation of nuclear and whole cell protein extracts
Nuclear protein extracts and whole cell protein extracts of PBMC, CD3+ cells and T cell clones were prepared as described previously (18, 19, 21), quantified by bicinchoninic acid assay (BCA, Pierce Biotechnology, Rockford, IL), aliquoted and stored at −70°C until use.
Electrophoretic mobility shift assay and super-shift assay
To measure binding of transcription factors in nuclear protein extracts to the IFN-γ proximal promoter (−48 to −73 bp), electrophoretic mobility shift assays (EMSA) were performed as described previously, using [γ-32P] dATP-labeled proximal promoter of IFN-γ as a probe (21, 22). For EMSA competition assays, the nuclear protein extracts were incubated with labeled probe on ice for 30 min with 50 molar excess of unlabeled DNA oligonucleotides of IFN-γ proximal promoter, wild-type CREB consensus binding site (wt-CRE), mutant CREB consensus binding site (mt-CRE) and NF-κB consensus binding site, respectively. For supershift assays, control immunoglobulin (Ig)G, or Abs to CREB, cyclic AMP response element modulator (CREM), ATF-2, c-Jun, c-Fos, JunB (all from Santa Cruz Biotechnologies, San Jose, CA), and ATF-1 (Novus Biologicals, Inc., Littleton, CO) were incubated with nuclear protein extracts on ice for 30 min, then incubated with labeled probe for 25 min at room temperature. The DNA-protein complexes were resolved by electrophoresis on a 5% non-denaturing polyacrylamide gel and detected by autoradiography.
Co-immunoprecipitation and Western blotting
Co-immunoprecipitation was performed as previously described (22). Briefly, 200 μg of nuclear protein extracts were precleared by incubation with 30 μl of a 50% slurry of protein G-sepharose beads (Sigma) in a total volume of 250 μl, reconstituted with co-precipitation buffer (0.1% Triton X-100, 100 mM NaCl, 15 mM EGTA, phenylmethanesulfonyl fluoride and a proteinase inhibitor cocktail) at 4°C for 1 h with rotation. After centrifugation at 1800g for 3 min, the supernatants were removed and incubated with 2 μg of Abs to CREB, ATF-2 or c-Jun, at 4°C overnight with rotation. Thirty μl of a 50 % slurry of protein G-sepharose beads was added and incubated at room temperature for 1 h with rotation. The beads were washed with wash buffer (0.1 % Triton X-100, 50 mM Tris-Cl, pH 7.4, 300 mM NaCl, 5 mM EDTA) three times. SDS-PAGE sample loading buffer was added to the beads, and the samples were boiled for 5 min. Proteins were separated by SDS-PAGE, followed by Western blotting with relevant Abs, and the protein bands were visualized by chemiluminescence (GE Health Care, Buckinghamshire, United Kingdom).
Promoter pull-down assay
Promoter pull-down assays to detect proteins bound to the proximal promoter of IFN-γ were performed according to published methods (23–25). Briefly, chemically synthesized wild type or mutant proximal IFN-γ promoter sequences (−71 to −40 bp) were biotinylated at the 5′ end of the sense strand and annealed to the anti-sense sequence. The wild type and mutant biotinylated IFN-γ proximal promoter sequences were 5′Bio-AAA ACT TGT GAA AAT ACG TAA TCC TCA GGA GA-3′ and 5′Bio-AAA ACT TGT GAA AAT CGC TAA TCC TCA GGA GA-3,′ respectively. Two μg of biotinylated dsDNA was conjugated to 100 μl of Ultra Link immobilized streptavidin gel (Pierce Biotechnology) in binding/washing buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 0.1 M NaCl) for 45 min at room temperature with rotation. Free unconjugated DNA was removed by extensive washing with binding-washing buffer. DNA-conjugated beads were blocked with 0.5% BSA and 5 μg/ml sheared herring sperm DNA in TGEDN buffer (120 mM Tris-HCl, pH 8.0, 1 mM EDTA, 0.1 M NaCl, 1 mM DTT, 0.1% Triton X-100, 10% glycerol) at room temperature for 1 h. The beads were then washed three times with TGDEN buffer and resuspended in 100 μl of the same buffer. Thirty μl of these beads were incubated with 250 μg of nuclear protein extracts and 5 μg/ml of sheared herring sperm DNA in 250 μl at 4 °C for 2 h with rotation. The beads were washed 3 times with TGEDN buffer and bound proteins were eluted with the same buffer, supplemented with 0.5 % SDS and 1 M NaCl. Eluted proteins were resolved by 10% SDS-PAGE, and proteins pulled down by the proximal promoter of IFN-γ were analyzed by immunoblotting with specific Abs for relevant transcription factors. To evaluate the specificity of binding, a 5-fold excess of non-biotinylated proximal promoter of IFN-γ, or non-biotinylated wild-type CREB consensus binding site (wt-CRE, 5′-AGAGATTGCCTGACGTCAGAGAGCTAG-3′) or mutant CREB consensus binding site (mt-CRE, 5′-AGAGATTGCCTGTGGTCAGAGAGCTAG-3′) was added to some of the reaction mixtures.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed as described previously (19). Briefly, M. tuberculosis-stimulated or unstimulated PBMC were treated with formaldehyde, followed by glycine neutralization, DNA shearing by microtip sonication, and preparation of chromatin supernatants. Abs to CREB, ATF-1, ATF-2, CREM and c-Jun were used to immunoprecipitate chromatin supernatants, from which DNA was purified and amplified by PCR with relevant primer sets to detect specific DNA sequences. PCR products were analyzed by electrophoresis on 1.5% agarose gels, and visualized with ethidium bromide. The following primer pairs were used (19, 26): IFN-γ forward, 5′-AATGCCACAAAACCTTAGTTATTAA-3′, IFN-γ reverse, 5′-ACTTAACTGATCTTTCTCTTCTAAT-3′; c-Fos forward, 5′-TCCTACATGCCGAGGTCCAGGAGAC-3′; c-Fos reverse, 5′-GAGTAGTAGGCGCCTCAGCTGGCCG-3′, rRNA forward, 5′-TTGAAAATCCGGGGGAGAG-3′; rRNA reverse, 5′-ACATTGTTCCAACATGCCAG-3′.
Nucleofection of T cells with an ATF-2 peptide-expressing plasmid, followed by stimulation with heat-killed M. tuberculosis
Negatively selected CD3+ T cells from PBMC of tuberculin-positive subjects were nucleofected with an empty pcDNA3 plasmid or pcDNA3 containing an insert encoding a 50 amino acid-ATF-2 peptide corresponding to amino acids 50–100 of the N-terminal domain (27), using the Human T Cell Nucleofector Kit (Amaxa Biosystems, Cologne, Germany), as previously described (19). Forty-eight h after nucleofection, the CD3+ cells were counted, and 2×105 cells were cultured with 5×104 autologous macrophages in 200 μl of RPMI with 10% human serum in a 96-well flat-bottom plate. Heat-killed M. tuberculosis (2.5 μg/ml) was added to some wells, and the cells were cultured at 37°C in 5% CO2. After an additional 72 h, supernatants were collected and stored at −70°C for measurement of IFN-γ levels. The cells were collected, washed, lysed with SDS-PAGE sample loading buffer and boiled for 10 min. Proteins were separated by SDS-PAGE, and expression of ATF-2, CREB, c-Jun, and β-actin was determined by Western blotting.
Introduction of ATF-2 siRNA into CD3+ T cells
Small interfering RNA (siRNA) for ATF-2 was generated, as described (19), using the Dicer siRNA kit (Gene Therapy Systems). Briefly, total RNA of CD3+ cells was reverse transcribed to cDNA with random hexamers, and ATF-2 cDNA was amplified by PCR with specific primers. The expected product was confirmed by sequencing, and reamplified with T7 primer-tagged ATF-2 primers (forward: 5′-TAATACGACTCACTATAGGGAGATCGTCCAGCATCATTACAGG-3, reverse: 5′-TAATACGACTCACTATAGGGAGAAAGACTGAACCCAGACTTTC-3′). This yielded a 647-bp product of ATF-2752–1399. ATF-2 RNA was then generated by in vitro transcription with T7 RNA polymerase. The RNA was digested with Dicer at 37°C for 16 h, siRNA was purified with spin columns, and the purity and size of siRNA was confirmed by electrophoresis on 3% agarose gels. GFP siRNA was generated in parallel, using a GFP expression plasmid as PCR template for GFP DNA and T7-tagged primers for the GFP gene.
Nucleofection of negatively selected peripheral blood CD3+ cells with siRNA was performed, as described above for the plasmid expressing the ATF-2 peptide. Forty eight h post-nucleofection, the cells were collected, counted and cultured with autologous monocyte-derived macrophages at a ratio of four T cells (2 × 105) to 1 macrophage (5 ×104), with or without 2.5 μg/ml of heat-killed M. tuberculosis in a 96-well flat-bottom plate in triplicate. After 72 h of additional incubation, supernatants were collected for measurement of IFN-γ, and the cells were collected for detection ATF-2, CREB, c-Jun, and β-actin by Western blotting.
Measurement of IFN-γ concentrations
IFN-γ levels in supernatants were measured by ELISA (BD Pharmingen, San Diego, CA). The lower limit of detection was 5 pg/ml.
Measurement of IFN-γ mRNA by real-time PCR
To measure IFN-γ mRNA levels, total cellular RNA was extracted with TRIzol LS reagent (Invitrogen Life Technologies, Gaithersburg, MD) and 250 ng of total RNA was treated with DNAase I and reverse transcribed, as described previously (18). Expression of IFN-γ and 18S rRNA was measured by real-time PCR, using primers and probes (Applied Biosystems, Foster City, CA). Reactions were performed with the ABI Prism 7700 Sequence Detection System (Applied Biosystems). Expression of IFN-γ mRNA was calculated using the ΔCt method, after normalization for 18S rRNA, and expressed as a ratio of M. tuberculosis-stimulated cells/cells cultured in medium alone: ΔCt=CtIFN-γ −Ct18srRNA; ΔΔCt sample= ΔCtsample− ΔCt IFNγ medium alone; relative expression = 2 −ΔΔCt sample.
Results
Binding of CREB/ATF/AP-1 transcription factors to the IFN-γ proximal promoter, using EMSA, supershift assays and promoter pull-down assays
We previously showed that stimulation of PBMC with heat-killed M. tuberculosis yields increased binding of a nuclear protein complex containing CREB to the IFN-γ proximal promoter (19). To elucidate the composition of these complexes, we performed EMSA with a labeled IFN-γ proximal promoter and nuclear protein extracts of PBMC from healthy tuberculin reactors that had been cultured in medium alone or stimulated with heat-killed M. tuberculosis (Fig. 1A). Baseline levels of DNA-binding protein complexes were present in unstimulated cells, and stimulation with M. tuberculosis induced increased binding of two major protein complexes to the IFN-γ proximal promoter (designated A and B for the slower and faster mobility complexes, respectively). Binding of these protein complexes after stimulation was paralleled by increased expression of mRNA and maximal secretion of IFN-γ (data not shown). The specificity of this binding was evaluated by competitive EMSA. Binding of these two major complexes was abrogated by excess unlabeled IFN-γ proximal promoter and wild-type CRE, but not by the mutated CRE or NF-κB binding site (Fig. 1B). Similar results were observed when CD3+ cells were stimulated with either PMA and ionomycin, or anti-CD3 plus anti-CD28 (data not shown), confirming the existence of these IFN-γ proximal promoter binding protein complexes in purified primary human T cells upon stimulation.
Figure 1. Binding of CREB/ATF/AP-1 transcription factors to the proximal promoter of IFN-γ.

A. Antigen-induced binding of protein complexes to the proximal promoter of IFN-γ. EMSA was performed using the [γ-32P]ATP-labeled proximal promoter of IFN-γ and nuclear protein extracts of PBMC from 8 healthy tuberculin reactors, cultured with medium alone for 48 h or in the presence of 2.5 μg/ml of heat-killed M. tuberculosis (TB) for 48. Specific bands are indicated by arrows. Three representative results are shown. B. Specific binding of the protein complexes to the proximal promoter of IFN-γ. EMSA was performed as described in panel A, with nuclear protein extracts of PBMC from 6 healthy tuberculin reactors, cultured in the presence of 2.5 μg/ml of heat-killed M. tuberculosis for 48 h and labeled proximal promoter of IFN-γ, in the absence or presence of unlabeled oligonucleotides representing the IFN-γ proximal promoter, the NF-κB consensus binding site, the wild-type CREB consensus binding site and the mutant CREB consensus binding site, as indicated. A representative result is shown. C. Supershift analysis of the protein complexes binding to the proximal promoter of IFN-γ with specific Abs against CREB/ATF/AP-1 transcription factors. EMSA was performed as described in panel A, with nuclear protein extracts from 5 healthy tuberculin reactors, cultured with 2.5 μg/ml of heat-killed M. tuberculosis for 48 h and labeled proximal promoter of IFN-γ, in the absence or presence of the Abs indicated. A representative result is shown.
To dissect the protein composition of these complexes, supershift assays were performed with the labeled IFN-γ proximal promoter as a probe and nuclear protein extracts of M. tuberculosis-stimulated PBMC from healthy tuberculin reactors, using Abs to CREB/ATF/AP-1 transcription factors (Fig. 1C). Although the effects of these Abs on protein complexes from different individuals was variable, anti-CREB consistently reduced the intensity of complex B anti-ATF-1 reduced the intensity of complex A, and anti-ATF-2 supershifted both complexes. The effects of the other antibodies were not clear.
To use a more definitive alternative technique to identify CREB/ATF/AP-1 transcription factors that bind to the IFN-γ proximal promoter, we used a promoter pull-down assay with a biotinylated IFN-γ proximal promoter, followed by Western blotting for relevant transcription factors. To optimize experimental conditions, we incubated nuclear protein extracts of M. tuberculosis-stimulated PBMC from a healthy tuberculin reactor with a biotinylated IFN-γ proximal promoter, immobilized on beads through conjugation to avidin. The proteins that bind to the beads were eluted and immunoblotted for CREB, since it is known to bind to the IFN-γ proximal promoter. We found that CREB binds to the proximal promoter in a stimulation-dependent manner, recruitment of CREB increased over time until 72 h (Fig. 2A), and binding was reduced by excess soluble proximal promoter and the wildtype CRE, but not by mutant CRE (Fig. 2B), indicating the specificity of binding. Having optimized experimental conditions, we then used the promoter pull-down assay to identify transcription factors that bind to the IFN-γ proximal promoter, using nuclear extracts from healthy tuberculin reactors. Western blotting demonstrated that CREB, ATF-2 and c-Jun bind to the proximal promoter of IFN-γ in a stimulation-dependent manner. In addition, these transcription factors failed to bind to a probe with changes in 4 nucleotides that are essential for IFN-γ proximal promoter activity, further demonstrating the specificity of the transcription factor/IFN-γ proximal promoter interactions in this system. The promoter pull-down assay showed no binding of ATF-1, CREM and c-Fos to the IFN-γ proximal promoter (Fig. 2C), although these Abs detected their respective proteins in nuclear extracts in standard Western blotting assays (data not shown).
Figure 2. Recruitment of CREB, ATF-2 and c-Jun to the proximal promoter of IFN-γ.
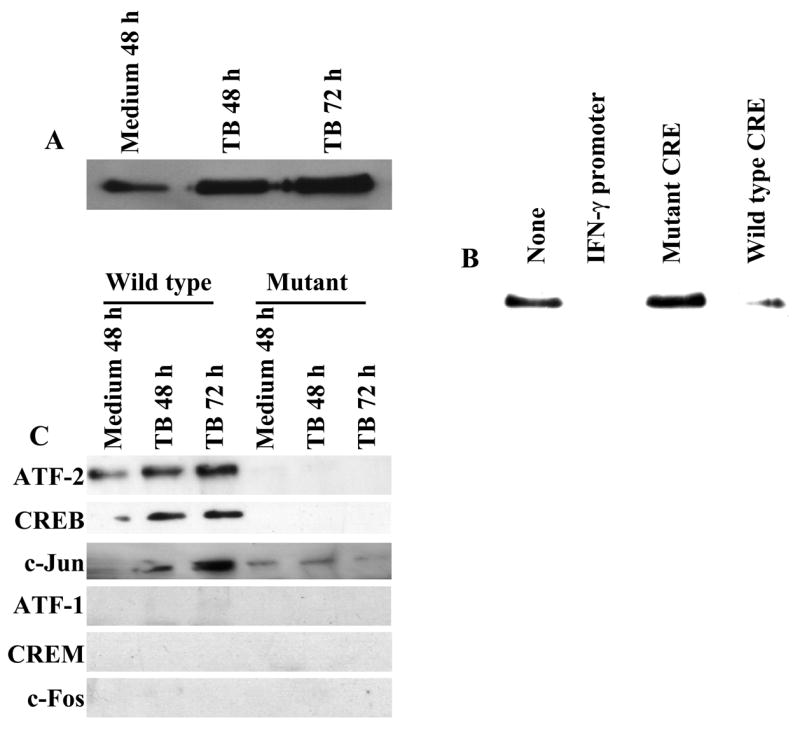
A. Antigen stimulation increases binding of CREB to the proximal promoter of IFN-γ. Promoter pull-down assays were performed with a 5′-biotinylated proximal promoter of IFN-γ and nuclear protein extracts of PBMC from three healthy tuberculin reactors, cultured in medium alone or with 2.5 μg/ml of heat-killed M. tuberculosis (TB). The eluents were subjected to immunoblotting with anti-CREB. A representative result is shown. B. Specific binding of CREB to the proximal promoter of IFN-γ. Promoter pull-down assays were performed, as described in panel A, with nuclear protein extracts of PBMC from three healthy tuberculin reactors, cultured with 2.5 μg/ml of heat-killed M. tuberculosis for 48 h with the 5′-biotinylated IFN-γ proximal promoter, either alone or with excess unlabeled proximal promoter of IFN-γ, mutated CRE or wild type CRE. The eluents were subjected to immunoblotting for CREB. A representative result is shown. C. Recruitment of CREB, ATF-2 and c-Jun to the proximal promoter of IFN-γ. Promoter pull-down assays were performed, as described in panel A, using a 5′-biotinylated wild type or mutant IFN-γ proximal promoter, and nuclear protein extracts of PBMC from 6 healthy tuberculin reactors, cultured with medium alone or 2.5 μg/ml heat-killed M. tuberculosis (TB). The eluents were blotted for transcription factors as indicated. A representative result is shown.
In summary, both EMSAs and promoter pull-down assays showed that CREB, ATF-2 and c-Jun in human T-cells bind to the proximal promoter of IFN-γ, and that binding is enhanced by antigenic stimulation.
Antigen-induced recruitment of CREB, ATF-2 and c-Jun to the proximal promoter of IFN-γ in live cells
EMSAs and promoter pull-down assays demonstrated binding of transcription factors to a double stranded DNA sequence in vitro. To determine if CREB, ATF-2 and c-Jun are recruited to bind to the IFN-γ proximal promoter ex vivo, we performed chromatin immunoprecipitation on chromatin supernatants from M. tuberculosis-stimulated PBMC from 5 healthy tuberculin reactors. CREB, ATF-2 and c-Jun were recruited to the proximal promoter of IFN-γ in live cells, but CREM and ATF-1 were not (Fig. 3A). This was not due to failure of the Abs to ATF-1 and CREM to immunoprecipitate DNA bound to these transcription factors, as these Abs immunoprecipitated the c-Fos promoter, to which these transcription factors are known to bind (26). As a specificity control, PCR for the rRNA promoter, which does not have a CRE consensus site, yielded negative results after immunoprecipitation with all Abs. Next, we determined if recruitment of transcription factors to the proximal promoter of IFN-γ in live cells is stimulation-dependent. Chromatin supernatants from unstimulated and M. tuberculosis stimulated PBMC from 3 healthy tuberculin reactors were immunoprecipated with Abs against CREB, ATF-2 and c-Jun. As shown in Fig 3B, CREB, ATF-2 and c-Jun were recruited to the proximal promoter of IFN-γ in antigen-stimulated PBMC but not in PBMC cultured in medium alone. Taken together with the results of EMSAs and promoter pull-down assays, these results indicate that CREB, ATF-2 and c-Jun were recruited and bind to the proximal promoter of IFN-γ in live antigen-stimulated cells.
Figure 3. Recruitment of CREB, ATF-2 and c-Jun to the proximal promoter of IFN-γ in antigen-stimulated live cells.
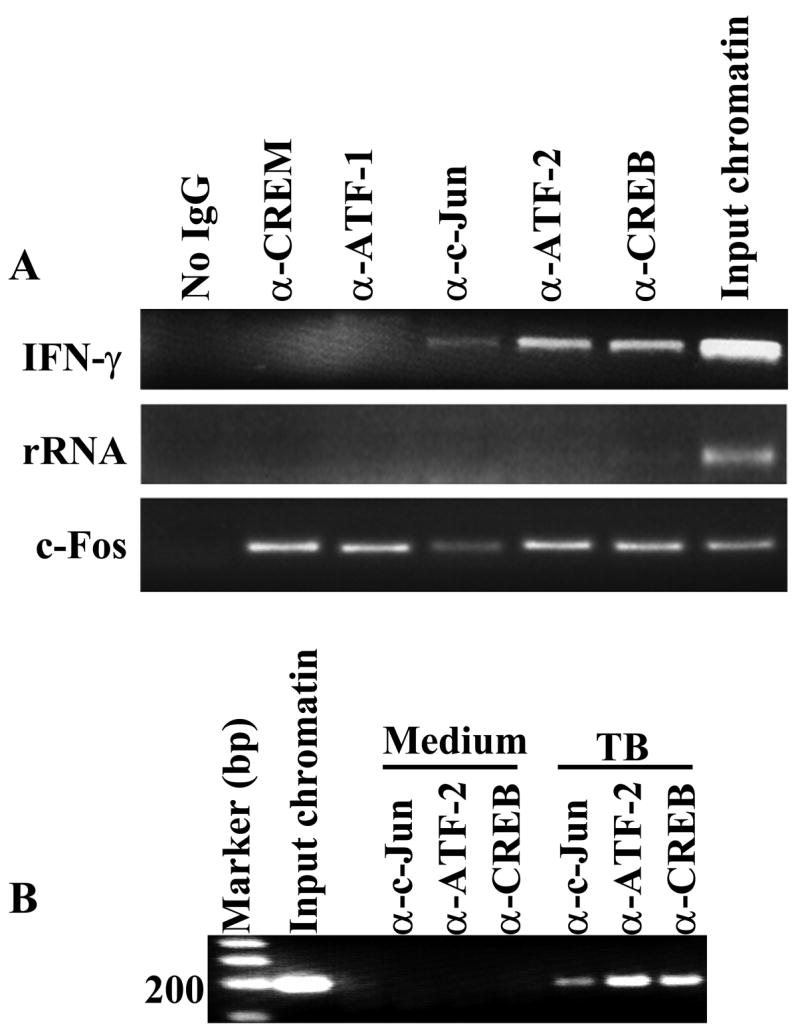
A. Recruitment of CREB, ATF-2 and c-Jun to the proximal promoter of IFN-γ. Chromatin immunoprecipitation was performed on formaldehyde-cross-linked chromatin supernatants of PBMC from five healthy tuberculin reactors, cultured with 2.5 μg/ml of heat-killed M. tuberculosis for 48 h, using Abs against the indicated transcription factors. PCR was performed with primers for the IFN-γ proximal promoter, the rRNA promoter and the c-Fos promoter. A representative result is shown. B. Antigen stimulation-induced recruitment of CREB, ATF-2 and c-Jun to the proximal promoter of IFN-γ in live cells. Chromatin immunoprecipitation was performed on formaldehyde-cross-linked chromatin of PBMC from three healthy tuberculin reactors, cultured in medium alone or with 2.5 μg/ml of heat-killed M. tuberculosis for 48 h, using Abs against the transcription factors indicated. PCR was performed with primers for the IFN-γ proximal promoter. A representative result is shown.
Activation of CREB, ATF-2 and c-Jun by stimulation with M. tuberculosis antigens
CREB/ATF/AP-1 transcription factors are constitutively expressed and become phosphorylated upon cellular activation. Phosphorylation of CREB and ATF-2 increases the affinity for their target gene promoters, and facilitates recruitment of CREB binding protein and P300, transcriptional enhancers with histone acetyltransferase activity, favoring initiation of transcription of the target genes (28). We therefore wished to determine if stimulation of peripheral blood T cells by antigen induces phosphorylation of these transcription factors. Using phospho-specific Abs for CREB, ATF-2 and c-Jun, we evaluated the activation of these transcription factors in PBMC from 8 healthy tuberculin reactors, cultured with heat-killed M. tuberculosis for different periods (Fig. 4). Stimulation of PBMC with M. tuberculosis induced phosphorylation of CREB, ATF-2 and c-Jun after 24 h, with increased expression at 48–72 h. Stimulation with M. tuberculosis markedly increased levels of total c-Jun, and this may account in part for the increase in phosphorylated c-Jun that was observed. When purified peripheral blood CD3+ cells were stimulated with PMA plus ionomycin or anti-CD3 plus anti-CD28, phosphorylation of these same transcription factors was observed, although with much faster kinetics (data not shown).
Figure 4. Stimulation-induced phosphorylation of CREB, ATF-2 and c-Jun.
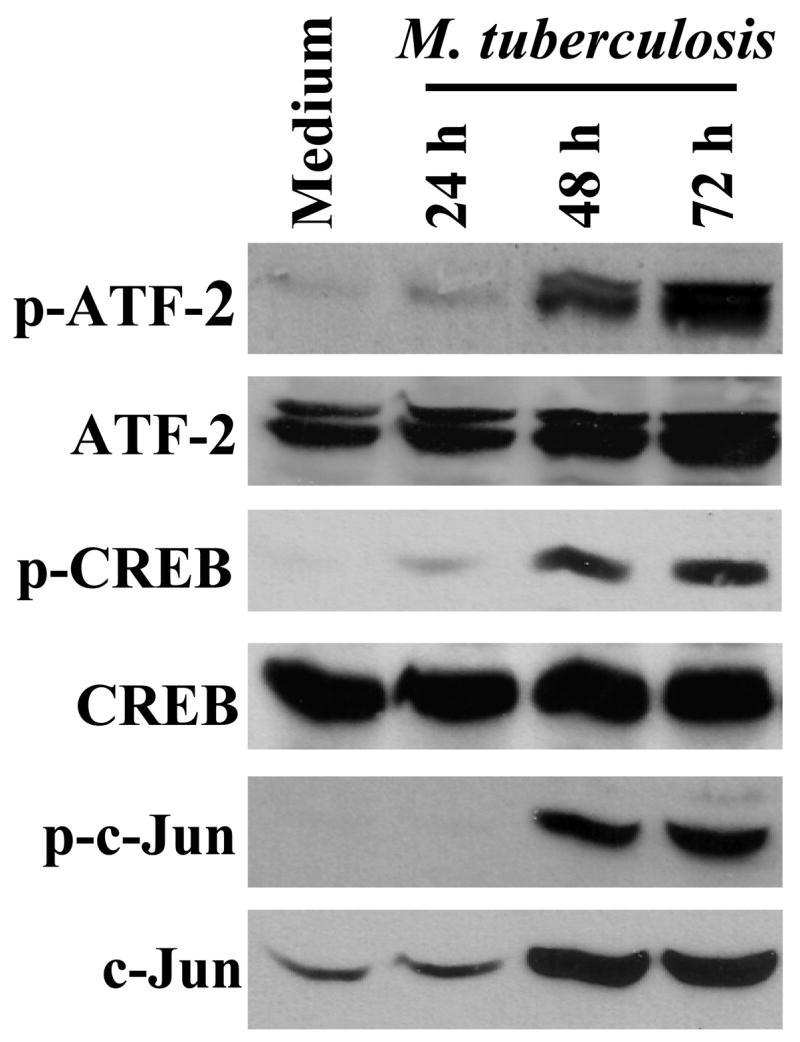
PBMC from 8 healthy tuberculin reactors were cultured with 2.5 μg/ml of heat-killed M. tuberculosis. At the time points indicated, whole cell protein extracts were subjected to Western blotting with Abs to phophorylated CREB, ATF-2 and c-Jun. After stripping, the nitrocellulose membrane was reblotted with Abs to total CREB, ATF-2 and c-Jun. Representative results are shown.
Protein-protein interactions among CREB, ATF-2 and c-Jun in response to antigenic stimulation
Our results demonstrate that T cell activation results in phosphorylation and binding of CREB, ATF-2 and c-Jun AP-1 to the proximal promoter of IFN-γ. Because CREB/ATF and AP-1 transcription factors bind to the CRE sequence by forming homo- or heterodimers (29, 30), we used co-immunoprecipitation to determine if these transcription factors form a complex in cell extracts, and if these interactions are stimulation-dependent. When nuclear extracts from M. tuberculosis-stimulated PBMC from four healthy tuberculin reactors were immunoprecipitated with anti-CREB, both ATF-2 and c-Jun were detected by immunoblotting, and this interaction was more marked in M. tuberculosis-stimulated cells than in those cultured in medium alone (Fig. 5A). Similarly, c-Jun and CREB were detected after immunoprecipitation with anti-ATF-2, suggesting that the interaction of CREB, ATF-2, and c-Jun was stimulation-dependent.
Figure 5. CREB, ATF-2 and c-Jun form a complex in response to T cell activation.

Co-immunoprecipitation was performed using Abs to CREB or ATF-2 and nuclear protein extracts of (A) PBMC from four healthy tuberculin reactors, in the presence or absence of 2.5 μg/ml of heat-killed M. tuberculosis; (B) purified CD3+ cells from three donors cultured with 50 ng/ml PMA plus 1 μM/L ionomycin for different time points; or (C), a human M. tuberculosis-reactive T-cell clone cultured with antigen-presenting Bare Lymphocyte Syndrome cells, cultured with medium alone or with its cognate peptide. The experiment with the T-cell clone was performed twice. For each panel, nuclear extracts were immunoprecipitated with Abs to either ATF-2 or CREB, the immunoprecipitate were resolved by SDS-PAGE and transfered to nitrocellulose. Western blotting was performed with anti-c-Jun Abs. The blots were stripped and reblotted with anti-ATF-2, then stripped again and blotted with anti-CREB. A representative result is shown in each panel.
Since PBMC contain cells other than T-cells, we performed co-immunoprecipitation to study the interaction of these transcription factors in protein extracts of purified CD3+ T cells stimulated with PMA plus ionomycin. CREB, ATF-2 and c-Jun formed a complex upon stimulation and this interaction was increased over 60 min of stimulation (Fig. 5B). To confirm these findings at the level of single T-cells, we cultured a M. tuberculosis-reactive human T cell clone with APCs and either medium alone or its cognate peptide. Nuclear extracts were then prepared, and immunoprecipitation with anti-ATF-2 revealed CREB and c-Jun (Fig. 5C), indicating that these three transcription factors formed a complex.
In summary, the results of coimmunoprecipitation and chromatin immunoprecipitation suggest that, upon activation of T cells, CREB, ATF-2 and c-Jun are phosphorylated and recruited to form part of a protein complex that binds to the IFN-γ proximal promoter.
Effect of neutralization of ATF-2 on M. tuberculosis-stimulated IFN-γ secretion
Our previous work demonstrated that binding of CREB to the IFN-γ proximal promoter enhances M. tuberculosis-induced IFN-γ transcription (19). To determine the physiologic effects of ATF-2 on IFN-γ gene expression, we used a plasmid that expresses the ATF-250–100 peptide, which binds to ATF-2 and behaves as a dominant negative form of ATF-2 by blocking its transcriptional activity in melanoma cells (27). Nucleofection of this plasmid into CD3+ cells from 6 healthy tuberculin reactors that were cultured with autologous monocytes and heat-killed M. tuberculosis reduced IFN-γ secretion in a dose-dependent manner by up to 80%, compared to cells nucleofected with the empty vector (Fig. 6A). Nucleofection of the ATF-250–100 peptide did not affect IFN-γ production by CD3+ cells cultured with monocytes and medium alone, mean IFN-γ levels ranging from 16–30 pg/ml (data not shown). Inhibition of ATF-2 also reduced IFN-γ mRNA expression by up to 99% (Fig. 6B), suggesting that this effect was mediated through transcription of IFN-γ. Nucleofection of CD3+ cells with the plasmid expressing the ATF-250–100 peptide significantly reduced expression, not only of ATF-2, but also of CREB and c-Jun in a dose-dependent manner, compared to cells nucleofected with the empty plasmid (Fig. 6C). To confirm the effect of the ATF-250–100–expressing plasmid with a stably transfected cell line, we used nucleofection to introduce the empty pcDNA3 plasmid or the pcDNA3 plasmid encoding ATF-250–100 into the human HUT-78 T cell line. The cells expressing ATF-250–100 had reduced expression of CREB, c-Jun and ATF-2 (data not shown). These findings indicate that ATF-2 positively regulates antigen-induced IFN-γ transcription and that ATF-2 also controls the expression of CREB and c-Jun.
Figure 6. Effect of inhibiting ATF-2 on expression of IFN-γ, CREB, ATF-2 and c-Jun.
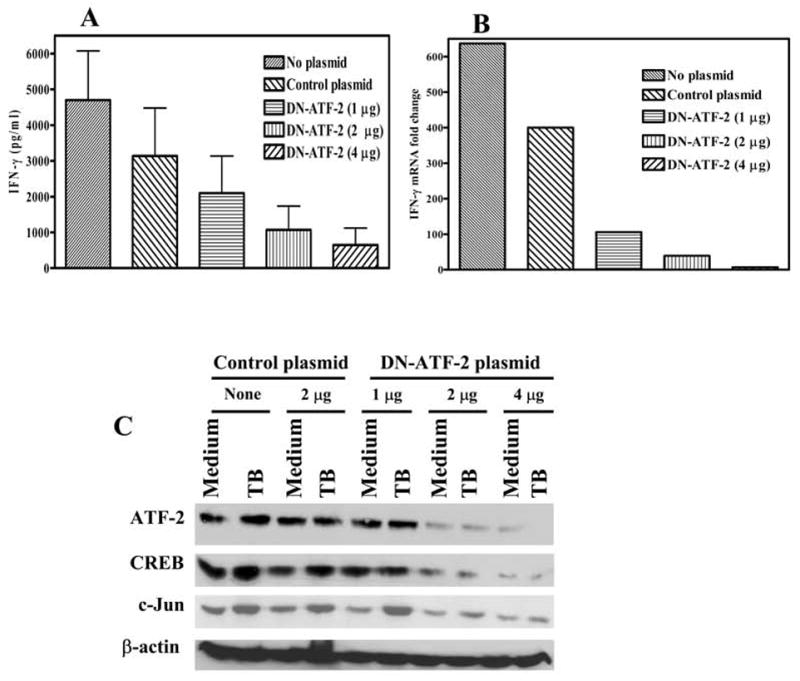
A. IFN-γ production. Purified CD3+ cells from 6 healthy tuberculin reactors were nucleofected with different amounts of the dominant-negative ATF-2 plasmid (DN-ATF-2), a control plasmid (2 μg) or no plasmid. Forty-eight h post-nucleofection, the cells were co-cultured with autologous monocytes in the presence of 2.5 μg/ml of heat-killed M. tuberculosis. After 72 h of incubation, culture supernatants were collected and IFN-γ levels were quantified by ELISA. Mean values and standard errors are shown. B. IFN-γ mRNA expression. Purified CD3+ cells from 3 healthy tuberculin reactors were nucleofected and cultured with autologous monocytes in the absence or presence of heat-killed M. tuberculosis, as in panel A. After 48 h of incubation, cells were collected and mRNA for IFN-γ was quantified by realtime PCR. A representative experiment is shown of the fold-change in IFN-γ mRNA induced by stimulation with M. tuberculosis, relative to medium alone. C. Effect of the dominant-negative ATF-2 peptide on expression of CREB, ATF-2 and c-Jun. Purified CD3+ cells from 6 healthy tuberculin reactors were nucleofected with the plasmids shown, and cultured with autologous monocytes and heat-killed M. tuberculosis, as in panel A. After 72 h of incubation, cells were collected and blotted for ATF-2, CREB, c-Jun and β-actin. A representative result is shown.
Effect of inhibiting ATF-2 in CD3+ T cell with siRNA
Given the unexpected result that ATF-2 may contribute to expression of CREB and c-Jun, we wished to confirm this effect by an alternative method, and used the RNAi technique to inhibit ATF-2 expression in human T cells. We nucleofected CD3+ cells from three healthy tuberculin reactors with ATF-2 siRNA, cultured them with autologous monocytes stimulated with heat-killed M. tuberculosis, and then measured expression of CREB, ATF-2 and c-Jun (Figs. 7A and 7B). Control siRNA to the irrelevant gene encoding GFP did not affect expression of these transcription factors, whereas ATF-2 siRNA reduced expression of CREB and c-Jun by more than 70 %. ATF-2 siRNA also decreased M. tuberculosis -induced secretion of IFN-γ by more than 60 %, whereas the GFP control siRNA had no effect (Fig. 7C). Thus, these findings confirmed our results with the dominant-negative ATF-2 plasmid.
Figure 7. Effect of ATF-2 siRNA on expression of ATF-2, CREB, c-Jun and IFN-γ.

A. Effect of ATF-2 siRNA on expression of transcription factors. Purified CD3+ cells from three healthy tuberculin reactors were nucleofected with siRNA to ATF-2 or to GFP, or no siRNA. Forty-eight h post-nucleofection, the cells were co-cultured with autologous monocytes in the presence of 2.5 μg/ml of heat-killed M. tuberculosis for 72 h. Cells were collected and blotted for ATF-2, CREB, c-Jun and β-actin. A representative result is shown. B. Densitometry analysis of ATF-2, CREB, and c-Jun bands in panel A. For the experiment shown in panel A, band intensity was quantified by densitometry, after normalization for intensity of the β-actin band. C. Effect of ATF-2 siRNA on M. tuberculosis-induced IFN-γ secretion. Cells from three healthy tuberculin reactors were treated as in panel A. After culture of CD3+ cells with autologous monocytes in the presence of M. tuberculosis for 72 h, supernatants were collected and IFN-γ concentrations were measured by ELISA. A representative result is shown.
Discussion
IFN-γ is pivotal for human defenses against M. tuberculosis and other intracellular pathogens, and the proximal promoter of IFN-γ is necessary and sufficient for its transcription in activated T cells (31). Previous studies of the molecules that bind to the IFN-γ proximal promoter have utilized IFN-γ promoter constructs in mitogen-stimulated Jurkat T cells and in transgenic mice (17, 31–34), but it is unclear if these findings reflect events in primary human T cells during a physiologic response to microbial antigen. In the current report, we studied the binding and regulatory effects of CREB/ATF and AP-1 transcription factors on the proximal promoter of IFN-γ in human T cells stimulated with M. tuberculosis antigens. Using EMSAs, supershift assays and promoter pull-down assays, in combination with Western blotting, we demonstrated that CREB, ATF-2 and c-Jun, but not CREM, ATF-1 or c-Fos, bind to the proximal promoter of IFN-γ upon stimulation with M. tuberculosis, and coimmunoprecipitation indicated that these transcription factors form a complex upon stimulation in T cells. Chromatin immunoprecipitation assay confirmed these results in live antigen-activated T cells. Inhibition of ATF-2 by a dominant-negative plasmid or by siRNA reduced M. tuberculosis-induced expression of IFN-γ mRNA and protein, and decreased levels of CREB, ATF-2 and c-Jun. We previously showed that siRNA to CREB reduced expression of IFN-γ, but did not have effect on expression of ATF-2 and c-Jun. These findings suggest that CREB, ATF-2 and c-Jun are recruited to and bind to the proximal promoter of IFN-γ as part of a protein complex, and positively regulate IFN-γ transcription in response to microbial antigen. In addition, ATF-2 controls expression of CREB and c-Jun during T cell activation.
IFN-γ gene regulation is complex, involving multiple enhancer and repressor elements upstream of the transcription start site (12, 31, 33, 35–38) as well as regulatory regions in the introns (32). We focused our study on the proximal (−73 to −48 bp) IFN-γ promoter for several reasons. First, the sequence is highly conserved in mammals, and is necessary and sufficient for transcription of IFN-γ in activated human T-cells (31). Second, activation of T cells through the T-cell receptor and the IL-12 receptor, a process that parallels production of IFN-γ in response to M. tuberculosis and other intracellular pathogens, increased activation of the proximal but not the distal IFN-γ promoter element (33). Finally, methylation is a widespread mechanism used to control mammalian gene expression, and the CpG motif at −53 bp in the proximal promoter of IFN-γ is hypermethylated in non-T cells, naïve T cells and Th2 cells that do not produce IFN-γ upon stimulation, whereas this motif is hypomethylated in Th1 cells that produce IFN-γ (14).
The proximal promoter of IFN-γ contains the sequence ACGT, which is a non-canonical low-affinity CRE half site that forms the central portion of the full high-affinity octamer CRE site, TGACGTCA (39). CRE sites are bound by CREB/ATF and AP-1 transcription factors, and previous studies have shown that CREB, ATF-1, ATF-2 and c-Jun bind to the IFN-γ proximal promoter (13, 31), but the effect of binding has been reported to inhibit or enhance IFN-γ transcription. Studies in Jurkat T cells and transgenic mice suggested that CREB inhibited transcription of IFN-γ (31, 33). In contrast, chromatin immunoprecipitation demonstrated increased recruitment of CREB to the IFN-γ proximal promoter in human Th1 but not Th2 cells (14) and we found that downregulation of CREB by siRNA and intracellular Abs reduced IFN-γ production by primary human T cells in response to M. tuberculosis (19), indicating that CREB enhances IFN-γ production. In addition, some authors found that c-Jun positively regulates transcription of IFN-γ by mitogen-stimulated Jurkat T-cells (31), whereas others suggested that c-Jun does not affect IFN-γ expression, because c-Jun levels were similar in Th1 and Th2 cells (33).
Using several techniques, we found that activation of peripheral blood T cells by M. tuberculosis increased phosphorylation of CREB, ATF-2 and c-Jun. Coimmunoprecipation demonstrated that these three transcription factors interact with each other in stimulated T cells, and chromatin immunoprecipitation showed that these same transcription factors bind to the IFN-γ proximal promoter in live antigen-stimulated T cells. This combination of findings strongly suggests that CREB, ATF-2 and c-Jun bind as part of a complex to the IFN-γ proximal promoter during T cell activation. Our conclusions are supported by recent work demonstrating that hypermethylation of the proximal promoter of IFN-γ inhibits binding by CREB, ATF-2 and c-Jun in nuclear extracts of a Th1 cell line (14). Furthermore, the current study and our previous work (16) show that ATF-2 and CREB are both required for optimal transcription of IFN-γ, and that ATF-2 controls expression of CREB and c-Jun in primary human T-cells.
Transcription of IFN-β, IL-2 and TNF-α are controlled through an enhanceosome, consisting of a complex of transcription factors that directly bind DNA and transcriptional coactivators that form a scaffold to stabilize interactions between DNA-binding proteins and the transcriptional machinery, such as RNA polymerase. These coactivators can also acetylate histones at the site of transcript initiation, opening up the chromatin structure and facilitating transcription, as has been demonstrated for expression of IFN-γ during Th1 development (40, 41). We speculate that CREB, ATF-2 and c-Jun form at least part of such a complex that increases the stability of binding to the IFN-γ proximal promoter and facilitates recruitment of transcriptional co-activators, such as CREB binding protein and p300, that enhance efficient transcription of IFN-γ. Our results are consistent with genome-wide promoter analysis which demonstrated that gene transcription is not generally activated by phosphorylated CREB and transcriptional coactivators, and that additional CREB-associated partners are required to mediate this process (42). This is especially important during interactions with non-canonical CRE regulatory elements, such as that present in the IFN-γ proximal promoter.
We found that activation of T cells with M. tuberculosis induced phosphorylation of CREB, ATF-2 and c-Jun. Previous studies demonstrated that Ser133 phoshorylation does not affect binding of CREB to the high-affinity full CRE site, but increased binding of CREB to the non-canonical low-affinity CRE site in the IL-2 Rα promoter (43). We speculate that phosphorylation of CREB may similarly enhance binding to the low-affinity CRE site in the IFN-γ proximal promoter. When T cells were stimulated with M. tuberculosis, reduction of ATF-2 expression by a dominant-negative plasmid or by siRNA inhibited expression of protein for CREB and c-Jun, whereas CREB siRNA did not affect levels of ATF-2 or c-Jun (19). The distal AP-1 binding site of the c-Jun promoter binds heterodimers of ATF-2 and c-Jun but not CREB (44). In addition, ATF-2 regulates c-Jun expression in a teratocarcinoma cell line (41) and in murine thymocytes (45), whereas heterodimers of CREB and c-Jun do not activate transcription of target genes (29). These published data and our current findings suggest that ATF-2 and c-Jun heterodimers may increase c-Jun mRNA expression in T-cells that respond to M. tuberculosis, whereas CREB does not.
Although we are not aware of published data directly demonstrating that heterodimers of ATF-2 and c-Jun bind to the CREB promoter, the latter contains CRE sites (46). Our results suggest that ATF-2 positively regulates transcription of CREB or increases stability of CREB mRNA and/or protein. Further studies are needed to address this question.
In summary, we found that CREB, ATF-2 and c-Jun are recruited to the proximal promoter of IFN-γ and regulate IFN-γ transcription in response to microbial antigen. In addition, ATF-2 enhances expression of CREB and c-Jun through transcriptional or post-transcriptional mechanisms.
Acknowledgments
We thank Dr. Ze’ev Ronai for providing the ATF-2 dominant-negative peptide and for critical comments on an early draft of the manuscript, Dr. Patrick Brennan for provision of heat-killed M. tuberculosis Erdman, and Ortho Biotech for providing anti-CD3 monoclonal Abs.
Footnotes
This work was supported by the National Institute of Health (A1063514), and the Margaret E. Byers Cain Chair for Tuberculosis Research (P. F. Barnes).
Abbreviations used in this paper: CREB, cyclic adenosine monophosphate response element binding protein; ATF, activating transcription factor; CREM, cyclic adenosine monophosphate response element modulator; CRE, cyclic adenosine monophosphate response element; AP-1, activating protein 1.
References
- 1.Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J Exp Med. 1993;178:2243–2247. doi: 10.1084/jem.178.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med. 1993;178:2249–2254. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossol S, Marinos G, Carucci P, Singer MV, Williams R, Naoumov NV. Interleukin-12 induction of Th1 cytokines is important for viral clearance in chronic hepatitis B. J Clin Iinvest. 1997;99:3025–3033. doi: 10.1172/JCI119498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou P, Sieve MC, Bennett J, Kwon-Chung KJ, Tewari RP, Gazzinelli RT, Sher A, Seder RA. IL-12 prevents mortality in mice infected with Histoplasma capsulatum through induction of IFN-gamma. J Immunol. 1995;155:785–795. [PubMed] [Google Scholar]
- 5.Heinzel FP, Sadick MD, Holaday BJ, Coffman RL, Locksley RM. Reciprocal expression of interferon gamma or interleukin 4 during the resolution or progression of murine leishmaniasis. Evidence for expansion of distinct helper T cell subsets. J Exp Med. 1989;169:59–72. doi: 10.1084/jem.169.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nature Rev. 2006;6:836–848. doi: 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- 7.Bajwa EK, Ayas NT, Schulzer M, Mak E, Ryu JH, Malhostra A. Interferon-gamma1b therapy in idiopathic pulmonary fibrosis.: a metaanalysis. Chest. 2005;128:203–206. doi: 10.1378/chest.128.1.203. [DOI] [PubMed] [Google Scholar]
- 8.Shigehara K, Shijubo N, Ohmichi M, Takahashi R, Kon S, Okamura H, Kurimoto M, Hiraga Y, Tatsuno T, Abe S, et al. IL-12 and IL-18 are increased and stimulate IFN-gamma production in sarcoid lungs. J Immunol. 2001;1666:642–649. doi: 10.4049/jimmunol.166.1.642. [DOI] [PubMed] [Google Scholar]
- 9.Millward JM, Caruso M, Campbell IL, Gauldie J, Owens T. IFN-gamma-induced chemokines synergize with pertussis toxin to promote T cell entry to the central nervous system. J Immunol. 2007;178:8175–8182. doi: 10.4049/jimmunol.178.12.8175. [DOI] [PubMed] [Google Scholar]
- 10.Hodge DL, Martinez A, Julias JG, Taylor LS, Young HA. Regulation of nuclear gamma interferon gene expression by interleukin 12 (IL-12) and IL-2 represents a novel form of posttranscriptional control. Mol Cell Biol. 2002;22:1742–1753. doi: 10.1128/MCB.22.6.1742-1753.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hatton RD, Harrington LE, Luther RJ, Wakefield T, Janowski KM, Oliver JR, Lallone RL, Murphy KM, Weaver CT. A distal conserved sequence element controls Ifng gene expression by T cells and NK cells. Immunity. 2006;25:717–729. doi: 10.1016/j.immuni.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 12.Penix L, Weaver WM, Pang Y, Young HA, Wilson CB. Two essential regulatory elements in the human interferon gamma promoter confer activation specific expression in T cells. J Exp Med. 1993;178:1483–1496. doi: 10.1084/jem.178.5.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cippitelli M, Sica A, Viggiano V, Ye J, Ghosh P, Birrer MJ, Young HA. Negative transcriptional regulation of the interferon-gamma promoter by glucocorticoids and dominant negative mutants of c-Jun. J Biol Chem. 1995;270:12548–12556. doi: 10.1074/jbc.270.21.12548. [DOI] [PubMed] [Google Scholar]
- 14.Jones B, Chen J. Inhibition of IFN-gamma transcription by site-specific methylation during T helper cell development. EMBO J. 2006;25:2443–2452. doi: 10.1038/sj.emboj.7601148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Young HA, Ghosh P, Ye J, Lederer J, Lichtman A, Gerard JR, Penix L, Wilson CB, Melvin AJ, McGurn ME, et al. Differentiation of the T helper phenotypes by analysis of the methylation state of the IFN-gamma gene. J Immunol. 1994;153:3603–3610. [PubMed] [Google Scholar]
- 16.Yano S, Ghosh P, Kusaba H, Buchholz M, Longo DL. Effect of promoter methylation on the regulation of IFN-gamma gene during in vitro differentiation of human peripheral blood T cells into a Th2 population. J Immunol. 2003;171:2510–2516. doi: 10.4049/jimmunol.171.5.2510. [DOI] [PubMed] [Google Scholar]
- 17.Fitzpatrick DR, Shirley KM, McDonald LE, Bielefeldt-Ohmann H, Kay GF, Kelso A. Distinct methylation of the interferon gamma (IFN-gamma) and interleukin 3 (IL-3) genes in newly activated primary CD8+ T lymphocytes: regional IFN-gamma promoter demethylation and mRNA expression are heritable in CD44(high)CD8+ T cells. J Exp Med. 1998;188:103–117. doi: 10.1084/jem.188.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Samten B, Ghosh P, Yi AK, Weis SE, Lakey DL, Gonsky R, Pendurthi U, Wizel B, Zhang Y, Zhang M, Gong J, Fernandez M, Safi H, Vankayalapati R, Young HA, Barnes PF. Reduced expression of nuclear cyclic adenosine 5′-monophosphate response element-binding proteins and IFN-gamma promoter function in disease due to an intracellular pathogen. J Immunol. 2002;168:3520–3526. doi: 10.4049/jimmunol.168.7.3520. [DOI] [PubMed] [Google Scholar]
- 19.Samten B, Howard ST, Weis SE, Wu S, Shams H, Townsend JC, Safi H, Barnes PF. Cyclic AMP response element-binding protein positively regulates production of IFN-gamma by T cells in response to a microbial pathogen. J Immunol. 2005;174:6357–6363. doi: 10.4049/jimmunol.174.10.6357. [DOI] [PubMed] [Google Scholar]
- 20.Shams H, Klucar P, Weis SE, Lalvani A, Moonan PK, Safi H, Wizel B, Ewer K, Nepom GT, Lewinsohn DM, Andersen P, Barnes PF. Characterization of a Mycobacterium tuberculosis peptide that is recognized by human CD4+ and CD8+ T cells in the context of multiple HLA alleles. J Immunol. 2004;173:1966–1977. doi: 10.4049/jimmunol.173.3.1966. [DOI] [PubMed] [Google Scholar]
- 21.Samten B, Wizel B, Shams H, Weis SE, Klucar P, Wu S, Vankayalapati R, Thomas EK, Okada S, Krensky AM, Barnes PF. CD40 ligand trimer enhances the response of CD8+ T cells to Mycobacterium tuberculosis. J Immunol. 2003;170:3180–3186. doi: 10.4049/jimmunol.170.6.3180. [DOI] [PubMed] [Google Scholar]
- 22.Ganster RW, Guo Z, Shao L, Geller DA. Differential effects of TNF-alpha and IFN-gamma on gene transcription mediated by NF-kappaB-Stat1 interactions. J Interferon Cytokine Res. 2005;25:707–719. doi: 10.1089/jir.2005.25.707. [DOI] [PubMed] [Google Scholar]
- 23.Brasier AR, Jamaluddin M, Casola A, Duan W, Shen Q, Garofalo RP. A promoter recruitment mechanism for tumor necrosis factor-alpha-induced interleukin-8 transcription in type II pulmonary epithelial cells. Dependence on nuclear abundance of Rel A, NF-kappaB1, and c-Rel transcription factors. J Biol Chem. 1998;273:3551–3561. doi: 10.1074/jbc.273.6.3551. [DOI] [PubMed] [Google Scholar]
- 24.Liu J, Cao S, Herman LM, Ma X. Differential regulation of interleukin (IL)-12 p35 and p40 gene expression and interferon (IFN)-gamma-primed IL-12 production by IFN regulatory factor 1. J Exp Med. 2003;198:1265–1276. doi: 10.1084/jem.20030026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Masternak K, Muhlethaler-Mottet A, Villard J, Zufferey M, Steimle V, Reith W. CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Genes & Development. 2000;14:1156–1166. [PMC free article] [PubMed] [Google Scholar]
- 26.Colmone A, Li S, Wang CR. Activating transcription factor/cAMP response element binding protein family member regulated transcription of CD1A. J Immunol. 2006;177:7024–7032. doi: 10.4049/jimmunol.177.10.7024. [DOI] [PubMed] [Google Scholar]
- 27.Bhoumik A, Ivanov V, Ronai Z. Activating transcription factor 2-derived peptides alter resistance of human tumor cell lines to ultraviolet irradiation and chemical treatment. Clin Cancer Res. 2001;7:331–342. [PubMed] [Google Scholar]
- 28.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 29.Benbrook DM, Jones NC. Heterodimer formation between CREB and JUN proteins. Oncogene. 1990;5:295–302. [PubMed] [Google Scholar]
- 30.Hai T, Curran T. Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc Natl Acad Sci U S A. 1991;88:3720–3724. doi: 10.1073/pnas.88.9.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Penix LA, Sweetser MT, Weaver WM, Hoeffler JP, Kerppola TK, Wilson CB. The proximal regulatory element of the interferon-gamma promoter mediates selective expression in T cells. J Biol Chem. 1996;271:31964–31972. doi: 10.1074/jbc.271.50.31964. [DOI] [PubMed] [Google Scholar]
- 32.Sica A, Tan TH, Rice N, Kretzschmar M, Ghosh P, Young HA. The c-rel protooncogene product c-Rel but not NF-kappa B binds to the intronic region of the human interferon-gamma gene at a site related to an interferon-stimulable response element. Proc Natl Acad Sci U S A. 1992;89:1740–1744. doi: 10.1073/pnas.89.5.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang F, Wang DZ, Boothby M, Penix L, Flavell RA, Aune TM. Regulation of the activity of IFN-gamma promoter elements during Th cell differentiation. J Immunol. 1998;161:6105–6112. [PubMed] [Google Scholar]
- 34.Aune TM, Penix LA, Rincon MR, Flavell RA. Differential transcription directed by discrete gamma interferon promoter elements in naive and memory (effector) CD4 T cells and CD8 T cells. Mol Cell Biol. 1997;17:199–208. doi: 10.1128/mcb.17.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ye J, Ghosh P, Cippitelli M, Subleski J, Hardy KJ, Ortaldo JR, Young HA. Characterization of a silencer regulatory element in the human interferon-gamma promoter. J Biol Chem. 1994;269:25728–25734. [PubMed] [Google Scholar]
- 36.Zhu H, Yang J, Murphy TL, Ouyang W, Wagner F, Saparov A, Weaver CT, Murphy KM. Unexpected characteristics of the IFN-gamma reporters in nontransformed T cells. J Immunol. 2001;167:855–865. doi: 10.4049/jimmunol.167.2.855. [DOI] [PubMed] [Google Scholar]
- 37.Carter LL, Murphy KM. Lineage-specific requirement for signal transducer and activator of transcription (Stat)4 in interferon gamma production from CD4(+) versus CD8(+) T cells. J Exp Med. 1999;189:1355–1360. doi: 10.1084/jem.189.8.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sweetser MT, Hoey T, Sun YL, Weaver WM, Price GA, Wilson CB. The roles of nuclear factor of activated T cells and ying-yang 1 in activation-induced expression of the interferon-gamma promoter in T cells. J Biol Chem. 1998;273:34775–34783. doi: 10.1074/jbc.273.52.34775. [DOI] [PubMed] [Google Scholar]
- 39.Montminy MR, Sevarino KA, Wagner JA, Mandel G, Goodman RH. Identification of a cyclic-AMP-responsive element within the rat somatostatin gene. Proc Natl Acad Sci U S A. 1986;83:6682–6686. doi: 10.1073/pnas.83.18.6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Avni O, Lee D, Macian F, Szabo SJ, Glimcher LH, Rao A. T(H) cell differentiation is accompanied by dynamic changes in histone acetylation of cytokine genes. Nature Immunol. 2002;3:643–651. doi: 10.1038/ni808. [DOI] [PubMed] [Google Scholar]
- 41.Aune TM. Transcriptional reprogramming during T helper cell differentiation. Immunol Res. 2001;23:193–204. doi: 10.1385/IR:23:2-3:193. [DOI] [PubMed] [Google Scholar]
- 42.Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G, Best J, Chen H, Jenner R, Herbolsheimer E, Jacobsen E, Kadam S, Ecker JR, Emerson B, Hogenesch JB, Unterman T, Young RA, Montminy M. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci U S A. 2005;102:4459–4464. doi: 10.1073/pnas.0501076102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yeh JH, Lecine P, Nunes JA, Spicuglia S, Ferrier P, Olive D, Imbert J. Novel CD28-responsive enhancer activated by CREB/ATF and AP-1 families in the human interleukin-2 receptor alpha-chain locus. Mol Cell Biol. 2001;21:4515–4527. doi: 10.1128/MCB.21.14.4515-4527.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Dam H, Wilhelm D, Herr I, Steffen A, Herrlich P, Angel P. ATF-2 is preferentially activated by stress-activated protein kinases to mediate c-jun induction in response to genotoxic agents. EMBO J. 1995;14:1798–1811. doi: 10.1002/j.1460-2075.1995.tb07168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reimold AM, Kim J, Finberg R, Glimcher LH. Decreased immediate inflammatory gene induction in activating transcription factor-2 mutant mice. Internat immunol. 2001;13:241–248. doi: 10.1093/intimm/13.2.241. [DOI] [PubMed] [Google Scholar]
- 46.Walker WH, Fucci L, Habener JF. Expression of the gene encoding transcription factor cyclic adenosine 3′,5′-monophosphate (cAMP) response element-binding protein (CREB): regulation by follicle-stimulating hormone-induced cAMP signaling in primary rat Sertoli cells. Endocrinol. 1995;136:3534–3545. doi: 10.1210/endo.136.8.7628390. [DOI] [PubMed] [Google Scholar]