

Though there is growing evidence for a critical connection between inflammation and carcinogenesis, the mechanistic links between the two are just beginning to emerge. Nuclear factor-κB (NF-κB) transcription factors are important in integrating multiple stress stimuli and regulating innate and adaptive immune responses seen in states of inflammation1. With the recognition that inflammatory conditions are often associated with or preceed cancer, it was natural to suspect a link between NF-κB and cancer, as was first suggested several years ago2. Since that time, experimental evidence revealing specific mechanisms by which NF-κB influences cancer initiation, promotion, and progression has been mounting at a dizzying pace. This review will focus on new data that has emerged over the last couple of years implicating NF-κB its signaling pathways and downstream targets in carcinogenesis.
One of the founding fathers of modern pathology, Rudolf Virchow, observed leukocytes in neoplastic tissue over a hundred years ago, and first suspected that inflammation might support or promote cancer3. This notion has re-emerged in the last decade in part because of the recognition that many chronic infectious diseases are associated with development of cancer. Approximately 15% of the global cancer burden has been attributed to chronic infections and the accompanying inflammatory reaction4, and 15–20% of cancer deaths arise from preventable infections5. Likewise, many non-infectious conditions of chronic inflammation increase the risk and accelerate the progression of diverse cancers6. The common denominator in these conditions is the presence of chronic inflammation, invariably associated with activation of NF-κB and its effector pathways.
NF-κB was first discovered as a protein bound to the kappa immunoglobulin gene enhancer in the nuclei of B cells7, and was thought to be restricted to these cells. Ironically, the name “NF-κB” applied to these transcription factors is no longer descriptive: the factors generally reside in the cytoplasm of resting cells, when activated bind to a large array of enhancer sequences (over 150 genes), and are present in most (if not all) cells. A detailed description of NF-κB regulation is beyond the scope of this article (see recent reviews8, 9, and Figure 1). Briefly, mammalian NF-κB transcription factors consist of 5 homologous subunits (RelA/p65, c-Rel, RelB, p50/ NF-κB1, and p52/ NF-κB2) which dimerize and are held in the cytoplasm by specific proteins, the inhibitors of NF-κBs (IκBs). Immediately upstream from the IκB-bound NF-κB dimers is the IκB kinase (IKK) complex, comprised of two catalytic (IKKα and IKKβ) and one regulatory (IKKγ/NEMO) subunits10. Several pathways of cell stimulation converge to activate the IKK complex, which then phosphorylates the IκBs, targeting them for ubiquitination and degredation by the 26S proteosome11. The liberated NF-κBs travel to the nucleus and engage transcriptional programs.
Figure 1.
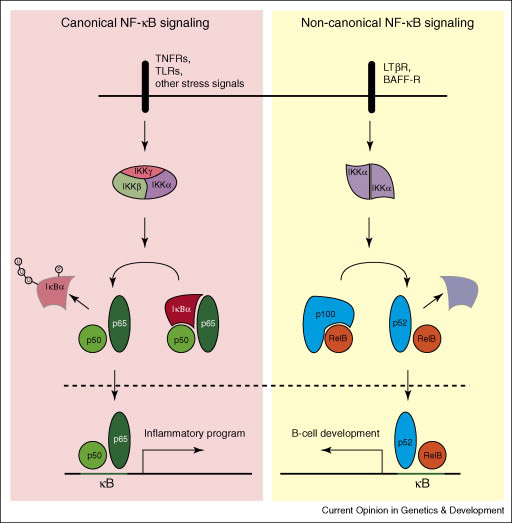
NF-κB signaling pathways
Though there is a broadening complexity to NF-κB signaling, the two most recognized pathways are the so-called “canonical” and “non-canonical.” The former depends on NEMO, IKKβ activation, nuclear localization of RelA/p50 dimers, and is associated with inflammation, while the latter depends on IKKα activation probably via the upstream kinase NIK, nuclear localization of p52/RelB dimers, and is important in lymphoid organogenesis8. Both pathways of NF-κB activation have now been implicated in carcinogenesis12, 13.
Activation of NF-κB (usually assessed by presence of nuclear RelA) has been observed in many cancers, including but not limited to breast cancer14, melanoma15, lung cancer16, colon cancer17, multiple myeloma13, pancreatic cancer18, esophageal adenocarcinoma19, and various types of leukemia20–22 and lymphoma23, 24. The presence of activated NF-κB in tumors does not, however, establish a causal link. Only with the advent of recent advances in experimental mouse models of cancer have investigators been able to tie specific functions of NF-κB activation to the carcinogenesis process, as well as tumor progression and metastatogenesis. A synopsis of cellular processes which contribute to cancer development and progression include self-sufficiency in growth signals, insensitivity to growth-inhibitors, evasion of apoptosis, limitless replicative potential, tissue invasion and metastasis, and sustained angiogenesis25. NF-κB activation has been linked to most of these cancer cell intrinsic processes, but most importantly has also been established to be a major mediator of influences that are extrinsic to cancer cells but nonetheless are critical to most aspects of tumorgenesis. This review will focus on the role of NF-κB in these processes and the development or progression of cancer.
NF-κB in malignant proliferation
Promotion of cell growth is a necessary feature of any cancer, and can be achieved either through abnormally activated or deregulated signaling pathways involved in cell cycle regulation, or quite often (but usually neglected) abnormal growth signals outside the malignant cell. NF-κB target genes regulating proliferation include CyclinD1, CyclinE, CDK2, and c-Myc, while growth signals include GM-CSF and interleukin-6 (IL-6)26. Activating mutations involving NF-κB pathways which lead to cancer are thought to be rare, but this may be a result of simply not understanding what to look for. A recent survey of 155 patient samples of multiple myeloma found 20% of them to have NF-κB-activating mutations, the most common of which was inactivation of TRAF312. A second study confirmed the presence of activating NF-κB mutations in 368 patient samples, and found that an IKKβ inhibitor induced death in cell lines established from these samples13. A similar study found that IKKβ inhibition in human multiple myeloma blocked cell cycle progression27. Cancer growth inhibition via NF-κB inhibition has similarly been shown in several flavors of human neoplasia, including breast28, lung16, melanoma15, colon29, and B-cell lymphoma23.
An insightful study of metastatic colon cancer revealed that stimulation of the inflammatory system by lipopolysaccharide (LPS) injection caused increased growth of the metastatic cancer, dependent on NF-κB signaling, which, when abolished, paved the way for TRAIL-dependent tumor regression30. This study confirmed that NF-κB-induced inflammation is directly linked to growth stimulation of malignant cells.
Genetic manipulation of NF-κB signaling in mice has proved most illuminating in dissecting the mechanisms which link inflammation to carcinogenesis. In a mouse model of colitis-associated cancer, genetic deletion of IKKβ in enterocytes, which are the cells that undergo malignant progression in this model, significantly reduced tumor multiplicity compared to controls, directly implicating NF-κB signaling in early tumor promotion during the initiation stage31. Interestingly, when IKKβ was deleted in myeloid cells (which control the inflammatory response but do not undergo any genetic alterations), tumor size was markedly reduced, suggesting that the inflammatory cells were elaborating growth signals which aided in the promotion of neoplastic growth31. Indeed, known growth factors such as IL-6 were dramatically decreased when NF-κB signaling was disrupted in myeloid cells. Although one study confirmed the contribution of IL-6 to colitis-associated cancer32, we find that the actual role of IL-6 in this malignancy is rather complex, and that canonical NF-κB activation in myeloid cells is likely to act via additional growth factors (unpublished results). Using the same model of colitis-associated cancer Rigby et al. found that deletion of SOCS3 (suppressors of cytokine signaling-3) led to increased NF-κB signaling, an increase in intestinal crypt cell proliferation, and more and larger tumors than wildtype (WT) counterparts33. Both of these studies point to a direct role for NF-κB-directed increased intestinal cell proliferation leading ultimately to cancer.
A third model of intestinal cancer directly linked the Toll-like receptor (TLR) adaptor protein MyD88 to increased spontaneous tumorigenesis in APCmin mutant mice34. MyD88 is an adaptor used by several TLRs as well as IL-1 receptor (IL-1R), and its activation integrates signals from the innate immune system. Genetic ablation of MyD88 in this study significantly reduced the number of intestinal tumors. Not surprisingly, the transcription factors through which MyD88 signals are NF-κBs, and several NF-κB target genes, including COX-2 and IL-6 were down-regulated in the MyD88-deficient mice who demonstrated reduced carcinogenesis34.
Hepatocyte-specific deletion of IKKβ in a murine model of chemically-induced hepatocellular carcinoma (HCC) resulted in a remarkable increase in the number of tumors35. Molecular mechanisms underlying increased HCC development included augmented hepatocyte injury (due to accumulation of ROS, which prolongs JNK activation in the absence of NF-κB signaling36), leading to increased compensatory proliferation and hepatocarcinogenesis35, 37. In a remarkable turnabout, when IKKβ was deleted in both hepatocytes and immune cells in the liver, the number and size of HCC tumors was greatly diminished, lower even than in WT littermates35. The mechanism was found to be through reduction of Kupffer cell (liver macrophage)-produced and NF-κB-regulated IL-6, which limited both liver injury and compensatory proliferation38.
In a follow-up study using the same model, canonical NF-κB regulation of IL-6 at the level of the Kupffer cell was confirmed to be necessary for development of HCC38. Genetic ablation of IL-6 markedly attenuated chemically-induced liver injury, subsequent compensatory proliferation, and development of HCC. These 2 studies35, 38 in which NF-κB-regulated IL-6 production contributes to hepatic injury suggest a mechanism by which chronic inflammation in the liver leads to perpetuation of injury, compensatory proliferation, and ultimately HCC development.
Hepatocyte-specific deletion of the regulatory IKKγ/NEMO subunit abolishes all NF-κB signaling in the liver, and induces a state of chronic steatohepatitis, leading ultimately to HCC in all male mice at 12 months of age39. However, mice with hepatocyte-specific IKKβ deletion develop normally, without features of steatohepatitis or any signs of liver damage40. Although ablation of IKKγ/NEMO results in more complete inhibition of canonical NF-κB signaling than IKKβ ablation41, it is possible that the hepatic damage in the conditional IKKγ/NEMO knockout mice is due to a yet-to-be-identified infection. Indeed, hepatic injury in hepatocyte-specific IKKγ/NEMO-deleted mice is dependent on accumulation of ROS, and is completely blocked by administration of an antioxidant, previously found to inhibit DEN-induced injury in hepatocyte-specific IKKβ knockout mice35. Together these studies paint a picture in which NF-κB signaling in hepatocytes prevents ROS-induced damage, thereby reducing compensatory hepatocyte proliferation and diminishing HCC development. Kupffer cell NF-κB signaling on the other hand (which can be activated by products of hepatocyte death, among other things38) creates inflammatory signals which amplify liver damage as well as increase compensatory proliferation and hepatocarcinogenesis. (Figure 2 outlines inflammation-associated hepatocarcinogenesis and NF-κB involvement)
Figure 2.
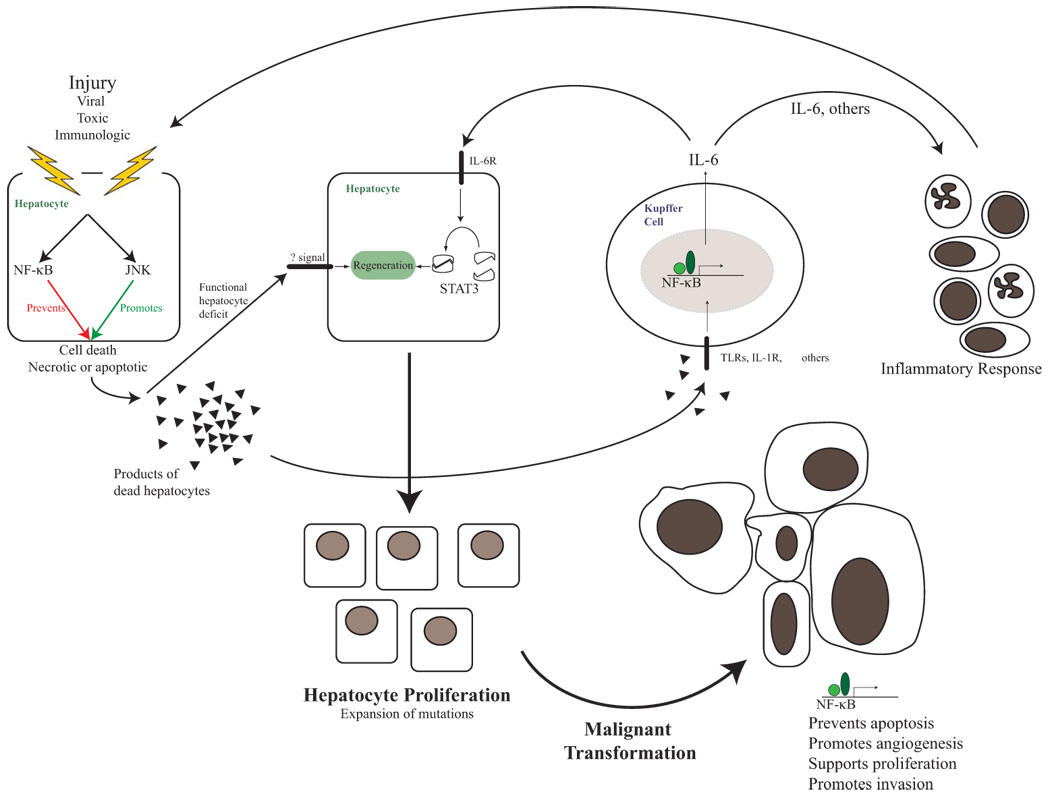
NF-κB in hepatocarcinogenesis
In the breast, mammary gland development and proliferation through cyclinD1 expression is dependent on IKKα activation2. Genetic introduction of a non-activateable IKKα mutant into the Neu/ErbB2-driven mouse model of breast cancer significantly retarded breast tumor development42. Of interest, inactive IKKα inhibited breast cancer development only in the ErbB2/Her2 model, but had no effect in a breast cancer model driven by the Ha-Ras oncogene. This finding is important, as the subset of human breast cancers which are estrogen receptor (ER)-negative (and unresponsive to hormonal therapies) often depend on ErbB2/Her2 upregulation, and were shown to be responsive to treatment with NF-κB inhibitors in vitro43. Interestingly, the inactivation of IKKα blocks the ability of Neu/ErbB2-induced tumors to generate secondary tumors upon orthotopic transplantation, as it inhibits the self-renewal capacity of breast cancer progenitors42. This suggests that IKKα may be targeted in ErbB2/Her2-dependent breast cancer to prevent emergence of metastases after resection or chemo-ablation of primary tumors.
NF-κB and prevention of apoptosis
Inhibition of apoptosis is perhaps the most obvious way through which NF-κB signaling promotes the development of cancer, and NF-κB activation has long been known to suppress apoptosis44. Numerous NF-κB target genes prevent apoptosis, a normal mechanism by which immune and genomic surveillance mechanisms can eliminate pre-malignant or malignant cells. Some of these genes include Bcl-2 family members such as Bcl-xL, IAPs (inhibitors of apoptosis), and c-FLIP6. NF-κB also indirectly prevents mitochondrially-mediated apoptosis though neutralization of ROS (through induction of manganese superoxide dismutase or ferritin heavy chain). Several of the studies discussed above have documented decreased apoptosis in response to NF-κB activation (in addition to promoting proliferation).
Many investigators have observed that resistance to apoptosis in human cancer cell lines may be dependent on activation of NF-κB because when NF-κB is inhibited apoptosis can be triggered more readily28, 43, 45–50. NF-κB-induced apoptosis resistance has been implicated in chemotherapeutic failures in cancer treatment, and this topic is currently under intense investigation.
An elegant study linking NF-κB activation to inhibition of apoptosis (and carcinogenesis) utilized a mouse model with genetic deletion of the Mdr2 gene. Mdr2 knockout mice develop cholestatic hepatitis and ultimately HCC51. Suppression of NF-κB signaling early in Mdr2−/− mice had no effect on HCC initiation, but suppression of late NF-κB signaling diminished dramatically the size of HCC. The differences seen in HCC development were directly related to NF-κB-mediated prevention of apoptosis in transformed hepatocytes52. The difference between this model and the DEN model in which NF-κB inhibition in hepatocytes accelerates HCC development35 is due to the fact that DEN-induced carcinogenesis is highly dependent on hepatocyte death which is prevented by NF-κB, whereas the Mdr2−/− model is mainly dependent on chronic inflammation which is promoted by NF-κB. Indeed, as long as NF-κB is activated for only 48 hours after DEN application HCC development is suppressed (Y. Ben-Neriah, personal communication).
A causal link between NF-κB activation and prevention of apoptosis in B-cell lymphomagenesis was recently shown: transgenic introduction of a mutant NF-κB2 (“p80HT”, a mutant form of p100 found in a human lymphoma) into mice caused high level TRAF1 expression, suppressing apoptosis and leading to B cell lymphomas24. Sodium salicylate (asprin) has long been known to inhibit NF-κB activation53, and use of this agent decreases NF-κB activation and high levels of cFLIP expression in leukemic cells, causing apoptosis20.
Genotoxic stress, an event that can lead to cancer-initiating mutations, was recently found to activate the NF-κB pathway through activation of IKKγ/NEMO, causing resistance to apoptosis54, and this mechanism of IKK activation was found to inhibit apoptosis in the myelodysplastic syndrome, leading to acute myeloid leukemia49. Lastly, ROS have been implicated in genetic alterations which could lead to cancer, and NF-κB signaling was found to decrease ROS accumulation within cells and diminish associated apoptotic and necrotic cell death35, 36.
NF-κB and metastasis
Progression from local to metastatic cancer signifies late stage tumor progression, and the molecular mechanisms underlying this important process are just coming to light. Canonical NF-κB activation was recently implicated55 in epithelial-mesenchymal transition (EMT), a change thought to herald tissue invasion and prophesize metastatic potential. Activation of a so-called mesenchymal program (involving genes such as MMP2/9, VCAM-1, ICAM-1, Cathepsins B and Z56) was found to be dependent on NF-κB activation in a breast cancer model, and reversal of EMT was triggered by NF-κB inhibition55. E-cadherin was newly added to the list of genes regulated by NF-κB, and its repression by NF-κB enhances EMT in breast cancer14. NF-κB activation of Bcl2 was also been shown to promote EMT in breast cancer, leading to a more malignant phenotype57. Another important regulator of EMT is the transcription factor TWIST58. It was recently suggested that NF-κB may promote EMT and metastasis through transcriptional activation of TWIST59.
The most specific linkage of IKK signaling to cancer metastatic potential comes from a model of prostate cancer, in which IKKα activation represses expression of the metastasis suppressor gene maspin60. Genetic inhibition of IKKα kinase activation was shown to result in higher levels of maspin expression in advanced prostate adenocarcinomas where its expression is normally repressed during metastatic progression by activated IKKα via an NF-κB-independent mechanism. This repression correlates with accumulation of activated IKKα in nuclei of prostatic carcinoma cells, something that is also seen in advanced human prostate cancer60. Altough the mechanism responsible for activation of nuclear IKKα remains to be identified, it correlates with infiltration of prostate cancers with T cells that express RANK ligand (RANKL), a known activator of IKKα. A knockdown of maspin expression in IKKα mutant tumors restores metastatic potential60.
NF-κB and angiogenesis
Formation of new blood vessels is essential for tumor progression, as the growing tumor mass quickly exceeds the capacity of the native blood supply. Many of the signals which orchestrate angiogenesis are elaborated by tumor-associated macrophages (TAM), most of which are dependent on NF-κB signaling3. The NF-κB-coordinated inflammatory cytokines TNFα, IL-1, and IL-6 can stimulate expression of vascular endothelial growth factor (VEGF), one of the main regulators of angiogenesis, and VEGF is itself an NF-κB target gene, along with other angiogenic regulators such as CXCL 1, 8 and IL-856. Study of the relationship between NF-κB and angiogenesis has probably been impaired by the near-universal decrease in tumor growth upon NF-κB inhibition. In the setting of reduced cancer growth, independent reduction in angiogenesis is difficult to evaluate. However, more solid evidence tying NF-κB activation to angiogenesis was just provided in a study identifying JunB upregulation as the hypoxia-induced NF-κB activation target which induces VEGF expression61. Deletion of JunB in teratocarcinomas severely impaired angiogenesis. Interestingly, oxygen tension controls the extent of IKK activation such that normoxia limits IKKβ activity through proline hydroxylation whereas hypoxia favours it by preventing IKKβ proline hydroxylation62. These results suggest that macrophages and even epithelial cells in the hypoxic core of tumors are more likely to undergo IKK and NF-κB activation.
Summary
The perceived hypothetical role of inflammation in carcinogenesis has been bolstered by epidemiological observations linking infections and chronic inflammatory conditions to cancer. Given their place as master regulators at the center of inflammation, NF-κB transcription factors were natural suspects in providing a mechanistic link between inflammation and carcinogenesis. Indeed, the inflammation-cancer field has moved quickly from descriptive observations that activated NF-κB is present in many tumors to biochemical and genetic studies performed to elucidate the causal effects of NF-κB signaling in specific processes considered hallmarks of carcinogenesis (Figure 3 outlines potential NF-κB targets for the prevention and treatment of cancer). Of major importance is the recognition that various effects of NF-κB on cancer initiation, promotion, and progression are cell-type, tissue and context specific, ascertainment of which was only possible due to recent advances in genetically dissectable mouse models of inflammation-linked cancer. These advances are rapidly paving the way to new insights into the origin and treatment of human cancers.
Figure 3.
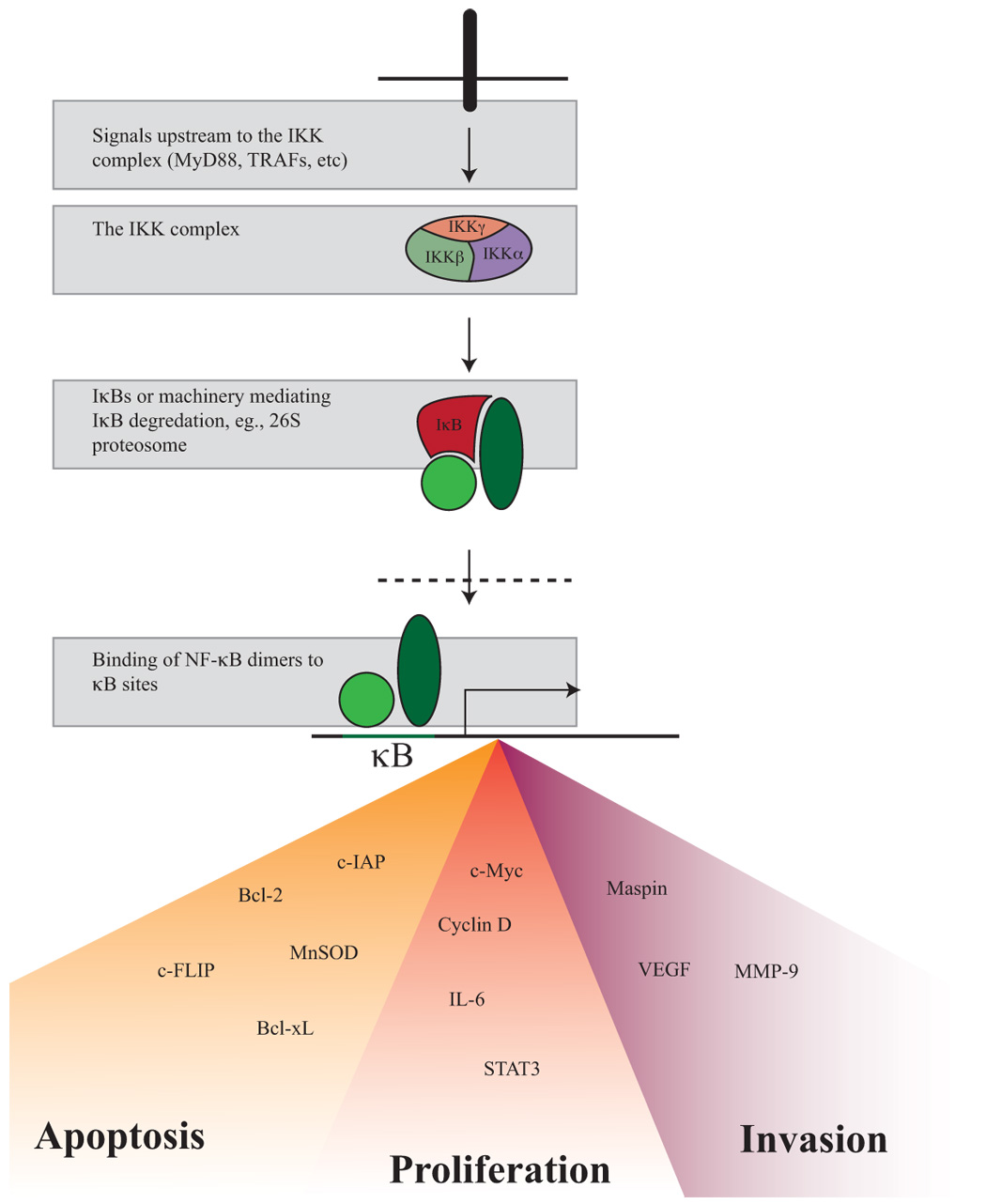
Targeting potential NF-κB contributors to carcinogenesis
Annotations to references for COGEDE-D-07-00059
1 NF-κB discovered and first described, thought to act solely in B cells
2 First suggestion that NF-κB may be a mechanistic link between cancer development to inflammation
3 Tumor progression directly linked to NF-κB activation for the first time
4 First study to show that NF-κB may have differing (and opposing) effects on carcinogenesis depending on the cell type (myeloid versus epithelial) in which it is active
5 Important study showing that innate immune mechanisms (as exemplified by the TLR adaptor MyD88) act upstream of NF-κB to promote cancer
6 Mechanism revealed by which NF-κB opposes JNK activation and cell death
7 NF-κB regulated IL-6 production in liver macrophages found to increase hepatic injury
8 Seminal study showing that TNF-alpha-induced apoptosis is opposed by NF-κB activation
9 NF-κB pathway (through IKKβ) found to oppose necrotic and apoptotic cell death by decreasing ROS accumulation
10, 11 Well-done studies showing that mutations involving the NF-κB signaling pathway occur and are responsible for a subset of human multiple myelomas
12 Excellent review on the processes of cancer development which gives framework for mechanistically studying carcinogenesis
13 NF-κB found to be important for promotion, but not initiation of inflammationassociated liver cancer
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124(4):823–835. doi: 10.1016/j.cell.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 2.Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2(4):301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 3.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357(9255):539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 4.Parkin DM, Bray F, Ferlay J, Pisani P. Estimating the world cancer burden: Globocan 2000. International Journal of Cancer. 2001;94(2):153–156. doi: 10.1002/ijc.1440. [DOI] [PubMed] [Google Scholar]
- 5.Kuper H, Adami HO, Trichopoulos D. Infections as a major preventable cause of human cancer. J Intern Med. 2000;248(3):171–183. doi: 10.1046/j.1365-2796.2000.00742.x. [DOI] [PubMed] [Google Scholar]
- 6.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441(7092):431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 7.Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46(5):705–716. doi: 10.1016/0092-8674(86)90346-6. [DOI] [PubMed] [Google Scholar]
- 8.Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25(6):280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 9.Hoffmann A, Baltimore D. Circuitry of nuclear factor kappaB signaling. Immunol Rev. 2006;210:171–186. doi: 10.1111/j.0105-2896.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- 10.Rothwarf DM, Zandi E, Natoli G, Karin M. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature. 1998;395(6699):297–300. doi: 10.1038/26261. [DOI] [PubMed] [Google Scholar]
- 11.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 12.Keats JJ, Fonseca R, Chesi M, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12(2):131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Annunziata CM, Davis RE, Demchenko Y, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12(2):115–130. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chua HL, Bhat-Nakshatri P, Clare SE, Morimiya A, Badve S, Nakshatri H. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene. 2007;26(5):711–724. doi: 10.1038/sj.onc.1209808. [DOI] [PubMed] [Google Scholar]
- 15.Yang J, Pan WH, Clawson GA, Richmond A. Systemic targeting inhibitor of kappaB kinase inhibits melanoma tumor growth. Cancer Res. 2007;67(7):3127–3134. doi: 10.1158/0008-5472.CAN-06-3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tew GW, Lorimer EL, Berg TJ, Zhi H, Li R, Williams CL. SmgGDS regulates cell proliferation, migration, and NF-kappa B transcriptional activity in non-small cell lung carcinoma. J Biol Chem. 2007 doi: 10.1074/jbc.M707526200. [DOI] [PubMed] [Google Scholar]
- 17.Scartozzi M, Bearzi I, Pierantoni C, et al. Nuclear factor-kB tumor expression predicts response and survival in irinotecan-refractory metastatic colorectal cancer treated with cetuximab-irinotecan therapy. J Clin Oncol. 2007;25(25):3930–3935. doi: 10.1200/JCO.2007.11.5022. [DOI] [PubMed] [Google Scholar]
- 18.Weichert W, Boehm M, Gekeler V, et al. High expression of RelA/p65 is associated with activation of nuclear factor-kappaB-dependent signaling in pancreatic cancer and marks a patient population with poor prognosis. Br J Cancer. 2007;97(4):523–530. doi: 10.1038/sj.bjc.6603878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Izzo JG, Malhotra U, Wu TT, et al. Clinical biology of esophageal adenocarcinoma after surgery is influenced by nuclear factor-kappaB expression. Cancer Epidemiol Biomarkers Prev. 2007;16(6):1200–1205. doi: 10.1158/1055-9965.EPI-06-1083. [DOI] [PubMed] [Google Scholar]
- 20.Rae C, Langa S, Tucker SJ, MacEwan DJ. Elevated NF-kappaB responses and FLIP levels in leukemic but not normal lymphocytes: reduction by salicylate allows TNF-induced apoptosis. Proc Natl Acad Sci U S A. 2007;104(31):12790–12795. doi: 10.1073/pnas.0701437104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vilimas T, Mascarenhas J, Palomero T, et al. Targeting the NF-kappaB signaling pathway in Notch1-induced T-cell leukemia. Nat Med. 2007;13(1):70–77. doi: 10.1038/nm1524. [DOI] [PubMed] [Google Scholar]
- 22.Fabre C, Carvalho G, Tasdemir E, et al. NF-kappaB inhibition sensitizes to starvation-induced cell death in high-risk myelodysplastic syndrome and acute myeloid leukemia. Oncogene. 2007;26(28):4071–4083. doi: 10.1038/sj.onc.1210187. [DOI] [PubMed] [Google Scholar]
- 23.Zou P, Kawada J, Pesnicak L, Cohen JI. Bortezomib induces apoptosis of Epstein-Barr virus (EBV)-transformed B cells and prolongs survival of mice inoculated with EBV-transformed B cells. J Virol. 2007;81(18):10029–10036. doi: 10.1128/JVI.02241-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang B, Wang Z, Li T, Tsitsikov EN, Ding HF. NF-kappaB2 mutation targets TRAF1 to induce lymphomagenesis. Blood. 2007;110(2):743–751. doi: 10.1182/blood-2006-11-058446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 26.Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18(49):6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 27.Jourdan M, Moreaux J, Vos JD, et al. Targeting NF-kappaB pathway with an IKK2 inhibitor induces inhibition of multiple myeloma cell growth. Br J Haematol. 2007;138(2):160–168. doi: 10.1111/j.1365-2141.2007.06629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rahman KW, Ali S, Aboukameel A, et al. Inactivation of NF-{kappa}B by 3,3′-diindolylmethane contributes to increased apoptosis induced by chemotherapeutic agent in breast cancer cells. Mol Cancer Ther. 2007;6(10):2757–2765. doi: 10.1158/1535-7163.MCT-07-0336. [DOI] [PubMed] [Google Scholar]
- 29.Fernandez-Majada V, Aguilera C, Villanueva A, et al. Nuclear IKK activity leads to dysregulated notch-dependent gene expression in colorectal cancer. Proc Natl Acad Sci U S A. 2007;104(1):276–281. doi: 10.1073/pnas.0606476104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo JL, Maeda S, Hsu LC, Yagita H, Karin M. Inhibition of NF-kappaB in cancer cells converts inflammation- induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Cancer Cell. 2004;6(3):297–305. doi: 10.1016/j.ccr.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 31.Greten FR, Eckmann L, Greten TF, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118(3):285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 32.Becker C, Fantini MC, Schramm C, et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004;21(4):491–501. doi: 10.1016/j.immuni.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 33.Rigby RJ, Simmons JG, Greenhalgh CJ, Alexander WS, Lund PK. Suppressor of cytokine signaling 3 (SOCS3) limits damage-induced crypt hyper-proliferation and inflammation-associated tumorigenesis in the colon. Oncogene. 2007;26(33):4833–4841. doi: 10.1038/sj.onc.1210286. [DOI] [PubMed] [Google Scholar]
- 34.Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science. 2007;317(5834):124–127. doi: 10.1126/science.1140488. [DOI] [PubMed] [Google Scholar]
- 35.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta Couples Hepatocyte Death to Cytokine-Driven Compensatory Proliferation that Promotes Chemical Hepatocarcinogenesis. Cell. 2005;121(7):977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 36.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNF-alpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120(5):649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 37.Sakurai T, Maeda S, Chang L, Karin M. Loss of hepatic NF-{kappa}B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc Natl Acad Sci U S A. 2006 doi: 10.1073/pnas.0603499103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naugler WE, Sakurai T, Kim S, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317(5834):121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 39.Luedde T, Beraza N, Kotsikoris V, et al. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11(2):119–132. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 40.Maeda S, Chang L, Li ZW, Luo JL, Leffert H, Karin M. IKKbeta is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFalpha. Immunity. 2003;19(5):725–737. doi: 10.1016/s1074-7613(03)00301-7. [DOI] [PubMed] [Google Scholar]
- 41.Makris C, Godfrey VL, Krahn-Senftleben G, et al. Female mice heterozygous for IKK gamma/NEMO deficiencies develop a dermatopathy similar to the human X-linked disorder incontinentia pigmenti. Mol Cell. 2000;5(6):969–979. doi: 10.1016/s1097-2765(00)80262-2. [DOI] [PubMed] [Google Scholar]
- 42.Cao Y, Luo JL, Karin M. IkappaB kinase alpha kinase activity is required for self-renewal of ErbB2/Her2-transformed mammary tumor-initiating cells. Proc Natl Acad Sci U S A. 2007;104(40):15852–15857. doi: 10.1073/pnas.0706728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singh S, Shi Q, Bailey ST, et al. Nuclear factor-kappaB activation: a molecular therapeutic target for estrogen receptor-negative and epidermal growth factor receptor family receptor-positive human breast cancer. Mol Cancer Ther. 2007;6(7):1973–1982. doi: 10.1158/1535-7163.MCT-07-0063. [DOI] [PubMed] [Google Scholar]
- 44.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274(5288):787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 45.Chen H, Li M, Campbell RA, et al. Interference with nuclear factor kappa B and c-Jun NH2-terminal kinase signaling by TRAF6C small interfering RNA inhibits myeloma cell proliferation and enhances apoptosis. Oncogene. 2006;25(49):6520–6527. doi: 10.1038/sj.onc.1209653. [DOI] [PubMed] [Google Scholar]
- 46.Chauhan D, Catley L, Li G, et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell. 2005;8(5):407–419. doi: 10.1016/j.ccr.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 47.Mi J, Zhang X, Liu Y, et al. NF-kappaB inhibition by an adenovirus expressed aptamer sensitizes TNFalpha-induced apoptosis. Biochem Biophys Res Commun. 2007;359(3):475–480. doi: 10.1016/j.bbrc.2007.05.125. [DOI] [PubMed] [Google Scholar]
- 48.Tapia MA, Gonzalez-Navarrete I, Dalmases A, et al. Inhibition of the canonical IKK/NF kappa B pathway sensitizes human cancer cells to doxorubicin. Cell Cycle. 2007;6(18):2284–2292. doi: 10.4161/cc.6.18.4721. [DOI] [PubMed] [Google Scholar]
- 49.Carvalho G, Fabre C, Braun T, et al. Inhibition of NEMO, the regulatory subunit of the IKK complex, induces apoptosis in high-risk myelodysplastic syndrome and acute myeloid leukemia. Oncogene. 2007;26(16):2299–2307. doi: 10.1038/sj.onc.1210043. [DOI] [PubMed] [Google Scholar]
- 50.Bernal-Mizrachi L, Lovly CM, Ratner L. The role of NF-{kappa}B-1 and NF-{kappa}B-2-mediated resistance to apoptosis in lymphomas. Proc Natl Acad Sci U S A. 2006;103(24):9220–9225. doi: 10.1073/pnas.0507809103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mauad TH, van Nieuwkerk CM, Dingemans KP, et al. Mice with homozygous disruption of the mdr2 P-glycoprotein gene. A novel animal model for studies of nonsuppurative inflammatory cholangitis and hepatocarcinogenesis. Am J Pathol. 1994;145(5):1237–1245. [PMC free article] [PubMed] [Google Scholar]
- 52.Pikarsky E, Porat RM, Stein I, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431(7007):461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 53.Kopp E, Ghosh S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science. 1994;265(5174):956–959. doi: 10.1126/science.8052854. [DOI] [PubMed] [Google Scholar]
- 54.Janssens S, Tinel A, Lippens S, Tschopp J. PIDD mediates NF-kappaB activation in response to DNA damage. Cell. 2005;123(6):1079–1092. doi: 10.1016/j.cell.2005.09.036. [DOI] [PubMed] [Google Scholar]
- 55.Huber MA, Azoitei N, Baumann B, et al. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114(4):569–581. doi: 10.1172/JCI21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Basseres DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25(51):6817–6830. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- 57.Wang X, Belguise K, Kersual N, et al. Oestrogen signalling inhibits invasive phenotype by repressing RelB and its target BCL2. Nat Cell Biol. 2007;9(4):470–478. doi: 10.1038/ncb1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117(7):927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 59.Horikawa T, Yang J, Kondo S, et al. Twist and epithelial-mesenchymal transition are induced by the EBV oncoprotein latent membrane protein 1 and are associated with metastatic nasopharyngeal carcinoma. Cancer Res. 2007;67(5):1970–1978. doi: 10.1158/0008-5472.CAN-06-3933. [DOI] [PubMed] [Google Scholar]
- 60.Luo JL, Tan W, Ricono JM, et al. Nuclear cytokine-activated IKKalpha controls prostate cancer metastasis by repressing Maspin. Nature. 2007;446(7136):690–694. doi: 10.1038/nature05656. [DOI] [PubMed] [Google Scholar]
- 61.Schmidt D, Textor B, Pein OT, et al. Critical role for NF-kappaB-induced JunB in VEGF regulation and tumor angiogenesis. Embo J. 2007;26(3):710–719. doi: 10.1038/sj.emboj.7601539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cummins EP, Berra E, Comerford KM, et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci U S A. 2006;103(48):18154–18159. doi: 10.1073/pnas.0602235103. [DOI] [PMC free article] [PubMed] [Google Scholar]