

Abstract
Measles virus (MV) is one of the most infectious pathogens known. In spite of the existence of a vaccine, approximately 350,000 deaths/year result from MV or associated complications. Anti-measles compounds could conceivably diminish these statistics and provide a therapy that complements vaccine treatment. We recently described a high-throughput screening hit compound 1 (16677) against MV-infected cells with the capacity to eliminate viral reproduction at 250 nM by inhibiting the action of the virus’s RNA-dependent RNA polymerase complex (RdRp). The compound, 1-methyl-3-(trifluoromethyl)-N-[4-sulfonylphenyl]-1H-pyrazole-5-carboxamide, carries a critical CF3 moiety on the 1,2-pyrazole ring. Elaborating on the preliminary structure-activity (SAR) study, the present work presents the synthesis and SAR of a much broader range of low nanomolar non-peptidic MV inhibitors and speculates on the role of the CF3 functionality.1
Keywords: measles virus, high-throughput screening (HTS), pyrazoles
Introduction
Paramyxoviruses are negative stranded RNA viruses, most of which are highly contagious airborne pathogens that spread via the respiratory route. Members of this viral family include major human and animal pathogens such as measles virus (MV), human parainfluenza viruses (HPIV), mumps virus, respiratory syncytial virus and Newcastle disease virus.1 Despite the existence of an effective live-attenuated vaccine,2 MV remains a serious threat to human health globally, accounting for approximately 0.35 million deaths annually.3 While most of these cases occur in developing countries with limited access to vaccination, measles outbreaks still occur in some developed countries that have failed to maintain high vaccine coverage rates. According to recent estimates, 20 million cases of measles occur annually worldwide. Current outbreaks in the developed world have affected Japan in the Western Pacific Region, Nova Scotia, New Brunswick, Prince Edward Island, and Ontario, Canada, and several parts of Western Europe.4,5a-c
This is partially due to declining herd-immunity as a result of reduced vaccination coverage resulting from parental concerns about vaccination safety.6 Vaccination coverage below protective levels in areas of the developing world and continued viral activity in the developed world make desirable the development of novel therapeutics that can be used for the rapid control of local outbreaks and improved case management to limit severe outcomes of infection.
To date, Ribavirin, (1a) is the only drug available for the treatment of some paramyxovirus infections7,8 It has been used experimentally for the treatment of measles but with limited efficacy.9 More recently, benzimidazo-thiazole derivatives (1b) have been reported to be more potent and less cytotoxic compared with Ribavirin against the Leningrad 16 strain, when assessed in Vero (African green monkey kidney carcinoma) cells. The most active compound in this series demonstrated a selectivity ratio (CC50/EC50) of 245.5 compared with 14.4 for Ribavirin.10
In previous work we reported the structure-based development of a MV entry inhibitor, N-(5-amino-2-hydroxy-phenyl)-2-phenyl-acetamide (AS-4, Figure 1), with an EC50 of 260 nM against the MV-Edmonston (MV-Edm) strain.11, 12 Since this compound proved to be unstable, we developed AS-48 with an EC50 of 0.6-3.0 μM (Figure 1) as a shelf-stable alternative.11(b) Further attempts to increase the activity within this series of compounds proved to be problematic. As a consequence, we broadened our search by turning to cell-based high throughput screening (HTS) to capture small molecules capable of netting both entry inhibitors as well as compounds operating against other proteins critical for viral infection and reproduction. The exercise identified 1-methyl-3-(trifluoromethyl)-N-[4-(pyrrolidinylsulfonyl)phenyl]-1H-pyrazole-5-carboxamide 1 (16677) (Figure 1) (EC50 = 250 nM, ) as a well-behaved, target-specific inhibitor of MV replication.13 Bypassing the fusion protein, 1 represents a first-in-class non-nucleoside inhibitor of the MV RNA-dependent RNA polymerase (RdRp) complex. Following re-synthesis of the hit compound 1, by coupling 1-methyl-3-trifluoro-pyrazole-5-acetyl chloride with 4-amino-prolidinyl sulfonamide 7a in the presence of pyridine,14 previous preliminary structural modifications focused on the right side of the molecule as depicted in Figure 2a.15 In the present work, we describe optimization of antiviral activity for compound 1 scaffold by manipulation of the pyrazole (A) and pyrrolidine (D) rings.
Figure 1.

Structures of compounds AS-4, AS-48 and 1
Figure 2.

(a) Sectors of compound 1 utilized as an SAR template; (b) Aromatic ring analogs of the compound 1 pyrazole ring, 2a-o.
Preparation of analogs around the HTS hit compound 1
Modification of the pyrazole ring
The first stage of hit optimization introduced a variety of aromatic rings as 1-methyl-3-trifluoromethyl pyrazole replacements. Compounds 2a-o (Figure 2b, Table S1) can be readily prepared by coupling the corresponding acetyl chlorides with 4-amino-prolidinyl sulfonamide 7a in the presence of pyridine. 15 Preliminary efforts were also devoted to modifying the 1-methyl-3-trifluoromethyl-pyrazole ring. For example, the corresponding 4-bromo-1-methyl-3-(trifluoromethyl)-1H-pyrazole-5-carboxylic acid was prepared by the protocol reported by Schlosser and treated with oxalyl chloride to obtain acetyl chloride 3. The latter was further coupled with sulfonamide 7a to give the 4-bromo-substituted analog 4. (Scheme 1)
Scheme 1.

Synthesis of 1-methyl-3-(trifluoromethyl)-4-bromo-N-[4-(pyrrolidinylsulfonyl)-phenyl]-1H-pyrazole-5-carboxamide (4)a
In the present work, 3-trifluoromethyl pyrazole was treated with NaH in DMF, followed by regioselective benzylation and alkylation to obtain 1-benzyl or alkyl-3-trifluoromethylpyrazole. The compound was then subjected to a routine deprotonation-carboxylation sequence before coupling with aniline 7c to obtain the desired compounds 9. (Scheme 2)
Scheme 2.

Synthesis of 1-alkyl-3-(trifluoromethyl)-5-carboxamide 9.a
2-Methoxycarbonylphenyl and 2-carboxyphenyl analogs were also prepared as outlined in Scheme 3. The furan precursor 10 was obtained as previously described16 and converted to ester 11 via the acid chloride/methylation method. Oxidation of the furyl functionality afforded the desired carboxylic acid 12. The latter as its acid chloride was coupled to aniline 7a to afford compound 13. Saponification of ester 13 provided the corresponding acid 14.
Scheme 3.

Synthesis of 1-[2-carboxy-phenyl]-3-trifluoromethyl-pyrazole analogs
Syntheses of 23, 24, 28 and 29 proved to be straightforward by following standard procedures for sulfonamide analogs as illustrated in Schemes 4 and 5. For example, combination of 4-nitro-benzenesulfonyl chloride with pyrrolidine afforded 4-nitro-pyrrolidinyl sulfonamide 6. The latter was reduced either with SnCl2 in EtOAc or by hydrogenation over Pd/C (5%) to obtain 4-amino-prolidinyl sulfonamide 7 in almost quantitative yield. Other secondary amides behave similarly. Coupling of 7 with 1-methyl-3-(trifluoromethyl)-1H-pyrazole-5-carbonyl chloride obtained compound 23. De-protection of Boc-groups with a mixture of TFA and CH2Cl2 (3:1) at room temperature for about 4h delivers free amines 24e and 24f. Compounds 29e and 29f were obtained by hydrolysis of the corresponding esters 28 in 1N HCl. Analogs 15a-r15 (See Table 1) were prepared by using the same procedure as described in Scheme 4.
Scheme 4.

Synthesis of derivatives 23 and 24.
Scheme 5.

Synthesis of substituted piperidine derivatives 28 and 29.
Table 1.
Antiviral MV EC50’s, cytotoxicities and selectivity indices for 1-methyl-N-(4-(piperidin-1-ylsulfonyl)phenyl)-3-(trifluoromethyl)- 1H-pyrazole-5-carboxamide (15a) and other sulfonamide analogs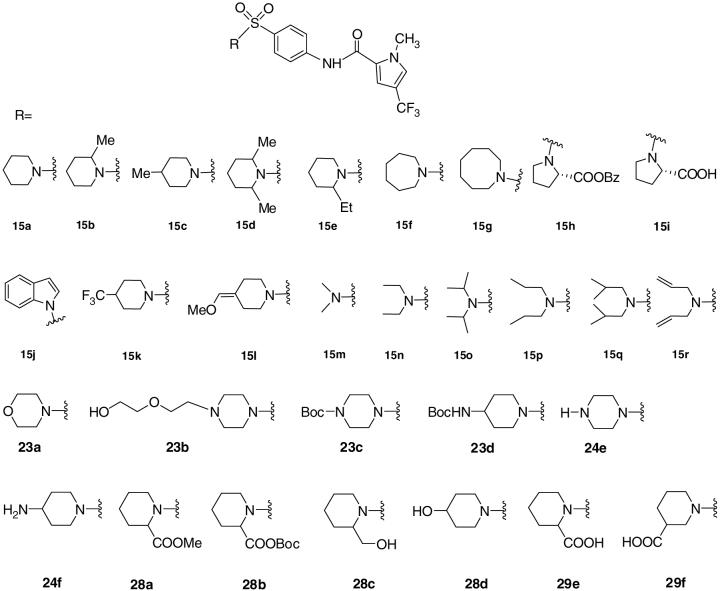
| Entry | EC50 (μM) (MV-Alaska) | CC50 (μM) (Vero cells) | SI (CC50/EC50) | |||
|---|---|---|---|---|---|---|
| ID # | CPE inhibitiona | virus titer reductionc | MTT cytotoxicityd | Trypan blue exclusion assaye | CPE + MTT | Titer + Trypan |
| 4 | >75 | ND | 95 ± 20 | ND | <1.3 | ND |
| 9a | <2.3 | 0.055±0.013 | 30±2.4 | ND | ND | ND |
| 9b | >75 | ND | >300 | ND | ND | ND |
| 9c | >19c | >19b | ND | 19 ± 1 | ND | ND |
| 13 | >75 | >75 | ND | > 300 | ND | ND |
| 14 | >75 | >75 | ND | > 300 | ND | ND |
| 15a f | < 2.3 | 0.014 ± 0.02 | > 300 | 199 ± 27 | > 130 | 14,214 |
| 15b | < 2.3 | 0.029 ± 0.031 | > 300 | 54 ± 3 | > 130 | 1,862 |
| 15c | < 2.3 | 0.035 ± 0.035 | > 300 | 53 ± 4 | > 130 | 1,514 |
| 15d | < 2.3 | 0.014 ± 0.013 | > 300 | 328 ± 28 | > 130 | 23,429 |
| 15e | < 2.3 | 0.087 ± 0.116 | > 300 | 14 ± 2 | > 130 | 160.9 |
| 15f | < 2.3 | 0.005± 0.0003 | > 300 | 425 ± 65 | >130 | 85,000 |
| 15g | < 2.3 | 0.045± 0.034 | 16 ± 0.8 | ND | >7 | ND |
| 15h f | 14 ± 2 | ND | 100 | ND | 7.1 | ND |
| 15i f | 23 ± 10 | ND | > 300 | ND | >13 | ND |
| 15j f | >13b | ND | 13 ± 0.7 | ND | ND | ND |
| 15k | 2.3 ± 0.7 | 0.02 ± 0.02 | 159 ± 12 | ND | 69.1 | ND |
| 15l | > 75 | ND | 92 ± 9.3 | ND | < 1.2 | ND |
| 15m | 6.3 ± 0.6 | ND | > 300 | ND | > 47.6 | ND |
| 15n | <2.3 | 0.019± 0.019 | > 300 | 280 ± 90 | >130 | 14,737 |
| 15o | 3.5± 0.4 | 0.53± 0.02 | >300 | ND | >85.7 | ND |
| 15p | < 2.3 | 0.19± 0.32 | >300 | ND | >130 | ND |
| 15q | >13.8b | ND | 13.8± 0.7 | ND | ND | ND |
| 15r | 3.3± 1.4 | ND | 34± 0.9 | ND | 10.3 | ND |
| 16 | > 15b | ND | 15 ± 0.6 | ND | ND | ND |
| 17 | > 75 | ND | > 300 | ND | ND | ND |
| 18 | > 75 | ND | 286 ± 17 | ND | < 3.8 | ND |
| 19 | > 75 | ND | 84 ± 23 | ND | < 1.1 | ND |
| 20 | > 75 | ND | > 300 | ND | ND | ND |
| 23a f | 43 ± 24 | ND | > 300 | ND | > 7 | ND |
| 23b | >75 | ND | >300 | ND | ND | ND |
| 23c | > 75 | ND | > 300 | ND | ND | ND |
| 23d | 14.1 ± 6.6 | ND | > 300 | ND | > 21.3 | ND |
| 24e | 28 ± 9 | ND | 126 ± 7 | ND | 4.5 | ND |
| 24f f | > 75 | ND | > 300 | ND | ND | ND |
| 28a | 10 ± 5.6 | ND | 136 ± 3 | ND | 13.6 | ND |
| 28b | > 38b | ND | 38 ± 1 | ND | ND | ND |
| 28c | < 2.3 | 0.85 ± 0.05 | 159 ± 40 | ND | > 69 | ND |
| 28d | 6.8 ± 0.9 | 0.57 ± 0.04 | 274 ± 19 | ND | 40.2 | ND |
| 29e | > 75 | ND | > 300 | ND | ND | ND |
| 29f | > 75 | ND | > 300 | ND | ND | ND |
EC50 not determined (ND) when CC50 ≤ 15 μM. Values represent averages of four experiments ± SD; highest concentration assessed 75 μM, lowest concentration assessed 2.3 μM.
No virus inhibition detected at CC50 concentration.
Determined only when CPE inhibition-based EC50 concentration < 2.3 μM. Values represent averages of two to four experiments ± SEM; highest concentration assessed 1 μM. (ND: not determined)
Values represent averages of at least three experiments ± SD; highest concentration assessed 300 μM.
Determined only when virus titer reduction was assessed and MTT-assay based cytotoxicity > 300 μM. (ND: not determined)
Published results15
Qualitative SAR for compound 1 analogs
Disappointedly, all attempts to replace the pyrazole ring with 5- or 6-membered aromatic rings15 or to alter substitutents on the pyrazole ring (e.g. 4, 13 and 14) led either to a decrease or to a complete loss of activity. A striking example is the inactivity of the series of N-alkyl analogs of 1 (EC50 30-150 nM15). Replacement of the N-methyl with N-isopropyl or N-benzyl (9b and c, respectively) leads to disppearance of potency in the virus titer reduction assay. While the N-ethyl pyrazole 9a demonstrates good activity in (EC50 = 55 nM), the compound is highly toxic. In a previous limited exploration of the SAR of 1, we learned that installing a piperidine ring instead of the pyrrolidine ring on the left side of the molecule (Figure 2a, D ring) provided a 100-fold boost in activity while simultaneously delivering very low toxicity.15 To exploit this potency advantage, a variety of alicyclic heterocyclic rings (15a-l) or dialkyl amines (15m-r) were employed as pyrrolidine replacements while retaining the remainder of compound 1 structure (see Table 1).
Piperidine analogs 15a-e and 15k exhibit significant potency improvements by comparison with the original hit. All yield EC50s in the low nM range. The 2-ethyl substitution on the piperidine ring in 15e elicits 2-6 fold less activity than methyl substitution (15b-15d). The seven membered ring variant 15f is 10-fold more potent than the azacyclooctane analog 15g and about 3-fold more active than the piperidine 15a. However, C2-substitution of the pyrrolidine ring with either an ester or a carboxylic acid (15h and 15i, respectively) greatly decreases activity by about 100-fold as does introduction of an indole ring (15j). Interestingly, while the open-chain diethyl amine compound 15n is highly active, the alkyl-enhanced di-isopropyl, dipropyl and diallyl variants 15o, and 15p and 15r lose 10-20 fold potency by comparison. Similarly, 15m and 15q, the dimethyl and di-isobutyl analogs, respectively, exhibit drops in activity.
Additional structural modifications of compound 1 included synthesis of 16-20, compounds that examine structural environments around a 6-membered ring. (Figure 3) Analog 16 moves the nitrogen out of the piperidine ring of 15a and introduces a secondary amine as an H-bond donor, while 17 incorporates a sulfonate moiety instead of a sulfonamide group. Compound 18 incorporates a methyl group on the amide and eliminates possible hydrogen bonding of NH with a nearby amino acid residue. All three compounds 16, 17 and 18 occasion a complete loss of activity. Compounds 19 and 20 switch the sulfonamide para-amide linker in sector C to the meta- and ortho-positions, respectively. Both compounds 19 and 20 similarly show no detectable activity below 75 μM. (Table 1)
Figure 3.

Variation scaffolds of piperidine analogs.
Discovery of four highly active piperidine analogs in the D-sector of 1 (15a-d, see Table 1 for biological data) encouraged us to further examine this center in an effort to retain nM potency while improving solubility within the series. Thus, morpholine derivative 23a, piperazines 23b-d and 24e and piperidine rings decorated with hydrophilic groups 24f, 28a-d and 29e-f were prepared. Unfortunately, with the exception of 28c bearing a CH2OH group alpha to nitrogen (EC50 850 nM), none of the compounds are significantly active. Clearly, the incorporation of a hydrophilic group in this sector is detrimental to MV blockade (Table 1). This may reflect impaired passive diffusion through the plasma membrane, since inhibitors of the RdRp complex activity need access to the cytosol of the infected cells.
Molecular Field Topology Analysis (MFTA) QSAR
The SAR discussion in the previous section is based on qualitative observations. In an effort to put this on a more quantitative footing, the MFTA method developed by Palyulin et al.17, 18 was applied to the MV RdRp complex inhibitor series to generate a quantitative structure-activity relationship (QSAR). MFTA performs a topological analysis for a compound series, generates a molecular supergraph with descriptor values for each compound mapped at each atom vertex, and finishes with PLS-based correlation statistics to generate predictive QSAR models. In the present case, the log(1/EC50) values for CPE inhibition without standard deviations were used as the numerical biological endpoints. To incorporate the virus titer data into the correlation, the titer values of compounds with CPE inhibitory activities less than 2.3 μM were scaled to an equivalent CPE EC50 by a conversion factor of 10 μM EC50 per 1 μM virus titer. The latter was based on the EC50s and virus titers for 15o and 28d. Where compounds exhibit activities greater than 75 μM, a value of 150 μM was assigned. Compounds with a cytoxicity greater than 300 μM or a theraputic index greater than 4 were selected for MFTA. To simplify the analysis within a congeneric series, only compounds with a scaffold similar to the pyrazole ring A, amide linker B, and sulfonamide linker C were included. Thus, the training set consisted of 26 compounds:1, 2j, 2o, 9a, 13, 14, 15a, 15c-15i, 15k, 15m, 15o, 15p, 15r, 23a, 23d, 28c, and 28d. Three compounds from a previous publication15 were also included because they are instructive members of this congeneric series (cf. S5, S7 and S8, Figure S1). On the other hand, 9b, 23c, 24f, 29e, and 29f were inactive and consistent outliers. Thus, these compounds were excluded from the training set. For the test set, 7 compounds were used 2n, 2k, 15b, 15n, 24e, 28a, and S6 (Figure S1).15
A variety of single local descriptors and sets of local descriptors were applied to the training set, generating a series of different models with varying predictive Q2 values based on MFTA’s leave-25%-cross-validation. The best results were obtained with descriptors for Gasteiger-Marsili atomic charge (Q), the effective environment van der Waal radius (Re), and group lipophilicity based on the sum of the Ghose-Crippen atomic contributions for an atom and attached hydrogens (Lg). This QSAR model generated a correlation (Figure 6) with the following statistics: N = 26, number of PLS factors NF = 2, R = 0.88, R2 = 0.77, RMSE = 0.49, and Q2 = 0.66. Adding additional descriptors did not substantially improve the correlation. To verify that MFTA has not simply fortuitously found a non-predictive model, a Y-randomization test38 was performed on the training set data in ten separate trials. In each trial the activity data was randomly re-ordered, and a QSAR model generated. Each random data set delivered the same mean, variance, and molecular supergraph, but no real correlation of activity to structure. The resulting QSAR models generally had a good correlation (Ravg = 0.83, Rmax = 0.92), but poor predictive ability (Q2avg = 0.21, Q2max = 0.42). This lends confidence that the experimental model with Q2 = 0.66 is not an artifact of the method.
Figure 6.

Descriptor impact on activity shown on the molecular supergraph. The QSAR model predicts that increasing the descriptor property at the red and blue positions results in an increase and decrease in activity, respectively.
Predictions for the seven compound test set with the present model were made by first using MFTA to map the test set compounds with assigned descriptor values to the previously generated molecular supergraph, followed by prediction with the PLS model based on the training set (Figure 5). Test set activities are predicted reasonably well with R = 0.67 and R2 = 0.45. For three of the compounds, the errors are somewhat substantial (about 30-40% of the training set activity span in log units), but for the other 4 compounds the error is less than 20%. The mean absolute error for the complete test set is 0.74 log units, and the RMS error of prediction is 0.86 log units.
Figure 5.
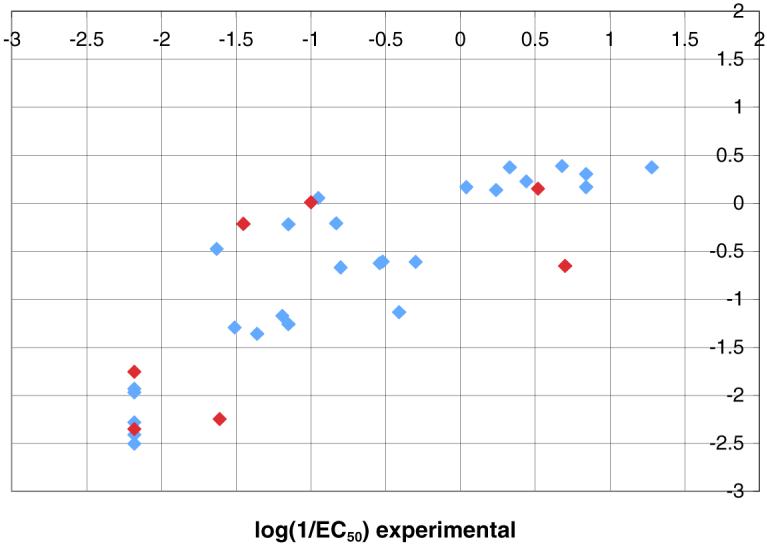
MFTA correlation for the 26 compounds of the training set (blue) and the 7 compounds of the test set (red) based on charge (Q), effective vdW radius (Re) and lipophilicity (Lg) descriptors.
MFTA was used to visualize the descriptors’ contributions to the correlation by means of a color-coded molecular supergraph (Figure 6). Positions colored brown and red suggest that an increase in descriptor property in that part of the molecule would increase activity, red positions having more effect than brown. Conversely, light blue and blue positions suggest that an increase in descriptor property would decrease activity, blue positions having more effect than light blue. At uncolored positions, either the descriptor property is not correlated to activity, or there is insufficient diversity in the training set for MFTA to develop correlation to activity. The reader should note that the attempt to display the range of substituents in 2D in sector D in Figure 6 leaves the impression of disorder around the sulfonyl amine group. This is inaccurate and misleading, since the correlation of descriptor property to activity is limited by the property range of the substituents in the training set. The graph accurately reflects how subtle changes in the atoms’ properties affect activity within the bounds of diversity in the training set.
Interpreting the descriptor impact graphs supports qualitative trends in the structure-activity data. Substituted rings and alkyl chains attached to the sulfonamide have a positive impact on activity if they increase the lipophilicity and decrease the charge on this part of the molecule. However, the QSAR model is able to parse some additional complexity in the structure-activity data by noting that increasing charge in the 3- and 4-positions of the pyrrolidine ring, as well as the para-position of the 6-membered ring, has a favorable effect on activity. In general, substituents connected to the pyrazole ring have a favorable impact on activity, if they contribute an increase in size and lipophilicity. Increasing charge at the pyrazole ring can either positively or negatively effect activity, depending on the substituent position.
Discussion
Mechanistic and functional characterization of the high-throughput screening hit 1 has demonstrated that the compound represents a first-in-class non-nucleoside inhibitor of MV RdRp complex activity.13 We have developed an SAR by structural manipulation of 1 within the four sectors highlighted in Figure 2a. Previously published work15 has been focused mainly on the phenyl sulfonamide unit, the amide linker and the pyrazole ring in sector A. A variety of modifications within sector A-C either essentially abolish anti-MV activity or result in high cytotoxicity. However, a highly potent analog has been identified by replacing the pyrrolidine ring in compound 1 with a piperidine. Subsequent SAR development of the current series was centered on modification of the heterocyclic rings in sectors A and C. Disappointedly, all attempts to replace the N-methyl group on the pyrazole ring (9a-c, 13 and 14) or to add other functionality on the same ring leads to complete loss of activity or high toxicity (9a). Furthermore, replacement of the pyrazole ring CF3 group with Me or t-Bu also causes significant loss of activity, a factor explained below by the beneficial electrostatic properties of the CF3 group.
Intriguingly, maintaining the structure of the hit molecule 1 in sectors A-C, while modifying the pyrrolidine ring in D, delivers highly active and non-cytotoxic compounds. (Table 1, 15a-g, 15k) The seven-membered ring analog 15f was the most potent series member with an EC50 around 5 nM and no detectable cytotoxicity at concentrations below 300 μM as determined by two independent assays (reflected by an approximate selectivity index (CC50/EC50) of 85,000). Further increases in the ring size, however, resulted in a reduction of antiviral activity by about 10-fold (Table 1, 15g). The open chain diethyl amine analog 15n also exhibited excellent antiviral activity. However, other amine substitutions (15o and 15q) caused a complete loss of activity.
Further optimization of the lead compound sought to increase solubility and improve cellular bioavailability. Such structural changes may, of course, reduce membrane-permeability and consequently compromise antiviral activity. In attempts to resolve the conflicting structural requirements, compounds with a piperazine ring (24e) and polar groups on the piperidine (e.g. 24d,f and 28a-f, Figure 2a, sector D) were prepared. Indeed, assessment of biological activity returned a correlation between increasing hydrophilicity and decreasing inhibitory potency. Within this series, all compounds were completely inactive with the exception of piperidines 28c and 28d, which harbor 2-CH2OH and 4-OH groups, respectively; both returning only moderate activity.
Interactions between ligand CF3 groups associated proteins
Compound 1, the most potent high throughput screening (HTS) inhibitor against MV13 identified from a 34,000 compound pool,19 carries a trifluoromethyl group on the pyrazole ring. Substitution of the CF3 with a methyl or a t-butyl group completely abrogates the inhibitory activity of the compound.15 In order to gain insight into the possible role of the pyrazole CF3 as an enhancer of MV inhibition and to investigate more generally the structural influence of CF3 groups in protein-bound ligands, we searched the Protein Data Bank (PDB) for such protein-ligand complexes using Relibase+ and ReliView. A total of 132 CF3-complemented ligands was returned from the search. Less than a dozen trifluoromethyl groups are located in hydrophobic environments, while 15 are directed into the aqueous layer surrounding the proteins. Carbon-fluorine bonds in 117 complexes were judged to participate in at least one CF---HX (X = O, N or S, r(F---H) 1.8-2.0 Å) interaction in which F---X distances range from 2.7-3.6 Å. It has been argued that fluorine rarely engages in hydrogen bonds in small molecule X-ray crystal structures.20 However, in protein pockets where ligands are immobilized by a variety of forces, they appear to be more common. F---X distances beyond 3.0 Å can be regarded as dipole-dipole interactions that most likely provide small stabilizing contributions (≤ 1 kcal/mol) for the observed binding poses. An example is presented in Figure 7 showing the heterocyclic terminus of the rhinovirus blocker pleconaril encased in a hydrophobic pocket (PDB code 1NCR21). One of the CF3 fluorine atoms experiences a short contact with the backbone NH of Tyr144.
Figure 7.
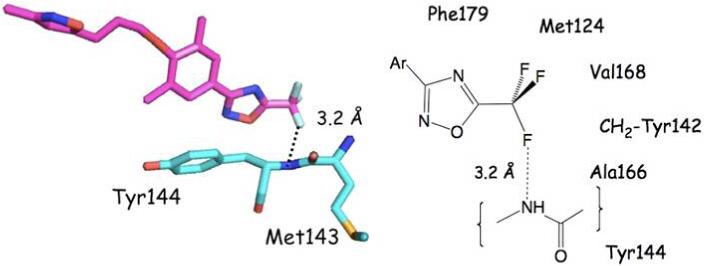
The 1,2,4-oxadiazole terminus of the rhinovirus blocker pleconaril sited in a hydrophobic pocket as determined by X-ray crystallography (1NCR). One of the CF3 fluorine atoms engages in a slightly elongated hydrogen bond with the backbone NH of Tyr144.
A subset of 28 cases involve cationic Arg, Lys and His residues that direct N-H bonds toward fluorine. One interesting CF3-protein association (1P2S22) locates the fluorinated moiety of 2,2,2-trifluoroethanol (CF3CH2OH) between Arg and Lys side chains in H-Ras 166 as shown in Figure 8.
Figure 8.
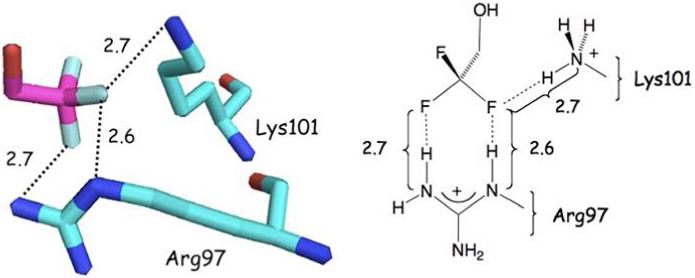
H-Ras 166 in 50% 2,2,2 triflouroethanol (1P2S) sequesters the CF3 group of a solvent molecule between Arg97 and Lys101 side chains by bidentate and monodentate hydrogen bonds, respectively. Distances are between F---N atoms. Units for numerical values are Å.
The topographical representation shows that Arg97 forms a bidentate contact with two fluorine atoms, while Lys101 associates with one fluorine in the same pair; all of which involve F---H distances of 2.6-2.7 Å. The latter are 0.3-0.4 Å smaller than the sum of the van der Waals radii (N 1.55, F 1.47 Å).23 These charge-dipole interactions are well known to operate as controlling forces in the conformational preferences of fluorinated piperidines. In particular, the fluorine in protonated 3-fluoropiperidines universally adopts an axial orientation contrary to intuition based on cyclohexane conformational analysis.24, 25
Figure 9 depicts a trifluoromethyl ketone inhibitor of human leukocyte elastase (1EAS26). The compound enjoys a weak interaction with the SH of Cys42 and a stronger association with His57. Intriguingly, four protein ligands incorporate CF3-subsituted pyrazole rings very similar to that found in compound 1. Three of these (1X7A,27 1CX228 and 6COX29) juxtapose the CF3 fluorines and a single Arg residue located among hydrophobic residues. This observation has been exploited to explain the activity of a series of anti-inflammatory COX inhibitors.30 Porcine Factor IXa complexed to 1-{3-[amino(imino)methyl]phenyl}-N-[4-(1H-benzimidazol-1-yl)-2-fluorophenyl]-3-(trifluoromethyl)-1H-pyrazole-5-carboxamide) (1X7A) is illustrative (Figure 10). The fourth CF3-substituted pyrazole ligand, a nanomolar inhibitor of carbonic anhydrase (1OQ530) positions the trifluoromethyl group in a hydrophobic cavity on the surface of the protein partially exposed to the aqueous layer surrounding the protein.
Figure 9.
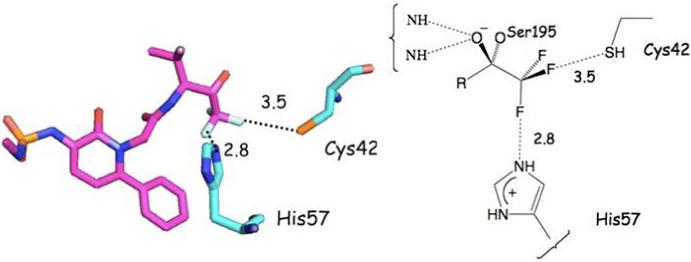
A trifluoromethyl ketone inhibitor of human leukocyte elastase (1EAS) illustrating an F---HS hydrogen bond and a charge dipole interaction between a ligand C-F bond and His57. Units for numerical values are Å.
Figure 10.
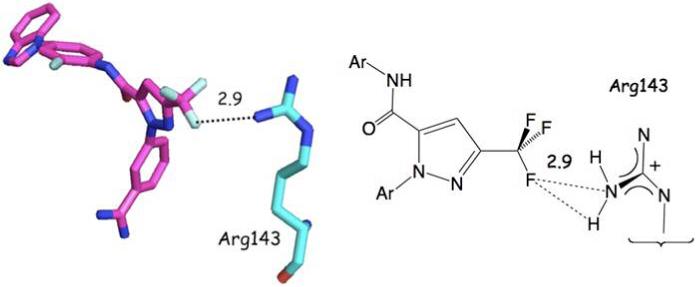
Porcine Factor IXa Complexed to 1-{3-[amino(imino)methyl]phenyl}-N-[4-(1H-benzimidazol-1-yl)-2-fluorophenyl]-3-(trifluoromethyl)-1H-pyrazole-5-carboxamide) (1X7A). The CF3 of the pyrazole ligand interacts tightly with the cationic terminus of the Arg143 side chain. Units for numerical values are Å.
If the binding site for pyrazole in the MV RNA-dependent RNA polymerase were hydrophobic or solvent exposed, then substitution of CF3 in compound 1 with CH3 (Figure 2b, 2n) is unlikely to cause complete loss of activity as observed. Consequently, we conclude that the pyrazole-CF3 in the highly active MV blockers examined in this work (Table 1, 15a-g, 15k&15n) is likely docked in a pocket that house either a cationic Arg, His or Lys. This prediction is being actively pursued to identify MV blockers which maintain potency but carry improved solubility and bioavailability properties.
Conclusions
High throughput screening followed by chemical synthesis has provided the first highly potent small molecule blockers of the measles virus accompanied by low cell toxicity. Synthetic modification of the 1 lead reveals that both potency gains and structural diversity resides primarily in sector D (Figure 2a). This behavior is captured by a three-descriptor QSAR model developed using molecular field topology analysis (MFTA) and implies the sector D sulfonyl amines reside in a rather tight hydrophobic cavity. Behavior at the other end of the molecule (sector A), suggests that the pyrazole-CF3 most likely sits in a pocket housing cationic Arg, His or Lys. These speculations are being actively pursued in an effort to identify MV blockers which maintain potency but carry improved solubility and bioavailability properties suitable for evaluating various MV strains and closely related viruses in animal models
Supplementary Material
Figure 4.

MFTA molecular supergraph (left) formed by the 26 compounds of the training set. The superposition of compound 1 (right) is highlighted to exemplify the alignment
Acknowledgements
This work was supported by a research grant from the American Lung Association, and Public Health Service grants AI056179 and AI071002 from NIH/NIAID (to RKP) and by the National Institutes of Health 1 U54 HG003918 (to JPS). We are grateful to Ernest Murray for synthetic assistance
Footnotes
- MV
- measles virus
- CPE
- cytopathic effect
- MTT
- [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]
- MV-Edm
- measles virus Edmonston strain
- RdRp
- RNA-dependent RNA polymerase
- CC50
- compound concentration that returns 50% cytotoxicity
- EC50
- compound concentration that returns 50% inhibition
- SI
- selectivity index, defined as CC50/EC50
- SAR
- structural activity relationship
- QSAR
- quantitative structural activity relationship
- MFTA
- molecular field topology analysis
References
- (1).Lamb RA, Kolakosky D. Paramyxoviruses: The viruses and their replication. In: Fields BN, Knipe DM, Howley PM, editors. Fundamental Virology. Lippencott-Raven; Philadelphia, PA: 1996. pp. 577–604. [Google Scholar]
- (2).Hilleman MR. Current overview of the pathogenesis and prophylaxis of measles with focus on practical implications. Vaccine. 2001;20:651–665. doi: 10.1016/s0264-410x(01)00384-x. [DOI] [PubMed] [Google Scholar]
- (3).CDC Progress in Reducing Measles Mortality - Worldwide, 1999-2003. MMWR. 2005;54:200–203. [PubMed] [Google Scholar]
- (4).Gans HA, Arvin AM, Galinus J, Logan L, DeHovitz R, Maldonado Y. Deficiency of the humoral immune response to measles vaccine in infants immunized at age 6 months. JAMA. 1998;280:527–532. doi: 10.1001/jama.280.6.527. [DOI] [PubMed] [Google Scholar]
- (5)(a).Polack FP, Lee SH, Permar S, Manyara E, Nousari HG, Jeng Y, Mustafa F, Valsamakis A, Adams RJ, Robinson HL, Griffin DE. Successful DNA immunization against measles: neutralizing antibody against either the hemagglutinin or fusion glycoprotein protects rhesus macaques without evidence of atypical measles. Nat. Med. 2000;6:776–781. doi: 10.1038/77506. [DOI] [PubMed] [Google Scholar]; (b) Wichmann O, Hellenbrand W, Sagebiel D, Santibanez S, Ahlemeyer G, Vogt G, Siedler A, van Treeck U. Large measles outbreak at a German public school, 2006. Pediatr. Infect. Dis. J. 2007;26:782. doi: 10.1097/INF.0b013e318060aca1. [DOI] [PubMed] [Google Scholar]; (c) Chironna M, Prato R, Sallustio A, Martinelli D, Germinario C, Lopalco P, Quarto M. Genetic characterization of measles virus strains isolated during an epidemic cluster in Puglia, Italy. Virol J. 2007;21:90. doi: 10.1186/1743-422X-4-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Jansen VA, Stollenwerk N, Jensen HJ, Ramsay ME, Edmunds WJ, Rhodes CJ. Measles outbreaks in a population with declining vaccine uptake. Science. 2003;301:804. doi: 10.1126/science.1086726. [DOI] [PubMed] [Google Scholar]
- (7).Shigeta Shiro, Shuichi Mori, Masanori Baba, Masahiko Ito, Honzumi Ken, Nakamura Kiyoto, Oshitani Hitoshi, Numazaki Yoshio, Matsuda Akira, et al. Antiviral activities of ribavirin, 5-ethynyl-1-beta -D-ribofuranosylimidazole-4-carboxamide, and 6′-(R)-6′-C-methylneplanocin A against several ortho- and paramyxoviruses. Antivir. Chem. Chemother. 1992;36:435–9. doi: 10.1128/aac.36.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Gabrielsen Bjarne, Phelan Michael J., Barthel-Rosa Luis, Cathy, Huggins John W., Kefauver Deborah F., Monath Thomas P., Ussery Michael A., Chmurny Gwendolyn N., et al. Synthesis and antiviral evaluation of N-carboxamidine-substituted analogs of 1-beta-D-ribofuranosyl-1,2,4-triazole-3-carboxamidine hydrochloride. J. Med. Chem. 1992;35:3231–3238. doi: 10.1021/jm00095a020. See. [DOI] [PubMed] [Google Scholar]
- (9).Barnard DL. Inhibitors of measles virus. Antivir. Chem. Chemother. 2004;15:111–9. doi: 10.1177/095632020401500301. [DOI] [PubMed] [Google Scholar]
- (10).Pokrovskii AG, Il’icheva TN, Kotovskaya SK, Romanova SA, Charushin VN, Chupakhin ON. Fluorinated Derivatives of Benz[4,5]imidazo[1,2-b][1,3] thiazole—Inhibitors of Reproduction of Measles Virus. Doklady Biochem. Biophys. 2004;398:285–287. doi: 10.1023/B:DOBI.0000046638.99138.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11)(a).Plemper RK, Erlandson KJ, Lakdawala AS, Sun A, Prussia AJ, Boonsombat J, Aki-Sener E, Yalcin I, Yildiz I, Temiz-Arpaci O, Tekiner B, Liotta DC, Snyder JP, Compans RW. Proc. Natl. Acad. Sci. USA. 2004;101:5628–5633. doi: 10.1073/pnas.0308520101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sun A, Prussia A, Zhan W, Murray EE, Doyle J, Cheng LT, Yoon JJ, Radchenko EV, Palyulin VA, Compans RW, Liotta DC, Plemper RK, Snyder JP. J Med. Chem. 2006;49:5080–5092. doi: 10.1021/jm0602559. [DOI] [PubMed] [Google Scholar]
- (12).Plemper RK, Lakdawala AS, Gernert KM, Snyder JP, Compans RW. Biochemistry. 2003;42:6645–6655. doi: 10.1021/bi034385k. [DOI] [PubMed] [Google Scholar]
- (13).White LK, Yoon J-J, Lee JK, Sun A, Du Y, Fu H, Snyder JP, Plemper RK. Nonnucleoside Inhibitor of Measles Virus RNA-Dependent RNA Polymerase Complex Activity. Antimic. Agents Chemother. 2007;51:2293–2303. doi: 10.1128/AAC.00289-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Schlosser M, Volle J, Leroux F, Schenk K. Switchable Reactivity: The site-Selective Functionalization of Trifluoromethyl Substituted Pyrazoles. Eur. J. Org. Chem. 2002:2913–20. [Google Scholar]
- (15).Sun A, Chandrakumar N, Yoon J-J, Plemper RK, Snyder JP. Nonnucleoside inhibitors of the measles virus RNA-Dependent RNA polymerase activity: Synthesis and in vitro evaluation. Bioorg. Med. Chem. Lett. 2007;17:5199–5203. doi: 10.1016/j.bmcl.2007.06.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Pruitt JR, Pinto DJP, Galemmo RA, Alexander RS, Rossi KA, Wells BL, Drummond S, Bostrom LL, Burdick D, Bruckner R, Chen H, Smallwood A, Wong PC, Wright MR, Bai S, Luettgen JM, Knabb R, Lam PYS, Wexler RR. Discovery of 1-(2-Aminomethylphenyl)-3-trifluoromethyl-N-[3-fluoro-2′-(aminosulfonyl)[1,1′-biphenyl)]-4-yl]-1H-pyrazole-5-carboxamide (DPC602), a Potent, Selective, and Orally Bioavailable Factor Xa Inhibitor. J. Med. Chem. 2003;46:5298–5315. doi: 10.1021/jm030212h. [DOI] [PubMed] [Google Scholar]
- (17).Palyulin VA, Radchenko EV, Zefirov NS. Molecular Field Topology Analysis Method in QSAR Studies of Organic Compounds. J. Chem. Inf. Model. 2000;40:659–667. doi: 10.1021/ci980114i. [DOI] [PubMed] [Google Scholar]
- (18).Mel’nikov AA, Palyulin VA, Zefirov NS. Generation of molecular graphs for QSAR studies. Dokl. Chem. 2005;402:81–85. doi: 10.1021/ci700156f. [DOI] [PubMed] [Google Scholar]
- (19).ChemDiv, Inc. 6605 Nancy Ridge Drive. San Diego, CA: 92121; http://www.chemdiv.com/en/products/screening/ [Google Scholar]
- (20).Dunitz JD, Taylor R. Organic Fluorine Hardly Ever Accepts Hydrogen Bonds. Chem. Eur. J. 1997;3:89–98. [Google Scholar]; Dunitz JD. Organic Fluorine: Odd Man Out. Chem. Bio. Chem. 2004;5:614–621. doi: 10.1002/cbic.200300801. [DOI] [PubMed] [Google Scholar]
- (21).Zhang Y, Simpson AA, Ledford RM, Bator CM, Chakravarty S, Skochko GA, Demenczuk TM, Watanyar A, Pevear DC, Rossmann MG. Structural and virological studies of the stages of virus replication that are affected by antirhinovirus compounds. J.Virol. 2004;78:11061–11069. doi: 10.1128/JVI.78.20.11061-11069.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Buhrman GK, de Serrano V, Mattos C. Organic Solvents Order the Dynamic Switch II in Ras Crystals. Structure. 2003;11:747–751. doi: 10.1016/s0969-2126(03)00128-x. [DOI] [PubMed] [Google Scholar]
- (23).Bondi A. van der Waals Volumes and Radii. J. Phys. Chem. 1964;68:441–451. [Google Scholar]
- (24).Lankin DC, Chandrakumar NS, Rao SN, Spangler DP, Snyder JP. Protonated 3-Fluoropiperidines: An Unusual Fluoro Directing Effect and a Test for Quantitative Theories of Solvation. J. Am. Chem. Soc. 1993;115:3356–3357. [Google Scholar]; Snyder JP, Chandrakumar NS, Sato H, Lankin DC. The Unexpected Diaxial Orientation of cis-3,5-Difluoropiperidine in Water: A Potent CF- - -NH Charge-Dipole Effect. J. Am. Chem. Soc. 2000;122:544–545. [Google Scholar]; Lankin DC, Grunewald GL, Romero FA, Oren IY, Snyder JP. The NH---FC Dipole Orientation Effect for Pendant Exocyclic CH2F. Org. Lett. 2002;4:3557–3560. doi: 10.1021/ol026358c. [DOI] [PubMed] [Google Scholar]; Sun A, Lankin DC, Hardcastle K, Snyder JP. 3-Fluoropiperidines and N-Methyl-3-fluoropiperidinium Salts: The Persistence of Axial Fluorine. Chem. Eur. J. 2005;11:1579–1591. doi: 10.1002/chem.200400835. [DOI] [PubMed] [Google Scholar]
- (25).Jensen HH, Lyngbye L, Jensen A, Bols M. Stereoelectronic Substituent Effects in Polyhydroxylated Piperidines and Hexahydropyridazines. Chem. Eur. J. 2002;8:1218–1226. doi: 10.1002/1521-3765(20020301)8:5<1218::aid-chem1218>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]; Jensen HH, Bols M. Stereoelectronic Substitutent Effects. Acc. Chem. Res. 2006;39:259–265. doi: 10.1021/ar050189p. [DOI] [PubMed] [Google Scholar]
- (26).Bernstein PR, Andisik D, Bradley PK, Bryant CB, Ceccrelli C, Damewood JR, Jr., Earley R, Edwards PD, Feeney S, Gomes BC, Kosmider BJ, Steelman GB, Thomas RM, Vacek EP, Veale CA, Williams JC, Wolanin DJ, Woolson SA. Nonpeptidic Inhibitors of Human Leukocyte Elastase. 3. Design, Synthesis, X-ray Crystallographic Analysis, and Structure-Activity Relationships for a Series of Orally Active 3-Amino-6-phenyl-2-pyridinyl Trifluoromethyl Ketones. J. Med. Chem. 1994;37:3313–3326. doi: 10.1021/jm00046a016. [DOI] [PubMed] [Google Scholar]
- (27).Smallheer JM, Alexander RS, Wang J, Wang S, Nakajima S, Rossi KA, Smallwood A, Barbera F, Burdick D, Luettgen JM, Knabb RM, Wexler RR, Jadhav PK. SAR and factor IXa crystal structure of a dual inhibitor of factors IXa and Xa. Bioorg.Med.Chem.Lett. 2004;14:5263–5267. doi: 10.1016/j.bmcl.2004.08.034. [DOI] [PubMed] [Google Scholar]
- (28).Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, Gildehaus D, Miyashiro JM, Penning TD, Seibert K, Isakson PC, Stallings WC. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996;384:644–648. doi: 10.1038/384644a0. [DOI] [PubMed] [Google Scholar]
- (29).Singh SK, Vobbalareddy S, Kalleda SR, Rajjak SA, Casturi SR, Datla SR, Mamidi RNVS, Mullangi R, Bhamidipati R, Ramanujam R, Akella V, Yeleswarapu KR. 2-Hydroxymethyl-4-[5-(4-methoxyphenyl)-3-trifluoromethyl-1H-1-pyrazolyl]-1-benzenesulfonamide (DRF-4367): an orally active COX-2 inhibitor identified through pharmacophoric modulation. Org. Biomol. Chem. 2004;2:2442–2450. doi: 10.1039/B402787F. [DOI] [PubMed] [Google Scholar]
- (30).Weber A, Casini A, Heine A, Kuhn D, Supuran CT, Scozzafava A, Klebe G. Unexpected nanomolar inhibition of carbonic anhydrase by COX-2-selective celecoxib: new pharmacological opportunities due to related binding site recognition. J. Med. Chem. 2004;47:550–557. doi: 10.1021/jm030912m. [DOI] [PubMed] [Google Scholar]
- (31). Reaction of 4-nitro-benzene sulfonyl chloride with pyrrolidine in the presence of pyridine afforded 4-nitro-prolidinyl sulfonamide. Reduction with tin dichloride dihydrate in ethyl acetate provided 4-amino-prolidinyl sulfonamide 7a in excellent yield (90%)
- (32).Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Hendlich M, Bergner A, Gunther J, Klebe G. Relibase - Design and Development of a Database for Comprehensive Analysis of Protein-Ligand Interactions. J. Mol. Biol. 2003;326:607–620. doi: 10.1016/s0022-2836(02)01408-0. [DOI] [PubMed] [Google Scholar]
- (34).Günther J, Bergner A, Hendlich M, Klebe G. Utilising Structural Knowledge in Drug Design Strategies: Applications Using Relibase. J. Mol. Biol. 2003;326:621–636. doi: 10.1016/s0022-2836(02)01409-2. [DOI] [PubMed] [Google Scholar]
- (35).Bergner A, Günther J, Hendlich M, Klebe G, Verdonk M. Use of Relibase for retrieving complex 3D interaction patterns including crystallographic packing effects. Biopolymers (Nucleic Acid Sci.) 2002;61:99–110. doi: 10.1002/1097-0282(2001/2002)61:2<99::AID-BIP10075>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- (36).Guex N, Diemand A, Peitsch MC, Schwede T. Swiss-PDB Viewer. http://www.expasy.org/spdbv/
- (37).DeLano WL. The PyMOL Molecular Graphics System (2002) DeLano Scientific. San Carlos; CA, USA: http://www.pymol.org. [Google Scholar]
- (38).Wold S, Eriksson L. In Chemometric Methods in Molecular Design. In: van de Waterbeemd H, editor. Methods and Principles in Medicinal Chemistry. Vol. 2. VCH Publishers, Inc.; New York, NY: 1995. pp. 309–317. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.