

Abstract
Studies related to the total synthesis of elisabethin C led to the discovery of a rhodium-catalyzed cascade sequence involving isoxazole ring expansion and a [4 + 3] cycloaddition. The scope of the isoxazole ring expansion was explored, resulting in the synthesis of a range of 4H-1,3-oxazines in 47–96% yield
Keywords: isoxazole ring expansion, N–O insertion, rhodium carbenoid, aryldiazoacetates
1. Introduction
The metal-catalyzed reactions of diazo compounds are capable of a diverse array of transformations.1 With the advent of new catalysts and the recognition that different classes of carbenoids can open up new vistas of reactivity, the field continues to expand.2 We have had a long-standing interest in developing new synthetic methods derived from the chemistry of donor/acceptor-substituted carbenoids. This paper describes the discovery of an unexpected but highly efficient ring expansion of isoxazoles by rhodium carbenoid intermediates.
A recent focus of our group has been the synthesis of biologically active marine natural products utilizing the enantiodivergent combined C–H activation/Cope rearrangement methodology.3 This reaction occurs during allylic C–H functionalization using rhodium-stabilized vinylcarbenoids. Recent total syntheses completed using this methodology include (+)-erogorgiaene (3),4 (−)-elisapterosin B (4),5 and (−)-colombiasin A (5).5 This strategy has been successful at rapidly introducing three of the stereocenters common in these natural products by differentiating between the enantiomers of the racemic dihydronaphthalene derivative 1. One enantiomer of the substrate undergoes the combined C–H activation/Cope rearrangement while the other is cyclopropanated (Scheme 1).
Scheme 1.
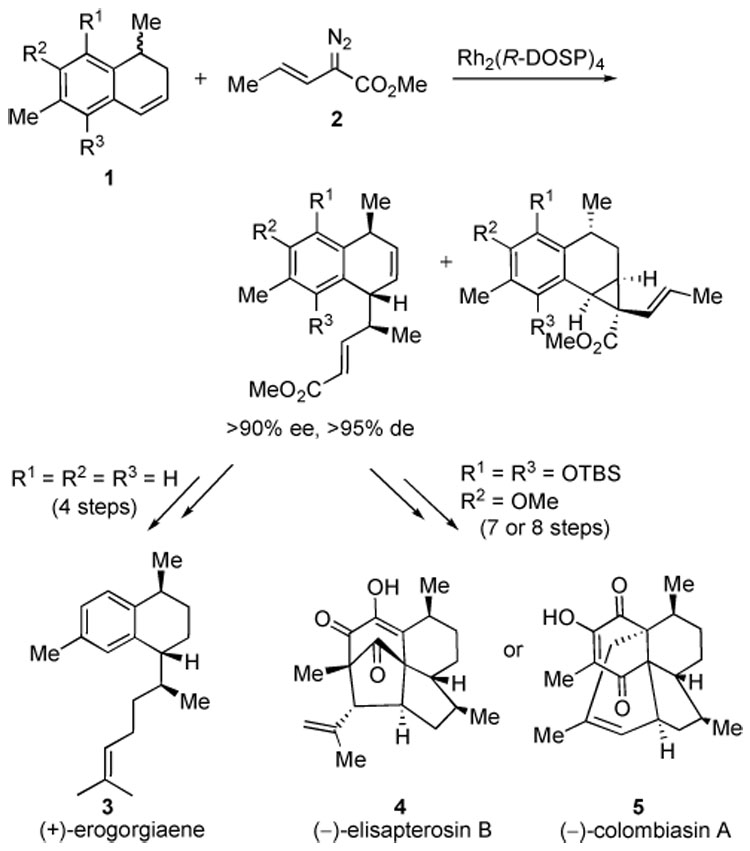
Synthesis of Marine Natural Products
In seeking to broaden the scope of this methodology, we focused our attention on the marine bisnor-diterpenoid elisabethin C (6),6,7 using the fused isoxazole 8 as the substrate for the combined C–H activation/Cope rearrangement (Scheme 2). The isoxazole subunit would be used as a protecting group for the diketone functionality of elisabethin C, to be unmasked at a late stage of the synthesis.
Scheme 2.
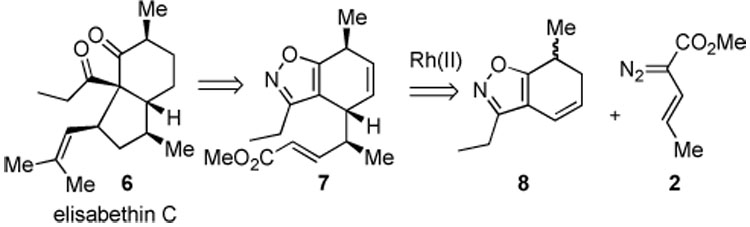
Retrosynthesis of Elisabethin C
2. Results and discussion
The synthesis of fused isoxazole 8 was achieved using a relatively straightforward approach starting from β-diketone 9, which in turn was readily prepared by literature procedures8,9 from the commercially available mono-protected 1,4-cyclohexadione (Scheme 3). If the isoxazole forming reaction was allowed to run to completion, the resulting regioisomeric mixture of isoxazoles proved inseparable by chromatography at all subsequent steps. Fortunately, if the reaction was stopped after 10 min, the regioisomeric oxime intermediates 10 and 11 could be chromatographically separated. Treatment of each isomer separately with warm acetic acid furnished the corresponding isoxazoles in isomerically pure form. Regioisomer 11 was chosen initially to carry through the sequence to test the key carbenoid reaction. Cyclization to form isoxazole 12 and subsequent acetal deprotection to ketone 13 proceeded smoothly. The remaining two steps to form vinyl triflate 15 using Comins’ reagent 1410 followed by a palladium-catalyzed reduction11 gave 8 in good overall yield.
Scheme 3.
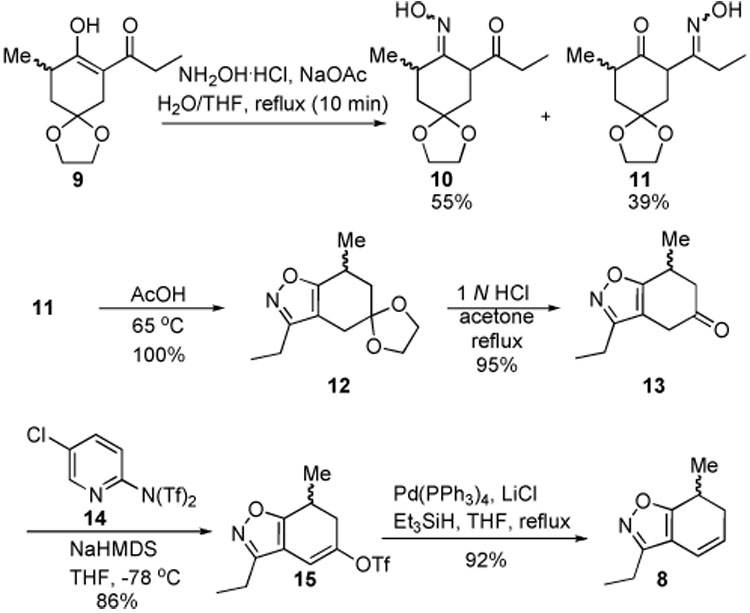
Synthesis of Dihydrobenzoisoxazole 8
The Rh2(R-DOSP)4-catalyzed reaction12 of the fused isoxazole 8 with vinyldiazoacetate 2 (3 equiv) gave a most unusual result (Scheme 4). Instead of the expected reaction to form 7, the major isolable product was an unprecedented tricyclic derivative 16, which was formed as a 1 : 1 mixture of diastereomers in 62% yield. Each diastereomer was formed with low enantioinduction (19% ee). This material has incorporated into isoxazole 8 two equivalents of the vinylcarbenoid derived from 2. Compound 16 can be considered as formally derived from a carbenoid insertion into the isoxazole N–O bond and a tandem cyclopropanation/Cope rearrangement between another carbenoid and the diene component of the substrate.
Scheme 4.
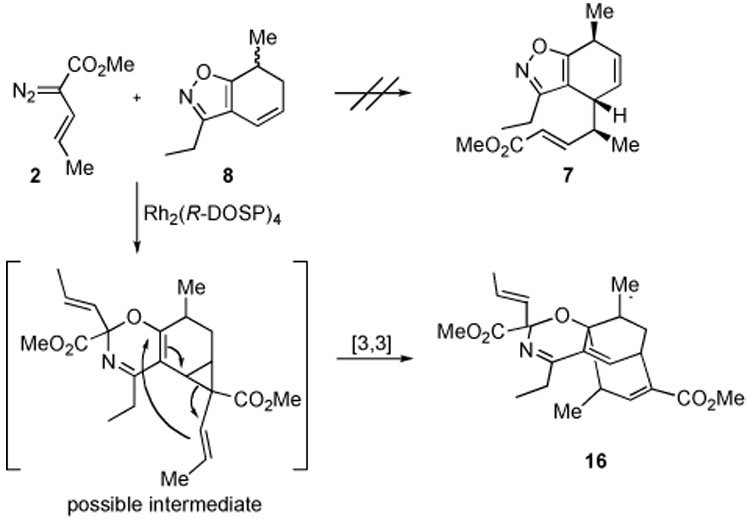
Reaction of 8 with a Vinylcarbenoid
The formation of 16 is an unusual transformation and we were intrigued by this novel carbenoid reactivity. Formal [4 + 3] cycloadditions between vinylcarbenoids and dienes by a tandem cyclopropanation/Cope rearrangement are well precedented,13 but the carbenoid insertion into the isoxazole N–O bond is not an established process. Rhodium carbenoids containing isoxazoles have been used in intermolecular cyclopropanations without any side reaction on the isoxazole ring.14 Additionally, intramolecular C–H insertion reactions have been successfully achieved on substrates containing an isoxazole ring.15 It is known, however, that isoxazolium ylides, typically generated by deprotonation of an isoxazolium salt, undergo rearrangements to generate either 4H-1,3-oxazines or 3-imino-2-en-1-ones, depending on the substitution of the isoxazolium salt.16 Consequently, we decided to explore further the scope of the N–O insertion chemistry using rhodium carbenoids and various isoxazole substrates.
The first series of experiments studied the effect of the carbenoid structure on the efficiency of the N–O insertion, using 3,5-dimethylisoxazole (18) as a reference substrate (Table 1). In recent years we have shown that donor/acceptor-substituted carbenoids are capable of higher selectivity than the conventional carbenoids lacking a donor group. In this case all three of the prototypical types of carbenoids, derived from 17a–c, induced an N–O insertion into the isoxazole, although the reaction with the donor/acceptor-substituted carbenoid (entry 3) was the most efficient (88% yield).
Table 1.
Reaction of Rhodium Carbenoids with 3,5-Dimethylisoxazole
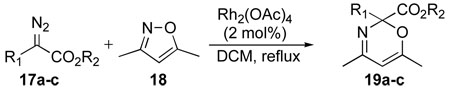 | ||||
|---|---|---|---|---|
| entry | R1 | R2 | 19 | yield, %a |
| 1 | H | Et | 19a | 56 |
| 2 | CO2Me | Me | 19b | 47 |
| 3 | Ph | Me | 19c | 88 |
Reported yields are of isolated products.
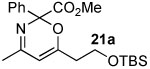
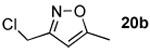
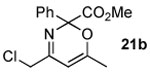
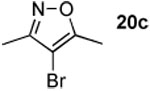
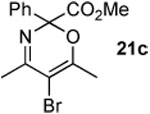
The next series of reactions were conducted to determine what types of functionality on the isoxazole would be compatible with the N–O insertion (Table 2). Methyl phenyldiazoacetate (17c) was used as the carbenoid source because it had resulted in the highest yield of product in the initial evaluation. Further studies were performed with a range of isoxazoles (Table 2). All four isoxazoles 20a–d produced the 4H-1,3-oxazine products 21a–d, respectively in high yield (67–96%). These studies demonstrate that siloxy, halo and even ester functionalities are compatible with this chemistry. The ester derivative 20d gave a tautomeric mixture of the ring expansion products 21d and 22d (Entry 5). Compound 21d was formed cleanly in the carbenoid reaction, but is prone to isomerization to 22d during silica gel chromatography, illustrating the relative mildness of the carbenoid reaction conditions.
Table 2.
Reaction of diazoacetate 17c with isoxazoles
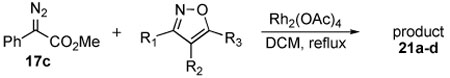
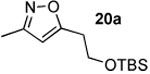
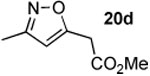
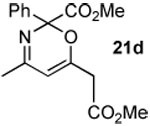
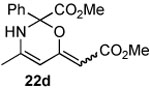
Reported yields are of isolated products.
During attempted purification, product formed a 1:1 inseparable mixture with its tautomer 22d.
Having discovered that isoxazoles could be effectively ring-expanded, it became of interest to determine if the reaction could be extended to other heterocyclic systems. Benzisoxazoles were found to be similarly reactive with carbenoids, although further transformations occurred in certain cases (Scheme 5). The reaction with 1,2-benzisoxazole 23 produced the ring expansion product 24 cleanly in 72% yield if 23 was used in excess, and aziridine 25 in 94% yield if 3 equiv of the diazo component was used. In the case of anthranil 26, aldehyde 27, formally the result of a 6π-electrocyclic ring-opening of the expected N–O insertion product, was isolated along with epoxide 28. Compound 28 could be obtained exclusively if 3 equiv of the diazoacetate was used in the reaction. 5-Chloro-3-phenylanthranil (29) cleanly produced ketone 30, which did not readily undergo epoxide formation.
Scheme 5.
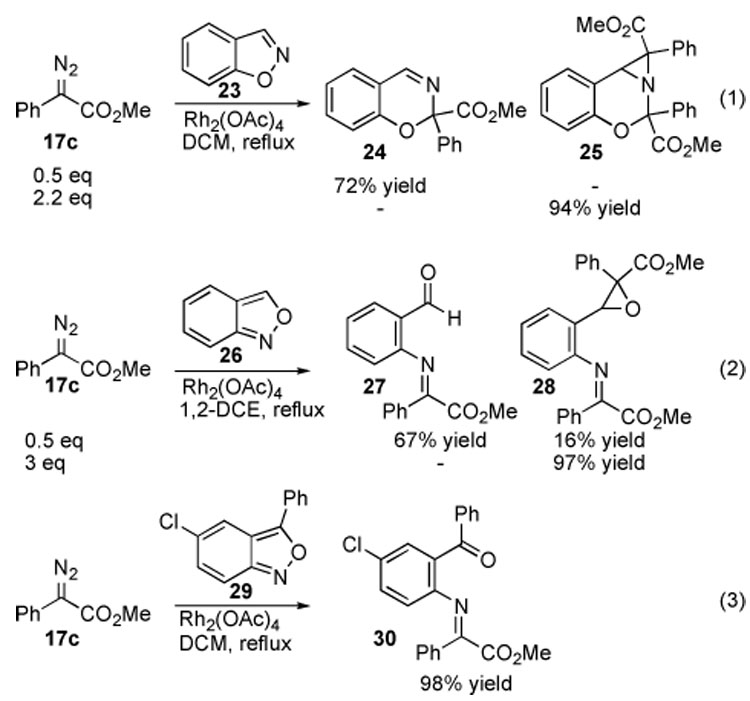
Reaction of Diazoacetate 17c with Benzisoxazoles
Another system worthy of study was benzoisothiazoles (Scheme 6). Previously it had been shown that the related 2-substituted isothiazol-3(2H)-ones undergo N–S insertion with carbenoids lacking an electron-donating group.17 The reaction of 17c with 3-chloro[d]benzoisothiazole (31) was a very efficient process resulting in the formation of 2H-1,3-benzothiazine 32 in 88% yield.
Scheme 6.
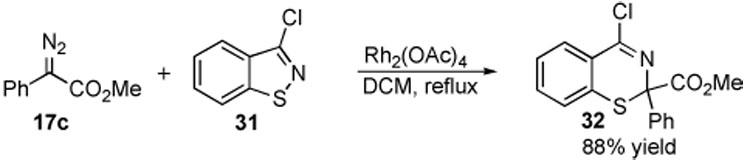
Ring Expansion of an Isothiazole
The reaction mechanism for the transformations described above likely proceeds through an isoxazolium ylide intermediate 33, formed by attack of the isoxazole nitrogen onto the rhodium carbenoid (Scheme 7). At this point, two reasonable pathways could lead to the ring-expanded product 35. The ylide 33 could undergo a 1,2-shift to generate 35 directly, as previously proposed for the ring expansion of 2-substituted isothiazol-3(2H)-ones.17 Alternatively, ylide 33 could undergo a ring opening to 34, followed by a 6π electrocyclization to give 35 as proposed for the ring expansion of isoxazolium ylides derived from deprotonation of isoxazolium salts.16c
Scheme 7.
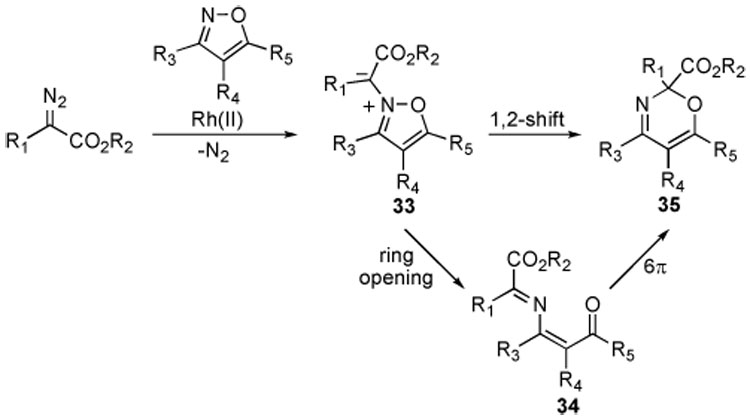
Possible Mechanistic Pathways for Isoxazole Ring Expansion
3. Conclusion
In summary, the reactions of various isoxazoles with rhodium carbenoids have been examined and found to produce 4H-1,3-oxazines through a ring expansion in good to excellent yields. These reactions are likely to proceed via ylide intermediates which then either expand through a 1,2-shift, or open up to 3-imino-2-en-1-ones which subsequently undergo a 6π electrocyclization to 4H-1,3-oxazines. Studies toward applications of this transformation in heterocycle synthesis are currently underway.
4. Experimental
4.1. General
All experiments were performed under anhydrous conditions in an atmosphere of argon except where stated, using flame dried glassware. 2,2-Dimethylbutane (DMB) was purified by distillation over sodium. DCM and THF were dried by a solvent purification system (passed through activated alumina). The vinyldiazoacetate 2,4 dimethyl diazomalonate 17b,18 methyl phenyldiazoacetate 17c,19 and methyl 2-(3-methylisoxazol-5-yl)acetate 20d8 were prepared by their respective literature procedures. The synthesis of compounds 9–13, 15, and 8 is described in the supplementary information. Unless otherwise noted, all other reagents were obtained from commercial sources and used as received. Mass spectral determinations were carried out by LC-MS (ESI), or electron impact ionization (EI). Melting points are uncorrected. Flash column chromatography was performed on silica gel 60Å (230–400 mesh) using a pentane/diethyl ether mixture as the eluent unless otherwise specified.
4.2. Synthesis of ring expansion/[4 + 3]-cycloaddition product 16
4.2.1. (16)
To a flame dried 25 mL round bottom flask under argon and charged with a stir bar was added 8 (0.0500 g, 0.306 mmol), Rh2(R-DOSP)4 (0.017 g, 0.0092 mmol, 0.03 eq) and 2,2 DMB (2 mL). A reflux condenser was attached to the flask and the solution was heated to reflux. A solution of 2 (0.172 g, 1.23 mmol) in 2,2-DMB (3 mL) was added via syringe pump addition over 20 min. The solution was refluxed for another 5 min, then allowed to cool to ambient temperature. The mixture was concentrated in vacuo and the residue was purified by flash chromatography (silica gel, 5:1-2:1 pentane:diethyl ether) to give the diastereomeric products 16a (0.038 g, 32% yield) and 16b (0.036 g, 30% yield) as clear oils.
16a
Rf 0.16 (2:1 pentane:diethyl ether); FTIR (neat): 2930, 1746, 1711, 1640, 1436, 1236, 1193, 1066, 1028, 1006, 970 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.89 (d, J = 8.0 Hz, 1H), 6.69 (d, J = 4.5 Hz, 1H), 5.77 (d, J = 15.5 Hz, 1H), 5.55 (dq, J = 15.5, 6.5 Hz, 1H), 3.81 (s, 3H), 3.74 (s, 3H), 3.55 (m, 1H), 2.67-2.60 (m, 3H), 2.29-2.20 (m, 2H), 1.68 (d, J = 6.5 Hz, 3H), 1.24 (t, J = 7.5 Hz, 3H), 1.19-1.15 (m, 1H), 0.91 (d, J = 6.5 Hz, 3H), 0.84 (d, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 170.8 (C), 167.3 (C), 165.4 (C), 145.5 (CH), 135.9 (CH), 132.9 (CH), 129.9 (C), 127.9 (CH), 126.1 (C), 89.0 (C), 77.6 (C), 52.9 (CH3), 52.1 (CH3), 48.5 (CH), 38.6 (CH2), 37.8 (CH), 31.2 (CH), 27.7 (CH2), 21.5 (CH3), 17.6 (CH3), 14.6 (CH3), 12.1 (CH3); LRMS (EI) m/z (relative intensity): 387 (24) [M]+, 328 (100) [M-CO2CH3]+; HRMS (EI) Calcd for [C22H29NO5]+ 387.2040, Found 387.2041; HPLC analysis: 19% ee (Chiralpak AD-H, 1% i-PrOH in hexanes, 0.8 mL/min, λ = 254 nm, tR = 17.3 min, major; 18.4 min, minor).
16b
Rf 0.31 (2:1 pentane:diethyl ether); FTIR (neat): 2919, 1741, 1710, 1641, 1435, 1234, 1065, 1026, 1003, 970 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.90 (d, J = 7.5 Hz, 1H), 6.69 (d, J = 5.0 Hz, 1H), 6.11 (dq, J = 15.5, 6.5 Hz, 1H), 5.91 (d, J = 15.5 Hz, 1H), 3.74 (s, 3H), 3.68 (s, 3H), 3.57 (m, 1H), 2.72-2.58 (m, 2H), 2.54-2.48 (m, 1H), 2.27-2.21 (m, 2H), 1.78 (d, J = 6.5 Hz, 3H), 1.27-1.20 (m, 4H), 0.80 (d, J = 6.0 Hz, 3H), 0.78 (d, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 170.2 (C), 167.3 (C), 165.0 (C), 145.3 (CH), 135.7 (CH), 131.6 (CH), 130.1 (C), 128.5 (CH), 126.0 (C), 87.8 (C), 77.9 (C), 52.3 (CH3), 52.1 (CH3), 48.4 (CH), 38.6 (CH2), 38.4 (CH), 31.2 (CH), 27.5 (CH2), 18.8 (CH3), 17.7 (CH3), 15.0 (CH3), 11.6 (CH3); LRMS (ESI) m/z (relative intensity): 388 (100) [M+H]+; HRMS (ESI) Calcd for [C22H30NO5]+ 388.2118, Found 388.2121; HPLC analysis: 19% ee (Regis R,R-Whelk, 0.5% i-PrOH in hexanes, 1.0 mL/min, λ = 254 nm, tR = 20.8 min, major; 24.7 min, minor).
4.3 Synthesis of isoxazole 20a
4.3.1 2-(3-Methylisoxazol-5-yl)ethanol
To a flame dried round bottom flask under argon and charged with a stir bar was added methyl 2-(3-methylisoxazol-5-yl)acetate 20d8 (0.62 g, 4.0 mmol) and THF (10 mL). The solution was cooled to 0 °C in an ice bath and a solution of LAH (2.0 M in THF, 1 mL) was added by syringe. The solution was stirred for 10 min and then slowly quenched with water. The solution was then extracted with ether (3 × 15 mL) and the combined organic extracts dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was purified by flash chromatography (silica gel, diethyl ether as eluent, visualized with KMnO4 stain) to give 2-(3-methylisoxazol-5-yl)ethanol as a clear oil (0.252 g, 50% yield). The characterization data were in agreement with the literature values.20
4.3.2 5-(2-(tert-Butyldimethylsilyloxy)ethyl)-3-methylisoxazole (20a)
To a flame dried round bottom flask under argon and charged with a stir bar was added 2-(3-methylisoxazol-5-yl)ethanol (0.105 g, 0.83 mmol), TBSCl (0.149 g, 0.99 mmol), DMAP (0.003 g, 0.025 mmol) and DCM (10 mL). Imidazole (0.062 g, 0.91 mmol) was then added and the solution stirred overnight. The solution was then washed with brine, dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was purified by flash chromatography (silica gel, 7:1 pentane:diethyl ether) to give the product as a clear oil (0.107 g, 54% yield). Rf 0.25 (7:1 pentane:diethyl ether); FTIR (neat): 2929, 1607, 1472, 1417, 1255, 1101, 835, 776 cm−1; 1H NMR (300 MHz, CDCl3) δ 5.89 (s, 1H), 3.88 (t, J = 6.5 Hz, 2H), 2.92 (t, J = 6.5 Hz, 2H), 2.26 (s, 3H), 0.87 (s, 9H), 0.02 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 170.4 (C), 159.5 (C), 102.5 (CH), 60.6 (CH2), 30.4 (CH2), 25.7 (CH3), 18.1 (C), 11.3 (CH3), −5.6 (CH3); LRMS (EI) m/z (relative intensity): 226 (9) [M]+, 184 100 [M-tBu]+; HRMS (EI) Calcd for [M-CH3]+ [C11H20NO2Si]+ 226.1258, Found 226.1249.
4.4. General Procedure for the rhodium catalyzed isoxazole/isothiazole ring expansion reactions
To a flame dried 25 mL round bottom flask under argon and charged with a stir bar was added the isoxazole substrate, Rh2(OAc)4 and solvent (DCM or 1,2-DCE, 5 mL). A water-cooled condenser was attached to the flask and the solution was heated to reflux. A solution of diazoacetate in solvent (DCM or 1,2-DCE, 5 mL) was then added by syringe pump over 45 min. The solution was refluxed another 15 min and then cooled to ambient temperature. The reaction mixture was concentrated in vacuo and the residue was purified by flash chromatography to give the product.
4.4.1. Ethyl 4,6-dimethyl-2H-1,3-oxazine-2-carboxylate (19a)
The reaction was performed with 3,5-dimethylisoxazole 18 (0.146 g, 1.5 mmol), Rh2(OAc)4 (6.6 mg, 0.015 mmol) and ethyl diazoacetate 17a (0.057 g, 0.5 mmol) in 1,2-DCE. Purified by flash chromatography (silica gel, 1:1 pentane:diethyl ether) to give 19a as a clear oil (0.051 g, 56% yield). Rf 0.13 (1:1 pentane:diethyl ether); FTIR (neat): 2983, 1743, 1619, 1575, 1443, 1385, 1202, 1096, 1023 cm−1; 1H NMR (500 MHz, CDCl3) δ 5.59 (s, 1H), 5.37 (s, 1H), 4.36-4.26 (m, 2H), 2.04 (s, 3H), 1.98 (s, 3H), 1.34 (t, J = 7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 168.1 (C), 165.8 (C), 162.9 (C), 100.7 (CH), 85.9 (CH), 61.8 (CH2), 23.7 (CH3), 19.0 (CH3), 14.0 (CH3); LRMS (ESI) m/z (relative intensity): 206 (100) [M+Na]+, 184 (36) [M+H]+; HRMS (ESI) Calcd for [C9H14NO3]+ 184.0968, Found 184.0970.
4.4.2. Dimethyl 4,6-dimethyl-2H-1,3-oxazine-2,2-dicarboxylate (19b)
The reaction was performed with 3,5-dimethylisoxazole 18 (0.049 g, 0.5 mmol), Rh2(OAc)4 (6.6 mg, 0.015 mmol), dimethyldiazomalonate 17b18 (0.111 g, 0.70 mmol) and 1,2-DCE. The solution was refluxed for 2 h after the diazo addition was complete. Purified by flash chromatography (silica gel, 1:5 pentane:diethyl ether) to give 19b as a clear oil (0.053 g, 47% yield). Rf 0.24 (diethyl ether); FTIR (neat): 2950, 1748, 1654, 1574, 1435, 1294, 1259, 1134, 1056, 784, 706 cm−1; 1H NMR (500 MHz, CDCl3) δ 5.39 (s, 1H), 3.86 (s, 6H), 2.10 (s, 3H), 2.02 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 166.6 (C), 166.2 (C), 162.1 (C), 100.5 (CH), 53.5 (CH3), 24.1 (CH3), 19.2 (CH3), missing C attributed to excessive peak broadening/quadrupolar effect due to N; LRMS (ESI) m/z (relative intensity): 477 (39) [2M+Na]+, 250 (51) [M+Na]+; HRMS (ESI) Calcd for [C10H13NO5Na]+ 250.0686, Found 250.0680.
4.4.3. Methyl 4,6-dimethyl-2-phenyl-2H-1,3-oxazine-2-carboxylate (19c)
The reaction was performed with 3,5-dimethylisoxazole 18 (0.049 g, 0.5 mmol), Rh2(OAc)4 (6.6 mg, 0.015 mmol), 17c (0.132 g, 0.75 mmol) and DCM. Purified by flash chromatography (silica gel, 1.5:1 pentane:diethyl ether) to give 19c as a white solid (0.109 g, 89% yield). mp = 72–73 °C, Rf 0.19 (1:1 pentane:diethyl ether); FTIR (neat): 2954, 1742, 1659, 1574, 1235, 729 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.74 (dd, J = 8.0, 1.5 Hz, 2H), 7.40-7.35 (m, 3H), 5.36 (s, 1H), 3.72 (s, 3H), 2.13 (s, 3H), 2.01 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 170.3 (C), 164.7 (C), 162.2 (C), 138.8 (C), 128.6 (CH), 127.9 (CH), 126.2 (CH), 100.7 (CH), 52.7 (CH3), 23.9 (CH3), 19.2 (CH3), missing C attributed to excessive broadening/quadrupolar effect due to N; LRMS (ESI) m/z (relative intensity): 246 (100) [M+H]+; Anal. Calcd for C14H15NO3: C, 68.56; H, 6.16; N, 5.71. Found: C, 68.42; H, 6.17; N, 5.60.
4.4.4. Methyl 6-(2-(tert-butyldimethylsilyloxy)ethyl)-4-methyl-2-phenyl-2H-1,3-oxazine-2-carboxylate(21a)
The reaction was performed with 20a (0.073 g, 0.30 mmol), Rh2(OAc)4 (4.0 mg, 0.009 mmol), 17c (0.064 g, 0.36 mmol) and DCM. Purified by flash chromatography (silica gel, 4:1-3:1 pentane:diethyl ether) to give 21a as a clear oil (0.098 g, 83% yield). Rf 0.11 (5:1 pentane:diethyl ether); FTIR (neat): 2954, 2928, 2856, 1745, 1658, 1574, 1235, 1098, 1006, 833, 776, 729, 696 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.73 (dd, J = 8.0, 1.5 Hz, 2H), 7.40-7.35 (m, 3H), 5.43 (s, 1H), 3.87-3.85 (m, 2H), 3.70 (s, 3H), 2.49 (t, J = 7.0 Hz, 2H), 2.15 (s, 3H), 0.86 (s, 9H), 0.03 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 170.5 (C), 164.8 (C), 162.9 (C), 138.8 (C), 128.8 (CH), 128.1 (CH), 126.4 (CH), 101.3 (CH), 78.9 (C), 59.4 (CH2), 52.8 (CH3), 37.0 (C), 25.7 (CH3), 24.1 (CH3), 18.1 (CH2), −5.5 (CH3); LRMS (ESI) m/z (relative intensity): 390 (100) [M+H]+; HRMS (ESI) Calcd for [C21H32NO4Si]+ 390.2095, Found 390.2091.
4.4.5. Methyl 4-(chloromethyl)-6-methyl-2-phenyl-2H-1,3-oxazine-2-carboxylate (21b)
The reaction was performed with 3-chloromethyl-5-methylisoxazole 20b (0.066 g, 0.5 mmol), Rh2(OAc)4 (6.6 mg, 0.015 mmol), 17c (0.115 g, 0.65 mmol) and DCM. Purified by flash chromatography (silica gel, 5:1 pentane:diethyl ether) to give 21b as a clear oil (0.130 g, 93% yield). Rf 0.14 (5:1 pentane:diethyl ether); FTIR (neat): 1743, 1655, 1572, 1434, 1369, 1238, 1031, 727, 697 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.73 (dd, J = 8.0, 1.5 Hz, 2H), 7.41-7.38 (m, 3H), 5.66 (s, 1H), 4.22 (s, 2H), 3.73 (s, 3H), 2.08 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 169.8 (C), 164.7 (C), 163.0 (C), 138.1 (C), 129.1 (CH), 128.2 (CH), 126.3 (CH), 98.0 (CH), 53.1 (CH3), 45.5 (CH2), 19.7 (CH3), missing C attributed to excessive broadening/quadrupolar effect due to N; LRMS (ESI) m/z (relative intensity): 581 (17) [2M+Na]+, 302 (100) [M+Na]+; HRMS (ESI) Calcd for [C14H14NO3ClNa]+ 302.0554, Found 302.0557.
4.4.6. Methyl 5-bromo-4,6-dimethyl-2-phenyl-2H-1,3-oxazine-2-carboxylate (21c)
The reaction was performed with 4-bromo-3,5-dimethylisoxazole (0.088 g, 0.5 mmol), Rh2(OAc)4 (4.4 mg, 0.01 mmol), 17c (0.115 g, 0.65 mmol) and DCM. Purified by flash chromatography (silica gel, 7:1 pentane:diethyl ether) to give 21c as a clear oil (0.155 g, 96% yield). Rf 0.21 (5:1 pentane:diethyl ether); FTIR (neat): 1746, 1638, 1564, 1432, 1376, 1309, 1241, 1137, 1011, 908, 726, 696 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.72 (dd, J = 7.5, 1.5 Hz, 2H), 7.40-7.38 (m, 3H), 3.72 (s, 3H), 2.34 (s, 3H), 2.22 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 169.9 (C), 163.1 (C), 160.2 (C), 137.9 (C), 129.0 (CH), 128.8 (C), 128.2 (CH), 126.3 (CH), 97.4 (C), 53.1 (CH3), 24.4 (CH3), 19.2 (CH3); LRMS (ESI) m/z (relative intensity): 324 (10) [M+H]+; HRMS (ESI) Calcd for [C14H15BrNO3]+ 324.0230, Found 324.0234.
4.4.7. (21d) and (22d)
The reaction was performed with 20d (0.078 g, 0.5 mmol), Rh2(OAc)4 (6.6 mg, 0.015 mmol), 17c (0.132 g, 0.75 mmol) and DCM. 1H NMR analysis of the crude reaction mixture in CD2Cl2 did not contain 22d. Purified by flash chromatography (silica gel, 1:1 pentane:diethyl ether) to give a 1:1 mixture of 21d and 22d as a sticky white solid (0.102 g, 67% yield). Rf 0.17 (1:1 pentane:diethyl ether); 1H NMR (500 MHz, CDCl3) δ 7.73-7.71 (m, 2H), 7.57-7.56 (m, 2H), 7.40-7.39 (m, 6H), 6.45 (s, 1H), 6.00 (s, 1H), 5.45 (s, 1H), 5.19 (s, 1H), 3.78 (s, 3H), 3.73 (s, 3H), 3.72 (s, 3H), 3.65 (s, 3H), 3.33 (s, 2H), 2.24 (s, 3H), 2.03 (s, 3H); LRMS (ESI) m/z (relative intensity): 629 (85) [2M+Na]+, 326 (100) [M+Na]+; HRMS (EI) Calcd for [C16H17NO5]+ 303.1101, Found 303.1111.
4.4.8. Methyl 2-phenyl-2H-benzo[e][1,3]oxazine-2-carboxylate (24)
The reaction was performed with freshly purified 1,2-benzisoxazole 23 (0.065 g, 0.55 mmol, purified by passing through a silica gel pipet column eluted with 5:1 pentane:diethyl ether), Rh2(OAc)4 (7 mg, 0.016 mmol), 17c (0.044 g, 0.25 mmol) and DCM. Purified by flash chromatography (silica gel, 2:1-1:1 pentane:diethyl ether) to give 24 as a clear oil (0.048 g, 72% yield). Rf 0.23 (1:1 pentane:diethyl ether); FTIR (neat): 2950, 1745, 1633, 1608, 1229, 1050, 1035, 1003, 759, 727, 697 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.41 (s, 1H), 7.76 (d, J = 7.0 Hz, 2H), 7.42-7.32 (m, 4H), 7.22 (dd, J = 7.5, 1.5 Hz, 1H), 7.08 (d, J = 8.5 Hz, 1H), 6.98 (appt t, J = 7.0 Hz, 1H), 3.76 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 169.1 (C), 157.5 (CH), 153.2 (C), 138.6 (C), 134.5 (CH), 128.8 (CH), 128.1 (CH), 127.4 (CH), 126.4 (CH), 122.0 (CH), 116.7 (C), 116.5 (CH), 91.7 (C), 53.1 (CH3); LRMS (ESI) m/z (relative intensity): 268 (27) [M+H]+; HRMS (ESI) Calcd for [C16H14NO3]+ 268.0968, Found 268.0969.
4.4.9. Aziridine 25
The reaction was performed with freshly purified 1,2-benzisoxazole 23 (0.065 g, 0.55 mmol, purified by passing through a silica gel pipet column eluted with 5:1 pentane:diethyl ether), Rh2(OAc)4 (7 mg, 0.016 mmol), 17c (0.291 g, 1.65 mmol) and DCM. Purified by flash chromatography (silica gel, 3:1–2:1 pentane:diethyl ether) to give 25 as a white solid (0.215 g, 94% yield). mp 147–149 °C; Rf 0.36 (1:1 pentane:diethyl ether); FTIR (neat): 2950, 1742, 1491, 1281, 1208, 1128, 1074, 1045, 1031, 986, 759, 727, 696 cm−1; 1H NMR (500 MHz, CDCl3) . 7.84 (d, J = 7.0 Hz, 2H), 7.80 (d, J = 7.0 Hz, 2H), 7.44-7.36 (m, 3H), 7.29-7.15 (m, 5H), 7.04 (d, J = 8.0 Hz, 1H), 6.95-6.92 (m, 1H), 3.83 (s, 3H), 3.60 (s, 3H), 3.36 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 168.6 (C), 167.4 (C), 149.5 (C), 137.5 (C), 137.0 (C), 128.9 (CH), 128.6 (CH), 128.3 (CH), 128.0 (CH), 127.7 (CH), 127.0 (CH), 122.8 (CH), 120.3 (C), 118.5 (CH), 89.5 (C), 53.0 (CH3), 52.7 (C), 52.3 (CH3), 44.4 (CH), 2 missing CH resonances attributed to overlapping signals; LRMS (ESI) m/z (relative intensity): 853 (100) [2M+Na]+, 438 (36) [M+Na]+; Anal. Calcd for C25H21NO5: C, 72.28; H, 5.10; N, 3.37. Found: C, 71.92; H, 5.09; N, 3.33.
4.4.10. Methyl 2-(2-formylphenylimino)-2-phenylacetate (27)
The reaction was performed with anthranil 26 (0.179 g, 1.5 mmol), Rh2(OAc)4 (6.6 mg, 0.015 mmol), 17c (0.085 g, 0.5 mmol) and 1,2-DCE. Purified by flash chromatography (silica gel, 5:1 pentane:diethyl ether) to give 27 (0.089 mg, 67% yield) and 28 (0.034 g, 16% yield) as yellow oils. 27: Rf 0.30 (2:1 pentane:diethyl ether); FTIR (neat): 1733, 1690, 1624, 1592, 1450, 1302, 1273, 1226, 1192, 1174, 1155, 1009, 764, 689, 669 cm−1; 1H NMR (400 MHz, CD2Cl2) δ 10.20 (s, 1H), 7.90 (d, J = 7.5 Hz, 2H), 7.86 (d, J = 7.5 Hz, 1H), 7.60-7.49 (m, 4H), 7.26 (t, J = 7.5 Hz, 1H), 6.85 (d, J = 8.0 Hz, 1H), 3.58 (s, 3H); 13C NMR (75 MHz, CD2Cl2) δ 190.3 (CH), 164.7 (C), 161.7 (C), 152.7 (C), 135.1 (CH), 133.5 (C), 132.8 (CH), 129.2 (CH), 128.6 (CH), 128.3 (CH), 127.0 (C), 125.5 (CH), 119.4 (CH), 52.4 (CH3); LRMS (ESI) m/z (relative intensity): 557 (52) [2M+Na]+, 290 (100) [M+Na]+, 268 (68) [M+H]+; HRMS (EI) Calcd for [C16H13NO3]+ 267.0890, Found 267.0903.
4.4.11. (E)-Methyl3-(2-(2-methoxy-2-oxo-1-phenylethylideneamino)phenyl)-2-phenyloxirane-2-carboxylate (28)
The reaction was performed with anthranil 26 (0.060 g, 0.5 mmol), Rh2(OAc)4 (6.6 mg, 0.015 mmol), 17c (0.255 g, 1.5 mmol) and 1,2-DCE. Purified by flash chromatography (silica gel, 5:1 pentane:diethyl ether) to give 28 as a yellow oil (0.201 g, 97% yield). Rf 0.24 (2:1 pentane:diethyl ether); FTIR (neat): 1734, 1449, 1434, 1298, 1234, 1197, 1171, 1009, 763, 739, 691 cm−1; 1H NMR (400 MHz, CD2Cl2) δ 7.87 (d, J = 7.0 Hz, 2H), 7.57-7.54 (m, 1H), 7.49-7.45 (m, 5H), 7.31-7.17 (m, 5H), 6.80 (d, J = 8.0 Hz, 1H), 4.33 (s, 1H), 3.64 (s, 3H), 3.48 (s, 3H); ); 13C NMR (75 MHz, CD2Cl2) δ 167.6 (C), 165.3 (C), 160.6 (C), 148.9 (C), 135.8 (C), 133.9 (C), 132.5 (CH), 129.2 (CH), 129.1 (CH), 128.9 (CH), 128.52 (CH), 128.48 (CH), 127.5 (CH), 126.5 (CH), 126.2 (C), 125.3 (CH), 117.4 (CH), 66.8 (C), 63.1 (CH), 52.4 (CH3), 52.3 (CH3); LRMS (ESI) m/z (relative intensity): 438 (100) [M+Na]+, 416 (41) [M+H]+; HRMS (EI) Calcd for [C25H21NO5]+ 415.1414, Found 415.1416.
4.4.12. Methyl 2-(2-benzoyl-4-chlorophenylimino)-2-phenylacetate (30)
The reaction was performed with 5-chloro-3-phenylanthranil 29 (0.115 g, 0.5 mmol), Rh2(OAc)4 (6.6 mg, 0.015 mmol), 17c (0.106 g, 0.6 mmol) and DCM. Purified by flash chromatography (silica gel, 5:1 pentane:diethyl ether) to give 30 as a sticky yellow oil (0.185 g, 98% yield). Rf 0.24 (5:1 pentane:diethyl ether); FTIR (neat): 1734, 1664, 1449, 1283, 1226, 1195, 1173, 1009, 688 cm−1; 1H NMR (500 MHz, C6D6) δ 7.81 (d, J = 7.0 Hz, 2H), 7.54 (d, J = 7.5 Hz, 2H), 7.50 (d, J = 2.5 Hz, 1H), 7.05-6.95 (m, 5H), 6.92-6.89 (m, 2H), 6.66 (d, J = 8.5 Hz, 1H), 3.10 (s, 3H); 13C NMR (75 MHz, CD2Cl2) δ 195.1 (C), 164.7 (C), 160.4 (C), 147.2 (C), 137.7 (C), 133.6 (C), 133.3 (CH), 132.9 (C), 132.5 (CH), 131.8 (CH), 130.7 (C), 130.1 (CH), 129.9 (CH), 128.9 (CH), 128.7 (CH), 128.3 (CH), 120.8 (CH), 52.5 (CH3); LRMS (ESI) m/z (relative intensity): 777 (100) [2M+Na]+, 400 (100) [M+Na]+, 378 (18) [M+H]+; HRMS (EI) Calcd for [C22H16NO3Cl]+ 377.0813, Found 377.0826.
4.4.13. Methyl 4-chloro-2-phenyl-2H-benzo[e][1,3]thiazine-2-carboxylate (32)
The reaction was performed with 3-chloro-1,2-benzisothiazole 31 (0.085 g, 0.5 mmol), Rh2(OAc)4 (6.6 mg, 0.015 mmol), 17c (0.115 g, 0.65 mmol) and DCM (5 mL). Purified by flash chromatography (silica gel, 3:1 pentane:diethyl ether) to give 32 as a clear oil (0.140 mg, 88% yield). Rf 0.25 (2:1 pentane:diethyl ether); FTIR (neat): 2966, 1736, 1446, 1434, 1234, 1007, 819, 764, 733, 693 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.65-7.62 (m, 2H), 7.55-7.54 (m, 2H), 7.51-7.46 (m, 2H), 7.34 (m, 3H), 3.85 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 167.6 (C), 137.8 (CH), 136.8 (C), 133.7 (CH), 133.2 (C), 132.3 (CH), 130.3 (CH), 129.2 (CH), 128.2 (CH), 127.0 (CH), 120.5 (C), 117.0 (C), 82.3 (C), 54.3 (CH3); LRMS (EI) m/z (relative intensity): 317 (5) [M]+, 121 (100); HRMS (EI) Calcd for [C16H12ClNO2S]+ 317.0272, Found 317.0265.
Supplementary Material
Synthesis of compounds 9–13, 15, and 8 and the method for structural assignment of 13 and its regioisomer. gHSQC and gHMBC correlations for 16. Supplementary data associated with this article can be found in the online version, at
Acknowledgments
The National Institutes of Health (GM080337) is gratefully acknowledged. J.R.M. thanks the National Institutes of Health for a Ruth L. Kirchstein predoctoral fellowship (DA019287).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Dedicated to Professor John Hartwig on his receipt of the 2007 Young Investigator Tetrahedron Award
1. References and notes
- 1.Doyle MP, McKervey M, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides. New York: John Wiley & Sons, Inc.; 1997. [Google Scholar]
- 2.a) Taber DF, Joshi PV. In: Modern Rhodium-Catalyzed Organic Reactions. Evans PA, editor. Weinheim: Wiley-VCH; 2005. pp. 357–377. [Google Scholar]; (b) Davies HML, Walji AM. In: Modern Rhodium-Catalyzed Organic Reactions. Evans PA, editor. Weinheim: Wiley-VCH; 2005. pp. 301–340. [Google Scholar]; (c) Doyle MP. In: Modern Rhodium-Catalyzed Organic Reactions. Evans PA, editor. Weinheim: Wiley-VCH; 2005. pp. 341–355. [Google Scholar]
- 3.(a) Davies HML, Jin Q. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5472. doi: 10.1073/pnas.0307556101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Davies HML, Jin Q. J. Am. Chem. Soc. 2004;126:10862. doi: 10.1021/ja047185k. [DOI] [PubMed] [Google Scholar]
- 4.Davies HML, Walji AM. Angew. Chem., Int. Ed. 2005;44:1733. doi: 10.1002/anie.200462227. [DOI] [PubMed] [Google Scholar]
- 5.Davies HML, Dai X, Long MS. J. Am. Chem. Soc. 2006;128:2485. doi: 10.1021/ja056877l. [DOI] [PubMed] [Google Scholar]
- 6.Isolation and structure determination Rodriguez AD, Gonzalez E, Huang SD. J. Org. Chem. 1998;63:7083. doi: 10.1021/jo981385v.
- 7.Previous total synthesis Miyaoka H, Honda D, Mitome H, Yamada Y. Tetrahedron Lett. 2002;43:7773.
- 8.Carcache DA, Cho YS, Hua Z, Tian Y, Li Y-M, Danishefsky SJ. J. Am. Chem. Soc. 2006;128:1016. doi: 10.1021/ja056980a. [DOI] [PubMed] [Google Scholar]
- 9.Kaiho T, San-nohe K, Kajiya S, Suzuki T, Otsuka K, Ito T, Kamiya J, Maruyama M. J. Med. Chem. 1989;32:351. doi: 10.1021/jm00122a012. [DOI] [PubMed] [Google Scholar]
- 10.Comins DL, Dehghani A. Tetrahedron Lett. 1992;33:6299. [Google Scholar]
- 11.Scott WJ, Stille JK. J. Am. Chem. Soc. 1986;108:3033. doi: 10.1021/ja00263a015. [DOI] [PubMed] [Google Scholar]
- 12.Davies HML. Eur. J. Org. Chem. 1999;9:2459. [Google Scholar]
- 13.(a) Davies HML, Stafford DG, Doan BD, Houser JH. J. Am. Chem. Soc. 1998;120:3326. [Google Scholar]; (b) Davies HML. Advances in Cycloaddition. Vol. 5. JAI Press, Inc; 1999. pp. 119–164. [Google Scholar]; (c) Reddy RP, Davies HML. J. Am. Chem. Soc. 2007;129:10312. doi: 10.1021/ja072936e. [DOI] [PubMed] [Google Scholar]
- 14.Davies HML, Townsend RJ. J. Org. Chem. 2001;66:6595. doi: 10.1021/jo015617t. [DOI] [PubMed] [Google Scholar]
- 15.(a) Ceccherelli P, Curini M, Marcotullio MC, Rosati O, Wenkert E. J. Org. Chem. 1994;59:2882. doi: 10.1021/jo951440p. [DOI] [PubMed] [Google Scholar]; (b) Padwa A, Dean DC, Osterhout MH, Precedo L, Semones MA. J. Org. Chem. 1994;59:5347. [Google Scholar]
- 16.(a) Kohler EP, Blatt AH. J. Am. Chem. Soc. 1928;50:1217. [Google Scholar]; (b) King JF, Durst T. Can. J. Chem. 1962;40:882. [Google Scholar]; (c) Kashima C, Tsuda Y, Imada S, Nishio T. J. Chem. Soc., Perkin Trans. 1. 1980:1866. [Google Scholar]
- 17.Crow WD, Gosney I, Ormiston RA. J. Chem. Soc., Chem. Commun. 1983:643. [Google Scholar]
- 18.Baum JS, Shook DA, Davies HML. Synth. Commun. 1987;17:1709. [Google Scholar]
- 19.Davies HML, Hansen T, Churchill MR. J. Am. Chem. Soc. 2000;122:3063. [Google Scholar]
- 20.Taddei M, Ricci I. Synthesis. 1986:633. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Synthesis of compounds 9–13, 15, and 8 and the method for structural assignment of 13 and its regioisomer. gHSQC and gHMBC correlations for 16. Supplementary data associated with this article can be found in the online version, at