Abstract
A series of α-amino acid derivatives containing the 2,3-dihydroindole or octahydroindole core have been chemoenzymatically synthesized in good overall yields and high enantiomeric purity under mild reaction conditions using lipases for the introduction of chirality. Candida antarctica lipase type A has shown excellent activity and high enantiodiscrimination ability towards the two cyclic amino esters used as substrates. The selectivity of the process proved to be greatly dependent on the alkoxycarbonylating agent. Thus, the enzymatic kinetic resolution of methyl indoline-2-carboxylate has been successfully achieved using 3-methoxyphenyl allyl carbonate, whereas (2R,3aR,7aR)-benzyl octahydroindole-2-carboxylate required the less reactive diallyl carbonate.
1. Introduction
The preparation of optically active compounds is a great challenge for organic chemists, and enantiomerically pure amines and amino acid derivatives represent one of the most important targets.1 Nowadays, enzyme-mediated processes are becoming usual techniques for enantioselective transformations and, among the biocatalysts of synthetic interest, lipases have gained much attention over the last two decades due to their ease of manipulation, low cost and wide substrate tolerance.2 Traditionally, most lipase-catalyzed enantioselective reactions have corresponded to the acylation of alcohols or hydrolysis of esters, whereas the preparation of chiral amides or carbamates have been much less investigated in spite of the fact that mild reaction conditions are required for the formation of these nitrogenated compounds.3 Examples of enzymatic kinetic or dynamic kinetic resolutions of cyclic secondary amines described in the literature are rare,4 even though they are present in a large number of biologically relevant systems and are useful building blocks for the preparation of many valuable products.
Among cyclic secondary amines, the indoline (2,3-dihydroindole) subunit is found in a wide range of natural products and biologically active compounds.5 Only a few catalytic methods for the production of optically active indolines, either through non-enzymatic6 or enzymatic4b,4g,4j methodologies, have been reported. Kurokawa and Sugai described the enzymatic kinetic resolution of methyl N-Boc-indoline-2-carboxylate through a Candida antarctica lipase B (CAL-B) enantioselective hydrolysis, which yielded both the substrate and the product in enantiopure form.4g In our ongoing research on the synthesis of optically active secondary amines, we observed4j a different reactivity of Candida antarctica lipase A (CAL-A) and CAL-B towards the N-protection of indolines. Thus, whereas CAL-A exhibited a high enantiopreference towards 2-substituted derivatives, CAL-B showed remarkable stereoselectivity towards 3-methyl indoline. This fact suggests that CAL-A prefers to act at sterically hindered positions,7 while the approach of indolines to the active site of CAL-B demands sterically non-hindered situations.8
Octahydroindole-2-carboxylic acid (Oic), the completely hydrogenated analogue of indoline-2-carboxylic acid, is the core structure of numerous compounds with applications in medicinal chemistry. For example, it is present in marine compounds with antithrombotic properties;9 in the dipeptide perindropil (a potent antihypertensive drug used in the prevention of cardiovascular disorders such as heart failure);10 and in the prolyl oligopeptidase inhibitor S 17092, a compound with antiamnesic and cognitive-enhancing properties.11 Recently, some of us reported12 the synthesis of enantiomerically pure (2S,3aS,7aS)- and (2R,3aR,7aR)-Oic derivatives using preparative chiral HPLC,12a and that of the (2R,3aS,7aS) isomer by selective formation of a trichloromethyloxazolidinone.12b The only enzymatic process for producing optically active Oic derivatives was described by Hirata13 and is based on the enzymatic hydrolysis of N-protected octahydroindole-2-carboxylates.
Our interest in indoline-2-carboxylic acid and octahydroindole-2-carboxylic acid to be used as proline analogues together with our experience in the enzymatic resolution of secondary amines, prompted us to apply procedures to the isolation of these compounds in enantiomerically pure form. Herein, we report the application of a straightforward and highly selective lipase-catalyzed methodology for the alkoxycarbonylation of the secondary amino group in these compounds.
2. Results and Discussion
Initially, we focused on the enzymatic resolution of methyl indoline-2-carboxylate rac-2. This compound was obtained in excellent yield by treatment of commercially available indoline-2-carboxylic acid rac-1 with thionyl chloride in refluxing methanol (Scheme 1). Prior to the enzymatic resolution assays, compound rac-2 was transformed into carbamate rac-3 by reaction with allyl chloroformate, in order to establish the appropriate analytical conditions for HPLC separation of the two enantiomers.
Scheme 1.

Chemical synthesis and enzymatic kinetic resolution of methyl indoline-2-carboxylate
The enzymatic kinetic resolution of rac-2 was then attempted using Candida antarctica lipase type A (CAL-A) from different sources and Candida antarctica lipase type B (CAL-B) as biocatalysts. The data are summarized in Table 1. Based on previous results with 2-substituted indoline derivatives,4j 3-methoxyphenyl allyl carbonate14 4a was initially selected as the alkoxycarbonylating reagent for enzyme activity screening, which was carried out at 30 °C using dry tert-butyl methyl ether (TBME) as the solvent and a 1:2.5 amino ester to carbonate ratio (entries 1–4). For all the tested CAL-A preparations, a complete enantioselectivity was observed in the alkoxycarbonylation process. Lipase from Biocatalytics showed a lower reaction rate than the ones from Roche or Codexis, although all of them reached 50% conversion (entries 1–3). An excellent selectivity was also observed for CAL-B, but low conversions were reached in longer reaction times (entry 4). Finally, the reaction was carried out using CAL-A from Codexis and commercially available diallyl carbonate 4b (entries 5 and 6). The use of this reagent avoids the need for the purification step to separate the 3-methoxyphenol released during the enzymatic process from the reaction crude. However, this change resulted in lower kinetics (entry 5), while higher temperatures led to a decrease in the enantioselectivity value (entry 6) probably due to the inactivation of the enzyme.
Table 1.
Lipase-catalyzed kinetic resolution of amino ester rac-2 using carbonates 4a–b (ratio 1:2.5) in TBME
| Entry | Enzyme | R | T (°C) | t (h) | eeP (%)a | eeS (%)a | c (%)b | Ec |
|---|---|---|---|---|---|---|---|---|
| 1 | CAL-A Biocatalytics | 3-MeO-C6H4 | 30 | 20 | >99 | 98 | 50 | >200 |
| 2 | CAL-A Roche | 3-MeO-C6H4 | 30 | 8 | >99 | 98 | 50 | >200 |
| 3 | CAL-A Codexis | 3-MeO-C6H4 | 30 | 4 | >99 (92)d | >99 (86)d | 50 | >200 |
| 4 | CAL-B | 3-MeO-C6H4 | 30 | 60 | 99 | 85 | 46 | >200 |
| 5 | CAL-A Codexis | CH2=CHCH2 | 30 | 74 | 92 | 16 | 15 | 29 |
| 6 | CAL-A Codexis | CH2=CHCH2 | 45 | 49 | 94 | 8 | 8 | 35 |
Calculated by HPLC
c = eeS/(eeS+eep)
E = ln[(1−c)×(1−eeP)]/ln[(1−c)×(1+eeP)]
Isolated yields in brackets.
Thus, under the optimal conditions, we were able to isolate one enantiomer of both the substrate and the product in enantiomerically pure form and good yield (Table 1, entry 4). The absolute configuration of the amino ester 2 remaining from the enzymatic kinetic resolution was determined by comparing its HPLC retention time15 with that of a sample of enantiopure (S)-2 obtained by esterification of commercially available (S)-indoline-2-carboxylic acid, (S)-1. This allowed us to assign an (R)-configuration to the unreacted amine 2 and an (S)-stereochemistry to the isolated carbamate 3 (Scheme 1).
We next undertook the development of adequate chemoenzymatic procedures to obtain Oic derivatives in enantiopure form (Scheme 2). Hydrogenation of racemic indoline-2-carboxylic acid rac-1 following our previously described methodology,12a afforded rac-5, which was transformed into the corresponding methyl rac-6a and benzyl rac-6b esters using standard procedures. Reaction with allyl chloroformate led to carbamates rac-7a and rac-7b, respectively, which were analyzed by HPLC to find adequate conditions for the separation of their enantiomers.
Scheme 2.

Chemical synthesis and enzymatic kinetic resolution of Oic derivatives
For the enzymatic kinetic resolution of rac-6a, CAL-A from Codexis was initially chosen as the biocatalyst, since it gave the best results in the resolution of 2, and different carbonates were tested. No reaction was observed when using dibenzyl carbonate, even when the temperature was raised to 45 °C. In contrast, the more reactive methoxyphenyl allyl carbonate 4a led to the formation of racemic carbamate 7a (Table 2, entry 1). The use of diallyl carbonate 4b, which is known to exhibit intermediate reactivity, allowed the formation of 7a with excellent selectivity, although only 20% conversion was reached after 23 h at 30 °C (entry 2). An increase in the temperature (entry 3) did not result in a higher conversion but rather in a loss of enzyme activity, as observed before for 2.
Table 2.
Lipase-catalyzed kinetic resolution of amino esters rac-6a–b using carbonates 4a–b (ratio 1:2.5) in TBME
| Entry | Enzyme | R1 | R2 | T (°C) | t (h) | eeP (%)a | eeS (%)a | c (%)b | Ec |
|---|---|---|---|---|---|---|---|---|---|
| 1 | CAL-A Codexis | Me | 3-MeO-C6H4 | 30 | 23.5 | ---- | ---- | 100 | ---- |
| 2 | CAL-A Codexis | Me | CH2=CHCH2 | 30 | 23 | >99 | 25 | 20 | >200 |
| 3 | CAL-A Codexis | Me | CH2=CHCH2 | 45 | 48 | >99 | 13 | 12 | >200 |
| 4 | CAL-A Codexis | Bn | CH2=CHCH2 | 30 | 57 | 88 | >99 | 53 | 121 |
| 5 | CAL-A Biocatalytics | Bn | CH2=CHCH2 | 30 | 72 | 98 (91)d | >99 (90)d | 50 | >200 |
Calculated by HPLC
c = eeS/(eeS+eep)
E = ln[(1−c)×(1−eeP)]/ln[(1−c)×(1+eeP)]
Isolated yields in brackets.
Our attention was then focused on the resolution of amino ester rac-6b that, with it being more sterically hindered than rac-6a, could be a better substrate for the active site of CAL-A.7 Indeed, the enzymatic kinetic resolution of rac-6b using CAL-A from Codexis was found to proceed with very high enantiopreference, thus allowing recovery of the remaining starting material in enantiopure form (entry 4). Alternatively, when CAL-A from Biocatalytics was used, a 50% conversion was reached, with both the product and the starting material being obtained in almost enantiomerically pure form and excellent isolated yields (entry 5). The absolute configuration of both compounds was assigned after transformation of the remaining substrate into a derivative of known stereochemistry. Thus, the unreacted enantiomerically pure amino ester 6b was subjected to N-tert-butoxycarbonyl protection, as previously described for the racemic compound,12a to afford 8b (Scheme 2); the HPLC retention time of the latter compound was found to be identical16 to that of a sample of (2R,3aR,7aR)-8b prepared in our previous work.12a Thereafter, we concluded that the unreacted amino ester 6b has a (2R,3aR,7aR)-configuration and the carbamate formed in the enzymatic process 7b is (2S,3aS,7aS).
3. Conclusions
In conclusion, the development of a practical synthetic route for the preparation of optically active indoline-2-carboxylic acid and octahydroindole-2-carboxylic acid derivatives has been successfully achieved through enzymatic kinetic resolution using lipases in organic solvents. The different lipases tested exhibited distinct behaviors in the alkoxycarbonylation process, depending on the structure of the starting materials. In all cases, CALA has been found to be the most efficient biocatalyst. The choice of the appropriate allyl carbonate also proved crucial to reach a good selectivity and conversion. Under the best conditions, all substrates and products were isolated in good yields and enantiomerically pure form (98% ee in one case). This methodology represents an excellent alternative to those previously described for the preparation of these biologically significant compounds.
4. Experimental
4.1. General
Candida antarctica lipase type B (CAL-B, Novozyme 435, 7300 PLU/g) was a gift from Novo Nordisk Co. Candida antarctica lipase type A was acquired from different commercial sources:17 CAL-A Chirazyme L-5, c–f, lyophilized, 1000 U/g using tributyrin from Roche, CAL-A immobilized NZL-101, 5.0 U/g using 1-butanol and ethyl laurate from Codexis; CAL-A IMB-104, 2.6 U/mg using tributyrin from Biocatalytics). All other reagents were purchased from Aldrich and used without further purification. Solvents were distilled over an adequate desiccant under nitrogen. Flash chromatography was performed using silica gel 60 (230–240 mesh). High performance liquid chromatography (HPLC) analyses were carried out at 20 °C in a Hewlett Packard 1100 chromatograph under the conditions specified for each substrate and with UV monitoring at 210 nm. IR spectra were recorded on using NaCl plates or KBr pellets in a Perkin-Elmer 1720-X FT. 1H, 13C NMR, DEPT, and 1H-13C heteronuclear experiments were obtained using AC-200 (1H, 200.13 MHz and 13C, 50.3 MHz), AC-300 (1H, 300.13 MHz and 13C, 75.5 MHz), DPX-300 (1H, 300.13 MHz and 13C, 75.5 MHz) or AV-400 (1H, 400.13 MHz and 13C, 100.6 MHz) Bruker spectrometers. The chemical shifts are given in delta (δ) values and the coupling constants (J) in Hertz (Hz).18 HP1100 chromatograph mass detector was used to record mass spectra experiments (MS) through APCI+ or ESI+ experiments. Measurement of the optical rotation was done in a Perkin-Elmer 241 polarimeter.
4.2. Synthesis of methyl indoline-2-carboxylate rac-2
To a solution of compound rac-1 (1.0 g; 6.13 mmol) in methanol (100 mL) under a nitrogen atmosphere, thionyl chloride (671 µL, 9.19 mmol) was added dropwise and the reaction mixture was heated at reflux for 2 h. The solvent was removed and the resulting residue was suspended in saturated aqueous NaHCO3 and extracted with CH2Cl2 (3 × 30mL). The organic fractions were combined, dried and evaporated to dryness. The crude product was purified by flash chromatography (30% EtOAc/hexane) to afford 1.01 g of rac-2 as an oil (97%). Rf (40% EtOAc/hexane): 0.44; IR (NaCl): ν 3335, 2950, 2356, 1732, 1684, 1652, 1609, 1531, 1487, 1436, 1313, 1257, 1207, 1151, 992, 773, 748 cm−1; 1H-NMR (CDCl3, 300.13 MHz): δ 7.02-7.11 (m, 2H, H4+H6), 6.71-6.79 (m, 2H, H5+H7), 4.39 (dd, J= 9.0, 6.0 Hz, 1H, H2), 3.80 (s, 3H, H10), 3.28-3.45 (m, 2H, 2H3); 13C-NMR (CDCl3, 75.5 MHz): δ 174.5 (C8), 149.9 (C7a), 127.6 (C5), 126.5 (C3a), 124.4 (C4), 119.4 (C6), 110.0 (C7), 59.7 (C2), 52.4 (C10), 33.6 (C3). MS (APCI+, m/z): 200 [(M+Na)+, 100%], 178 [(M+H)+, 12%]. HPLC elution times: (S)-2 15.7 min, (R)-2 17.1 min; conditions: 25 × 0.46 cm ID Chiralcel OD column, eluent n-hexane/ethanol (97:3) at 0.8 mL/min flow rate.
4.3. Synthesis of methyl N-(allyloxycarbonyl)indoline-2-carboxylate rac-3
Pyridine (14.5 µL, 0.18 mmol) was added to a solution of amino ester rac-2 (30 mg, 0.17 mmol) in dry CH2Cl2 (1.13 mL) under a nitrogen atmosphere and the mixture was cooled in an ice bath. Allyl chloroformate (15.3 µL, 0.18 mmol) was added dropwise and the system was allowed to warm up to room temperature. After stirring for 4 h, no starting material was detected by TLC analysis. The solvent was evaporated under reduced pressure and the residue obtained was purified by flash chromatography (30% EtOAc/hexane) to afford 40 mg of carbamate rac-3 as a pale yellow oil (90%). Rf (40% EtOAc/hexane): 0.51; IR (NaCl): ν 3336, 2954, 2365, 2342, 1756, 1701, 1648, 1604, 1530, 1487, 1400, 1268, 1205, 826, 749 cm−1; 1H-NMR (CDCl3, 400.13 MHz): δ 7.70 (t, J= 8.0 Hz, 1H, H7), 7.43 (d, J= 8.3 Hz, 1H, H4), 7.33 (t, J= 15.1 Hz, 1H, H5), 7.16 (t, J= 15.1 Hz, 1H, H6), 6.00-6.25 (m, 1H, H14), 5.25-5.60 (m, 2H, H15), 5.05-5.15 (m, 1H, H2), 4.70-5.00 (m, 2H, H13), 3.90 (s, 3H, H10), 3.69 (t, J = 12.0 Hz, 1H, H3), 3.30 (d, J = 15.0 Hz, 1H, H3); 13C-NMR (CDCl3, 100.6 MHz): δ 172 (C8), 152 (C11), 142.1 (C7a), 132.1 (2C, C14 and C3a), 127.9 (C5), 124.3 (C4), 122.9 (C6), 117.7 (C15), 114.7 (C7), 66.0 (C13), 59.9 (C2), 52.9 (C10), 32.8 (C3); MS (APCI+, m/z): 300 [(M+K)+, 34%], 284 [(M+Na)+, 100%], 262 [(M+H)+, 51%]. HPLC elution times: (S)-3 12.3 min, (R)-3 14.9 min; conditions: 25 × 0.46 cm ID Chiralcel OD column, eluent n-hexane/ethanol (97:3) at 0.8 mL/min flow rate.
4.4. Typical procedure for the enzymatic kinetic resolution of methyl indoline-2-carboxylate rac-2
To a suspension of compound rac-2 (50 mg, 0.28 mmol) and lipase (100 mg) in TBME (1.9 mL) kept under a nitrogen atmosphere was added the corresponding carbonate 4a–b (0.70 mmol) and the system was shaken at 30 °C and 250 rpm. The course of the reaction was followed by HPLC till conversions were around 50% (see Table 1). The mixture was then filtered and the filtrate was concentrated under reduced pressure. The crude product was purified by flash chromatography (eluent gradient 5–15% EtOAc/hexane) to give (R)-2 {[α]D 20 = −47.3 (c 0.28, CH2Cl2) for >99% ee} and (S)-3 {[α]D20 = −106.5 (c 1.2, CH2Cl2) for >99% ee}.
4.5. Synthesis of (2S*,3aS*,7aS*)-methyl octahydroindole-2-carboxylate rac-6a
Thionyl chloride (1.18 mL, 16.18 mmol) was added dropwise to a solution of rac-5 (1.32 g, 7.81 mmol) in dry methanol (20 mL) at 0° C. The resulting solution was stirred at room temperature for 24 h. The solvent was evaporated to dryness and the resulting residue was solved in water and lyophilized. The white solid obtained was partitioned between saturated aqueous NaHCO3 (25 mL) and CH2Cl2 (50 mL). The organic layer was separated and the aqueous phase was further extracted with CH2Cl2 (2 × 50 mL). The combined organic layers were dried over anhydrous MgSO4, filtered and evaporated to afford rac-6a as an oil (1.20 g; 6.56 mmol; 84%). Rf (5% MeOH/CH2Cl2): 0.24; IR (NaCl): ν 3319, 3033, 2927, 2856, 1733, 1454, 1191, 750 cm−1; 1HNMR (CDCl3, 400.13 MHz): δ 3.81 (dd, J = 10.0, 6.0 Hz, 1H, H2), 3.74 (s, 3H, 3H10), 3.09 (m, 1H, H7a), 2.38 (bs, 1H, NH), 2.17 (ddd, J= 12.8, 10.0, 6.8 Hz, 1H, H3), 2.03 (m, 1H, H3a), 1.74-1.60 (m, 3H, H3+2H7), 1.52-1.30 (m, 3H, H4+H5+H6), 1.30-1.15 (m, 3H, H6+H4+H5); 13C-NMR (CDCl3, 100.6 MHz): δ 176.2 (C8); 58.4 (C2); 58.0 (C7a); 52.0 (C10); 37.9 (C3a); 35.6 (C3); 27.9 (C7); 27.0 (C4); 23.4 (C5); 21.5 (C6); MS (APCI+, m/z): 184 [(M+H)+, 100%], 124 [(M-CO2Me)+, 32%]. HPLC elution times: first enantiomer 8.7 min, second enantiomer 10.7 min; conditions: 25 × 0.46 cm ID Chiralcel OD column, eluent n-hexane/2-propanol (95:5) at 0.8 mL/min flow rate.
4.6. Synthesis of (2S*,3aS*,7aS*)-benzyl octahydroindole-2-carboxylate rac-6b
To a solution of rac-5 (1.00 g, 5.92 mmol) in toluene (21 mL) p-toluenesulphonic acid monohydrate (1.63 g, 8.59 mmol) and benzyl alcohol (2.30 mL, 22.21 mmol) were added. The resulting system was heated at reflux for 4 h using a Dean-Stark trap. After evaporation to dryness, the residue was triturated with diisopropyl ether and the white solid formed was collected by filtration. It was then partitioned between saturated aqueous NaHCO3 (25 mL) and CH2Cl2 (50 mL). The organic layer was separated and the aqueous phase further extracted with CH2Cl2 (2 × 50 mL). The combined organic extracts were dried over anhydrous MgSO4, filtered and evaporated to afford rac-6b as an oil (1.380 g; 5.33 mmol; 90%). Rf (5% MeOH/CH2Cl2): 0.41; IR (NaCl): ν 3319, 3033, 2927, 2856, 1733, 1454, 1191, 750 cm−1; 1H-NMR (CDCl3, 400.13 MHz): δ 7.37-7.28 (m, 5H, Ph); 5.20 (s, 2H, H10); 3.88 (dd, J = 10.1, 5.9 Hz, 1H, H2); 3.12 (m, 1H, H7a); 2.52 (bs, 1H, NH); 2.20 (ddd, J = 12.8, 10.1, 6.9 Hz, 1H, H3); 2.04 (m, 1H, H3a); 1.77-1.65 (m, 3H, H3+2H7); 1.48 (m, 3H, H4+H5+H6); 1.39 (m, 1H, H6); 1.26 (m, 2H, H4+H5); 13C-NMR (CDCl3, 100.6 MHz): δ 175.7 (C8); 135.7 (CipsoPh), 128.5, 128.2, 128.1 (5C, Ph), 66.7 (C10); 58.6 (C2); 58.1 (C7a); 37.9 (C3a); 35.8 (C3); 27.9 (C7); 27.0 (C4); 23.5 (C5); 21.5 (C6). MS (APCI+, m/z): 260 [(M+H)+, 100]. HPLC elution times: (2S,3aS,7aS)-6b 8.2 min, (2R,3aR,7aR)-6b 9.6 min; conditions: 25 × 0.46 cm ID Chiralcel OD column, eluent n-hexane/2-propanol (90:10) at 0.8 mL/min flow rate.
4.7. Synthesis of carbamates rac-7a–b
Allylchloroformate (18.0 µL, 0.17 mmol) was added dropwise to a solution of the amino ester rac-6a–b (0.15 mmol) in dry CH2Cl2 (1.03 mL) kept under a nitrogen atmosphere at 0 °C and the system was allowed to warm up to room temperature. After stirring for 4 h, no starting material was detected by TLC analysis. The solvent was evaporated under reduced pressure and the residue obtained was purified by flash chromatography (20% EtOAc/hexane) to afford the corresponding carbamate rac-7a–b as an oil (40–49%).
4.7.1. (2S*,3aS*,7aS*)-Methyl N-(allyloxycarbonyl)-octahydroindole-2-carboxylate rac-7a
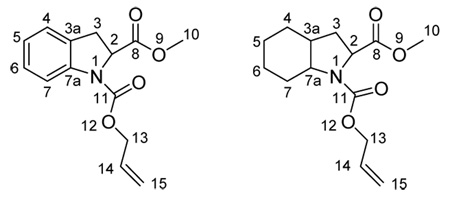
Rf (20% EtOAc/hexane): 0.25; IR (NaCl): ν 2929, 2857, 1753, 1705, 1407, 1345, 1303, 1262, 1176, 1123, 1010 cm−1; 1H-NMR (CDCl3, 300.13 MHz): δ 5.98-5.77 (m, 1H, H11), 5.31-5.13 (m, 2H, 2H15), 4.58-4.44 (m, 2H, H13), 4.33-4.25 (m, 1H, H2), 3.93-3.78 (m, 1H, H7a), 3.73 (d, J = 9.8 Hz, 3H, H10), 2.34-2.29 (m, 1H, H3a), 2.14-1.92 (m, 3H, 2H3+H7), 1.73-1.58 (m, 3H, H5+2H4), 1.50-1.36 (m, 2H, H7+H6), 1.34-1.08 (m, 2H, H5, H6); 13C-NMR (CDCl3, 75.5 MHz): δ (duplicate signals are observed for all carbon atoms) 173.6, 173,4 (C8), 154.4, 153.7 (C11), 133.0, 132.7 (C14), 117.1, 116.8 (C15), 65.7, 65.5 (C13), 59.0, 58.9 (C2), 57.7, 57.3 (C7a), 52.1, 52.0 (C10), 37.0, 36.4 (C3a), 32.5, 31.5 (C3), 27.8, 27.2 (C7), 25.7, 25.6 (C4), 23.6, 23.5 (C5), 20.4, 20.3 (C6); MS (APCI+, m/z): 268 [(M+H)+, 100%], 210 [(M-CO2Me+2H)+, 10%], 182 [(M-CO2CH2CHCH2)+, 31%], 124 [(M-CO2CH2CHCH2-CO2Me+H)+, 5%]. HPLC elution times: first enantiomer 9.6 min, second enantiomer 15.6 min; conditions: 25 × 0.46 cm ID Chiralcel OD column, eluent n-hexane/2-propanol (95:5) at 0.8 mL/min flow rate.
4.7.2. (2S*,3aS*,7aS*)-Benzyl N-(allyloxycarbonyl)-octahydroindole-2-carboxylate rac-7b
Rf (20% EtOAc/hexane): 0.29; IR (NaCl): ν 2929, 2857, 1750, 1703, 1407, 1347, 1303, 1262, 1172, 1124, 989 cm−1; 1H-NMR (CDCl3, 300.13 MHz): δ 7.43-7.28 (m, 5H, Ph), 5.98-5.68 (m, 1H, H14), 5.32-5.08 (m, 4H, 2H10+2H13), 4.67-4.50 (m, 1H, H15), 4.47-4.44 (m, 1H, H15), 4.40-4.30 (m, 1H, H2), 3.95-3.80 (m, 1H, H7a), 2.32-2.23 (m, 1H, H3a), 2.21-2.10 (m, 2H, H3), 2.06-1.96 (m, 1H, H7), 1.73-1.58 (m, 3H, H5+2H4), 1.50-1.38 (m, 2H, H7+H6), 1.32-1.14 (m, 2H, H5+H6); 13C-NMR (CDCl3, 75.5 MHz): δ (duplicate signals are observed for most carbon atoms) 173.0, 172.8 (C8), 154.4, 153.7 (C11), 135.8, 135.6 (Cipso Ph), 133.0, 132.7 (C14), 128.5, 128.4, 128.2, 128.0, 127.9 (5C, Ph), 117.1, 116.9 (C15), 66.6 (C14), 65.7, 65.6 (C13), 59.1, 59.0 (C2), 57.7, 57.3 (C7a), 37.0, 36.4 (C3a), 32.5, 31.4 (C3), 27.8, 27.2 (C7), 25.7, 25.6 (C4), 23.6, 23.5 (C5), 20.4, 20.3 (C6). MS (APCI+, m/z): 366 [(M+Na+H)+, 70%], 344 [(M+2H)+, 100%]. HPLC elution times: (2S,3aS,7aS)-7b 9.1 min, (2R,3aR,7aR)-7b 12.1 min; conditions: 25 × 0.46 cm ID Chiralcel OD column, eluent n-hexane/2-propanol (90:10) at 0.8 mL/min flow rate.
4.8. Typical procedure for the enzymatic kinetic resolution of (2S*,3aS*,7aS*)-methyl and benzyl octahydroindole-2-carboxylate rac-6a–b
To a suspension of the amino ester rac-6a–b (0.12 mmol) and lipase (1:2 in weight with respect to the amino ester) in TBME (1.8 mL) kept under a nitrogen atmosphere was added the corresponding carbonate 4a–b (0.30 mmol) and the system was shaken at the required temperature and 250 rpm. The course of the reaction was followed by HPLC (see Table 2). After filtration, the solution was concentrated under reduced pressure and the crude was purified by flash chromatography (eluent gradient 10–100% EtOAc/hexane) to afford 6a–b {(2R,3aR,7aR)-6b: [α]D20 = +23.1 (c 1.0, CHCl3) for >99% ee} and 7a–b {one enantiomer of 7a: [α]D20 = +30.4 (c 0.76, CHCl3) for >99% ee; (2S,3aS,7aS)-7b: [α]D20 = −42.8 (c 1.0, CHCl3) for 98% ee}.
Acknowledgments
We thank Novo Nordisk Co. for the generous gift of CAL-B (Novozyme 435). This work was supported by the Spanish Ministerio de Educación y Ciencia (Projects CTQ 2007-61126 and CTQ2007-62245) and Gobierno de Aragón (project PIP206/2005 and research group E40). V. G.-F. thanks the Spanish MEC for a personal grant (Ramón y Cajal Program), S. A.-S. thanks the Mexican CONACYT for a pre-doctoral fellowship and M. C. M thanks the Brazilian Agency CAPES for a post-doctoral fellowship. This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number N01-CO-12400. The content of this publication does not necessarily reflect the view of the policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organization imply endorsement by the U.S. Government. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Breuer M, Ditrich K, Habicher T, Hauer B, Kesseler M, Stuermer R, Zelinski T. Angew. Chem. Int. Ed. 2004;43:788–824. doi: 10.1002/anie.200300599. [DOI] [PubMed] [Google Scholar]; (b) Turner NJ, Carr R. In: Biocatalysis in the Pharmaceutical and Biotechnology Industries. Patel RN, editor. Boca Raton: CRC Press; 2007. pp. 743–755. [Google Scholar]
- 2.Gotor-Fernández V, Brieva R, Gotor V. J. Mol. Catal. B: Enzym. 2006;40:111–120. [Google Scholar]
- 3.Gotor-Fernández V, Gotor V. Curr. Org. Chem. 2006;10:1125–1143. [Google Scholar]
- 4.(a) Asensio G, Andreu C, Marco JA. Tetrahedron Lett. 1991;32:4197–4198. [Google Scholar]; (b) Orsat B, Alper PB, Moree W, Mak C–P, Wong C–H. J. Am. Chem. Soc. 1996;118:712–713. [Google Scholar]; (c) Chiou T–W, Chang C-C, Lai C–T, Tai D–F. Bioorg. Med. Chem. Lett. 1997;7:433–436. [Google Scholar]; (d) Wong C–H, Orsat B, Moree WJ, Takayama S. US-5981267. [Google Scholar]; (e) Morgan B, Zaks A, Dodds DR, Liu J, Jain R, Megati S, Njoroge FG, Girijavallabhan VM. J. Org. Chem. 2000;65:5451–5459. doi: 10.1021/jo991513v. [DOI] [PubMed] [Google Scholar]; (f) Liljeblad A, Lindborg J, Kanerva A, Katajisto J, Kanerva LT. Tetrahedron Lett. 2002;43:2471–2474. [Google Scholar]; (g) Kurokawa M, Sugai T. Bull. Chem. Soc. Jpn. 2004;77:1021–1025. [Google Scholar]; (h) Breen GF. Tetrahedron: Asymmetry. 2004;15:1427–1430. [Google Scholar]; (i) Hu S, Tat D, Martinez CA, Yazbeck DR, Tao J. Org. Lett. 2005;7:4329–4331. doi: 10.1021/ol051392n. [DOI] [PubMed] [Google Scholar]; (j) Gotor-Fernández V, Fernández-Torres P, Gotor V. Tetrahedron: Asymmetry. 2006;17:1933–1936. [Google Scholar]; (k) Paál TA, Forró E, Liljeblad A, Kanerva LT, Fülöp F. Tetrahedron: Asymmetry. 2007;18:1428–1433. [Google Scholar]; (l) Stirling M, Blacker J, Page MI. Tetrahedron Lett. 2007;48:1247–1250. [Google Scholar]
- 5.(a) Borschberg H-J. Curr. Org. Chem. 2005;9:1465–1492. [Google Scholar]; (b) Dunetz JR, Danheiser RL. J. Am. Chem. Soc. 2005;127:5776–5777. doi: 10.1021/ja051180l. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ganton MD, Kerr MA. Org. Lett. 2005;7:4777–4779. doi: 10.1021/ol052086c. [DOI] [PubMed] [Google Scholar]; (d) Cacchi S, Fabrizi G. Chem. Rev. 2005;105:2873–2920. doi: 10.1021/cr040639b. [DOI] [PubMed] [Google Scholar]; (e) Taber DF, Tian WJ. Am. Chem. Soc. 2006;128:1058–1059. doi: 10.1021/ja058026j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Davies HML, Manning JR. J. Am. Chem. Soc. 2006;128:1060–1061. doi: 10.1021/ja057768+. [DOI] [PubMed] [Google Scholar]; (g) Palimkar SS, Kumar PH, Lahoti RJ, Srinivasan KV. Tetrahedron. 2006;62:5109–5115. [Google Scholar]
- 6.(a) Yip K-T, Yang M, Law K-L, Zhu N-Y, Yang D. J. Am. Chem. Soc. 2006;128:3130–3131. doi: 10.1021/ja060291x. [DOI] [PubMed] [Google Scholar]; (b) Kuwano R, Kashiwabara M, Sato K, Ito T, Kaneda K, Ito Y. Tetrahedron: Asymmetry. 2006;17:521–535. [Google Scholar]; (c) Arp FO, Fu GC. J. Am. Chem. Soc. 2006;128:14264–14265. doi: 10.1021/ja0657859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Domínguez de María P, Carboni-Oerlemans C, Tuin B, Bargeman G, Van de Meer A, Van Gemert R. J. Mol. Catal. B: Enzym. 2005;37:36–46. [Google Scholar]
- 8.Busto E, Gotor-Fernández V, Gotor V. Adv. Synth. Catal. 2006;348:797–812. [Google Scholar]
- 9.(a) Hanessian S, Del Valle JR, Xue Y, Blomberg N. J. Am. Chem. Soc. 2006;128:10491–10495. doi: 10.1021/ja0625834. [DOI] [PubMed] [Google Scholar]; (b) Ersmark K, Del Valle JR, Hanessian S. Angew. Chem. Int. Ed. 2008;47:1202–1223. doi: 10.1002/anie.200605219. [DOI] [PubMed] [Google Scholar]
- 10.(a) Alfakih K, Hall AS. Expert Opin. Pharmacother. 2006;7:63–71. doi: 10.1517/14656566.7.1.63. [DOI] [PubMed] [Google Scholar]; (b) Curran MP, McCormack PL, Simpson D. Drugs. 2006;66:235–255. doi: 10.2165/00003495-200666020-00010. [DOI] [PubMed] [Google Scholar]; (c) ASCOT Investigators Lancet 2005366895–906.16154016 [Google Scholar]; (d) PROGRESS Collaborative Group. Lancet. 2001;358:1033–1041. doi: 10.1016/S0140-6736(01)06178-5. [DOI] [PubMed] [Google Scholar]; (e) Todd PA, Fitton A. Drugs. 1991;42:90–114. doi: 10.2165/00003495-199142010-00006. [DOI] [PubMed] [Google Scholar]
- 11.(a) Gass J, Khosla C. Cell. Mol. Life Sci. 2007;64:345–355. doi: 10.1007/s00018-006-6317-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Brandt I, Scharpé S, Lambeir A-M. Clin. Chim. Acta. 2007;377:50–61. doi: 10.1016/j.cca.2006.09.001. [DOI] [PubMed] [Google Scholar]; (c) García-Horsman JA, Männistö PT, Venäläinen JI. Neuropeptides. 2007;41:1–24. doi: 10.1016/j.npep.2006.10.004. [DOI] [PubMed] [Google Scholar]; (d) Bellemère G, Vaudry H, Morain P, Jégou S. J. Neuroendocrinol. 2005;17:306–313. doi: 10.1111/j.1365-2826.2005.01308.x. [DOI] [PubMed] [Google Scholar]; (e) Schneider JS, Giardiniere M, Morain P. Neuropsychopharmacology. 2002;26:176–182. doi: 10.1016/S0893-133X(01)00307-4. [DOI] [PubMed] [Google Scholar]; (f) Morain P, Lestage P, De Nanteuil G, Jochemsen R, Robin JL, Guez D, Boyer PA. CNS Drug Rev. 2002;8:31–52. doi: 10.1111/j.1527-3458.2002.tb00214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Morain P, Robin JL, De Nanteuil G, Jochemsen R, Heidet V, Guez D. Br. J. Clin. Pharmacol. 2000;50:350–359. doi: 10.1046/j.1365-2125.2000.00270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Barelli H, Petit A, Hirsch E, Wilk S, De Nanteuil G, Morain P, Checler F. Biochem. Biophys. Res. Commun. 1999;257:657–661. doi: 10.1006/bbrc.1999.0366. [DOI] [PubMed] [Google Scholar]; (i) Portevin B, Benoist A, Rémond G, Hervé Y, Vincent M, Lepagnol J, De Nanteuil G. J. Med. Chem. 1996;39:2379–2391. doi: 10.1021/jm950858c. [DOI] [PubMed] [Google Scholar]
- 12.(a) Sayago FJ, Jiménez AI, Cativiela C. Tetrahedron: Asymmetry. 2007;18:2358–2364. doi: 10.1016/j.tetasy.2008.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sayago FJ, Calaza MI, Jiménez AI, Cativiela C. Tetrahedron. 2008;64:84–91. doi: 10.1016/j.tet.2007.10.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirata N. U.S. Pat. Appl. Publ. 2005106690 A1. CAN 142:462365
- 14.3-Methoxyphenyl allyl carbonate was prepared following the protocol described in reference 4j.
- 15.See Experimental for elution conditions.
- 16.HPLC retention times: (2R,3aR,7aR)-8b, 8.1 min; (2S,3aS,7aS)-8b, 13.3 min. Conditions: 25 × 0.46 cm ID Chiralpak IA column; eluent n-hexane/2-propanol (95:5) at 0.8 mL/min flow rate.
- 17.Three years ago, some companies commercialized CAL-A in different immobilized supports as for example Biocatalytics or Roche. Nowadays, this enzyme is not available from Roche any more. On the other hand, after Biocatalytics was purchased by Codexis on July 2007, this immobilized enzyme is produced in a different form.
-
18.Carbamates 3 and 7a are given as examples of the numerical locants used for NMR assignment: