

Abstract
The palladium-catalyzed cross-coupling of dialkylphosphite with aromatic electrophiles (Hirao coupling) was re-investigated. Some limitations in terms of palladium loadings and substrate reactivity are alleviated with the use of Pd(OAc)2 complexed to 1,1′-bis(diphenylphosphino)ferrocene (dppf) as a ligand. Various aryl and heteroaryl halides are employed to deliver both known and novel substituted phosphonates. The first examples of aryl chloride couplings are also reported.
Keywords: cross-coupling, phosphorus, palladium, heterocycles, aryl halides
Introduction
Hirao and co-workers reported in the early eighties the palladium-catalyzed cross-coupling of dialkylphosphites with aromatic halides (Eq. 1).[1] Since that time, the reaction has been widely employed to prepare functionalized phosphonates.[2] While some transition-metal (Ni, Cu) couplings forming phosphonates were known much earlier,[3] the Hirao reaction has established itself as a fixture in phosphorus-carbon bond formation.[4] More recent work by Buchwald[5] and Fu[6] has aimed at replacing palladium with copper, and some useful results were obtained. One of the reasons for avoiding palladium is its high cost, especially because Hirao’s conditions employ 5 mol% Pd.[1,2] In an extensive survey of the reaction, we could not find any example in which the catalyst loading was lower than 3 mol%, and unfortunately, we were not able to reproduce the yield reported for the latter conditions.[2d] This prompted the present study.

In connection with our ongoing work on the development of phosphorus-carbon bond-forming reactions,[7] as well as the need for heterocyclic phosphonic acids for the development of metal-organic frameworks (MOFs),[8] we decided to re-investigate the Hirao coupling seeking conditions which would expand the scope with respect to the aromatic electrophile substrates, as well as reduce the quantity of palladium catalyst.
Our own cross-coupling investigations with hypophosphorous derivatives[7d,e] had indicated that the ligand plays a crucial role, and 1,3-bis(diphenylphosphino)propane (dppp) or 1,1′-bis(diphenylphosphino)ferrocene (dppf)[9] were found to be best suited. Of course, hypophosphorous compounds also feature the added complication of the potential transfer hydrogenation pathways.[7a,b] While Hirao reported marginal problems with this side-reaction,[1] dialkylphosphites are much less prone to transfer hydrogenation since they are weaker reducing agents than hypophosphorous compounds. Initially, we wanted to prepare pyrazine-2-phosphonic acid for use in the preparation of metal organic frameworks (MOFs), since pyrazine-2-COOH led to interesting results.[10] One requirement for this objective is the preparation of multi-gram quantities of the corresponding phosphonate diester precursor. Additionally, our work with pyridyl phosphonic acids[8a,b] revealed problems with 4-bromopyridine in spite of the literature precedent.[2d]
Reaction of 2-iodopyrazine, or of commercially available 2-chloropyrazine, failed under Hirao’s conditions. Fortunately, we soon discovered that when dppf is used as a ligand, the reaction takes place in good yield with 2-chloropyrazine, and with only 1 mol% Pd. Herein we report our investigations using these improved reaction conditions, which significantly expand upon the original Hirao coupling and its subsequent implementations.
Results and Discussion
Hirao prepared aryl phosphonates from aryl bromides or iodides using Pd(PPh3)4 as the catalyst and triethylamine as the base.[1] The best results were obtained in the absence of a solvent. Subsequent work generally employed similar conditions.[2] However, when we tried applying these conditions with a reduced amount of palladium, yields were significantly reduced. Using 5 mol% Pd is a major drawback especially on large, multi-gram scales. Additionally, some substrates gave low yields or no yield at all in the P-C bond-forming reaction. Several factors appeared important: the amount of palladium employed, as well as the partial triethylamine-promoted dealkylation of the diethyl phosphonate product and diethylphosphite reagent (Eq. 2), which reduced the yield when some product was formed. While Hirao reported isolated examples on the use of diisopropyl phosphite in his seminal work, the importance of this was not discussed.[1] Because dealkylation takes place via SN2, a secondary ester would be better suited, and a more hindered base was also expected to reduce this side-reaction. We therefore investigated the direct cross-coupling reaction of aryl and heteroaryl electrophiles with N,N-diisopropylethylamine and diisopropyl phosphite (i-PrO)2P(O)H[11] since this combination is expected to minimize dealkylation (Eq. 2). The test reaction was the synthesis of dialkyl 2-pyrazine phosphonate. Under Hirao’s conditions, no product formed even with 2-iodopyrazine. After screening various catalyst/ligand systems, Pd(OAc)2 (1 mol%) complexed to dppf gave the most satisfactory result (67% yield) when reacted with 2-chloropyrazine and diethylphosphite [(EtO)2P(O)H] in the presence of triethylamine. However, some dealkylation (ca. 20%) was also observed. The lower amount of catalyst was an encouraging element from an economic point of view, but the reaction still required some improvement. Although Et3N can be used successfully, i-Pr2NEt was superior in order to suppress unwanted dealkylation.


The solvent choice also impacted the reaction. Most of the reactions proceeded well in acetonitrile but in some cases, the use of N,N-dimethylformamide (DMF) provided better results. Once we had determined the best reaction conditions (Eq. 3) for the modified Hirao coupling, we investigated the scope of this process on a wide range of aromatic and non-aromatic halides. The reactions were carried out by adding the dialkylphosphite, aryl halide, base, and then Pd(OAc)2 with dppf. The reaction mixture was then heated at reflux in CH3CN, or at 110 °C in DMF, for 24 h under nitrogen.
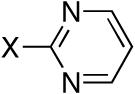
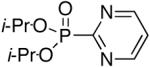
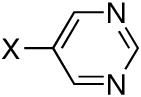
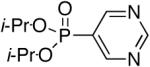
The conversion of nitrogen-containing heteroaromatic substrates proceeded in moderate to excellent isolated yields (Table 1). Gooßen and Dezfuli also tried to improve the Pd-catalyzed cross-coupling reaction of diethyl phosphite with various aryl bromides by using 1.5 equivalent (c-Hex)2NMe as a base, 2 mol% of Pd(OAc)2, and 6 mol% PPh3 in refluxing ethanol.[12] These conditions were limited to some substrates and they were not able to form the corresponding phosphonate from 3-bromopyridine.[12] On the other hand, Hirao obtained this compound in 77% isolated yield using 5 mol% Pd(PPh3)4[1a]. 4-Bromopyridine hydrochloride was reported in the literature to give the desired product in 70% isolated yield with 3 mol% Pd,[2d] but under identical conditions, we only obtained 30%. Following our new protocol with 1 mol% Pd(OAc)2/dppf, in DMF at 110 °C, the product was obtained in good isolated yield (entry 3). However, when reacting 1 equivalent of 2,6-dibromopyridine, only 30% isolated yield of the monobromopyridine phosphonate was obtained due to the expected competing disubstitution (Table 1, entry 4). 2-Chloropyrimidine reacted as well as the bromo analog affording the desired product in good yield (Table 1, entry 5). In the literature, the thermal Arbuzov reaction of 2-halopyrimidine was described,[13] but when we performed this reaction, violent decomposition took place. Fortunately, 2-chloropyrazine (which was our initial entry into this study) reacted very well under our conditions to provide diisopropyl pyrazine phosphonate in 97% isolated yield.
Table 1.
Scope of the cross-coupling with heterocyclic halides
| Entry | Substrate | X | Solvent | Temperature | Product | 31P NMR (ppm) | Isolated yielda (%) |
|---|---|---|---|---|---|---|---|
| 1 |
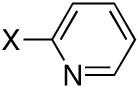
|
Br | CH3CN | Reflux |

|
9.9 | 85 |
| 2 |

|
Br | CH3CN DMF |
Reflux 110 °C |

|
14.6 | 61 48 |
| 3b |
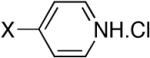
|
Br | DMF | 110 °C |

|
13.6 | 63 |
| 4 |
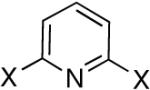
|
Br | DMF | 110 °C |
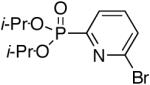
|
7.6 | 30 |
| 5 |
|
Br Cl |
CH3CN DMF |
Reflux 110 °C |
|
4.7 | 67 62 |
| 6 |
|
Br | CH3CN | Reflux |
|
10.9 | 83 |
| 7 |

|
Cl | CH3CN | Reflux |
|
7.7 | 97 |
Isolated yield of pure compounds after chromatography on silica gel.
3 equiv of Et3N used instead of 1.3 equiv of i-Pr2NEt.
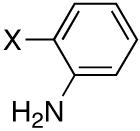
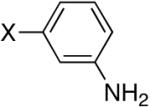
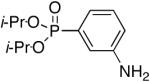
Next, we turned our attention to aniline derivatives. Interestingly, unprotected 2-iodoaniline (Table 2, entry 1) gave satisfactory results in DMF at 110 °C. Similarly, the iodo aniline isomers provided satisfactory results (entries 2 and 3). Savignac and co-workers reported on the failure of the Hirao coupling with unprotected iodoanilines, and prepared the corresponding products using a photostimulated nucleophilic substitution.[14] On the other hand, Buchwald obtained the dibutylphosphonate analog in 86% yield, using CuI (5 mol%) and N,N’-dimethylethylenediamine (20 mol%) as the ligand with 2 equivalents of Cs2CO3 as the base.[5]
Table 2.
Scope reaction on unprotected and Boc-protected anilines
| Entry | Substrate | X | Solvent | Temperature | Product | 31P NMR (ppm) | Isolated yielda (%) |
|---|---|---|---|---|---|---|---|
| 1b |
|
I | DMF | 110 °C |

|
20.2 | 70 |
| 2 |
|
I | CH3CN | Reflux |
|
18.5 | 72 |
| 3 |

|
I I |
CH3CN DMF |
Reflux 110 °C |
|
19.2 | 70 92 |
| 4 |
|
Br | CH3CN | Reflux |

|
17.4 | 81 |
| 5 |

|
Br | CH3CN | Reflux |

|
18.1 | 82 |
Isolated yield of pure compounds after silica gel chromatography.
Reaction conducted in a sealed tube.
Two Boc-protected anilines reacted uneventfully affording the products in good yields (entries 4 and 5) and multi-gram scale syntheses were also performed easily. In addition, the 2-Boc-protected aniline phosphonate (entry 4) was easily isolated by simply washing the solid obtained after work-up.
More examples of cross-coupling were investigated. A variety of electron-poor and electron-rich substrates were also suitable and the aryl phosphonate products were generally isolated in good to excellent yields (Table 3). Reaction with iodobenzene and a reduced amount of palladium (0.1 mol% Pd) still gave an 82% isolated yield after 48 h (compare with Table 3, entry 6). Gratified with the overall results, we then investigated our conditions on a few aryl chlorides. Apparently, there is no literature example on the successful Pd-catalyzed cross-coupling reaction of a dialkyl phosphite with any aryl chloride. We found that reacting diisopropyl phosphite in DMF, at 110 °C, with an activated (electron-poor) ArCl led to the formation of the desired product in low to moderate yields (Scheme 2) using our conditions (1 mol% Pd). Chlorobenzene only gave a 22% NMR yield and the product was not isolated. These results represent our first attempts using the standard conditions reported herein, and although promising, further investigations need to be conducted in order to further improve those yields.
Table 3.
Scope of the reaction with activated and deactivated aryl halides
| Entry | Substrate | X | Solvent | Temperature | Product | 31P NMR (ppm) | Isolated yielda (%) |
|---|---|---|---|---|---|---|---|
| 1 |
|
Br | CH3CN | Reflux |
|
17.8 | 99 |
| 2 |
|
Br | CH3CN | Reflux |

|
18.3 | 61 |
| 3 |

|
Br | DMF | 110 °C |
|
13.9 | 60 |
| 4 |
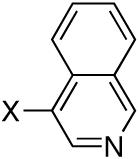
|
Br | DMF | 110 °C |
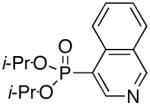
|
19.3 | 46 |
| 5b |
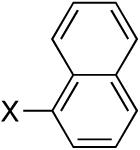
|
TfO | DMF | 110 °C |

|
17.8 | 86 |
| 6 |
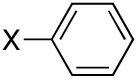
|
I TfO |
CH3CN DMF |
Reflux 110 °C |
|
17.6 | 93 47 |
| 7 |

|
I I Br Br |
CH3CN DMF CH3CN DMF |
Reflux 110 °C Reflux 110 °C |
|
19.9 | 27 51 27 76 |
Isolated yield of pure compounds after chromatography on silica gel.
reaction performed in a sealed tube.
Scheme 2.

Preparation of some phosphonic acids
Finally, the multi-gram scale implementation of the cross-coupling was employed in the preparation of some heterocyclic phosphonic acids (Scheme 2) as potential metal organic framework (MOFs) precursors. Deprotection of the phosphonate diesters using McKenna’s protocol[15] gave excellent results. The phosphonate diester precursors were prepared either using (i-PrO)2P(O)H/i-Pr2NEt, or (EtO)2P(O)H/Et3N, with 0.5-1.0 mol% Pd. Since the compounds are nitrogen-containing heterocycles, these intermediates can be purified by acid-base extraction instead of chromatography. The low yield observed in the case of the 2-pyrimidyl ester intermediate is due to incomplete protonation of the pyrimidine phosphonate during work-up (compare with Table 1, entry 5), but no attempt was made at optimizing this particular compound. Interestingly, while the deprotected pyridine compounds such as 7 generally exist as the zwitterion, the 5-pyrimidine-derived phosphonic acid 5 exists as a mixture of the neutral compound and the zwitterion, and the pyrazine- or 2-pyrimidine phosphonic acids (4 and 6) exist as the neutral compounds since the nitrogen atoms are very weak bases.[16]
Conclusion
We have developed an efficient, versatile and economically attractive alternative to the original Hirao cross-coupling. A wide variety of aryl and heteroaryl phosphonates, including many new compounds, were synthesized using Pd(OAc)2/dppf (1 mol%) as the catalyst. This cross-coupling is successful with various substrates which gave low yields or entirely failed using Hirao’s and other related conditions. The reaction is appropriate for multi-gram scale implementation. In addition, the first examples of palladium-catalyzed P-C bond formation between an activated aryl chloride and a phosphite are reported. While the yields are low to moderate, this indicates the possibility for further improvement. Because dppf is widely available and relatively inexpensive, the present cross-coupling should find some use in the preparation of a variety of phosphonate diesters, and appears competitive to the emerging copper-based systems.
Experimental Section
Representative procedure for cross-coupling of aryl halide substrates
To a solution of diisopropyl phosphite (4.8 mmol, 1.2 equiv) in CH3CN or DMF (both previously dried over 4 Å molecular sieves) (see Tables 1, 2, 3) (15 mL), was added an aryl or heteroaryl halide (4 mmol, 1.0 equiv), N,N-diisopropylethylamine (5.2 mmol, 0.9 mL, 1.3 equiv), Pd(OAc)2 (0.04 mmol, 1 mol%) and dppf (0.044 mmol, 1.1 mol%) at room temperature. The solution was heated for 24 h at reflux in CH3CN, or at 110 °C in DMF, under nitrogen. After cooling to room temperature, the crude mixture was concentrated in vacuo and the residue was partitioned between de-ionized water and EtOAc, followed by extraction of the aqueous phase with EtOAc (3 x). The organic fractions were combined and washed with brine (1 x). Drying and concentration furnished the crude compound, which was purified by radial or column chromatography using mixtures hexanes/EtOAc, unless otherwise specified.
Diisopropyl-2-pyridylphosphonate (Table 1, Entry 1)[17]
Yield: 85%. 1H NMR (CDCl3, 300 MHz) δ 8.81 (d, J = 4.5 Hz, 1H), 7.98 (dd, J = 7.2 Hz, J = 6.6 Hz 1H), 7.84-7.76 (m, 1H), 7.44-7.39 (m, 1H), 4.91-4.78 (m, 2H), 1.40 (d, J = 6.3 Hz, 6H), 1.28 (d, J = 6.3 Hz, 6H); 13C NMR (CDCl3, 75.45 MHz) δ 152.7 (d, JPC = 225 Hz), 150.1 (d, JPCNC = 23 Hz), 135.8 (d, JPCCC = 12 Hz), 127.5 (d, JPCC = 27 Hz), 125.6, 71.1 (d, JPOC = 6 Hz, 2C), 23.7 (2C), 23.5 (2C); 31P NMR (CDCl3, 121.47 MHz) δ 9.88 (s).
Diisopropyl-3-pyridylphosphonate (Table 1, Entry 2)
Yield: 48-61%. 1H NMR (CDCl3, 300 MHz) δ 9.20-8.86 (m, 1H), 8.78-8.70 (m, 1H), 8.18-8.02 (m, 1H), 7.42-7.36 (m, 1H), 4.81-4.68 (m, 2H), 1.40 (d, J = 6.3 Hz, 6H), 1.26 (d, J = 6.3 Hz, 6H); 13C NMR (CDCl3, 75.45 MHz) δ 152.8, 152.3 (d, JPCC = 13 Hz), 139.4 (d, JPCC = 8 Hz), 126.6 (d, JPC = 190 Hz), 123.4 (d, JPCCC = 12 Hz), 71.4 (d, JPOC = 6 Hz, 2C), 24.1 (d, JPOCC = 4 Hz, 2C), 23.9 (d, JPOCC = 5 Hz, 2C); 31P NMR (CDCl3, 121.47 MHz) δ 14.4 (s); HRMS (EI+) calcd for C11H18NO3P, (M)+ 243.1024, found 243.1022.
Diisopropyl-4-pyridylphosphonate (Table 1, Entry 3)[18]
Yield: 63%. 1H NMR (CDCl3, 300 MHz) δ 8.78-8.73 (m, 2H), 7.67 (dm, J= 12.9 Hz, 2H), 4.82-4.70 (m, 2H), 1.40 (d, J= 6.2 Hz, 6H), 1.27 (d, J= 6.2 Hz, 6H); 13C NMR (CDCl3, 75.45 MHz) δ 150.1 (d, JPCCC = 12 Hz, 2C), 139.1 (d, JPC = 187 Hz), 125.4 (d, JPCC = 8 Hz), 71.9 (d, JPOC = 6 Hz), 24.2 (d, JPOCC = 4 Hz), 24.0 (d, JPOCC = 4 Hz); 31P NMR (CDCl3, 121.47 MHz) δ 13.5 (s).
Diisopropyl-2-bromo-pyridyl-6-phosphonate (Table 1, Entry 4)
After purification by column chromatography over silica gel, the solid was recrystallized with hot hexanes. White crystal needles were formed after cooling to room temperature and the crystals were filtered and washed several times with cold hexanes. Yield: 30%. M.p. 92-94 °C. 1H NMR (CDCl3, 300 MHz) δ 7.90 (t, J = 7.2 Hz, 1H), 7.66-7.55 (m, 2H), 4.92-4.76 (m, 2H), 1.39 (d, J = 6.0 Hz, 6H), 1.31 (d, J = 6.3 Hz, 6H); 13C NMR (CDCl3, 75.45 MHz) δ 154.7 (d, JPC = 227 Hz), 143.0 (d, JPCNC = 26 Hz), 138.4 (d, JPCCC = 12 Hz), 130.7 (d, JPCCCC = 3 Hz), 126.9 (d, JPCC = 24 Hz), 72.5 (d, JPOC = 6 Hz, 2C), 24.3 (d, JPOCC = 4 Hz, 2C), 24.0 (d, JPOCC = 5 Hz, 2C); 31P NMR (CDCl3, 121.47 MHz) δ 7.64 (s); HRMS (EI+) calcd for C11H17BrNO3P, (M+H)+ 322.0208, found 322.0201.
Diisopropyl-2-pyrimidylphosphonate (Table 1, Entry 5)[13]
Yield: 62-67%. M.p. 51-57 °C. 1H NMR (CDCl3, 300 MHz) δ 8.90 (d, J = 5.2 Hz, 2H), 7.45-7.40 (m, 1H), 5.02-4.88 (m, 2H), 1.44 (d, J = 6.2 Hz, 6H), 1.35 (d, J = 6.2 Hz, 6H); 13C NMR (CDCl3, 75.45 MHz) δ 164.1 (d, JPC = 273 Hz), 156.9 (d, JPCNC = 18 Hz, 2C), 122.5 (d, JPCNCC = 4 Hz), 72.4 (d, JPOC = 6 Hz, 2C), 24.0 (d, JPOCC = 4 Hz, 2C), 23.6 (d, JPOCC = 4 Hz, 2C); 31P NMR (CDCl3, 121.47 MHz) δ 4.7 (s).
Diisopropyl-5-pyrimidylphosphonate (Table 1, Entry 6)
Yield: 83%. M.p. 30-32 °C. 1H NMR (CDCl3, 300 MHz) δ 9.25 (s, 1H), 8.97 (d, J = 6.6 Hz, 2H), 4.76-4.66 (m, 2H), 1.32 (d, J = 6.0 Hz, 6H), 1.20 (d, J = 6.0 Hz, 6H); 13C NMR (CDCl3, 75.45 MHz) δ 160.8, 159.5 (d, JPCC = 5 Hz), 159.3 (d, JPCC = 5 Hz), 125.1 (d, JPC = 192 Hz), 71.9 (d, JPOC = 6 Hz, 2C), 23.9 (d, JPOCC = 4 Hz, 2C), 23.8 (d, JPOCC = 5 Hz, 2C); 31P NMR (CDCl3, 121.47 MHz) δ 10.9 (s); HRMS (EI+) calcd for C10H17N2O3P, (M)+ 244.0977, found 244.0975.
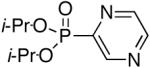
Diisopropyl-2-pyrazylphosphonate (Table 1, Entry 7)
Yield: 97%. 1H NMR (CDCl3, 300 MHz) δ 9.02 (s, 1H), 8.64 (d, J = 16.1 Hz, 2H), 4.86-4.75 (m, 2H), 1.33 (d, J = 6.2 Hz, 6H), 1.23 (d, J = 6.2 Hz, 6H); 13C NMR (CDCl3, 75.45 MHz) δ 148.9 (d, JPC = 226 Hz), 147.7 (d, JPCC = 26 Hz), 146.7 (d, JPCCNC = 3 Hz), 145.2 (d, JPCNC = 18 Hz), 72.2 (d, JPOC = 6 Hz, 2C), 24.0 (d, JPOCC = 4 Hz, 2C), 23.8 (d, JPOCC = 4 Hz, 2C); 31P NMR (CDCl3, 121.47 MHz) δ 7.73 (s); HRMS (EI+) calcd for C10H17N2O3P, (M)+ 244.0977, found 244.0977.
Diisopropyl-2-anilinylphosphonate (Table 2, Entry 1)
Yield: 70%. M.p. 46-47 °C. 1H NMR (CDCl3, 300 MHz) δ 7.47 (ddd, J = 7.8 Hz, J = 1.7 Hz, 1H), 7.28-7.23 (m, 1H), 6.71-6.60 (m, 2H), 5.19 (s, 2H, NH2), 4.70-4.58 (m, 2H), 1.38 (d, J = 6.2 Hz, 6H), 1.24 (d, J = 6.2 Hz, 6H); 13C NMR (CDCl3, 75.45 MHz) δ 151.3 (d, JPCC = 9 Hz), 133.7 (d, JPCCCC = 2 Hz), 133.4 (d, JPCC = 7 Hz), 116.5 (d, JPCCC = 14 Hz), 116.3 (d, JPCCC = 13 Hz), 109.3 (d, JPC = 184 Hz), 70.7 (d, JPOC = 5 Hz, 2C), 24.2 (d, JPOCC = 4 Hz, 2C), 23.8 (d, JPOCC = 5 Hz, 2C); 31P NMR (CDCl3, 121.47 MHz) δ 20.2 (s); HRMS (EI+) calcd for C12H20NO3P, (M)+ 257.1181, found 257.1178.
Diisopropyl-3-anilinylphosphonate (Table 2, Entry 2)[19]
Yield: 72%. M.p. 103-104 °C. 1H NMR (CDCl3, 300 MHz) δ 7.28-7.12 (m, 3H), 6.81 (d, J = 7.2 Hz, 1H), 4.71-4.60 (m, 2H), 3.86 (s, 2H, NH2), 1.36 (d, J = 6.0 Hz, 6H), 1.22 (d, J = 6.3 Hz, 6H); 13C NMR (CDCl3, 75.45 MHz) δ 146.9 (d, JPCCC = 18 Hz), 130.6 (d, JPC = 190 Hz), 129.5 (d, JPCCC = 17 Hz), 121.3 (d, JPCC = 9 Hz), 118.6 (d, JPCCCC = 3 Hz), 118.0 (d, JPCC = 12 Hz), 70.7 (d, JPOC = 5 Hz, 2C), 24.2 (d, JPOCC = 6 Hz, 2C), 24.0 (d, JPOCC = 5 Hz, 2C); 31P NMR (CDCl3, 121.47 MHz) δ 18.5 (s).
Diisopropyl-4-anilinylphosphonate (Table 2, Entry 3)
Yield: 70-92%. M.p. 113-115 °C. 1H NMR (CDCl3, 300 MHz) δ 7.58 (dd, J = 12.9 Hz, J = 8.4 Hz, 2H), 6.67 (dd, J = 8.4 Hz, J = 3.3 Hz, 2H), 4.69-4.54 (m, 2H), 4.10 (s, 2H, NH2), 1.35 (d, J = 6.0 Hz, 6H), 1.21 (d, J = 6.0 Hz, 6H); 13C NMR (CDCl3, 75.45 MHz) δ 150.6, 133.7 (d, JPCC = 11 Hz, 2C), 117.1 (d, JPC = 198 Hz), 114.2 (d, JPCCC = 16 Hz, 2C), 70.3 (d, JPOC = 5 Hz, 2C), 24.3 (d, JPOCC = 4 Hz, 2C), 24.0 (d, JPOCC = 5 Hz, 2C); 31P NMR (CDCl3, 121.47 MHz) δ 19.1 (s); HRMS (EI+) calcd for C12H20NO3P, (M)+ 257.1181, found 257.1183.
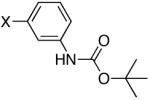
Diisopropyl-[(3-tert-butoxycarbonylamino)phenyl]phosphonate (Table 2, Entry 4)
After work-up, the solid was simply washed with Et2O, affording the desired product as a white powder. Yield: 81%. M.p. 168-169 °C. 1H NMR (CDCl3, 300 MHz) δ 7.82-7.78 (m, 1H), 7.62 (d, J = 14.4 Hz, 1H), 7.48-7.37 (m, 2H), 6.78 (s, 1H, OH), 4.73-4.62 (m, 2H), 1.52 (s, 9H), 1.36 (d, J = 6.3 Hz, 6H), 1.22 (d, J = 6.3 Hz, 6H); 13C NMR (CDCl3, 75.45 MHz) δ 152.9, 139.1 (d, JPCCC = 19 Hz), 130.1 (d, JPC = 189 Hz), 129.1 (d, JPCC = 16 Hz), 125.4 (d, JPCCC = 9 Hz), 122.0 (d, JPCCCC = 1 Hz), 121.9 (d, JPCC = 8 Hz), 80.4, 70.7 (d, JPOC = 6 Hz, 2C), 28.3, 24.0 (d, JPOCC = 4 Hz, 2C), 23.8 (d, JPOCC = 4 Hz, 2C); 31P NMR (CDCl3, 121.47 MHz) δ 17.4 (s); HRMS (EI+) calcd for C17H28NO5P, (M)+ 357.1705, found 357.1696.
Diisopropyl-[(4-tert-butoxycarbonylamino)phenyl]phosphonate (Table 2, Entry 5)
After purification by column chromatography over silica gel, the solid was washed with petroleum ether, affording the pure product as a white powder. Yield: 82%. 1H NMR (CDCl3, 300 MHz) δ 7.73 (dd, J = 12.9 Hz, J = 8.7 Hz, 2H), 7.45 (dd, J = 8.4 Hz, J = 3.3 Hz, 2H), 6.67 (s, 1H, NH), 4.72-4.57 (m, 2H), 1.52 (s, 9H), 1.35 (d, J = 6.3 Hz, 6H), 1.20 (d, J = 6.3 Hz, 6H); 13C NMR (CDCl3, 75.45 MHz) δ 152.7, 142.5 (d, JPCCCC = 3 Hz), 133.1 (d, JPCC = 11 Hz, 2C), 123.3 (d, JPC = 194 Hz), 117.8 (d, JPCCCC = 15 Hz, 2C), 81.2, 70.7 (d, JPOC = 5 Hz, 2C), 28.5, 24.3 (d, JPOCC = 4 Hz, 2C), 24.0 (d, JPOCC = 5 Hz, 2C); 31P NMR (CDCl3, 121.47 MHz) δ 18.1 (s); HRMS (EI+) calcd for C17H28NO5P, (M)+ 357.1705, found 357.1710.
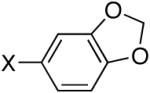
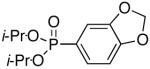
Diisopropyl-1,2-methylenedioxyphenyl-4-phosphonate (Table 3, Entry 1)
Yield: 99%. 1H NMR (CDCl3, 400 MHz) δ 7.39 (ddd, J = 7.6 Hz, J = 6.1 Hz, J = 1.4 Hz, 1H), 7.21 (dd, J = 12.9 Hz, J = 1.5 Hz, 1H), 6.87 (dd, J = 7.9 Hz, J = 3.5 Hz, 1H), 6.01 (s, 2H), 4.68-4.61 (m, 2H), 1.35 (d, J = 6.3 Hz, 6H), 1.22 (d, J = 6.2 Hz, 6H); 13C NMR (CDCl3, 75.45 MHz) δ 150.1(d, JPCCCC = 3 Hz), 147.8 (d, JPCCC = 22 Hz), 127.2 (d, JPCC = 11 Hz), 122.9 (d, JPC = 194 Hz), 111.1 (d, JPCC = 12 Hz), 108.4 (d, JPCCC = 19 Hz), 101.6, 70.6 (d, JPOC = 6 Hz, 2C), 24.0 (d, JPOCC = 4 Hz, 2C), 23.8 (d, JPOCC = 5 Hz, 2C); 31P NMR (CDCl3, 121.47 MHz) δ 17.8 (s); HRMS (EI+) calcd for C13H19O5P, (M)+ 286.0970, found 286.0972.
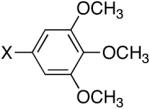
Diisopropyl-1,2,3-trimethoxyphenyl-5-phosphonate (Table 3, Entry 2)
Yield: 61%. M.p. 72-74 °C. 1H NMR (CDCl3, 300 MHz) δ 7.00 (d, J = 15.0 Hz, 2H), 4.73-4.60 (m, 2H), 3.86 (s, 9H), 1.35 (d, J = 6.3 Hz, 6H), 1.21 (d, J = 6.3 Hz, 6H); 13C NMR (CDCl3, 75.45 MHz) δ 153.2 (d, JPCCC = 22 Hz, 3C), 141.3, 124.6 (d, JPC = 189 Hz), 108.9 (d, JPCC = 4 Hz), 108.8 (d, JPCC = 4 Hz), 70.9 (2C), 56.3 (3C), 24.2 (2C), 23.8 (2C); 31P NMR (CDCl3, 121.47 MHz) δ 18.3 (s); HRMS (EI+) calcd for C15H25O6P, (M)+ 332.1389, found 332.1393.
Diisopropyl-4-nitrophenylphosphonate (Table 3, Entry 3)[6,20]
Yield: 60%. 1H NMR (CDCl3, 300 MHz) δ 8.30 (dd, J = 8.7 Hz, J = 3.0 Hz, 2H), 8.01 (dd, J = 12.6 Hz, J = 8.4 Hz, 2H), 4.82-4.71 (m, 2H), 1.40 (d, J = 6.3 Hz, 6H), 1.25 (d, J = 6.3 Hz, 6H); 13C NMR (CDCl3, 75.45 MHz) δ 150.1(d, JPCCCC = 3 Hz), 137.4 (d, JPC = 188 Hz), 132.9 (d, JPCC = 11 Hz, 2C), 123.3 (d, JPCCC = 15 Hz, 2C), 71.8 (d, JPOC = 6 Hz, 2C), 24.1 (d, JPOCC = 4 Hz, 2C), 23.9 (d, JPOCC = 5 Hz, 2C); 31P NMR (CDCl3, 121.47 MHz) δ 13.9 (s).
Diisopropyl-3-quinolinylphosphonate (Table 3, Entry 4)
Yield: 46%. 1H NMR (CDCl3, 300 MHz) δ 9.15 (dd, J = 4.5 Hz, J = 1.8 Hz, 1H), 8.72 (dd, J = 15.0 Hz, J = 1.5 Hz, 1H), 8.16 (d, J = 8.7 Hz, 1H), 7.92 (d, J = 7.8 Hz, 1H), 7.85 (dt, J = 7.2 Hz, J = 1.5 Hz, 1H), 7.64 (t, J = 7.8 Hz, 1H), 4.84-4.72 (m, 2H), 1.43 (d, J = 6.0 Hz, 6H), 1.26 (d, J = 6.0 Hz, 6H); 13C NMR (CDCl3, 75.45 MHz) δ 151.1(d, JPCC = 12 Hz), 149.4, 142.9 (d, JPCC = 9 Hz), 131.9, 129.7 (d, JPCCCC = 1 Hz), 128.9, 127.7, 126.9 (d, JPCCC = 14 Hz), 123.6 (d, JPC = 189 Hz), 71.7 (d, JPOC = 5 Hz, 2C), 24.3 (d, JPOCC = 4 Hz, 2C), 24.1 (d, JPOCC = 5 Hz, 2C); 31P NMR (CDCl3, 121.47 MHz) δ 19.3 (s); HRMS (EI+) calcd for C15H20NO3P, (M)+ 293.1181, found 293.1181.
Diisopropyl-1-naphthylphosphonate (Table 3, Entry 5)
Yield: 86%. 1H NMR (CDCl3, 300 MHz) δ 8.53 (d, J = 8.1 Hz, 1H), 8.30 (dd, J = 16.5 Hz, J = 6.9 Hz, 1H), 8.02 (d, J = 8.1 Hz, 1H), 7.87 (d, J = 8.1 Hz, 1H), 7.62-7.48 (m, 3H), 4.79-4.67 (m, 2H), 1.41 (d, J = 6.3 Hz, 6H), 1.14 (d, J = 6.3 Hz, 6H); 13C NMR (CDCl3, 75.45 MHz) δ 134.6 (d, JPCC = 9 Hz), 133.7 (d, JPCCC = 13 Hz), 133.5 (d, JPCCCC = 3 Hz), 132.8 (d, JPCCC = 11 Hz), 128.8 (d, JPCCCC = 1 Hz), 127.2, 127.1, 126.4, 126.3 (d, JPC = 183 Hz), 124.6 (d, JPCC = 17 Hz), 71.1 (d, JPOC = 5 Hz, 2C), 24.3 (d, JPOCC = 4 Hz, 2C), 23.9 (d, JPOCC = 5 Hz, 2C); 31P NMR (CDCl3, 121.47 MHz) δ 17.8 (s); HRMS (EI+) calcd for C16H21O3P, (M)+ 292.1228, found 292.1124.
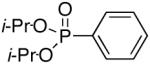
Diisopropyl-phenylphosphonate (Table 3, Entry 6)[6,20]
Yield: 47-93%. 1H NMR (CDCl3, 300 MHz) δ 7.82 (dd, J = 13.5 Hz, J = 6.6 Hz, 2H), 7.53-7.49 (m, 1H), 7.47-7.40 (m, 2H), 4.75-4.63 (m, 2H), 1.37 (d, J = 6.6 Hz, 6H), 1.22 (d, J = 6.6 Hz, 6H); 13C NMR (CDCl3, 75.45 MHz) δ 132.1, 131.7 (d, JPCC = 10 Hz, 2C), 130.0 (d, JPC = 188 Hz), 128.4 (d, JPCCC = 15 Hz, 2C), 70.7 (2C), 24.1 (d, JPOCC = 4 Hz, 2C), 23.8 (d, JPOCC = 5 Hz, 2C); 31P NMR (CDCl3, 121.47 MHz) δ 17.6 (s).
Diisopropyl-4-hydroxyphenylphosphonate (Table 3, Entry 7)
Yield: 27-76%. M.p. 125-127 °C. 1H NMR (CDCl3, 300 MHz) δ 9.79 (s, 1H, OH), 7.64 (dd, J = 12.9 Hz, J = 6.9 Hz, 2H), 7.02-6.99 (m, 2H), 4.71-4.56 (m, 2H), 1.36 (d, J = 6.3 Hz, 6H), 1.23 (d, J = 6.3 Hz, 6H); 13C NMR (CDCl3, 75.45 MHz) δ 161.9 (d, JPCCCC = 3 Hz), 133.9 (d, JPCC = 12 Hz, 2C), 117.9 (d, JPC = 198 Hz), 116.1 (d, JPCCC = 16 Hz, 2C), 71.2 (d, JPOC = 5 Hz, 2C), 24.2 (d, JPOCC = 4 Hz, 2C), 24.0 (d, JPOCC = 4 Hz, 2C); 31P NMR (CDCl3, 121.47 MHz) δ 19.9 (s); HRMS (EI+) calcd for C12H19O4P, (M)+ 258.1021, found 258.1022.
Diisopropyl-4-methylbenzoylphosphonate (Scheme 1, compound 1)
Scheme 1.

First cross-coupling with aryl chlorides
Yield: 44%. 1H NMR (CDCl3, 300 MHz) δ 8.14-8.09 (m, 2H), 7.94-7.86 (m, 2H), 4.77-4.66 (m, 2H), 3.95 (s, 3H), 1.38 (d, J = 6.2 Hz, 6H), 1.23 (d, J = 6.2 Hz, 6H); 13C NMR (CDCl3, 75.45 MHz) δ 166.4, 135.0 (d, JPC = 187 Hz), 133.4 (d, JPCCCC = 3 Hz), 131.8 (d, JPCC = 10 Hz, 2C), 129.4 (d, JPCCC = 15 Hz, 2C), 71.3 (d, JPOC = 5 Hz, 2C), 52.5, 24.2 (d, JPOCC = 4 Hz, 2C), 23.9 (d, JPOCC = 5 Hz, 2C); 31P NMR (CDCl3, 36.441 MHz) δ 14.7 (m); HRMS (EI+) calcd for C14H21O5P, (M)+ 300.1127, found 300.1120.
Diisopropyl-4-cyanophenylphosphonate (Scheme 1, compound 2)
Yield: 57%. 1H NMR (CDCl3, 300 MHz) δ 7.93 (dd, J = 12.9 Hz, J = 7.8 Hz, 2H), 7.78-7.73 (m, 2H), 4.82-4.67 (m, 2H), 1.40 (d, J = 6.3 Hz, 6H), 1.25 (d, J = 6.3 Hz, 6H); 13C NMR (CDCl3, 75.45 MHz) δ 135.7 (d, JPC = 188 Hz), 132.4 (d, JPCC = 10 Hz, 2C), 132.1 (d, JPCCC = 15 Hz, 2C), 118.2, 115.9 (d, JPCCCC = 3 Hz), 71.8 (d, JPOC = 6 Hz, 2C), 24.2 (d, JPOCC = 4 Hz, 2C), 24.1 (d, JPOCC = 5 Hz, 2C); 31P NMR (CDCl3, 36.441 MHz) δ 14.2 (s); HRMS (EI+) calcd for C13H18NO3P, (M)+ 267.1024, found 267.1025.
Diisopropyl-4-trifluorophenylphosphonate (Scheme 1, compound 3)[21]
Yield: 22%. 1H NMR (CDCl3, 300 MHz) δ 7.95 (dd, J = 12.9 Hz, J = 8.1 Hz, 2H), 7.74-7.70 (m, 2H), 4.80-4.68 (m, 2H), 1.40 (d, J = 6.2 Hz, 6H), 1.25 (d, J = 6.2 Hz, 6H); 13C NMR (CDCl3, 75.45 MHz) δ 134.6 (d, JPC = 187 Hz), 133.9 (dq, JFC = 33 Hz, JPCCCC = 3 Hz), 132.3 (d, JPCC = 10 Hz, 2C), 125.3 (d, JFC = 15 Hz, 4 Hz), 123.4 (d, JFC = 273 Hz), 71.5 (d, JPOC = 5 Hz), 24.2 (d, JPOCC = 4 Hz), 24.0 (d, JPOCC = 5 Hz); 31P NMR (CDCl3, 36.441 MHz) δ 16.5 (s); 19F NMR (CDCl3, 282.306 MHz) δ -63.7 (s).
General procedure for Scheme 2
The crude reaction mixture from the Pd-catalyzed cross-coupling was concentrated in vacuo. The residue was partitioned between concentrated aqueous HCl and CHCl3. The aqueous layer was washed once with CHCl3, then treated with concentrated aqueous NaOH. The resulting basic aqueous layer was extracted with CHCl3 (3 X), and the combined organic layers dried with MgSO4, and concentrated under reduced pressure. The resulting oil was dissolved in CH2Cl2 and treated with bromotrimethylsilane (2.2 equiv) at room temperature under N2. When silylation was complete (24 - 48 h, monitored by 31P-NMR), the solvent was removed in vacuo and MeOH was added. The phosphonic acids were obtained as solids either directly, or by precipitation from water/acetone or water/methanol.
Pyrazine phosphonic acid (Scheme 2, compound 4)
Yield: 69%. M.p. 171 - 173 °C. 1H NMR (D2O, 300 MHz) δ 8.63 (bs, 1H), 8.55 (bs, 1H), 8.45 (bs, 1H); 13C NMR (D2O, 75.45 MHz) δ 152.1 (d, JPC = 206 Hz), 145.2 (d, JPCC = 16 Hz), 144.5, 144.2; 31P NMR (D2O, 121.47 MHz) δ 3.64 (s); HRMS (ES+) calcd for C4H5N2O3P, (M + H)+ 161.0116, found 161.0111.
5-Pyrimidine phosphonic acid (Scheme 2, compounds 5)
Yield: 78%. M.p. 203 - 204 °C. 1H NMR (D2O, 300 MHz) δ (major, PyrimP(O)(OH)2) 9.13 (s, 1H), 8.92 (d, J = 7 Hz, 2H), (minor, PyrimH+P(O)(OH)(O-)) 8.09 (s, 1 H), 6.84 (d, J = 13 Hz, 1 H), 5.57 (d, J = 6 Hz, 1 H); 13C NMR (D2O, 75.45 MHz) δ (major) 158.6 (d, JPCC = 7 Hz), 155.7, 134.24 (d, JPC = 161 Hz); 31P NMR (D2O, 121.47 MHz) δ (minor, PyrimH+P(O)(OH)(O-)) 9.34 (s) and (major, PyrimP(O)(OH)2) 4.51 (s); HRMS (ES+) calcd for C4H5N2O3P, (M + H)+ 161.0116, found 161.0116.
2-Pyrimidine phosphonic acid (Scheme 2, compound 6)[13]
Yield: 89%. M.p. 207 - 208 °C. 1H NMR (D2O, 300 MHz) δ 8.97 (d, J = 5 Hz, 2H), 7.79 (dt, J = 5, 3 Hz, 1H); 13C NMR (D2O, 75.45 MHz) δ 164.1 (d, JPC = 224 Hz), 157.2 (d, JPCNC = 12 Hz), 123.12; 31P NMR (D2O, 121.47 MHz) δ - 1.31 (s).
3-Pyridine phosphonic acid (Scheme 2, compound 7)[2d, 14, 22]
Yield: 100%. M.p. 237 - 240 °C. 1H NMR (D2O, 300 MHz) δ 8.84 (dd, J = 7, 1 Hz, 1H), 8.75 (dd, J = 7, 1 Hz, 1H), 8.71 (ddt, J = 12, 7, 1 Hz, 1H), 8.01 - 8.07 (m, 1H) ; 13C NMR (CDCl3, 75.45 MHz) δ 148.4 (d, JPCC = 7 Hz), 142.37, 142.1 (d, JPCCC = 15 Hz), 136.5 (d, JPC = 175 Hz), 127.5 (d, JPCC = 11 Hz); 31P NMR (D2O, 121.47 MHz) δ 4.48 (t, JPCCH = 9 Hz).
Supplementary Material
Acknowledgments
We gratefully acknowledge the National Institute of General Medical Sciences/NIH (1R01 GM067610) for the financial support of this research.
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1](a).Hirao T, Masunaga T, Ohshiro Y, Agawa T. Tetrahedron Lett. 1980;21:3595. [Google Scholar]; (b) Hirao T, Masunaga T, Yamada N. Bull. Chem. Soc. Jpn. 1982;55:909. [Google Scholar]
- [2].For example: Kabachnik MM, Solntseva MD, Beletskaya IP. Russ. Chem. Bull. 1997;46:1491. Beletskaya IP, Neganova EG, Veits YA. Russ. J. Org. Chem. 2004;40:1782. Zhong P, Xiong ZX, Huang X. Synth. Commun. 2000;30:273. Ayyappan P, Evans OR, Foxman BM, Wheeler KA, Warren TH, Lin W. Inorg. Chem. 2001;40:5954. doi: 10.1021/ic010609g. Kalek M, Stawinski J. Organomet. 2007;26:5840. Chauhan SS, Varshney A, Verma B, Pennington MW. Tetrahedron Lett. 2007;48:4051. Montoneri E, Viscardi G, Bottigliengo S, Gobetto R, Chierotti MR, Buscaino R, Quagliotto P. Chem. Mater. 2007;19:2671. Vasylyev MV, Wachtel EJ, Popovitz-Biro R, Neumann R. Chem. Eur. J. 2006;12:3507. doi: 10.1002/chem.200501143. Suri JT, Steiner DD, Barbas CF., III Org. Lett. 2005;7:3885. doi: 10.1021/ol0512942. Vasylyev MV, Astruc D, Neumann R. Adv. Synth. Catal. 2005;347:39. Ngo HL, Hu A, Lin Wenbin. J. Mol. Catal. A. 2004;215:177. Oishi S, Kang S-U, Liu H, Zhang M, Yang D, Deschampsc JR, Burke TR., Jr. Tetrahedron. 2004;60:2971. Muthukumaran K, Loewe RS, Ambroise A, Tamaru S.-i., Li Q, Mathur G, Bocian DF, Misra V, Lindsey JS. J. Org. Chem. 2004;69:1444. doi: 10.1021/jo034945l. Evans OR, Manke DR, Lin W. Chem. Mater. 2002;14:3866. Guerrero G, Mutin PH, Dahan F, Vioux A. J. Organomet. Chem. 2002;649:113. Liu W-Q, Olszowy C, Bischoff L, Garbay C. Tetrahedron Lett. 2002;43:1417. Kant M, Bischoff S, Siefken R, Gründemann E, Köckritz A. Eur. J. Org. Chem. 2001:477. Machnitzkia P, Nickela T, Stelzer O, Landgrafeb C. Eur. J. Inorg. Chem. 1998:1029. Villemin D, Elbilali A, Simeon F, Jaffres P-A, Maheut G, Mosaddak M, Hakiki A. J. Chem. Res. (S) 2003:436. Prim D, Campagne J-M, Joseph D, Andrioletti B. Tetrahedron. 2002;58:2041.
- [3].For examples of Ni and Cu couplings, see: Ogawa T, Usuki N, Ono N. J. Chem. Soc. Perkin Trans. 1. 1998:2953. Fischer J, Schurmann M, Mehring M, Zachwieja U, Jurkschat K. Organometallics. 2006;25:2886. Yao Q, Levchik S. Tetrahedron Lett. 2006;47:277. Kong D, Clearfield A. Cryst. Growth Design. 2005;5:1767. Märkl von G., Gschwendner K, Rötzer I, Kreitmeier Peter. Helv. Chim. Acta. 2004;87:825. Balthazor TM, Grabiak RC. J. Org. Chem. 1980;45:5425. Tavs P. Chem. Ber. 1970;103:2428.
- [4](a).Engel R, I J. Cohen Synthesis of Carbon-Phosphorus Bonds. 2nd ed CRC Press; Boca Raton, FL: 2003. [Google Scholar]; (b) Schwan AL. Chem. Soc. Rev. 2004;33:218. doi: 10.1039/b307538a. [DOI] [PubMed] [Google Scholar]
- [5].Gelman D, Jiang L, Buchwald SL. Org. Lett. 2003;5:2315. doi: 10.1021/ol0346640. [DOI] [PubMed] [Google Scholar]
- [6](a).Huang C, Tang X, Fu H, Jiang Y, Zhao Y. J. Org. Chem. 2006;71:5020. doi: 10.1021/jo060492j. [DOI] [PubMed] [Google Scholar]; (b) Rao H, Jin Y, Fu H, Jiang Y, Zhao Y. Chem. Eur. J. 2006;12:3636. doi: 10.1002/chem.200501473. [DOI] [PubMed] [Google Scholar]
- [7].Reviews: Montchamp J-L. Specialty Chemicals Magazine. 2006;26:44. Montchamp J-L. J. Organomet. Chem. 2005;690:2388. Cross-Coupling: Montchamp J-L, Dumond YR. J. Am. Chem. Soc. 2001;123:510. Dumond YR, Montchamp J-L. J. Organomet. Chem. 2002;653:252. Bravo-Altamirano K, Huang Z, Montchamp J-L. Tetrahedron. 2005;61:6315. Bravo-Altamirano K, Montchamp J-L. Tetrahedron Lett. 2007;48:5755. doi: 10.1016/j.tetlet.2007.06.090. Bravo-Altamirano K, Montchamp J-L. Org. Lett. 2006;8:4169. doi: 10.1021/ol061828e. Coudray L, Bravo-Altamirano K, Montchamp J-L. Org. Lett. 2008;10:1123. doi: 10.1021/ol8000415. Hydrophosphinylation: Deprèle S, Montchamp J-L. J. Org. Chem. 2001;66:6745. doi: 10.1021/jo015876i. Deprèle S, Montchamp J-L. J. Am. Chem. Soc. 2002;124:9386. doi: 10.1021/ja0261978. Deprèle S, Montchamp J-L. Org. Lett. 2004;6:3805. doi: 10.1021/ol0484198. Ribière P, Bravo-Altamirano K, Antczak MI, Hawkins JD, Montchamp J-L. J. Org. Chem. 2005;70:4064. doi: 10.1021/jo050096l. Bravo-Altamirano K, Abrunhosa-Thomas I, Montchamp J-L. J. Org. Chem. 2008;73:2292. doi: 10.1021/jo702542a.
- [8](a).Fry JA, Samanamu CR, Montchamp J-L, Richards AF. Eur. J. Inorg. Chem. 2008:463. doi: 10.1021/ic800023d. [DOI] [PubMed] [Google Scholar]; (b) Samanamu CR, Olmstead MM, Montchamp J-L, Richards AF. Inorg. Chem. 2008;47:3879. doi: 10.1021/ic800023d. [DOI] [PubMed] [Google Scholar]; (c) Samanamu CR, Zamora EN, Montchamp J-L, Richards AF. J. Solid State Chem. 2008;181:1462. [Google Scholar]; (d) Samanamu CR, Zamora EN, Lesikar LA, Montchamp J-L, Richards AF. Cryst. Eng. Comm. 2008 in press. [Google Scholar]
- [9].Belabassi Y, Gouault-Bironneau S, Montchamp J-L. Encyclopedia of Reagents for Organic Synthesis (eEROS) 2006 update article. ( http://www.mrw.interscience.wiley.com/eros/articles/rb161/abstract-fs.html)
- [10].Samanamu CR, Richards AF. Polyhedron. 2007;26:923. [Google Scholar]
- [11].Diisopropyl phosphite was purchased from TCI America (catalog # P0629) and used as received.
- [12].Gooßen LJ, Dezfuli MK. Synlett. 2005:445. [Google Scholar]
- [13].Gennady GM, Roy CH. J. Org. Chem. 1961;26:1895. [Google Scholar]
- [14].Bulot JJ, Aboujaoude EE, Collignon N, Savignac P. Phosphorus and Sulfur. 1984;21:197. [Google Scholar]
- [15].McKenna CE, Schmidhuser J. J. Chem. Soc. Chem. Commun. 1979:739. [Google Scholar]
- [16].pKas of heterocycles: pyridine, 5.2; pyrimidine, 1.3; pyrazine, 0.65.
- [17](a).Conary GS, Russel AA, Paine RT, Hall JH, Ryan RR. Inorg. Chem. 1988;27:3242. [Google Scholar]; (b) Kiselev AS, Gakh AA, Kagramanov ND, Semenov VV. Mendeleev Communications. 1991;4:128. [Google Scholar]
- [18](a).Redmore D. J. Org. Chem. 1976;41:2148. [Google Scholar]; (b) Haase M, Gunther W, Gorls H, Anders E. Synthesis. 1999:2071. [Google Scholar]; (c) Haase M, Goerls H, Anders E. Synthesis. 1998:195. [Google Scholar]; (d) Redmore D. Phosphorus and Sulfur and the Related Elements. 1979;5:271. [Google Scholar]
- [19](a).Cates LA, Li V-S, Yakshe CC, Fadeyi MO, Andree TH, Karbon EW, Enna SJ. J. Med. Chem. 1984;27:654. doi: 10.1021/jm00371a017. [DOI] [PubMed] [Google Scholar]; (b) Cates LA, Rashed MS. Pharm. Res. 1984;6:271. doi: 10.1023/A:1016350119870. [DOI] [PubMed] [Google Scholar]
- [20].Huang C, Tang X, Fu H, Jiang Y, Zhao Y. J. Org. Chem. 2006;71:8328. doi: 10.1021/jo060492j. [DOI] [PubMed] [Google Scholar]
- [21].Hall CD, Beer PD, Powell RL, Naan MP. Phosphorus, Sulfur Silicon and Relat. Elem. 1995;105:145. [Google Scholar]
- [22].Bennett RD, Burger A, Volk WA. J. Org. Chem. 1958;23:940. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.