

SUMMARY
Excess levels of circulating amino acids (AAs) play a causal role in specific human pathologies, including obesity and type 2 diabetes. Moreover, obesity and diabetes are contributing factors in the development of cancer, with recent studies suggesting this link is in part mediated by AA activation of mammalian Target Of Rapamycin (mTOR) Complex1. AAs appear to mediate this response through class 3 PI3K, or hVps34, rather than through the canonical class 1 PI3K pathway used by growth factors and hormones. Here we show that AAs induce a rise in intracellular Ca2+ ([Ca2+]i), which triggers mTOR Complex1 and hVps34 activation. We demonstrate that the rise in [Ca2+]i increases the direct binding of Ca2+/CaM to an evolutionarily conserved motif in hVps34 that is required for lipid kinase activity and increased mTOR Complex1 signaling. These findings have important implications regarding the basic signaling mechanisms linking metabolic disorders with cancer progression.
Nutrient overload is a key contributing factor to the epidemic in obesity (Um et al., 2006), which until recently was largely confined to the Western world, but now is a world wide problem (Finkelstein et al., 2005). The morbidity of obesity not only extends to diabetes and cardiovascular disease, but recently has been shown to be linked to 20% of cancer deaths (Calle and Kaaks, 2004). A critical effector of nutrient signaling is the mTOR protein kinase, which exists in two distinct complexes (Wullschleger et al., 2006). The first, mTOR Complex1, is sensitive to rapamycin and includes three additional proteins; regulatory-associated protein of mTOR (raptor), G-protein β-subunit-like protein (GβL) and proline-rich PKB/Akt substrate 40 kDa (PRAS40) (Dann et al., 2007; Kim et al., 2002). In contrast, mTOR Complex2 is rapamycin insensitive and in addition to GβL consists of rapamycin-insensitive companion of mTOR (rictor) and mammalian stress-activated protein kinase (SAPK)-interacting protein-1 (mSin1) and protein observed with rictor (protor) (Dann et al., 2007; Pearce et al., 2007). Both complexes are regulated by hormones and growth factors, however only mTOR Complex1 is acutely regulated by nutrients, such as amino acids (AA) and glucose (Dann et al., 2007). The importance of the AA arm of mTOR Complex1 signaling is highlighted by the observation that circulating AAs, particularly branched-chain AAs (BCAAs), are elevated in obese humans and are known to drive mTOR Complex1 signaling (Krebs, 2005; Um et al., 2006).
AA activation of mTOR Complex1 increases growth through increased ribosome biogenesis and elevated rates of protein synthesis, while suppressing autophagy (Wullschleger et al., 2006). However, mTOR Complex1 also acts as a homeostatic regulator to attenuate insulin-induced uptake of nutrients under conditions of nutrient overload (Patti and Kahn, 2004; Tremblay et al., 2005a; Tzatsos and Kandror, 2006). These effects are in part attributed to mTOR Complex1 phosphorylation of IRS1 at sites which antagonize binding of either IRS1 to the insulin receptor or to class 1 phosphatidyl-inositide-3OH-kinase (PI3K), attenuating insulin action (Tzatsos and Kandror, 2006; Um et al., 2004). Moreover, recent studies show AAs can directly mediate these responses by phosphorylation of IRS1 by S6K1 at specific sites residing at the amino (Harrington et al., 2004) and carboxy (Tremblay et al., 2007) termini of IRS1, respectively. These observations have stimulated the need to elucidate the molecular mechanisms by which nutrient overload, through increased mTOR Complex1 activation, leads to the development of specific pathologies.
Hormones and growth factors mediate mTOR Complex1 activation through a canonical signaling cascade triggered by the activation of class 1 PI3K and protein kinase B (PKB), leading to the sequential activation of the small GTPase Ras homologue enriched in brain (Rheb) and mTOR Complex1 (Dann et al., 2007). In contrast, AAs stimulate mTOR Complex1 activation through class 3 PI3K, or human vacuolar protein sorting 34 (hVps34) (Byfield et al., 2005; Nobukuni et al., 2005). AA-induced activation of hVps34 leads to increased production of phosphatidylinositol 3 phosphate (PI(3)P), which acts to recruit Fab1/YOTB/-2K632.12/Vac1/EEA1 (FYVE) or PI(3)P-targeting phox homology (PX) domain-containing proteins to early endosomes (Nobukuni et al., 2007). These (PI(3)P)-rich domain protein complexes are known to function as intracellular signaling platforms (Nobukuni et al., 2007). Consistent with this model, depletion of hVps34 protein levels or ectopic expression of a FYVE domain construct, which sequesters PI(3)P, blunts AA-induced mTOR Complex1 signaling (Byfield et al., 2005; Nobukuni et al., 2005). In contrast, lowering hVps34 protein levels has no effect on insulin-induced PKB/Akt activation (Byfield et al., 2005; Nobukuni et al., 2005). However, despite the importance of AA signaling in controlling mTOR Complex1 signaling, we know little of the underlying mechanism which mediates the hVps34 response.
Here we set out to identify the pathway and mechanism by which AAs signal to hVps34 to stimulate mTOR Complex1 signaling. Unexpectedly, we find that AAs induce an increase in [Ca2+]i, leading to enhanced binding of calmodulin (CaM) to hVps34, an interaction that occurs through an evolutionarily conserved CaM-binding site, leading to the activation of mTOR Complex1 signaling.
RESULTS
AA induced mTOR Complex1 signaling is [Ca2+]i dependent
Although, like growth factors and hormones, AAs induce mTOR Complex1-mediated S6 kinase1 (S6K1) activation, they do not activate PKB, as judged by PKB S473 phosphorylation (Byfield et al., 2005; Hara et al., 1998; Nobukuni et al., 2005). Consistent with these findings, we observe no effect of AAs on either PKB S473 or T308 phosphorylation (Figure 1A and Supplemental Data, Figure S1A). Moreover, siRNA depletion of PKBα, the major PKB isoform present in HeLa cells, had no impact on AA-induced S6K1 T389 phosphorylation, but like AA withdrawal (Nobukuni et al., 2005) inhibited the insulin-induced response (Figure 1A). In addition, the extracellular signal-regulated kinases (Erks), which have been implicated in regulating mTOR Complex1 activation through phosphorylation of TSC2 (Um et al., 2006), are not affected by AA stimulation (Supplemental Data, Figure S1B). Like AAs, agents that increase [Ca2+]i levels have been reported to induce S6K1 activation without inducing PKB activation (Conus et al., 1998), raising the possibility that AA-induction of S6K1 is dependent on [Ca2+]i levels. To assess this possibility, AA-deprived HeLa cells were stimulated by the re-addition of AAs in the absence or presence of the [Ca2+]i chelator BAPTA-acetoxymethyl ester (BAPTA-AM). We found that re-addition of AAs led to increased S6K1 T389 phosphorylation, a response attenuated by BAPTA-AM (Figure 1B). Moreover, BAPTA-AM suppressed insulin-induced S6K1 T389 phosphorylation, but had no effect on PKB S473 phosphorylation, the phosphorylation of two PKB substrates, TSC2 S939 and FoxO1 T24, or on Erk T202 and Y204 phosphorylation (Figure 1B and Supplemental Data, Figures S1C and S1D, respectively). Importantly, pretreatment of AA-deprived cells with the cell impermeant Ca2+ chelator, ethylene glycol tetra-acetic acid (EGTA), also suppressed AA-induction of S6K1 T389 phosphorylation (Figure 1C, left panel), indicating that extracellular Ca2+ is also required for this response. In addition, these effects appeared to be mTOR Complex1-dependent as EGTA and BAPTA-AM blocked 4E binding protein 1 (4E-BP1) S65 phosphorylation (Figure 1C, left panel), a known mTOR Complex1 substrate (Gingras et al., 1999). Finally, earlier studies showed that leucine-induced GSK3-β phosphorylation is sensitive to rapamycin and PKB-independent (Peyrollier et al., 2000), and more recently it has been argued that GSK3-β S9 phosphorylation and c-Myc stability are mediated by S6K1 in TSC2 deficient cells (Zhang et al., 2006). Consistent with these results we find that AA-induce GSK3-β S9 phosphorylation and c-Myc stability are suppressed by EGTA and BAPTA-AM (Figure 1C, right panel). Moreover, the effects of these Ca2+ chelators appear to be mediated by inhibition of S6K1, as siRNA depletion of S6K1 attenuates both responses (Figure 1C). These results suggest that both AA- and insulin-induced mTOR Complex1 signaling require [Ca2+]i.
Figure 1. Ca2+chelators selectively block mTOR Complex1 signaling.

(A) HeLa cells were transfected with 16 nM PKBα siRNA for 48 hr (Experimental Procedures). After serum withdrawal overnight, cells were deprived of AAs for 2 hr and then stimulated with 2x the concentration of AAs present in DMEM for 30 min (left panel). Alternatively, after transfection, cells were deprived of serum overnight followed by stimulation with 200 nM insulin for 30 min (right panel). (B) HeLa cells were treated with AAs (left panel) or insulin (middle panel) as described in (A). Prior to stimulation cells were pretreated with 2 μM BAPTA-AM for 15 min where indicated. Quantification of Western blots from 3 independent experiments (right panel) was performed using ImageQuant TL software (Amersham). (C) HeLa cells were treated with AAs as in (A). Where indicated cells were pretreated with either 2 mM EGTA or 2 μM BAPTA-AM for 15 min (left panel). S6K1 siRNA (16 nM) transfection and AA-stimulation was performed as in (A) (right panel).
AAs induce an increase in [Ca2+]i
To discriminate between a permissive role for Ca2+ in mTOR Complex1 signaling versus an AA-induced increase in [Ca2+]i, cells were loaded with the Ca2+-sensitive indicator dye, Fluo-4, and [Ca2+]i levels were monitored by fluorescence microscopy (Thomas et al., 1996). The addition of AAs evoked a rapid increase in [Ca2+]i, which was largely inhibited by preincubation with BAPTA-AM (Figure 2A). Oscillations of [Ca2+]i are a common form of physiological Ca2+ signaling, and transition to a sustained [Ca2+]i rise is typical of maximal stimulation (Thomas et al., 1996). AA-evoked [Ca2+]i increases, detected by employing fura-2, included both [Ca2+]i oscillations (159/304 cells) and sustained [Ca2+]i increases (145/304 cells) (Figure 2B, first and second panels, respectively). A submaximal dose of the IP3-linked agonist histamine also evoked [Ca2+]i oscillations, albeit with a higher amplitude and frequency compared with the AA-induced [Ca2+]i responses (Figure 2B, first panel). The [Ca2+]i response to AAs was rapidly reversed by AA withdrawal (Figure 2B, third panel), or by either removal of extracellular Ca2+ or addition of EGTA (Figure 2B, second and fourth panels, respectively). In the presence of EGTA, addition of the endoplasmic reticulum (ER) Ca2+-ATPase inhibitor thapsigargin (Tg) (Thastrup et al., 1990) caused a rapid increase in [Ca2+]i, indicating that EGTA treatment had not depleted internal Ca2+ stores (Figure 2B, fourth panel). This suggests that the AA-induced [Ca2+]i signal requires entry of extracellular Ca2+. Other studies have shown the importance of the BCAA leucine in inducing mTOR Complex1 signaling, as compared with other essential AAs, such as phenylalanine, or other BCAAs, such as isoleucine (Hara et al., 1998). Consistent with earlier observations (Hara et al., 1998), we found that leucine, but not phenylalanine or isoleucine, evoked a significant increase in S6K1 T389 phosphorylation and [Ca2+]i (Supplemental Data, Figure S2A), and that these responses were reversed by removal of leucine (data not shown and Supplemental Data, Figure S2B). Thus, AAs, and leucine alone, induce an increase in [Ca2+]i, which is dependent on the influx of extracellular Ca2+.
Figure 2. AA stimulation evokes an increase in [Ca2+]i. (A).

Confocal images of Fluo-4 fluorescence stained HeLa cells treated with AAs as in Figure 1A for 5 min in the absence or presence of BAPTA-AM. (B) [Ca2+]i traces in fura2-loaded HeLa cells stimulated with AAs as in Figure 1A. First panel shows representative single-cell [Ca2+]i response to AAs followed by 0.5 μM histamine addition. Second panel shows effect of removing extracellular Ca2+ by replacement of medium with AA-containing buffer without added Ca2+. Third panel shows effect of AA withdrawal on single-cell [Ca2+]i response; this experiment was carried out in the presence of 10% dialyzed FCS. Fourth panel shows effect of extracellular Ca2+ chelation with 4 mM EGTA, followed by treatment with 2 μM Tg.. (C) Left panel, HeLa cells plated onto 35 mm tissue culture plates were deprived of serum and AAs, then loaded with Calcium Green-1/AM and [Ca2+]i responses were monitored by confocal microscopy before and after the addition of 150 μM Tg. The trace is the mean Tg-evoked [Ca2+]i increase (n = 332 cells). Right panel, HeLa cells treated with AAs as in Figure 1A, then with 150 μM Tg for 30 min in the absence or presence of either 2 mM EGTA or 2 μM BAPTA-AM. EGTA and BAPTA-AM were added 15 min prior to the addition of Tg. (D) HeLa cells treated with either EGTA or BAPTA-AM were stimulated with AAs or Tg for 15 min as described in Figures 1A and 2C, respectively. Cells were harvested, lysed, mTOR was immunoprecipitated and in vitro mTORC1 activity was assayed as described (Sancak et al., 2007).
A rise in [Ca2+]i is sufficient for mTOR Complex1 activation
To determine whether a rise in intracellular Ca2+ was sufficient to recapitulate AA-induced S6K1 T389 phosphorylation, AA-deprived cells were treated with Tg in the presence of external Ca2+. The results show that such treatment leads to a sustained increase in [Ca2+]i (Figure 2C) and in S6K1 T389 as well as 4E-BP1 S65 phosphorylation (Figure 2C), whereas Tg has no effect on ErK or PKB phosphorylation (Supplemental Data, Figure S1B). Consistent with the transient nature of the Tg-induced rise in [Ca2+]i in the presence of EGTA (Figure 2B, fourth panel), the increase in S6K1 T389 and 4E-BP1 S65 phosphorylation is blocked by pretreatment of cells with EGTA, as well as BAPTA-AM (Figure 2C). In addition, Tg alone did not increase the uptake of AAs, including leucine (Supplemental Data, Figure S2C). Recent studies have shown that immunoprecipitates of mTOR Complex1 from insulin-stimulated cells have increased in vitro kinase activity towards S6K1 and 4E-BP1 (Sancak et al., 2007). Following similar assay conditions, we find that immunoprecipitates of mTOR from AA-stimulated cells, versus AA-deprived cells, have increased in vitro mTOR Complex1 kinase activity towards S6K1 and 4E-BP1 as substrates (Figure 2D and Supplemental Data, Figure S2C). Moreover, the effect of AAs on the in vitro mTOR Complex1 kinase activity can be recapitulated by Tg and blocked by either EGTA or BAPTA-AM (Figure 2D and Supplemental Data, Figure S2D). Consistent with the rise in [Ca2+]i triggering increased mTOR Complex1 kinase signaling, neither rapamycin nor wortmannin block the AA-induced rise in [Ca2+]i (data not shown). These data strongly support a model whereby the AA-induced increase in [Ca2+]i leads to mTOR Complex1 activation.
AA activation of hVps34 is mediated by [Ca2+]i
To determine where [Ca2+]i acts in the mTOR Complex1 pathway, we first examined if Tg activation of S6K1 in the absence of AAs is sensitive to wortmannin. The results show that Tg-induced T389 S6K1 phosphorylation is abolished by pre-treatment with wortmannin (Figure 3A), similar to results observed with AAs (Figure 3A). Moreover, as might be expected from the apparently distinct mechanisms by which Tg and AAs induce increased [Ca2+]i, the two together caused an apparent additive increase in S6K1 T389 phosphorylation (Figure 3A). We also find that siRNA depletion of Rheb protein levels blocks Tg-induced S6K1 T389 phosphorylation (Figure 3B), as we previously reported for the AA response (Nobukuni et al., 2005). Consistent with these effects being mediated by hVps34, depletion of the lipid kinase with a previously described siRNA (Nobukuni et al., 2005) also attenuated Tg-induced S6K1 T389 phosphorylation (Figure 3B). To determine whether the effects of increased [Ca2+]i on mTOR Complex1 activation are mediated through hVps34, we analyzed the effect of BAPTA-AM on PI(3)P production and hVps34 activation, as previously described (Byfield et al., 2005; Nobukuni et al., 2005). The results show that pre-loading AA-deprived cells with BAPTA-AM was as effective as wortmannin in blocking the increase in AA-induced PI(3)P production (Figure 3C). Consistent with this finding pre-incubation of AA-deprived cells with BAPTA-AM blocks AA-stimulated hVps34 activation (Figure 3D), as scored for in an immune-complex activity assay (Byfield et al., 2005). Moreover, removal of leucine, which leads to a drop in both S6K1 T389 phosphorylation (data not shown) and [Ca2+]I (Supplemental Data, Figures S2A and B, respectively), inhibits hVps34 activity to the same extent as total AA withdrawal (Supplemental Data, Figure S3A). These findings support a model whereby Ca2+ mediates increased mTOR Complex1 signaling by regulating hVps34 activity.
Figure 3. Increased [Ca2+]i induces mTOR Complex1 signaling in an hVps34-dependent manner. (A).

HeLa cells, pre-treated with either DMSO or 100 nM wortmannin for 15 min, were stimulated with AAs or Tg as in Figure 2C. (B) HeLa cells were transfected with either 16 nMs of Rheb or hVps34 siRNA as in Figure 1A, then after transfection cells were treated with Tg as in Figure 2C. Where indicated 150 μM Tg was added for 30 min and Western blot analysis was conducted using indicated antibodies (Experimental Procedures). (C) HeLa cells were treated with AAs as in Figure 1A and where indicated either preincubated for 15 mins with 2 μM BAPTA-AM or 100 nM wortmannin. Immunostaining with PI(3)P antibody was carried out as previously described (Nobukuni et al., 2005). Quantification of immunostaining from 4 independent experiments (right panel) was performed as in Figure 1B. (D) 293T cells were first deprived of AAs then stimulated with AAs as previously described (Byfield et al., 2005; Nobukuni et al., 2005), in either the absence or presence of BAPTA-AM, as in Figure 1B. The hVps34 activity assay was performed on hVps34 immunoprecipitated from the same amount of protein extract (Experimental Procedures). Quantification of autoradiogram from 3 independent experiments (right panel) was performed as in Figure 1B. Note: The differential radioactivity at the origins is due to contaminating radioactive beads from the interphase (see Experimental Procedures).
CaM is required for mTOR Complex1 activation
Recently it was reported that Ca2+, through CaM, regulates hVps34 activity during bacterial infection of macrophages by M. tuberculosis (Vergne et al., 2003). To test the potential role of Ca2+/CaM in AA-induced mTOR Complex1 signaling, we pretreated cells with N-(6-aminohexyl)-5-chloro-1-naphthalenesulfonamide (W7), a cell-permeable CaM antagonist that binds to CaM, thus preventing the binding of Ca2+/CaM to target enzymes (Kahl and Means, 2003). Consistent with a role for CaM in the Ca2+ response, pretreatment of HeLa cells with W7 blocked the ability of AAs to induce S6K1 T389 phosphorylation (Figure 4A). To test this model further, we asked whether depletion of CaM had an impact on AA-induced mTOR Complex1 signaling. There are three distinct CaM genes, each encoding for the identical CaM protein (Means et al., 1982). Treatment of HeLa cells with a mixture of three distinct siRNAs, each specific for a single CaM gene product, resulted in depleted levels of CaM and inhibition of AA-induced S6K1 T389 phosphorylation (Figure 4B). In parallel, hVps34 immunoprecipitated from such cells exhibited reduced PtdIns kinase activity in vitro (Figure 4B). In contrast, treatment with a nonsilencing siRNA had no effect on either response (Figure 4B). To test whether Ca2+ affects the ability of CaM to interact with hVps34, cells were lysed in the presence of a mild detergent, 3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), and in the absence of Ca2+. The lysate was then incubated with CaM-linked agarose beads in the presence of either Ca2+ or EGTA. The beads were washed twice in binding buffer containing either Ca2+ or EGTA, and Ca2+-dependent interacting proteins were released by extensive washes of both sets of beads in EGTA. The results show that the beads initially incubated with Ca2+, in contrast to those incubated with EGTA, specifically bound hVps34 (Figure 4C). As we have previously noted (Nobukuni et al., 2005) immunoprecipitates of endogenous mTOR also contain hVps34 (Supplemental Data, Figure S3B). Here we find that CaM-agarose beads also bind mTOR in a Ca2+-dependent manner (Figure 4C). Moreover, this interaction was dependent on hVps34, as siRNA depletion of hVps34 protein blocked the ability of CaM-agarose beads to interact with mTOR (Figure 4C). However, the hVps34-mTOR interaction appears both Ca2+- and AA-independent, as the same amount of hVps34 co-immunoprecipitates with endogenous mTOR in the presence Ca2+ or EGTA and in the absence or presence of AAs (Figure 4C and Supplemental Data, Figure S3B). These results support a model in which the effects of Ca2+ on mTOR Complex1 signaling are mediated by Ca2+/CaM via hVps34 (see Discussion).
Figure 4. CaM is required for hVps34 activity and mTOR Compex1 signaling. (A).

HeLa cells were treated with AAs as in Figure 1A, in the absence or presence of 10 μM W7. W7 was added 15 min prior to the AA stimulation. (B) HeLa cells were transfected with either 45 nM nonsilencing (NS) siRNA or an siRNA mix containing 15 nM each of CaM1, CaM2, and CaM3 siRNA, as in Figure 1A (Experimental Procedures). Following transfection, cells were treated with AAs as in Figure 1A. The hVps34 activity assay was performed on hVps34 immunoprecipitated as described (Experimental Procedures). (C) HeLa cells were treated with AAs as in Figure 1A. Extracts were prepared in the absence of Ca2+, and then divided into 2 aliquots, with one brought to 0.5 mM CaCl2 and the other to 2 mM EGTA. CaM-agarose beads were added to each aliquot of extract, and the binding assays were performed as described (Vergne et al., 2003). Right panel, endogenous mTOR was immunoprecipitated from cells in the presence of either 2 mM EGTA or 0.5 mM CaCl2.
Ca2+/CaM binds to hVps34
To identify potential Ca2+/CaM target proteins in the mTOR Complex1 signaling pathway, we queried the CaM Target Database (http://calcium.uhnres.utoronto.ca). Most CaM targets contain one of two recognition motifs, a Ca2+-dependent motif, or a Ca2+-independent modified version of an IQ motif (Rhoads and Friedberg, 1997). Analysis of hVps34 itself revealed a potential Ca2+-dependent CaM-binding motif between residues 318 and 334, in the PI3K accessory domain (Nobukuni et al., 2007), which is conserved from yeast to man (Figure 5A). To test whether CaM interacts directly with hVps34, we resolved immunoprecipitated GST-hVps34 by SDS-PAGE and determined by far-Western analysis whether it interacted with CaM (Hall, 2004). The results show that CaM bound directly to GST-hVps34 in the presence of Ca2+, but not EGTA (Figure 5B). Moreover, this interaction was independent of AA stimulation (Figure 5B), suggesting that AA-induced posttranslational modifications of hVps34 were not required for Ca2+/CaM binding. These findings raised the possibility that the AA-induced rise in [Ca2+]i acts to stabilize binding of the Ca2+/CaM complex to hVps34, as suggested for the binding of the Ca2+/CaM complex to type II Ca2+/CaM -dependent protein kinase (Meyer et al., 1992). Consistent with this prediction, employing a cell-permeable bifunctional protein-protein crosslinking agent, we found an approximate two to three-fold increase in the amount of CaM bound to hVps34 following AA-stimulation, which was blocked by pretreatment with W7 (Figure 5C). Thus, the AA-induced rise in [Ca2+]i appears to stabilize an interaction between CaM and hVps34.
Figure 5. hVps34 contains Ca2+/CaM binding motif. (A).
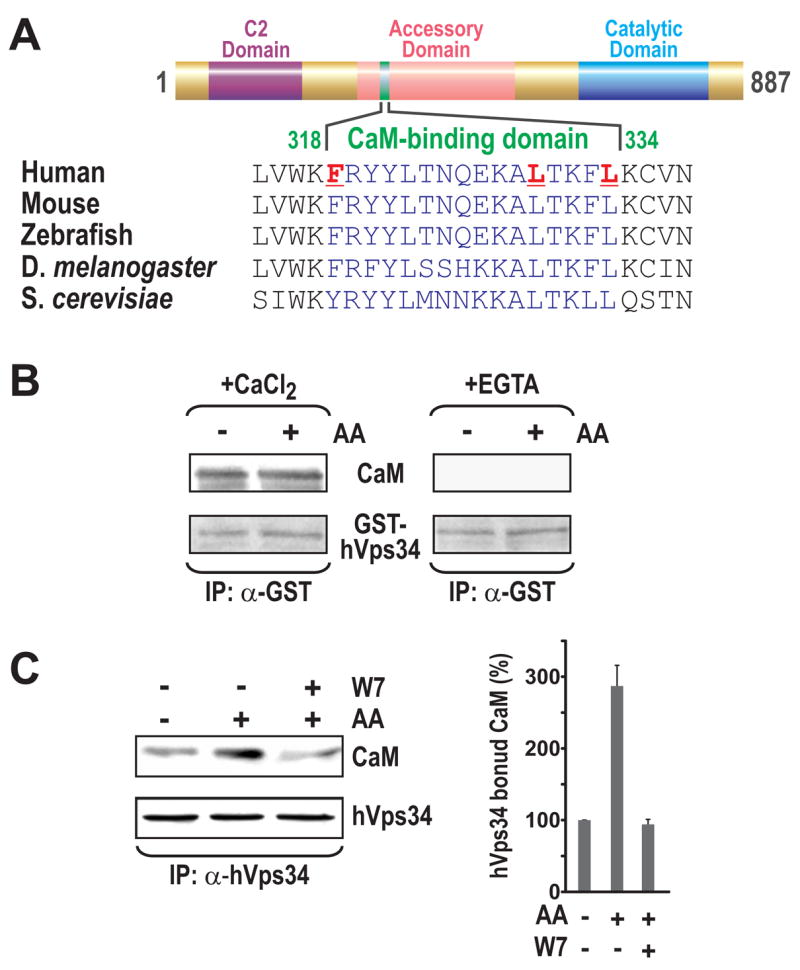
Alignment of the hVps34 CaM-binding site in different species. (B) Far Western blotting of GST-hVps34 from AA-deprived or AA-stimulated HEK 293 cells with purified CaM in the presence of CaCl2 or EGTA (Experimental Procedures). (C) HeLa cells were treated with AAs as in Figure 1A, then after 30 min, 1 mM dithiobis(succinimidyl) propionate (DSP) was added for an additional 30 min at 37ºC to either AA-deprived cells or to 2x AA-stimulated cells with or without W7 treatment, as in Figure 4A. Cells were then harvested and hVps34 was immunoprecipitated from lysates (Experimental Procedures). Quantification of Western blots from 3 independent experiments (right panel) was performed as in Figure 1B.
Ca2+/CaM is required for hVps34 activity
To elucidate the role of Ca2+/CaM in hVps34 activation, we took advantage of a GST epitope-tagged hVps34 construct, which we previously showed by ectopic expression drives T389 phosphorylation of a myc-tagged S6K1 reporter, but only In the presence of AAs (Nobukuni et al., 2005). Consistent with these findings we observe that when immunoprecipitated from cells in the presence of AAs this construct displays high hVps34 activity, which is sharply reduced by AA or leucine withdrawal as well as incubation in the presence of BAPTA-AM (Supplemental Data, Figure S4A). To determine whether the Ca2+/CaM-hVps34 interaction is mediated through the presumed Ca2+/CaM-binding motif, we generated two GST epitope-tagged hVps34 mutants, either F318R or L330R and L334R (see Figure 5A). The mutants, as well as GST-tagged wild-type (WT) and kinase-dead (KD) hVps34, were expressed at comparable levels, but only WT and KD GST-hVps34 interacted strongly with CaM-agarose beads in the presence of Ca2+ (Figure 6A and Supplemental Data, Figure S4B). Furthermore, like the KD GST-hVps34, both mutants were inactive in vitro, when compared to WT GST-hVps34 (Figure 6B). The absence of lipid kinase activity in the Ca2+/CaM–binding mutants did not appear to be simply attributable to protein misfolding, as the ability hVps34 mutants to bind to known hVps34 interacting proteins, hVps15 and beclin (Nobukuni et al., 2007) was unaffected (Supplemental Data, Figure S4C). In agreement with these findings, co-expression of the Ca2+/CaM-binding-motif mutants or GST-KD hVps34 with Myc-tagged GST-S6K1, in contrast to WT GST-hVps34, suppressed S6K1 reporter T389 phosphorylation, suggesting all three mutants acted dominant-negative (Figure 6C). These findings imply that hVps34 requires Ca2+/CaM for production of PI(3)P. Compatible with this hypothesis, extensive washes with EGTA and/or W7, as compared to extensive washes in the presence of Ca2+, removed Ca2+/CaM from immunoprecipitated endogenous hVps34 (Supplemental Data, Figure S4D), and significantly reduced hVps34 in vitro lipid kinase activity (Figure 6D). Importantly, re-addition of Ca2+/CaM restored PI3K activity, an effect abolished by the presence of W7 (Figure 6D). These findings show that hVps34 activity is modulated by Ca2+/CaM, and that AAs appear to induce hVps34 activation by increasing intracellular Ca2+ levels.
Figure 6. Ca2+/CaM regulates hVps34 lipid kinase activity.
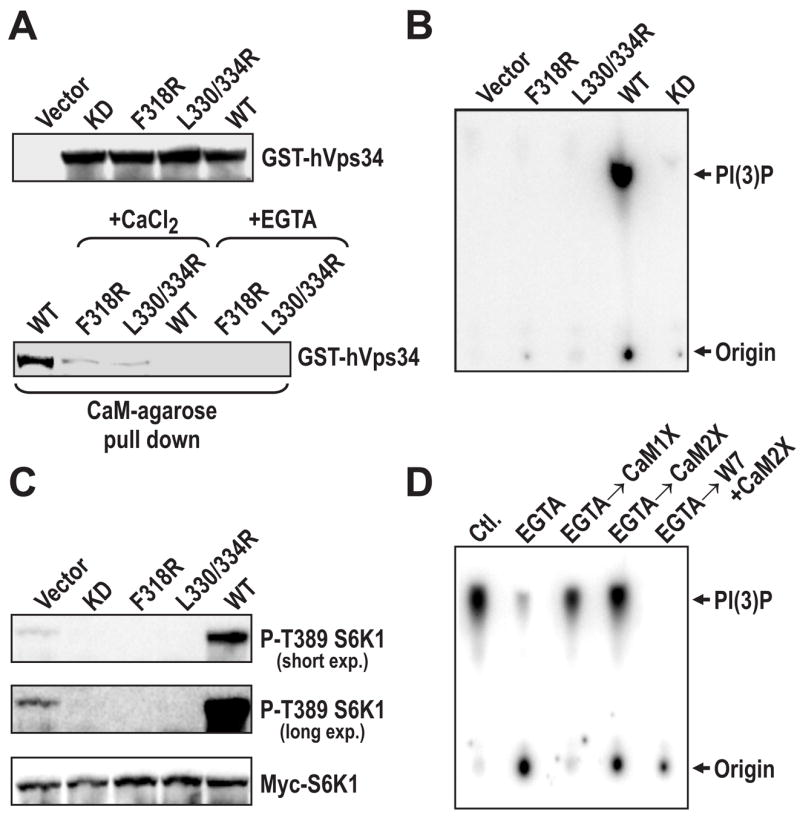
(A) HEK 293 cells were transfected with vectors expressing WT or CaM-binding mutants of hVps34 (Experimental Procedures). Twenty-four hours post-transfection, cells were treated as in Figure 1A, then cell lysates were prepared and used to measure the expression levels of each construct (upper panel) or for Ca2+/CaM-binding assays (lower panel), as in Figure 4C. (B) Equal amounts of WT, KD, and CaM-binding mutants of hVps34 expressed in HEK 293 cells, treated with AAs (Figure 6A), were immunoprecipitated with an anti-GST antibody and lipid kinase assays were performed on the immune complex in the presence of 0.5 mM CaCl2, as previously described (Nobukuni et al., 2005). (C) HEK 293 cells were transfected with either with WT, KD or the CaM-binding hVps34 mutants with myc-S6K1, treated with AAs as in Figure 1A, then cell lysates were prepared and probed with indicated antibodies. (D) HEK 293 cells were treated with AAs as in Figure 1A, endogenous hVps34 was immunoprecipitated from cell extracts in the presence of 0.5 mM CaCl2 or 2 mM EGTA. The resulting hVps34 immunoprecipitate was then extensively washed in either 0.5 mM CaCl2 or 2 mM EGTA, as in Supplemental Data, Figure S4C. The hVps34 immunoprecipitates were either directly assayed for lipid kinase activity as in Figure 3D or after the indicated additions (Experimental Procedures), as previously described (Nobukuni et al., 2005).
DISCUSSION
The role of AAs in mTOR Complex1 signaling was initially uncovered in studies on AA inhibition of autophagy, which revealed that AAs in parallel induce an increase in S6K1 activation (Dann et al., 2007). With the identification of mTOR Complex1 it was found that AAs and glucose, but not growth factors, cause a conformational change in mTOR Complex1, which through raptor allows mTOR to access and phosphorylate S6K1 (Kim et al., 2002). Consistent with such a model it was subsequently found that the ability Rheb to interact with mTOR Complex1 is inhibited in the absence of AAs (Long et al., 2005). This led to the hypothesis that AAs induce a conformational change in mTOR Complex1, potentially releasing an inhibitor and allowing Rheb to interact with and activate mTOR Complex1 (Long et al., 2005). Recent studies have revealed a potential inhibitor, FKBP38, which binds to the FKBP12-rapamycin binding site on mTOR Complex1, and which is released by AA treatment and interaction with Rheb-GTP (Bai et al., 2007 and Figure 7).
Figure 7. Model depicting the role of AAs in regulating mTOR Complex1 signaling through Ca2+/CaM and hVps34.

AA stimulation leads to an increase in [Ca2+]i, which increases the interaction of Ca2+/CaM with the inactive mTOR Complex1 signalosome. Ca2+/CaM interacts with hVps34, through its conserved CaM binding motif, resulting in hVps34 activation, the production of PI(3)P and a conformational change in the hVps34 associated mTOR Complex1 signalosome. Increased PI(3)P would be predicted to recruit an unknown PX or FYVE-domain containing protein(s), which may also participate in generating the conformationally-altered form of the mTOR Complex1 signalosome. This conformational change is required for the displacement of FKBP38 from the conformationally-altered mTOR Complex1 signalosome by insulin-induced Rheb-GTP. The model also indicates that Rheb-GTP may independently drive activation of the mTOR Complex1 signalosome, through its reported direct interaction with mTOR (Long et al., 2005).
The data presented here are consistent with the model above. Here we show that AAs induce an increase in [Ca2+]i, which acts to enhance the binding of Ca2+/CaM to hVps34, resulting in increased PI(3)P levels and enhanced mTOR Complex1 signaling. We have previously demonstrated that activation of S6K1 is associated with a Ca2+-dependent shift of the kinase into a larger protein complex (Hannan et al., 2003). Such data would be compatible with PI(3)P acting to recruit an unknown FYVE or PX domain containing protein(s) to endosomes (Byfield et al., 2005; Nobukuni et al., 2005), which is required to build a mTOR Complex1 signalosome. That hVps34 is part of such a complex was unexpected, however, it has been shown that that small Rab GTPases recruit the hVps34/hVps15 complex to specific cellular compartments, where FYVE or PX domain containing proteins serve as Rab effectors (see Nobukuni et al., 2007). In the studies presented here we do not see an alteration the mTOR-hVps34 interaction as a function of AA stimulation, suggesting that mTOR is recruited to the signalosome though its interaction with hVps34. We would hypothesize that such an interaction results in the described conformational change in mTOR Complex1, which allows Rheb-GTP then to interact with mTOR (Long et al., 2005) and/or FKBP38 (Bai et al., 2007), allowing activation of downstream effectors, such as S6K1 (Figure 7).
Earlier it was shown that autophagy, which is triggered by AA-withdrawal, also requires hVps34 and Ca2+ (Gordon et al., 1993). However, in this case the Ca2+ requirement was initially argued to be for the maintenance of high Ca2+ levels within the ER (Gordon et al., 1993), although a recent report suggests increased [Ca2+]i as well as depletion of Ca2+ from the ER triggers an autophagic response (Hoyer-Hansen et al., 2007). In contrast to the effects on autophagy, which takes days, AA-induced activation of mTOR signaling occurs within minutes, with no apparent requirement for the emptying of ER Ca2+ stores. These observations are consistent with a potential role for hVps34 in distinct functions of early endosome signaling versus targeting of late endosomes to autophagic vesicles (Nobukuni et al., 2005). This model is compatible with studies in yeast where at least two distinct Vps34 protein complexes were identified (Kihara et al., 2001b) and the finding that whereas all the cellular beclin is associated with hVps34, 50% of the hVps34 is beclin-free (Kihara et al., 2001a). Consistent with these two distinct hVps34 complexes, immunofluorescence microscopy show that the beclin/hVps34 complex is localized to the TGN, but the beclin free-hVps34 complex is localized to endosomes (Kihara et al., 2001a). Thus, different hVps34 complexes maybe involved in regulating distinct responses, such as endosome signaling and autophagy. Given the ability of mTOR Complex1 to drive cell growth and to suppress autophagy, it will be critical to identify the mechanisms by which these two pathways interact at the cellular level.
In humans it is known that circulating AAs are often elevated in the obese and the insulin resistant state (Um et al., 2006). Consistent with these observations it has been demonstrated in human clamp studies that an approximate two-fold rise in plasma AA concentrations is paralleled by a 25% reduction in insulin sensitivity in skeletal muscle (Krebs, 2005). Moreover, the effects of AAs on insulin resistance appear to be mediated by activation of mTOR Complex1 signaling followed by phosphorylation of IRS1 at sites which antagonize insulin action (Tremblay et al., 2007; Tremblay et al., 2005b). The importance of these observations extends beyond obesity and insulin resistance, as a recent record-linked case-controlled study showed that metformin, a widely prescribed anti-diabetic, which increases insulin sensitivity and blunts mTOR Complex1 signaling, reduces the risk of cancer in diabetic patients in a dose dependent manner (Dann et al., 2007). With respect to a potential role for Ca2+ in these pathologies, it is interesting to note that in humans increased skeletal muscle [Ca2+]i is associated with insulin resistance (Resnick, 1999). It will now be critical to determine both the mechanism by which AAs cause a rise in [Ca2+]I and the identity of the signalosome which mediates mTOR Complex1 activation. In summary, these findings point to the importance of mTOR Complex1 in insulin resistance and cancer, potentially offering novel avenues to develop new therapeutic strategies.
EXPERIMENTAL PROCEDURES
Reagents
Rabbit anti-CaM and rabbit anti-hVps34 antibodies are from Invitrogen; antibodies to mTOR, phospho T-389 S6K-1, phospho S-473 PKB, phospho T308 PKB, phospho S-65 4E-BP1, phospho T37/46 4E-BP1, phospho 44/42 Erk, PKB, phospho-S9 GSK3, and S6K-1 are from Cell Signaling Technologies; anti-GST antibodies is from Bethyl; anti-PI(3)P antibody is from Echelon; anti-tubulin antibody is from Neomarkers; antibodies to FRAP (N19) and c-Myc is from Santa Cruz Biotechology; and mouse monoclonal anti-CaM and rabbit anti-raptor antibodies from Upstate Biotechology; rabbit anti-hVps15 antibody from Abgent; mouse monoclonal anti-beclin antibody from Novus; rabbit anti-Rheb antibody from ProSci. Pan anti-4E-BP1 antibody was a kind gift from Nahum Sonenberg. Rabbit brain CaM was a gift from John Dedman. Recombinant 4E-BP1 is from A.G. Scientific Inc. BAPTA-AM, EGTA, W7, Tg, histamine, and wortmannin is from Calbiochem. Fluo-4, and Fura-2 are from Molecular Probes; DSP, chemiluminescence kit is from Pierce; PreScission protease, Protein A- and G-Sepharose and HRP-conjugated secondary antibodies is from Amersham. All other reagents were from Sigma.
Cell Culture, Western blotting, and Immunostaining
Maintenance of HeLa and HEK 293 cells as well as AA deprivation, SDS-PAGE, and immunostaining were performed as described ((Nobukuni et al., 2005)), except we have recently found that the responses to either AA withdrawal or addition works as well in dialyzed serum as in no serum.
Transfections and hVps34 CaM-binding-site mutagenesis
[Ca2+]i Measurements
HeLa cell [Ca2+]i measurements were either made directly on tissue culture plates, or the cells were plated onto 25 mm round glass coverslips, then cultured for 24 to 48 hrs prior to experimentation. Note that the high background autofluorescence of plastic necessitated the use of glass coverslips in the measurement of fura-2 fluorescence intensities. While the percentage of cells responding to AAs decreased when cells were cultured on glass (80% vs 25–40%), similar [Ca2+]i response patterns were observed on both types of substratum. Cells were loaded with Ca2+-sensitive indicator dyes during the 2-hour AA-deprivation protocol. Cell loading was achieved by incubation with the acetoxymethylester form of the dye (5 μM) and pluronic acid (0.02% v/v) for 1 h in serum-and AA-free DMEM. Cells were then washed and transferred to a thermostatically regulated microscope chamber (37°C). Ca2+-imaging experiments were performed in AA-free DMEM or a HEPES-buffered physiological saline solution (HBSS) composed of (in mM) 25 HEPES (pH 7.4 at 37 °C), 121 NaCl, 5 NaHCO3, 4.7 KCl, 1.2 KH2PO4, 1.2 MgSO4, 2.0 CaCl2, 25 glucose, and 1 pyruvate. A 5% CO2/95% O2 atmosphere was maintained above the microscope chamber when DMEM was used for Ca2+ measurements. Fura-2 fluorescence images (excitation, 340 and 380 nm, emission 420–600 nm) were acquired at 3 s intervals with a cooled charge-coupled device (CCD) camera, as previously described (Rooney et al., 1989). Values of [Ca2+]i were calculated from the 340/380 nm ratio after correcting for autofluorescence. Autofluorescence values were obtained at the end of each experiment by permeabilizing the HeLa cells with digitonin in the presence of 2 mM EGTA. The fura-2 calibration parameters were determined as previously described (Morgan and Thomas, 1999). Fluo-4 fluorescence intensity changes were determined by confocal microscopy. The indicator was excited with the 488 nm argon laser line, and emitted fluorescence was collected using a 515 nm long-band-pass filter and manufacturer-supplied software.
Far-Western Blot Analysis
GST-hVps34 was immunoprecipitated from cells and then blotted on a nitrocellulose membrane. The membrane was blocked for 30 min with 1% BSA prepared in PBS with 0.1% Tween-20. The membrane was then incubated with purified CaM (1μg/ml) in the presence of either 0.5 mM CaCl2 or 2 mM EGTA in the blocking buffer at 4ºC overnight. The next day the membrane was washed 3 times (10 min each) in PBS with 0.1% Tween-20. Afterwards, the membrane was immunostained with anti-CaM antibody. Membranes were then stripped and probed with anti-hVps34 antibody.
Immunoprecipitation and kinase assays
GST-hVps34 was immunoprecipitated by adding 5 μg of goat anti-GST antibody (overnight at 4ºC) per 0.8 to1.0 mg of protein lysate from cells transfected with GST-hVps34 expression plasmid. Afterwards 50 μl of a 50% slurry of protein-G sepharose beads was added and incubation was continued for 2 more hours. Further, the immunocomplex was washed 3 times with the lysis buffer, as previously described (Kim et al., 2002). Endogenous hVps34 was immunoprecipitated using 2.5 μg of anti-hVps34 antibody (anti-rabbit polyclonal antibody generated in house against a peptide corresponding to the 19 C-terminal AA residues of hVps34) For hVps34 activity assays, immunopricipitation was performed in lysis buffer as previously described (Nobukuni et al., 2005), except the buffer contained 1mM CaCl2 and 1mM MgCl2 and the cell extracts were incubated in the lysis buffer for 20 min. at 4ºC prior to centrifugation. The hVps34 immune complex initial washes were done as described (Nobukuni et al., 2005) except (i) the final 3 washes in TNE buffer were replaced by 3x 20 min washes with either EGTA, CaCl2, or W7 where indicated, and (ii) all assays were performed in assay buffer containing 0.5 mM CaCl2, 10 mM Tris and 100 mM NaCl. In some samples the higher radioactivity at the origin is due to contaminating ATP from the aqueous phase, i.e. when the capillary tube is passed through the radioactive Sepahrose A bead containing interphase, to recover the PI(3)P containing organic phase, some beads stick to the tip of the capillary tube, and are loaded on to the TLC plate (Nobukuni et al., 2005). This does not interfere with the chromatographic separation of PI(3)P.
mTOR immunoprecipitation, in vitro kinase activity assays and S6K1 substrate preparation was performed as recently described (Sancak et al., 2007), except that mTOR was immunoprecipitated in the presence of 150 mM NaCl and during the preparation of S6K1 substrate, after removing the affinity tag, S6K1 was purified on a glutathione agarose column.
Supplementary Material
Acknowledgments
We are indebted to A. Means for his critical reading of the manuscript, as well as Drs. P. Dennis, P. D. Plas, J. Dedman and J. Moscat for both reading the manuscript and numerous discussions. We are thankful to M. Wymann for the hVps34 construct and D. Sabatini for the pRK5 HA-GST-PreSc S6K1 construct. We are also grateful to J. Backer for his kind gift of 293T cells and for guiding us through the hVps34 activity assay. Finally we thank J. Meller and G. Doerman for assisting us in bioinformatic searches and computer graphics, respectively. G.T., S. K. and P. G. are supported by the NIH Mouse Models for Human Cancer Consortium, U01 CA84292-06, and NIH Grant DK73802. G. T. is separately supported by the Strauss Chair in Cancer Research. A.P.T. is supported by NIH Grant DK38422 and the Thomas P. Infusino Endowment. S. D. is sponsored by an ORISE Fellowship from US Department of Defense.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bai X, Ma D, Liu A, Shen X, Wang QJ, Liu Y, Jiang Y. Rheb activates mTOR by antagonizing its endogenous inhibitor, FKBP38. Science. 2007;318:977–980. doi: 10.1126/science.1147379. [DOI] [PubMed] [Google Scholar]
- Byfield MP, Murray JT, Backer JM. hVps34 Is a Nutrient-regulated Lipid Kinase Required for Activation of p70 S6 Kinase. J Biol Chem. 2005;280:33076–33082. doi: 10.1074/jbc.M507201200. [DOI] [PubMed] [Google Scholar]
- Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4:579–591. doi: 10.1038/nrc1408. [DOI] [PubMed] [Google Scholar]
- Conus NM, Hemmings BA, Pearson RB. Differential regulation by calcium reveals distinct signaling requirements for the activation of Akt and p70S6k. J Biol Chem. 1998;273:4776–4782. doi: 10.1074/jbc.273.8.4776. [DOI] [PubMed] [Google Scholar]
- Dann SG, Selvaraj A, Thomas G. mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol Med. 2007 doi: 10.1016/j.molmed.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Finkelstein EA, Ruhm CJ, Kosa KM. Economic causes and consequences of obesity. Annu Rev Public Health. 2005;26:239–257. doi: 10.1146/annurev.publhealth.26.021304.144628. [DOI] [PubMed] [Google Scholar]
- Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R, Sonenberg N. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev. 1999;13:1422–1437. doi: 10.1101/gad.13.11.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon PB, Holen I, Fosse M, Rotnes JS, Seglen PO. Dependence of hepatocytic autophagy on intracellularly sequestered calcium. J Biol Chem. 1993;268:26107–26112. [PubMed] [Google Scholar]
- Hall RA. Studying protein-protein interactions via blot overlay or Far Western blot. Methods Mol Biol. 2004;261:167–174. doi: 10.1385/1-59259-762-9:167. [DOI] [PubMed] [Google Scholar]
- Hannan KM, Thomas G, Pearson RB. Activation of S6K1 (p70 ribosomal protein S6 kinase 1) requires an initial calcium-dependent priming event involving formation of a high-molecular-mass signalling complex. Biochem J. 2003;370:469–477. doi: 10.1042/BJ20021709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem. 1998;273:14484–14494. doi: 10.1074/jbc.273.23.14484. [DOI] [PubMed] [Google Scholar]
- Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, et al. The TSC1–2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–223. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25:193–205. doi: 10.1016/j.molcel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Kahl CR, Means AR. Regulation of cell cycle progression by calcium/calmodulin-dependent pathways. Endocr Rev. 2003;24:719–736. doi: 10.1210/er.2003-0008. [DOI] [PubMed] [Google Scholar]
- Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2001a;2:330–335. doi: 10.1093/embo-reports/kve061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara A, Noda T, Ishihara N, Ohsumi Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001b;152:519–530. doi: 10.1083/jcb.152.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- Krebs M. Amino acid-dependent modulation of glucose metabolism in humans. Eur J Clin Invest. 2005;35:351–354. doi: 10.1111/j.1365-2362.2005.01506.x. [DOI] [PubMed] [Google Scholar]
- Long X, Ortiz-Vega S, Lin Y, Avruch J. Rheb binding to mammalian target of rapamycin (mTOR) is regulated by amino acid sufficiency. J Biol Chem. 2005;280:23433–23436. doi: 10.1074/jbc.C500169200. [DOI] [PubMed] [Google Scholar]
- Means AR, Tash JS, Chafouleas JG. Physiological implications of the presence, distribution, and regulation of calmodulin in eukaryotic cells. Physiol Rev. 1982;62:1–39. doi: 10.1152/physrev.1982.62.1.1. [DOI] [PubMed] [Google Scholar]
- Meyer T, Hanson PI, Stryer L, Schulman H. Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science. 1992;256:1199–1202. doi: 10.1126/science.256.5060.1199. [DOI] [PubMed] [Google Scholar]
- Morgan AJ, Thomas AP. Single cell and subcellular measurement of intracellular Ca2+ concentration ([Ca2+]i) Methods Mol Biol. 1999;114:93–123. doi: 10.1385/1-59259-250-3:93. [DOI] [PubMed] [Google Scholar]
- Nobukuni T, Joaquin M, Roccio M, Dann SG, Kim SY, Gulati P, Byfield MP, Backer JM, Natt F, Bos JL, et al. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc Natl Acad Sci U S A. 2005;102:14238–14243. doi: 10.1073/pnas.0506925102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobukuni T, Kozma SC, Thomas G. hvps34, an ancient player, enters a growing game: mTOR Complex1/S6K1 signaling. Curr Opin Cell Biol. 2007;19:135–141. doi: 10.1016/j.ceb.2007.02.019. [DOI] [PubMed] [Google Scholar]
- Patti ME, Kahn BB. Nutrient sensor links obesity with diabetes risk. Nat Med. 2004;10:1049–1050. doi: 10.1038/nm1004-1049. [DOI] [PubMed] [Google Scholar]
- Pearce LR, Huang X, Boudeau J, Pawlowski R, Wullschleger S, Deak M, Ibrahim AF, Gourlay R, Magnuson MA, Alessi DR. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem J. 2007;405:513–522. doi: 10.1042/BJ20070540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyrollier K, Hajduch E, Blair AS, Hyde R, Hundal HS. L-leucine availability regulates phosphatidylinositol 3-kinase, p70 S6 kinase and glycogen synthase kinase-3 activity in L6 muscle cells: evidence for the involvement of the mammalian target of rapamycin (mTOR) pathway in the L-leucine-induced up-regulation of system A amino acid transport. Biochem J. 2000;350(Pt 2):361–368. [PMC free article] [PubMed] [Google Scholar]
- Resnick LM. Therapeutic implications of calcium intake in hypertension: more analyses, a little progress, and a different approach. Am J Hypertens. 1999;12:82–83. [PubMed] [Google Scholar]
- Rhoads AR, Friedberg F. Sequence motifs for calmodulin recognition. Faseb J. 1997;11:331–340. doi: 10.1096/fasebj.11.5.9141499. [DOI] [PubMed] [Google Scholar]
- Rooney TA, Sass EJ, Thomas AP. Characterization of cytosolic calcium oscillations induced by phenylephrine and vasopressin in single fura-2-loaded hepatocytes. J Biol Chem. 1989;264:17131–17141. [PubMed] [Google Scholar]
- Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–915. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci U S A. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas AP, Bird GS, Hajnoczky G, Robb-Gaspers LD, Putney JW., Jr Spatial and temporal aspects of cellular calcium signaling. Faseb J. 1996;10:1505–1517. [PubMed] [Google Scholar]
- Tremblay F, Brule S, Hee Um S, Li Y, Masuda K, Roden M, Sun XJ, Krebs M, Polakiewicz RD, Thomas G, Marette A. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc Natl Acad Sci U S A. 2007;104:14056–14061. doi: 10.1073/pnas.0706517104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay F, Jacques H, Marette A. Modulation of insulin action by dietary proteins and amino acids: role of the mammalian target of rapamycin nutrient sensing pathway. Curr Opin Clin Nutr Metab Care. 2005a;8:457–462. doi: 10.1097/01.mco.0000172589.55434.03. [DOI] [PubMed] [Google Scholar]
- Tremblay F, Krebs M, Dombrowski L, Brehm A, Bernroider E, Roth E, Nowotny P, Waldhausl W, Marette A, Roden M. Overactivation of S6 kinase 1 as a cause of human insulin resistance during increased amino acid availability. Diabetes. 2005b;54:2674–2684. doi: 10.2337/diabetes.54.9.2674. [DOI] [PubMed] [Google Scholar]
- Tzatsos A, Kandror KV. Nutrients suppress phosphatidylinositol 3-kinase/Akt signaling via raptor-dependent mTOR-mediated insulin receptor substrate 1 phosphorylation. Mol Cell Biol. 2006;26:63–76. doi: 10.1128/MCB.26.1.63-76.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um SH, D'Alessio D, Thomas G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab. 2006;3:393–402. doi: 10.1016/j.cmet.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- Vergne I, Chua J, Deretic V. Tuberculosis toxin blocking phagosome maturation inhibits a novel Ca2+/calmodulin-PI3K hVPS34 cascade. J Exp Med. 2003;198:653–659. doi: 10.1084/jem.20030527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Zhang HH, Lipovsky AI, Dibble CC, Sahin M, Manning BD. S6K1 Regulates GSK3 under Conditions of mTOR-Dependent Feedback Inhibition of Akt. Mol Cell. 2006;24:185–197. doi: 10.1016/j.molcel.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.