

Abstract
The NF-κB/Rel family of transcription factors plays pivotal roles in multiple biologic processes, including innate and adaptive immunity and regulation of cell proliferation and death processes. In many human malignancies, NF-κB is often dysregulated leading to its constitutive activation. The two most well-known NF-κB activation mechanisms, canonical and noncanonical, which are mediated by degradation or processing of NF-κB inhibitor proteins by 26S proteasomes, are often implicated in its constitutive activation. Less well-studied NF-κB activation mechanisms also exist, and one of them is referred to as the “proteasome inhibitor resistant” (PIR) pathway, which was originally described as the principle mechanism behind constitutive NF-κB activation in certain murine B lymphoma cell lines as well as the human multiple myeloma (MM) cell line RPMI8226. Here we report that constitutive NF-κB activity in ten out of fourteen primary MM patient samples is refractory to inhibition by high doses of a proteasome inhibitor, bortezomib (Velcade/PS341). Moreover, when MM cells were co-cultured with patient-derived bone marrow stromal cells (BMSCs), microenvironment components critical for MM growth and survival, further increases in NF-κB activity were observed that were also resistant to bortezomib. Similarly, constitutive PIR NF-κB activity observed in RPMI8226 cells was augmented by the presence of BMSCs, resulting in increased NF-κB-dependent transcription and resistance to bortezomib-induced apoptosis. Thus, we propose that the PIR mechanism of NF-κB regulation is likely relevant to the often devastating biology of multiple myeloma.
Keywords: NF-κB, Myeloma, bortezomib, bone marrow stroma, apoptosis
Introduction
The nuclear factor -κB (NF-κB)/Rel family of transcription factors is composed of five members, p65 (RelA), RelB, cRel, p100/p52, and p105/p50, which bind one another to form homo- or heterodimers. NF-κB dimers are normally held inactive in the cytoplasm due to association with inhibitor proteins, IκBs. The best characterized mechanism of NF-κB activation, so called “canonical,” involves signaling events leading to activation of the cytoplasmic IκB kinase (IKK) complex that causes site specific phosphorylation on IκBs to create a docking site for the β-TrCP ubiquitin ligase complex. IκBs are then polyubiquitinated and degraded by the ubiquitin-dependent 26S proteasome. The newly liberated NF-κB then migrates into the nucleus, binds to its cognate -κB elements and regulates transcription of genes involved in embryogenesis, innate and adaptive immunity, and other physiologic processes (1).
There are also other distinct mechanisms of NF-κB activation known. The so called “noncanonical” pathway targets p100, the precursor of p52, by phosphorylation and ubiquitination-mediated limited processing of the C-terminal IκB domain by the 26S proteasome to liberate the N-terminal p52 subunit, thus selectively activating the p52/RelB complex (1). Previous studies have also demonstrated that the IκBα inhibitor protein can be inducibly phosphorylated on tyrosine 42 upon hypoxia/reoxygenation stress, which results in its dissociation from NF-κB (2). Ultraviolet irradiation has been shown to cause C-terminal IκBα phosphorylation by casein kinase II, resulting in its degradation without the involvement of IKK complex (3). Calcium-dependent proteases, calpains, can also play a role in mediating degradation of IκBα to release active NF-κB (4–6). This latter mechanism has been linked to limb girdle muscular dystrophy type 2A, where calpain 3 deficiency due to genetic mutations in the patients results in loss of IκBα degradation and constitutive NF-κB activation in muscle precursor cells contributing to myocyte apoptosis and muscular dystrophy (7). Calpain-mediated activation of NF-κB has also been linked to constitutive activation in certain B cell lines (8). Moreover, mitochondria have been shown to harbor the NF-κB/IκB system, in which calpain-mediated degradation of IκB leads to mitochondrial NF-κB activation and regulation of mitochondrial genes (9).
There is yet another mechanism of NF-κB activation that is highly resistant to over ten different proteasome inhibitors examined, including those that potently block both calpain and proteasome activities (e.g., calpain inhibitor 1) and bortezomib (Velcade/PS341), the first proteasome-selective inhibitor approved for clinical use (10, 11). This pathway was originally described in murine B cell lines as a mechanism responsible for constitutive IκBα degradation and NF-κB activation (12). It requires the selective degradation of IκBα in a manner dependent on calcium and calmodulin, and is sensitive to inhibition by the anticancer agent perillyl alcohol (POH), which among other effects has been shown to inhibit a specific L-type calcium channel (11, 13). Experimental evidence suggested that several major intracellular proteolytic systems, including calpains, caspases, lysosomal proteases and the giant/tricorn protease, the latter of which can complement proteasome deficiency (14), are not involved in PIR IκBα degradation and NF-κB activation (11). Moreover, cis-element analysis has demonstrated that IκBα degradation in the PIR pathway involves distinct amino acid structures and does not require the canonical phosphorylation event or polyubiquitination mediated by β-TrCP (10, 11, 15), thereby partly explaining the lack of proteasome requirement. Although the precise proteolytic system involved in the PIR pathway remains to be determined, it has been referred to as the “proteasome inhibitor resistant” (PIR) pathway to distinguish this pathway from other known mechanisms (10). In addition to certain murine B lymphoma cell lines mentioned above (e.g., WEHI231), normal murine splenic B cells, the human breast cancer cell line MDA-MB-463, and the human multiple myeloma cell line RPMI8226 have also been found to harbor constitutive NF-κB activity that is maintained by the PIR mechanism (10, 11, 13, 15).
The proteasome inhibitor bortezomib potently blocks chymotryptic-like activity within the 26S proteasome (16). Phase I and II clinical trials showed a significant overall response rate for bortezomib as a single agent against refractory multiple myeloma (MM) (17). Since proteasomes are involved in a wide variety of intra- and intercellular events, the precise molecular pathway(s) of bortezomib that give rise to favorable clinical outcomes are unclear; however, modulation of many important pathways such as p53/MDM2, c-Jun NH2-terminal kinase (JNK), endoplasmic reticulum associated degradation (ERAD), and NF-κB within the malignant cells have been implicated (18–21). Since MM pathogenesis also involves the critical participation of bone marrow microenvironment components, such as bone marrow stromal cells (BMSCs), endothelial cells and osteoclasts, among others (22), bortezomib effects on these non-malignant cells have also been implicated in clinical efficacy (22, 23). Recent studies in mantle cell lymphoma, for which bortezomib is also approved for clinical use, demonstrated the critical involvement of the Noxa/Mcl/Bak pathway in bortezomib-induced apoptosis (24). Noxa upregulation has also been associated with bortezomib-induced apoptosis in MM (25), suggesting that this may also be an important pathway in MM response to bortezomib. The exact mechanisms of bortezomib sensitivity and resistance in vivo in MM patients, however, remains incompletely understood.
Since constitutive NF-κB activity is observed by immunofluorescence in virtually all primary MM samples analyzed (26, 27) and is implicated in chemoresistance of MM cells, here we investigated (i) the frequency of constitutive PIR NF-κB activity in a cohort of primary MM patient samples and (ii) whether PIR NF-κB activation in MM cells is modulated by the presence of BMSCs derived from MM patients. Below we describe a surprising finding that a bortezomib-resistant pathway of constitutive NF-κB activation is frequently detected in primary MM cells and that BMSCs have the capacity to further increase this activity. This increase in constitutive NF-κB activity due to BMSCs is also seen in RPMI8226 cells, which correlated with increased bortezomib resistance of MM cells in vitro. Our data provide the first insight into the possible involvement of the PIR pathway of NF-κB activation in MM pathobiology.
Results
Constitutive bortezomib-resistant NF-κB activity is observed in primary and established human MM cells
Figure 1A shows EMSA analysis of total cell extracts prepared from RPMI8226 cells along with supershift analysis using antibodies selective for NF-κB family members. Based on reactivity to these antibodies, the two major NF-κB complexes were assigned to be p50/p65(RelA) and p52/RelB dimers (Figure 1A, lanes 3–8). Increasing concentrations of bortezomib resulted in corresponding inhibition of the proteasomal activity in the RPMI8226 cells within 2 hrs (Figure 1B); however, a high concentration could not block constitutive NF-κB activity (Figure 1A). As a positive control to evaluate the efficacy of bortezomib, we tested its effect on TNF-α-induced canonical activation and found, as expected, that 100nM bortezomib nearly completely blocked this activation (Figure 1C). In contrast to bortezomib, treatment with a calcium chelator (BAPTA-AM) or POH, agents that selectively block the PIR activation pathway, effectively inhibited constitutive NF-κB activity (Figure 1D).
Figure 1. Constitutive NF-κB activity in the RPMI8226 cells is resistant to high-dose bortezomib.

(A) Representative EMSA analysis of RPMI8226 cell extract using a 32P-labeled Ig-B (κ B) probe with or without cold probe competition (Cold) or using Oct-1 DNA probes is shown. Where indicated, the DNA-binding assay was performed in the presence of antibodies (Antibody) against the five NF-κ B family members, or a non-specific IgG antibody control. RPMI8226 cells were treated with 4 hours of 100nM bortezomib (Bort) and analyzed by EMSA where indicated. Probe not bound by protein in the DNA-binding assay migrates at the bottom of the gel (Free). Supershift analysis (Supershift) indicates that constitutive NF-κ B activity in RPMI8226 cells is comprised of two main complexes of interest; a p65:p50 dimer (*), and a RelB:p52 dimer (**). Non-specific binding is also indicated. (B) Analysis of percent proteasome inhibition by increasing doses of bortezomib in RPMI8226 using Proteo-Glo (Biorad) assay. Basal proteasomal activity for each cell type is assigned as 100% activity. (C) EMSA of extracts isolated from RPMI8226 cells exposed to with 100nM bortezomib for 4 hours or 20ng/mL TNFα for 30 minutes where indicated. (D) EMSA of extracts isolated from RPMI8226 cells treated with 4 hours of 1.0mM POH or 30μM Bapta-AM (Bap).
Previous studies have demonstrated that NF-κB is constitutively active in nearly all primary MM samples examined (26, 27). However, whether such activities are highly sensitive to bortezomib has not been reported. The existence of constitutive PIR NF-κB activity in RPMI8226 MM cells prompted us to test if constitutive NF-κB activity in a certain fraction of primary MM cells is also resistant to bortezomib. Because of the limited number of primary cells available, we developed a “mini-EMSA” that is reproducible with at least 50,000 cells per sample with a variance of ~10% (Figure 2A). When we isolated CD138-posititve MM cells from 14 patients, we found that all patients harbored some level of constitutive NF-κB activity. Treatment with 100nM bortezomib for four hours induced near-complete proteasome inhibition in several primary MM cell samples tested (Figure 2B); nevertheless, we were surprised to find that in many cases (10 of 14), this high-dose bortezomib failed to inhibit constitutive NF-κB activity, as measured by EMSA, in these primary cells (Figure 2C). Paradoxically, in 8 of these cases, NF-κB DNA-binding was enhanced by treatment with bortezomib (Figure 2C and D). Thus, these studies demonstrated that bortezomib-resistant (or even -augmented) NF-κB activity is frequently detectable in primary MM samples.
Figure 2. Primary MM cells display constitutive NF-κ B activity that is largely resistant to high concentrations of bortezomib.

(A) Representative mini-EMSA, using whole cell lysates from the indicated total cell numbers, fold-induction as determined by phosphoimager is labeled below the gel. (B) Analysis of percent proteasome inhibition by increasing doses of bortezomib in CD138+ cells obtained from two MM patients using Proteo-Glo (Biorad) assay. Basal proteasomal activity for each cell type is assigned as 100% activity. (C) EMSA analysis of lysates derived from CD138+ cells isolated from the indicated patients and then treated with 100nM bortezomib for 4 hours where indicated. Fold-change in NF-κ B activity corrected for Oct-1 DNA-binding is displayed below the gels. (D) Fold-change NF-κ B DNA-binding, as measured by phosphoimage quantification corrected for Oct-1 DNA-binding, induced by treatment with 4 hours 100nM bortezomib in CD138+ cells from the indicated MM patients.
Constitutive bortezomib-resistant NF-κB activity is further enhaced by BMSCs in primary MM cells
Although a considerable fraction of primary MM cell samples examined displayed bortezomib-resistant constitutive NF-κB activity, this observation was made in the suspension culture conditions in the absence of bone marrow microenvironment components. It was unclear how these primary MM cells would behave in the presence of patient-derived BMSCs. Thus, we established BMSC cultures from mononuclear cell preparations of MM patient bone marrow aspirates using the standard protocol (28). Next we incubated primary MM cells on top of BMSCs and evaluated the consequent sensitivity of NF-κB activity to bortezomib in primary MM cells. Placing primary MM cells in co-culture with MM-BMSCs caused further enhancement in NF-κB activity in most cases (Figure 3, lanes 2, 5, 8, 10). Significantly, we were unable to block this BMSC-enhanced NF-κB activity using up to 100 nM bortezomib in certain primary MM cells (Figure 3, lanes 3 and 11). However, this was not a universal phenotype of MM cells, since in some cases, bortezomib was able to effectively inhibit BMSC-induced NF-κB activity in MM cells (lanes 6 and 9). These data suggest that depending on the MM samples, BMSC-induced NF-κB activity could be sensitive or insensitive to high dose bortezomib treatment.
Figure 3. MM-BMSCs induce NF-κ B activation in primary MM and RPMI8226 MM cells a largely bortezomib-resistant manner.
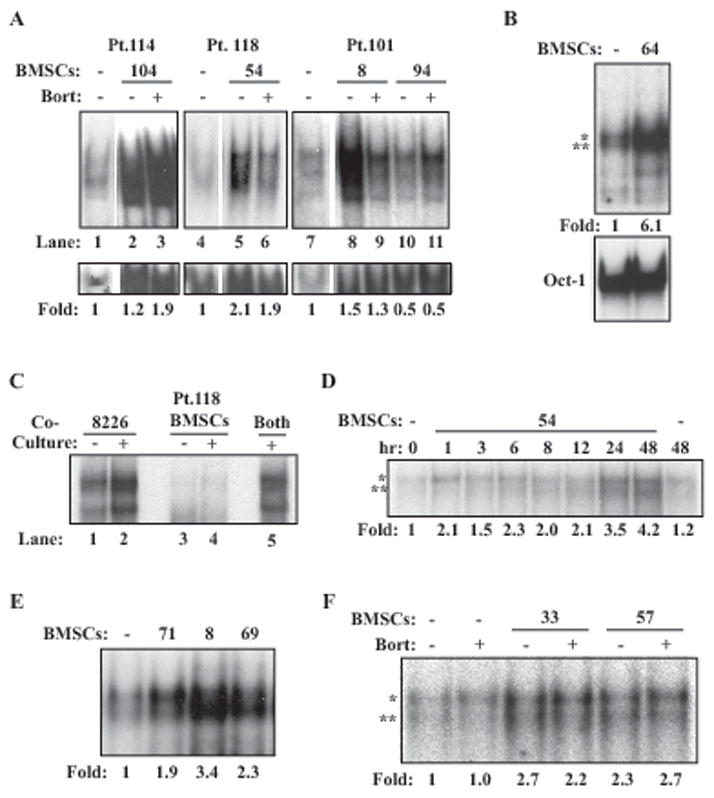
(A) EMSA analysis of CD138+ cells from the indicated MM patients cultured alone or with MM-BMSCs for 24 hours. 100nM bortezomib was added 4 hours before termination of co-culture where indicated. Fold change in NF-κ B activation is calculated and displayed as before. (B) EMSA of RPMI8226 cells cultured alone or with Pt. 64 BMSCs for 24 hours. Oct-1 DNA-binding was measured as a loading control. (C) EMSA of RPMI8226 cells and MM-BMSCs culture alone (lanes 1 and 3), cultured together but separated for analysis (lanes 2 and 4), or cultured together and analyzed together (lane 5). (D) EMSA of RPMI8226 cells cultured with MM-BMSCs for the indicated times. Fold-change in NF-κB activity was measured as before and is displayed below the gel. (E) EMSA of RPMI8226 cells cultured alone or with the indicated MM patients for 24 hours. (F) EMSA of RPMI8226 cells cultured alone or with the indicated MM patients for 24 hours with 100nM bortezomib added for the last 4 hours of co-culture where indicated.
BMSCs also enhance PIR NF-κB activation in RPMI8226 MM cells
To gain insight into the significance of BMSC-induced NF-κB activation, we next turned to the more experimentally amenable RPMI8226 MM cell model. We placed RPMI8226 cells on top of BMSCs for 24 hours as in the case with the primary MM cells and then performed EMSA analysis using cell extracts obtained from the RPMI8226 cells. This analysis revealed enhanced NF-κB activity (up to six-fold over basal activity) (Figure 3B). Oct-1 DNA-binding was used as a loading control for all experiments (Figure 3B, not shown in subsequent subpanels). Because removal of MM cells from BMSCs can be incomplete or possibly contaminated with BMSCs, we performed a control experiment in which RPMI8226 cells and BMSCs either alone or in co-culture for 24 hours were collected either separately by mechanically tapping the plate to free RPMI8226 cells or by scraping all the cells together. We then performed EMSA on RPMI8226 cells, BMSCs left behind, and those collected together. The NF-κB activity was observed in MM cell fractions and those collected together, but not in BMSC fractions (Figure 3C). Thus, the bulk of the NF-κB activity comes from MM cells, which is not contaminated with that derived in BMSCs.
Time course analysis indicated that NF-κB DNA-binding activity was induced as early as one hour but the peak activity was seen after 24–48 hours of co-culture (Figure 3D). Interestingly, we observed variable degrees of increased NF-κB activity in RPMI8226 cells when they were co-cultured with BMSCs derived from different MM patients (Figure 3E). This indicated that different patient-derived BMSCs have differential NF-κB-inducing activity. Importantly, 100nM bortezomib failed to appreciably inhibit BMSC-enhanced NF-κB activity (Figure 3F). Two other human MM cell lines, U266 and H929, also displayed inducible NF-κB activation when co-cultured with MM-BMSCs, but the Jurkat human T cell leukemia line did not respond to MM BMSCs with respect to NF-κB activation (not shown), suggesting the possibility of cell type selectivity of BMSC-inducible NF-κB activation.
To determine if the observed increases in NF-κB DNA-binding activity in RPMI8226 cells when co-cultured with BMSCs were functionally relevant, we transiently transfected a -κB-GFP reporter gene into RPMI8226 cells, co-cultured them with BMSCs, and evaluated NF-κB-dependent GFP expression (Figure 4). GFP expression was significantly increased by the presence of BMSCs derived from MM patients (Figure 4B, p=0.01). To evaluate if this NF-κB-dependent reporter gene induction is sensitive to inhibition by bortezomib, we next established the dose of bortezomib required to abrogate TNFα-induced activation. Incubation of MM cells with 7.5 nM bortezomib was sufficient to nearly completely block activation (Figure 4A). This concentration of bortezomib failed to significantly inhibit NF-κB-driven GFP expression in RPMI8226 cells grown on top of BMSCs (p=0.33), with the exception of Pt.147 BMSCs where transcriptional activity was blocked by about 50% (Figure 4B). This patient BMSCs also induced NF-κB activation as measured by EMSA that was partially bortezomib sensitive (not shown). These findings together demonstrated that certain MM patient-derived BMSCs have the capacity to induce NF-κB activation in RPMI8226 cells as measured by both EMSA and a -κB-driven reporter assay.
Figure 4. MM-BMSCs induce expression of an NF-κB-dependent GFP reporter gene in RPMI8226 cells in a bortezomib-resistant manner.

(A, B) 10x fluorescent images of -κB-GFP reporter-transfected RPMI8226 cells cultured alone (RMPMI8226 Cells Alone, 4A) or with BMSCs from the indicated patients (Co-culture, 4B). Cells were treated with 1ng/mL TNFα and/or 7.5nM bortezomib where indicated. Phase contrast images of the same fields are shown below each fluorescent image. (C) Percentage of green cells over total viable cells displayed on the y-axis for RPMI8226 cells alone or co-cultured with BMSCs. Standard deviation is represented by error bars, and the statistical significance of the change in -κB-reporter expression was determined by Mann-Whitney test with a 95% confidence interval.
BMSC-enhanced NF-κB activity correlates with increased bortezomib resistance in RPMI8226 cells
Because NF-κB activity can increase drug resistance in a number of human cancer cell lines, including MM cells (29), we evaluated the relationship between the extent of NF-κB activation and that of bortezomib resistance induced by BMSCs. We exposed RPMI8226 cells to varying doses of bortezomib for 24 hrs in the presence or absence of five patient BMSCs with different NF-κB-inducing potentials and analyzed the extent of apoptosis by determining the fraction of cells that displayed nuclear apoptotic morphology (Figure 5A). Our results show that BMSC-enhanced NF-κB activity strongly correlated with increased resistance of RPMI8226 to bortezomib-induced apoptosis (Figure 5B). Rank-based statistical analysis of this correlation shows a highly statistically significant p-value of 0.00000752. Thus, we conclude that BMSCs obtained from MM patients induced bortezomib resistance in RPMI8226 cells in part by inducing bortezomib-resistant NF-κB activation.
Figure 5. The degree of BMSC-induced NF-κB activity correlates with that of BMSC-induced protection of RPMI8226 MM cells against bortezomib-induced apoptosis.
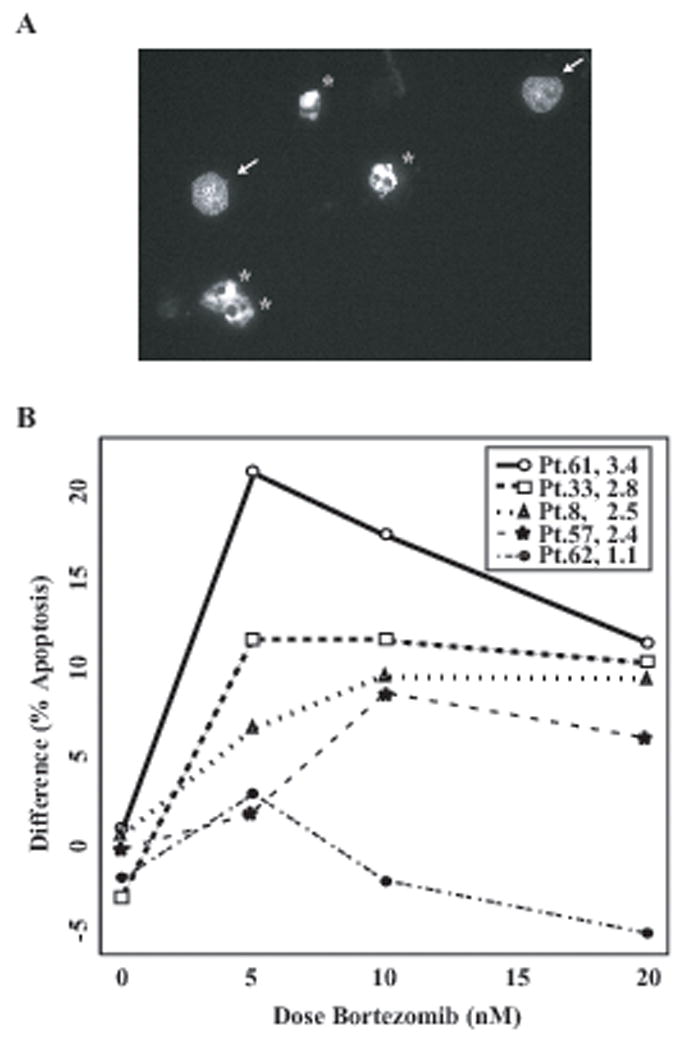
(A) Representative 40x fluorescent micrograph of slides created in apoptosis assays quantified in Figure 5B. Cells are stained with Hoechst 33342 and visualized through a DAPI filter; arrows indicate representative cells that were considered non-apoptotic, and stars indicate representative cells that were considered apoptotic. (B) The difference between % apoptosis induced in RPMI8226 cells alone and that induced in RPMI8226 cells co-cultured with BMSCs (y = % apoptosis (RPMI8226 Alone) - % apoptosis (RPMI8226 + BMSC)) from the indicated patients is plotted on the y-axis versus increasing doses of bortezomib. The figure legend represents the patient numbers as well as fold change in NF-κB activity induced by each BMSC. There is a highly statistically significant correlation between BMSC-induced NF-κB activity and protection against bortezomib-induced apoptosis (p=0.00000752).
Discussion
Many different molecular mechanisms of NF-κB activation have been described thus far, with the canonical and noncanonical pathways being the most widely studied (1). Indeed, in the context of MM, two recent reports found that ~20% of MM patients harbor one or more genetic aberrations in components of the NF-κB pathway, leading to constitutive canonical and/or noncanonical NF-κB activation (30, 31). These studies found amplification or rearrangement of genes including NIK, LTBR, TACI, NFKB1, NFKB2, and CD40, as well as deletion or loss-of-function mutations in genes including CYLD, BIRC2/BIRC3 (cIAP1/cIAP2), TRAF2, and TRAF3, each of which results in the activation of NF-κB associated gene expression. Both groups began their studies with an unbiased, genetic screening approach to identify genetic aberrations in MM, and both uncovered targets primarily within the NF-κB signaling pathway. These seminal studies thus solidly implicated the NF-κB signaling system as one of the major targets of deregulation in MM pathogenesis.
The proteasome inhibitor bortezomib is approved for the treatment of refractory MM patients and demonstrates overall response rates ranging from about 30% as a single agent to nearly 70% in combination with other targeted traditional therapies (32). Given the extensive role of proteasomes in regulating intracellular processes, the precise mechanism (or mechanisms) that mediate clinical efficacy of bortezomib in MM is not clearly defined. For example, previous metabolic labeling studies have demonstrated that the vast majority of intracellular proteins are degraded by proteasomes (33). Many signal transduction pathways that regulate the activity of important transcription factors are also dependent on the activity of proteasomes. These include p53/MDM2, JNK, HIF1, c-Myc, Notch, and others (34–37), pathways that along with NF-κB have been implicated in mediating bortezomib effects on malignant cells (20, 38). A recent study has implicated Noxa/Bcl2 pathway in response to bortezomib in MCL patients, by inducing Noxa upregulation thus freeing Bak to induce mitochondrial apoptosis pathways. Thus, Noxa upregulation appears to be one of the major mechanisms of bortezomib resistance in MCL patients. Whether a similar mechanism is relevant for bortezomib resistance in MM patients remains to be determined. Interestingly, Keats et al correlated the presence of constitutive NF-κB activation associated with a TRAF3 deletion to clinical bortezomib response (31).
Unique among the various NF-κB activation mechanisms, the PIR pathway is highly resistant to bortezomib and other proteasome inhibitors (11, 15). If MM cells harbor constitutive NF-κB activity maintained by the PIR pathway, it is theoretically possible that this could lead to increased resistance of MM cells to bortezomib in patients. To gain initial insight into this possibility, we examined the presence of constitutive NF-κB activity in primary MM samples. We were surprised to find that a significant fraction of the samples analyzed demonstrated the lack of inhibition of constitutive NF-κB activity by bortezomib, even though bortezomib (i) nearly completely blocked proteasome activity in patient samples and (ii) NF-κB activation by a canonical inducer, TNFα. Thus, we favor the notion that the lack of constitutive NF-κB inhibition in primary MM cells by bortezomib is not due to ineffective inhibition of proteasome activity in MM cells but rather this NF-κB activity is maintained by a mechanism that is independent of proteasomes. Alternatively, in the presence of a potent proteasome inhibitor, a bypass pathway may be invoked to overcome the inhibition of proteasome activity. The PIR mechanism might represent one such bypass mechanism in MM cells. Moreover, in multiple cases, we observed paradoxical increases in NF-κB activity after treatment with bortezomib. Although NF-κB proteins have been shown to be degraded by proteasomes (39–41), our limited Western blot analyses did not reveal any increases in p50, p65 and RelB protein levels in several MM patient samples analyzed (unpublished observations). Thus, there could also be a possible compensatory increase in the PIR mechanism in the presence of high bortezomib, leading to increased NF-κB activation in some cases. Given that bortezomib-resistant NF-κB activation has also been documented in endometrial, colon, and other cancer cell lines and/or samples (42, 43), bortezomib-resistant (or even inducible) NF-κB activation might be more prevalent than currently perceived.
Since the role of the bone marrow microenvironment in providing a setting for MM tumor cell growth and survival is well documented (23), we also evaluated the impact of the presence of MM patient-derived BMSCs on the regulation of NF-κB. Previous studies have demonstrated that these cells secrete factors, such as IL-6, TNFα, SDF-1 and IGF-1, among others, which can mediate the protective effect on MM cells. Indeed some of these factors have been demonstrated to cause NF-κB activation in MM cells (23). Similarly, MM cells also secrete factors that can induce NF-κB activity in BMSCs to in turn produce MM supporting factors (23). IMiD’s, such as thalidomide and revlimid, are believed to modulate these MM-stromal interactions to induce clinical efficacy (23). Bortezomib has also been shown to block these mutual interactions by blocking NF-κB activation in BMSCs by primarily interfering with canonical and noncanonical pathways of NF-κB activation. Our current results introduce yet another possibility that involves the PIR pathway. We found that BMSCs derived from MM patients were capable of activating NF-κB in MM cell lines and primary cells to variable degrees in a manner resistant to bortezomib. Significantly, this activation was largely resistant to treatment with high concentrations of bortezomib. Our preliminary studies suggest that the critical mediator (or mediators) of this inducible PIR activity is soluble and likely a peptide, based on the presence of the activity in the soluble fraction of BMSC conditioned media following 100,000xg ultracentrifugation for 2 hrs and its sensitivity to proteinase K and heat. We have also tentatively eliminated the involvement of aforementioned factors by means of neutralizing antibodies. Further studies are thus required to elucidate the molecular mediator(s) of BMSC-induced PIR NF-κB activity. Identification of such a factor may provide deeper insight into both the signaling mechanism and biologic role of the PIR pathway in MM pathobiology and other PIR involving systems in general.
Materials and Methods
Cell lines, antibodies, and chemicals
RPMI8226, U266, Jurkat, and H929 cell lines (American Type Cell Culture, Rockville, MD) were cultured in 37ºC incubators with 5% CO2, using either Dulbecco’s Modified Eagle’s Medium (DMEM, Mediatech, Inc., Herndon, VA) (RPMI8226) supplemented with 10% fetal bovine serum (FBS), 1250 U of penicillin G (Sigma), 0.5mg of streptomycin sulfate (Sigma), and other supplements as per ATCC recommendations. DMEM with 20% FBS, 1% L-glutamine (“BMSC media”) was used in co-culture assays. p65 (C20) and RelB (C19) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), p50 (06–886) and p52 (06–413) antibodies were purchased from Upstate Biotechnologies (Lake Placid, NY), and the cRel (SA-172) antibody was obtained from Biomol (Plymouth Meeting, PA). Bortezomib was commercially obtained from Millennium Pharmaceuticals, Inc. (Cambridge, MA) for experimental purposes. Human recombinant TNFα was purchased from Calbiochem (San Diego, CA). POH was obtained from Sigma-Aldrich Inc. (St. Louis, MO). Bapta-AM was purchased from Merck (Darmstadt, Germany).
Primary BMSCs
Primary human BMSCs were derived from de-identified fresh whole bone marrow aspirates. The aspirates were collected under the University of Wisconsin Institutional Review Board (IRB) exemption protocol #M-2004–1315, and thus were accompanied only by the patients’ diagnosis of active myeloma (>10% plasma cells on Wright Giemsa stained aspirate smears, analysis provided by Dr. Leith and other UW Staff Pathologists). Disease stage, treatment history and/or response, and other identifying patient characteristics are not available for the samples analyzed in this study. Mononuclear cells were isolated from aspirates as detailed in the Miltenyi MidiMACS bone marrow mononuclear cell protocol. Briefly, whole bone marrow is diluted with IMDM media supplemented with heparin sulfate and DNAse I, and centrifuged with lymphocyte separation media (Cellgro, Manassas, VA) to isolate the mononuclear population. Mononuclear cells were plated in BMSC media, and cultured at 37°C/5% CO2. After two weeks non-adherent cells were washed off and one-half volume media exchange was performed every three days, until a confluent layer of adherent stromal cells had formed. These cells were expanded, and cryopreserved in liquid nitrogen. Repeated analysis of cryopreserved BMSCs from 22 patients, the NF-κB-inducing activity on RPMI8226 cells remained relatively constant over 7 passages.
Primary MM Cells
To obtain primary MM cells, mononuclear cells isolated from patient bone marrow aspirates as described above were positively sorted using anti-CD138 magnetic microbeads and the MidiMACS cell sorting system following manufacturer’s protocol (Miltenyi Biotec, Auburn, CA) to >95% purity as determined by anti-CD138-PE staining and FACS analysis, and cultured in 37°C/5%CO2 incubators in BMSC media.
Co-culture assays
6x104 BMSCs per well were plated in 6-well dishes and allowed to form an adherent monolayer over 24 hours. Media were aspirated, and 106 RPMI8226 cells or 5x105 primary MM cells were plated over the monolayer. Where appropriate, 100nM bortezomib was added for the last 4 hours of co-culture. After 24 hours, MM cells were mechanically detached from stromal cells, leaving ~5% of MM cells behind, mostly along the perimeter of the dish (as determined by immunofluorescence staining with anti-CD138 antibody (Miltenyi Biotec, Auburn, CA)). The BMSC monolayer was undisturbed. For apoptosis assays, after 24 hours of co-culture, cells were treated with various doses of bortezomib for an additional 24 hours before MM cells were removed for analysis.
Proteasome Activity Assay
Chymotryptic proteasomal activity was measured using the Proteasome-Glo™ Assay (Promega Corporation, Madison, WI). 10,000 cells per well were plated in a black-walled 96-well tissue culture dish, allowed to rest for three hours, and treated with increasing doses of bortezomib for 105 minutes before addition of the luminescent reagent as directed by the manufacturer. Luminescence was read on a Victor3V 1420 Multilabel Counter from Perkin Elmer (Walther, MA). Relative light units (RLU) for the no drug control was designated 100% proteasome activity, and the ratio of RLU for each dose of bortezomib over control was used to determine the percentage decrease in chymotryptic activity.
DNA-binding analysis
Electrophoretic mobility shift assays (EMSA) for cell lines were performed as described previously (10). For primary MM cells, miniaturized EMSA (“mini-EMSA”) by using the Mini-Protean 3 system (Bio-Rad Laboratories, Hercules, CA), 1mm thickness gel, and 15-well comb to accommodate small amounts of protein obtained from primary MM cells. Briefly, CD138+ cells were washed in 1xPBS, centrifuged in 200μL PCR tubes at 5,000 rpm for 3 minutes at room temperature, and pellets frozen at -70°C for later analysis. Frozen cell pellets were thawed on ice, lysed in Totex buffer with protease inhibitors (10) at 2.0x104 cells/μL, incubated on ice for 30 minutes with occasional tapping, and spun at 13,000 rpm for 10 minutes at 4°C. The resultant extracts in the supernatants were used for mini-EMSAs. 1.5μL of extract were incubated with 5x binding buffer, 0.1mM dithiothreitol, and 0.5μL of radioactive probe on ice for 20 minutes as described in (10). 32P-labeled probe was added and samples incubated at room temperature for 20 minutes. For each sample, one reaction with Ig-κ-κB probe, and one with Oct-1 probe were run, using Oct-1 binding levels as a loading control. Gels were run at 120 volts for ~45 minutes, dried onto Whatman filters under vacuum, exposed to phosphoimage screen and quantified using Image Quant software. The volume of intensity using local area average background correction was calculated for cumulative NF-κB bands, and the Oct-1 band.
GFP-reporter assay
5x106 RPMI8226 cells were transiently transfected using nucleofection (Amaxa Inc., Gaithersburg, MD) with 3μg of the 3x-κB-GFP reporter construct as described previously (44) with solution T, program G15. After a 24 hour rest period, cells were split equally into culture alone or on top of 3x104 BMSC cells/mL plated 24 hours previously and treated with 7.5nM bortezomib and/or 1ng/mL TNFα where indicated. GFP expression was visualized 24 hours later. Three random 10x fields of view were recorded through a GFP filter on a Zeiss Axiovert 200M microscope, using Axiovision 3.1 software and a Zeiss Axiocam to produce images (Thornwood, NY). A 486ms exposure time was used for each condition. The contrast of each of the images was adjusted to the exact same value using Adobe Photoshop (Adobe Systems Incorporated, San Jose, CA) to visualize GFP expression above untreated control levels. The remaining fluorescent cells were counted and displayed as a percentage of the total live cells counted in phase contrast images of the same field. Representative 10x images were colored and adjusted with identical Photoshop methods for each, eliminating Photoshop bias.
Apoptosis assay
Following culture and treatment as described in the Co-culture assay section, MM cells were cytospun onto microscope slides, fixed with 4% paraformaldehyde, and stained with Hoechst 33342 to label DNA. Slides were visualized through a DAPI filter. 1800 cells were counted for each condition (3 × 200 for each of 3 slides), and the percentage of apoptotic nuclei among them was quantified. Examples of what was considered normal versus apoptotic nuclei are presented.
Statistical analysis
Fold-induction of NF-κB activity was determined by phosphoimage quantification of enhanced NF-κB activity over constitutive activity, which was assigned an arbitrary value of one. The change in NF-κB-dependent GFP expression induced by BMSC co-culture was tested for significance using a two-sided Mann-Whitney test with a 95% confidence interval. Significant changes in GFP expression with and without bortezomib were determined by the student’s t-test using a 95% confidence interval. The difference in bortezomib-induced apoptosis between RPMI8226 cells alone and co-culture with different patient BMSCs is plotted as a function of bortezomib dose. When the permutation test was done using the sum of the squares of rank difference to the observed BMSC-induced NF-κB activation level as a test statistic, the p-value was 0.00000752.
Acknowledgments
Predoctoral NIH Fellowship in Hematology Research T32 HL07899, Predoctoral National Institute of Aging Fellowship F30-AG029714–01 (S. Markovina), NIH grant R01–CA08106, University of Wisconsin Trillium Foundation Grant, Shaw Scientist Award from Milwaukee Foundation (S. Miyamoto).
We thank the Miyamoto lab members for helpful discussion, Dr. Alice Garry-McCoy for expert opinion and help with initiation of the project, and the Department of Hematology Clinical Trials Staff for help with IRB protocols and sample acquisition.
References
- 1.Perkins ND. Integrating cell-signaling pathways with NF-kappaB and IKK function. Nature reviews. 2007;8(1):49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 2.Fan C, Li Q, Ross D, Engelhardt JF. Tyrosine phosphorylation of I kappa B alpha activates NF kappa B through a redox-regulated and c-Src-dependent mechanism following hypoxia/reoxygenation. The Journal of biological chemistry. 2003;278(3):2072–80. doi: 10.1074/jbc.M206718200. [DOI] [PubMed] [Google Scholar]
- 3.Kato T, Jr, Delhase M, Hoffmann A, Karin M. CK2 Is a C-Terminal IkappaB Kinase Responsible for NF-kappaB Activation during the UV Response. Molecular cell. 2003;12(4):829–39. doi: 10.1016/s1097-2765(03)00358-7. [DOI] [PubMed] [Google Scholar]
- 4.Lee FY, Kim DW, Karmin JA, et al. mu-Calpain regulates receptor activator of NF-kappaB ligand (RANKL)-supported osteoclastogenesis via NF-kappaB activation in RAW 264.7 cells. The Journal of biological chemistry. 2005;280(33):29929–36. doi: 10.1074/jbc.M414600200. [DOI] [PubMed] [Google Scholar]
- 5.Romieu-Mourez R, Landesman-Bollag E, Seldin DC, Sonenshein GE. Protein kinase CK2 promotes aberrant activation of nuclear factor-kappaB, transformed phenotype, and survival of breast cancer cells. Cancer research. 2002;62(22):6770–8. [PubMed] [Google Scholar]
- 6.Schaecher K, Goust JM, Banik NL. The effects of calpain inhibition on IkB alpha degradation after activation of PBMCs: identification of the calpain cleavage sites. Neurochemical research. 2004;29(7):1443–51. doi: 10.1023/b:nere.0000026410.56000.dd. [DOI] [PubMed] [Google Scholar]
- 7.Baghdiguian S, Richard I, Martin M, et al. Pathophysiology of limb girdle muscular dystrophy type 2A: hypothesis and new insights into the IkappaBalpha/NF-kappaB survival pathway in skeletal muscle. Journal of molecular medicine (Berlin, Germany) 2001;79(5–6):254–61. doi: 10.1007/s001090100225. [DOI] [PubMed] [Google Scholar]
- 8.Shen J, Channavajhala P, Seldin DC, Sonenshein GE. Phosphorylation by the protein kinase CK2 promotes calpain-mediated degradation of IkappaBalpha. J Immunol. 2001;167(9):4919–25. doi: 10.4049/jimmunol.167.9.4919. [DOI] [PubMed] [Google Scholar]
- 9.Cogswell PC, Kashatus DF, Keifer JA, et al. NF-kappa B and I kappa B alpha are found in the mitochondria. Evidence for regulation of mitochondrial gene expression by NF-kappa B. The Journal of biological chemistry. 2003;278(5):2963–8. doi: 10.1074/jbc.M209995200. [DOI] [PubMed] [Google Scholar]
- 10.O’Connor S, Shumway SD, Amanna IJ, Hayes CE, Miyamoto S. Regulation of constitutive p50/c-Rel activity via proteasome inhibitor-resistant IkappaBalpha degradation in B cells. Molecular and cellular biology. 2004;24(11):4895–908. doi: 10.1128/MCB.24.11.4895-4908.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shumway SD, Miyamoto S. A mechanistic insight into a proteasome-independent constitutive inhibitor kappaBalpha (IkappaBalpha) degradation and nuclear factor kappaB (NF-kappaB) activation pathway in WEHI-231 B-cells. The Biochemical journal. 2004;380(Pt 1):173–80. doi: 10.1042/BJ20031796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyamoto S, Seufzer BJ, Shumway SD. Novel IkappaB alpha proteolytic pathway in WEHI231 immature B cells. Molecular and cellular biology. 1998;18(1):19–29. doi: 10.1128/mcb.18.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berchtold CM, Chen KS, Miyamoto S, Gould MN. Perillyl alcohol inhibits a calcium-dependent constitutive nuclear factor-kappaB pathway. Cancer research. 2005;65(18):8558–66. doi: 10.1158/0008-5472.CAN-04-4072. [DOI] [PubMed] [Google Scholar]
- 14.Yao T, Cohen RE. Giant proteases: beyond the proteasome. Curr Biol. 1999;9(15):R551–3. doi: 10.1016/s0960-9822(99)80352-2. [DOI] [PubMed] [Google Scholar]
- 15.O’Connor S, Markovina S, Miyamoto S. Evidence for a phosphorylation-independent role for Ser 32 and 36 in proteasome inhibitor-resistant (PIR) IkappaBalpha degradation in B cells. Experimental cell research. 2005;307(1):15–25. doi: 10.1016/j.yexcr.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 16.Adams J, Palombella VJ, Sausville EA, et al. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer research. 1999;59(11):2615–22. [PubMed] [Google Scholar]
- 17.Richardson PG, Barlogie B, Berenson J, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. The New England journal of medicine. 2003;348(26):2609–17. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 18.Hideshima T, Mitsiades C, Akiyama M, et al. Molecular mechanisms mediating antimyeloma activity of proteasome inhibitor PS-341. Blood. 2003;101(4):1530–4. doi: 10.1182/blood-2002-08-2543. [DOI] [PubMed] [Google Scholar]
- 19.Mitsiades N, Mitsiades CS, Poulaki V, et al. Biologic sequelae of nuclear factor-kappaB blockade in multiple myeloma: therapeutic applications. Blood. 2002;99(11):4079–86. doi: 10.1182/blood.v99.11.4079. [DOI] [PubMed] [Google Scholar]
- 20.Nencioni A, Grunebach F, Patrone F, Ballestrero A, Brossart P. Proteasome inhibitors: antitumor effects and beyond. Leukemia. 2007;21(1):30–6. doi: 10.1038/sj.leu.2404444. [DOI] [PubMed] [Google Scholar]
- 21.Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107(12):4907–16. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson KC. Targeted therapy of multiple myeloma based upon tumor-microenvironmental interactions. Experimental hematology. 2007;35(4 Suppl 1):155–62. doi: 10.1016/j.exphem.2007.01.024. [DOI] [PubMed] [Google Scholar]
- 23.Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer. 2007;7(8):585–98. doi: 10.1038/nrc2189. [DOI] [PubMed] [Google Scholar]
- 24.Perez-Galan P, Roue G, Villamor N, Montserrat E, Campo E, Colomer D. The proteasome inhibitor bortezomib induces apoptosis in mantle-cell lymphoma through generation of ROS and Noxa activation independent of p53 status. Blood. 2006;107(1):257–64. doi: 10.1182/blood-2005-05-2091. [DOI] [PubMed] [Google Scholar]
- 25.Gomez-Bougie P, Wuilleme-Toumi S, Menoret E, et al. Noxa up-regulation and Mcl-1 cleavage are associated to apoptosis induction by bortezomib in multiple myeloma. Cancer research. 2007;67(11):5418–24. doi: 10.1158/0008-5472.CAN-06-4322. [DOI] [PubMed] [Google Scholar]
- 26.Bharti AC, Shishodia S, Reuben JM, et al. Nuclear factor-kappaB and STAT3 are constitutively active in CD138+ cells derived from multiple myeloma patients, and suppression of these transcription factors leads to apoptosis. Blood. 2004;103(8):3175–84. doi: 10.1182/blood-2003-06-2151. [DOI] [PubMed] [Google Scholar]
- 27.Podar K, Hideshima T, Chauhan D, Anderson KC. Targeting signalling pathways for the treatment of multiple myeloma. Expert opinion on therapeutic targets. 2005;9(2):359–81. doi: 10.1517/14728222.9.2.359. [DOI] [PubMed] [Google Scholar]
- 28.Gregoretti MG, Gottardi D, Ghia P, et al. Characterization of bone marrow stromal cells from multiple myeloma. Leukemia research. 1994;18(9):675–82. doi: 10.1016/0145-2126(94)90067-1. [DOI] [PubMed] [Google Scholar]
- 29.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441(7092):431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 30.Annunziata CM, Davis RE, Demchenko Y, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer cell. 2007;12(2):115–30. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keats JJ, Fonseca R, Chesi M, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer cell. 2007;12(2):131–44. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Merchionne F, Perosa F, Dammacco F. New therapies in multiple myeloma. Clinical and experimental medicine. 2007;7(3):83–97. doi: 10.1007/s10238-007-0134-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rock KL, Gramm C, Rothstein L, et al. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78(5):761–71. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 34.Ehebauer M, Hayward P, Martinez-Arias A. Notch signaling pathway. Sci STKE. 2006;2006(364):cm7. doi: 10.1126/stke.3642006cm7. [DOI] [PubMed] [Google Scholar]
- 35.Hunter T, Karin M. The regulation of transcription by phosphorylation. Cell. 1992;70(3):375–87. doi: 10.1016/0092-8674(92)90162-6. [DOI] [PubMed] [Google Scholar]
- 36.Sears R, Leone G, DeGregori J, Nevins JR. Ras enhances Myc protein stability. Molecular cell. 1999;3(2):169–79. doi: 10.1016/s1097-2765(00)80308-1. [DOI] [PubMed] [Google Scholar]
- 37.Wei W, Yu XD. Hypoxia-inducible factors: crosstalk between their protein stability and protein degradation. Cancer letters. 2007;257(2):145–56. doi: 10.1016/j.canlet.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 38.Richardson PG, Mitsiades C, Hideshima T, Anderson KC. Proteasome inhibition in the treatment of cancer. Cell cycle (Georgetown, Tex. 2005;4(2):290–6. [PubMed] [Google Scholar]
- 39.Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature. 2005;434(7037):1138–43. doi: 10.1038/nature03491. [DOI] [PubMed] [Google Scholar]
- 40.Ryo A, Suizu F, Yoshida Y, et al. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Molecular cell. 2003;12(6):1413–26. doi: 10.1016/s1097-2765(03)00490-8. [DOI] [PubMed] [Google Scholar]
- 41.Tanaka T, Grusby MJ, Kaisho T. PDLIM2-mediated termination of transcription factor NF-kappaB activation by intranuclear sequestration and degradation of the p65 subunit. Nature immunology. 2007;8(6):584–91. doi: 10.1038/ni1464. [DOI] [PubMed] [Google Scholar]
- 42.Dolcet X, Llobet D, Encinas M, et al. Proteasome inhibitors induce death but activate NF-kappaB on endometrial carcinoma cell lines and primary culture explants. The Journal of biological chemistry. 2006;281(31):22118–30. doi: 10.1074/jbc.M601350200. [DOI] [PubMed] [Google Scholar]
- 43.Nagy K, Szekely-Szuts K, Izeradjene K, et al. Proteasome inhibitors sensitize colon carcinoma cells to TRAIL-induced apoptosis via enhanced release of Smac/DIABLO from the mitochondria. Pathol Oncol Res. 2006;12(3):133–42. doi: 10.1007/BF02893359. [DOI] [PubMed] [Google Scholar]
- 44.Wuerzberger-Davis SM, Chang PY, Berchtold C, Miyamoto S. Enhanced G2-M arrest by nuclear factor-{kappa}B-dependent p21waf1/cip1 induction. Mol Cancer Res. 2005;3(6):345–53. doi: 10.1158/1541-7786.MCR-05-0028. [DOI] [PubMed] [Google Scholar]