

SUMMARY
Macrophage infiltration and activation in metabolic tissues underlie obesity-induced insulin resistance and type 2 diabetes. While inflammatory activation of resident hepatic macrophages potentiates insulin resistance, the functions of alternatively activated Kupffer cells in metabolic disease remain unknown. Here we show that, in response to the Th2 cytokine interleukin-4 (IL-4), peroxisome proliferator activated receptor δ (PPARδ) directs expression of the alternative phenotype in Kupffer cells and adipose tissue macrophages of lean mice. However, adoptive transfer of PPARδ null bone marrow into wild type mice only diminishes alternative activation of hepatic macrophages, causing hepatic dysfunction and systemic insulin resistance. Suppression of hepatic oxidative metabolism is recapitulated by treatment of primary hepatocytes with conditioned media from PPARδ null macrophages, indicating direct involvement of Kupffer cells in liver lipid metabolism. Taken together, these data suggest an unexpected beneficial role for alternatively activated Kupffer cells in metabolic syndrome and type 2 diabetes.
INTRODUCTION
Chronic inflammation is causally linked to obesity, insulin resistance, and type 2 diabetes (T2D) (Hotamisligil, 2006; Shoelson et al., 2006). Increased adiposity promotes macrophage infiltration into adipose tissue (Weisberg et al., 2003; Xu et al., 2003), perpetuating local inflammation and causing insulin resistance (Kamei et al., 2006; Kanda et al., 2006; Weisberg et al., 2006). However, not all adipose tissue macrophages (ATMs) exhibit the inflammatory phenotype. For instance, macrophages resident in the adipose tissue of lean animals are alternatively activated and have decreased inflammatory potential (Lumeng et al., 2007; Odegaard et al., 2007a). Consistent with this observation, we recently showed that alternatively activated ATMs exert beneficial effects during times of caloric excess. Specifically, absence of alternatively activated ATMs, as in macrophage-specific PPARγ (Mac-PPARγ) null mice, promotes mitochondrial dysfunction, leading to obesity and insulin resistance (Odegaard et al., 2007a). While these studies have elucidated anti-diabetic activities of alternatively activated ATMs in metabolic syndrome, the functions of these cells in other metabolic tissues remain poorly understood.
Inflammatory activation of Kupffer cells, the resident macrophages in liver, has been implicated in both obesity-induced insulin resistance and fatty liver disease. Genetic ablation of IκB kinase β (IKKβ), an upstream kinase required for activation of nuclear factor κB (NF-κB), in myeloid cells reduces macrophage-mediated inflammation, and improves systemic and hepatic insulin sensitivity (Arkan et al., 2005). Conversely, inflammatory activation of Kupffer cells by lipopolysaccharide (LPS) promotes hepatoxicity in obese mice (Li and Diehl, 2003). While these and other studies have delineated the pathogenic activities of inflammatory Kupffer cells, resident hepatic macrophages display tremendous plasticity in their activation programs, ranging from the pro-inflammatory classical state to the anti-inflammatory alternative state (Gordon, 2003; Herbert et al., 2004). Despite the great therapeutic potential of alternatively activated Kupffer cells, their functions in metabolic disease remain enigmatic.
PPARs, members of the nuclear receptor superfamily (Chawla et al., 2001), control systemic fatty acid metabolism by transcriptional activation of target genes (Evans et al., 2004; Kliewer et al., 2001). Since a variety of fatty acids and fatty acid metabolites can bind and activate these receptors (Xu et al., 1999), PPARs act as fatty acid sensors to alter metabolic pathways in response to changes in fuel availability (Kliewer et al., 2001). Surprisingly, in alternatively activated murine macrophages, PPARγ is required for mitochondrial biogenesis and β-oxidation of fatty acids (Odegaard et al., 2007a). Despite an absolute requirement for oxidative metabolism in alternative macrophage activation (Vats et al., 2006), macrophage-specific disruption of PPARγ primarily effects alternative activation of ATMs, while sparing Kupffer cells (Odegaard et al., 2007a). This observation suggests that distinct transcriptional regulators and signaling pathways control depot-specific maturation of alternatively activated macrophages; however, the identity of these factors is unknown.
In response to IL-4, macrophages enact a coordinated program of oxidative metabolism and mitochondrial biogenesis necessary for alternative macrophage activation (Vats et al., 2006). One critical feature of this metabolic switch is the substantial increase in fatty acid influx. Because fatty acids and fatty acid-enriched lipoproteins can transcriptionally activate the nuclear receptor PPARδ in macrophages (Chawla et al., 2003; Evans et al., 2004; Xu et al., 1999), we investigated its functions in alternative macrophage activation. We report here that PPARδ is absolutely required for full expression of the effector phenotype of alternatively activated macrophages. Genetic deletion of PPARδ in bone marrow cells not only impairs alternative activation of tissue macrophages, but also predisposes animals to the development of insulin resistance and metabolic syndrome. Surprisingly, PPARδ deficiency has minimal impact on ATM biology. In contrast, Kupffer cells deficient in PPARδ display marked impairment in alternative activation, resulting in hepatic dysfunction and insulin resistance. Together, our findings have identified an unexpected role for PPARδ in Kupffer cell biology, thereby delineating a molecular mechanism by which this receptor and alternatively activated macrophages ameliorate obesity-induced insulin resistance.
RESULTS
PPARδ regulates arginase I expression in alternatively activated macrophages
To understand the role of PPARδ in alternative macrophage activation, bone marrow-derived macrophages (BMDMs) from wild type and PPARδ deficient mice were stimulated with IL-4 for 48–72 hours, a time period that promotes maximal expression of the alternatively activated phenotype (Munder et al., 1999). Notably, induction of arginase I mRNA and protein, a signature gene induced during alternative macrophage activation (Gordon, 2003), was dramatically reduced in PPARδ null macrophages (Figures 1A, B). Consistent with impaired induction of arginase I, cytosolic arginase activity was reduced by ~75% in PPARδ null macrophages (Figure 1C). To assess whether PPARδ can directly regulate arginase I, transient transfections were performed with the arginase I reporter construct containing the identified PPAR response element (Odegaard et al., 2007a; Pauleau et al., 2004). Strikingly, co-transfection of PPARδ greatly augmented IL-4’s transcriptional activity on this promoter (~4-fold), which was not further potentiated by GW501516, a synthetic agonist of PPARδ (Oliver et al., 2001) (Figure 1D). However, deletion of the PPAR binding site completely abolished the ability of IL-4 and PPARδ to activate this promoter (Figure 1D), indicating that PPARδ regulates arginase I in alternatively activated macrophages via this site.
Figure 1. PPARδ regulates expression of arginase I in alternatively activated macrophages.

(A–C) Decreased levels of arginase I mRNA (A), protein (B), and enzymatic activity (C) in IL-4-stimulated PPARδ −/−BMDMs. (D) Activation of the arginase I promoter by PPARδ/RXR heterodimers and IL-4. Mutation of PPAR response element in the arginase I enhancer abolishes transcriptional activation by PPARδ and IL-4. (E–F) Attenuated induction of arginase I in PPARδ/γ double knockout macrophages. (E) Real time analysis of PPARδ and PPARγ expression in BMDMs generated from control, macrophage-specific PPARδ null and macrophage-specific PPARδ/γ null mice. Primers specific for exon 4 of PPARδ or exon 1 of PPARγ were used to quantify excision efficiency in BMDMs. (F) qRT-PCR analysis of arginase I mRNA in various genotypes. Data presented as mean ± s.e.m. *P < 0.05, **P<0.01.
We have previously shown that macrophage-specific PPARγ plays an essential role in the regulation of arginase I expression during alternative macrophage activation (Odegaard et al., 2007a). Since our current data also indicate a requirement for PPARδ in the regulation of this facet of alternative activation, we next investigated the hierarchical relationship between these two nuclear receptors. For these experiments, we generated BMDMs from control (PPARδflox/flox), macrophage-specific PPARδ null (PPARδflox/flox; LsyMCre), and macrophage-specific PPARδ/γ null (PPARδ/γflox/flox; LsyMCre) mice, strains recently generated in the laboratory on the C57Bl/6J background (Figure 1E). As shown in Figures 1F, IL-4 failed to stimulate arginase I mRNA expression in macrophages deficient in both PPARγ and δ, indicating that both nuclear receptors are required for optimal expression of this gene during alternative macrophage activation.
PPARδ regulates immunologic phenotype of alternatively activated macrophages
To further evaluate the requirement of PPARδ in expression of the alternative phenotype, we examined other characteristics of alternative activation in PPARδ null macrophages. As shown in Figure 2A, IL-4 failed to induce mRNAs encoding Retnla (resistin like alpha), Mrc1 (mannose receptor), Chi3l3 (acidic chitinase), and Pdcd1lg2 (PD-L2) in PPARδ null macrophages (Gordon, 2003; Loke and Allison, 2003; Raes et al., 2002). This was further substantiated by markedly lower cell surface expression of dectin-1 (Clec7a) and mannose receptor in PPARδ null macrophages (Figure S1a). Supporting a critical role for PPARδ in alternative macrophage activation, macrophages deficient in PPARδ were also refractory to IL-13 mediated alternative activation (Figure S2).
Figure 2. PPARδ is required for expression of the immune phenotype of alternative macrophage activation.
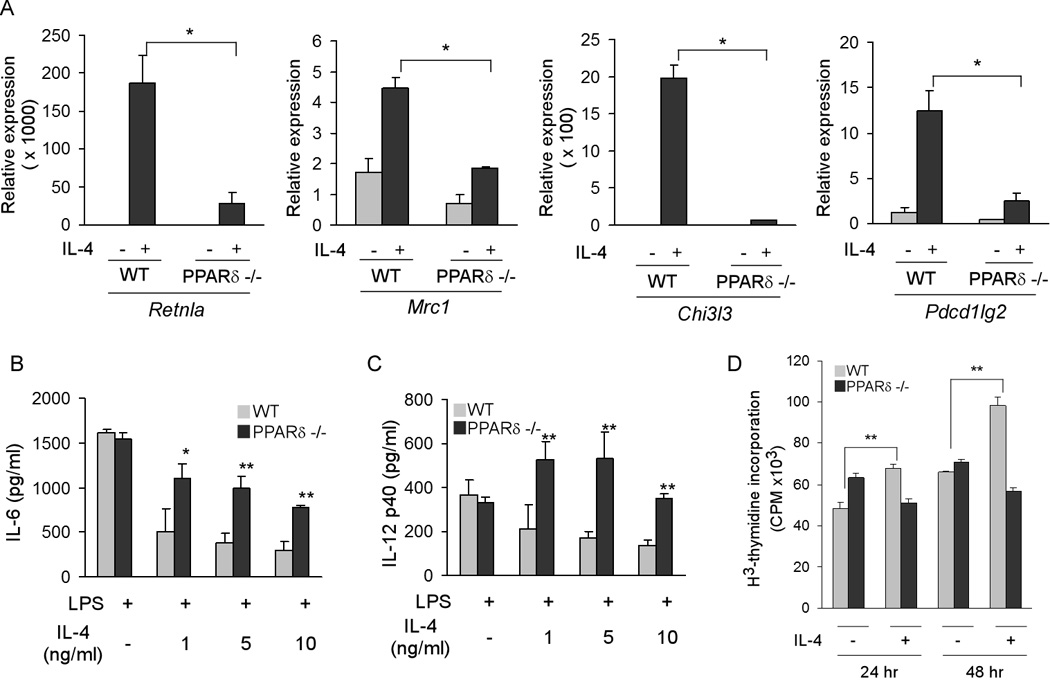
(A) Decreased expression of markers of alternative activation in IL-4-stimulated PPARδ −/− BMDM. Relative expression of alternative activation mRNAs was quantified using by qRT-PCR. Retnla, resistin-like a; Mrc1, mannose receptor; Chi3l3, chitinase 3-like 3; Pdcd1lg2, programmed cell death 1 ligand 2. (B, C) PPARδ is required for suppression of IL-6 and IL-12 production in alternatively activated BMDMs. (D) PPARδ is required for the mitogenic response to IL-4 in BMDMs. Data presented as mean ± s.e.m. *P < 0.05, **P < 0.01.
Because treatment of macrophages with IL-4 dampens their inflammatory response (Herbert et al., 2004; Odegaard et al., 2007b), we next investigated whether PPARδ was also required for this facet of alternative activation. In a dose-dependent manner, pretreatment with IL-4 suppressed lipopolysaccharide (LPS)-induced secretion of inflammatory cytokines IL-6 and IL-12 in wild type, but not PPARδ null, macrophages (Figures 2B, C). Consistent with a global decrement in the acquisition of the alternative phenotype, macrophages lacking PPARδ had a markedly lower mitogenic response to IL-4, as assessed by incorporation of 3H-thymidine (Figure 2D). Lastly, in support of the central role of PPARδ and γ in acquisition of the alternative phenotype, comparative analyses of BMDMs from control, macrophage-specific PPARδ null, and macrophage-specific PPARδ/γ null mice, revealed that both receptors are required for optimal alternative macrophage activation (Figure S3). Taken together with our previous findings, these results suggest that both PPARδ and γ coordinate the macrophage’s transcriptional response to the Th2 cytokines IL-4 and IL-13. PPARγ primarily regulates metabolic programs in alternatively activated macrophages (Odegaard et al., 2007a), whereas PPARδ is required for the full expression of their immune phenotype, including expression of pattern recognition receptors (Mrc1 and Clec7a) and co-stimulatory molecules (Pdcd1lg2), and suppression of macrophage-inflammatory response.
To further explore whether PPARδ ligands can initiate or potentiate alternative macrophage activation, we treated wild type macrophages with PPARδ activators under three different experimental settings. First, to determine whether PPARδ activation can induce a Th2 bias in an elicited macrophage population, wild type mice were injected (i.p.) with vehicle or the synthetic PPARδ agonist GW0742 for 5 days (Graham et al., 2005), and markers of alternative activation were quantified in bio-gel elicited macrophages. As shown in Figure S4A, GW0742 treatment led to a dramatic increase in the expression of signature genes for alternative macrophage activation. Second, to determine whether chronic treatment with PPARδ agonist induces an alternative bias in BMDMs, wild type bone marrow progenitor cells were treated with the PPARδ agonist GW501516 (100 nM) for 7 days. Consistent with the Th2 polarizing effects of PPARδ in macrophages, stimulation with GW501516 induced markers of alternative activation in BMDMs (Figure S4B). Lastly, to explore whether PPARδ agonist can synergize with Th2 cytokines in promoting alternative macrophage activation, wild type BMDMs were stimulated with vehicle, IL-4 (2ng/ml), GW501516 (100 nM), or a combination of GW501516 (100 nM) and IL-4 (2 ng/ml). Figure S4C shows that BMDMs co-stimulated with GW501516 and IL-4 had markedly higher levels of transcripts encoding alternative activation signature genes. In aggregate, these data suggest that synthetic ligands for PPARδ can work alone or in combination with endogenous Th2 signals to promote an alternative bias in macrophage activation.
Unsaturated fatty acids potentiate alternative macrophage activation via PPARδ
Tissue macrophages, specifically those present in liver and adipose tissue, are exposed to high levels of fatty acids. Because a large influx of fatty acids accompanies alternative macrophage activation, we reasoned that these incoming fatty acids might activate PPARδ, a genetic sensor of fatty acids (Chawla et al., 2001; Evans et al., 2004). To test this hypothesis, BMDMs were stimulated with varying doses of IL-4 in the presence of bovine serum albumin (BSA) or BSA conjugated to 100 µM oleic acid. Figure 3A shows that BSA:oleic acid conjugate synergizes with IL-4 to induce arginase activity in macrophages. Notably, this synergistic effect of oleic acid is completely abolished in macrophages lacking PPARδ, indicating that monounsaturated fatty acids might potentiate the broader program of alternative activation. Indeed, in a PPARδ-dependent manner, oleic acid enhanced alternative macrophage activation, as evidenced by the induction of Arg1, Clec7a, Pdcd1lg2, and Chi3l3 mRNAs (Figure 3B–E). Taken together with recent findings of Shi et. al. (Shi et al., 2006), these data indicate that local availability of fatty acids can alter the activation potential of macrophages. While saturated fatty acids, such as lauric and palmitic acids, ligate toll-like receptor 4 (TLR4) to promote pro-inflammatory activation of macrophages (Kim et al., 2007; Shi et al., 2006), monounsaturated fatty acids enhance expression of the anti-inflammatory alternative state by activating the nuclear receptor PPARδ.
Figure 3. Conjugated oleic acid potentiates alternative macrophage activation.
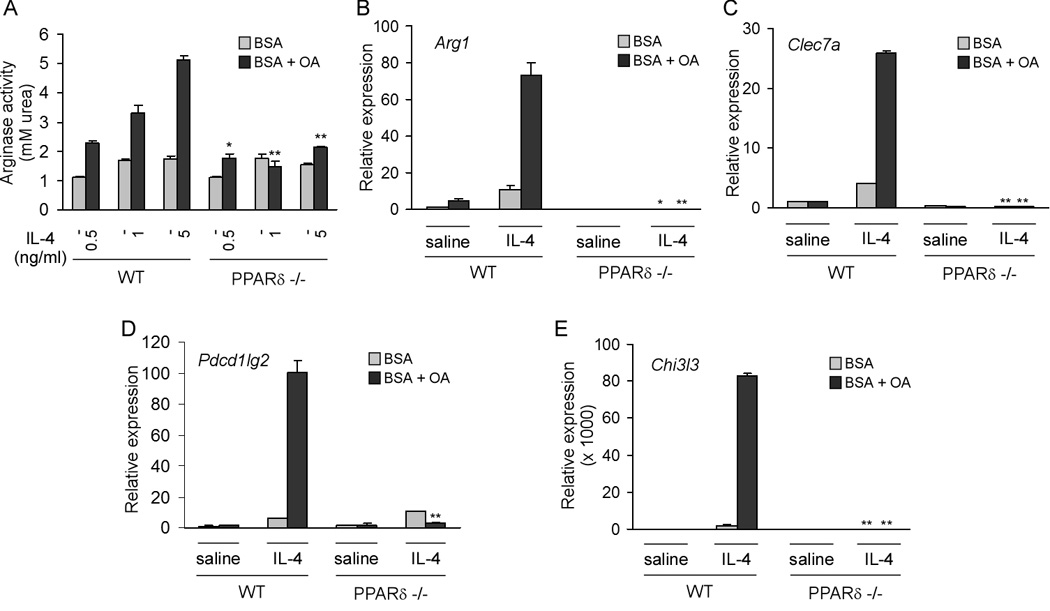
(A) Oleic acid potentiates IL-4 induction of arginase activity in wild type, but not PPARδ−/− BMDM. (B–E) PPARδ is required for potentiation of alternative activation by oleic acid. BMDMs from 129/SvJ wild type and PPARδ null mice were stimulated IL-4 for 48 hours in the presence of BSA or BSA:oleic acid conjugate. qRT-PCR analysis were performed on mRNAs for Arg1 (arginase I), Clec7a (dectin-1), Pdcd1lg2 (programmed cell death 1 ligand 2), and Chi3l3 (chitinase 3-like 3). Data presented as mean ± s.e.m. *P < 0.05, **P < 0.01.
PPARδ is required for alternative activation of resident macrophages
To verify whether PPARδ is essential for alternative activation of resident macrophages, we analyzed macrophage signatures in metabolic tissues of lean mice. As expected, the signature genes of alternatively activated macrophages, including Clec7a, Retnla, Tgfb1, Jag1, and Mrc1, were dramatically reduced in white adipose tissue (WAT) of PPARδ null mice (Figure S5A). Surprisingly, genetic deletion of PPARδ also impaired alternative activation of hepatic macrophages, the Kupffer cells, as evidenced by decreased expression of Arg1, Clec7a, Jag1, Pdcd1lg2, and Chia (Figure S5B). Furthermore, treatment of lean wild type mice with GW0742, a highly selective agonist for PPARδ, increased expression of alternative macrophage activation markers in both WAT and liver (Figure S5 C, D). Thus, in contrast to PPARγ (Odegaard et al., 2007a), PPARδ transcriptional signaling is required for the maintenance of alternatively activation of ATMs and Kupffer cells in lean mice.
Hematopoietic PPARδ protects against diet-induced insulin resistance
Having established that PPARδ regulates the immune program of alternative macrophage activation, we investigated whether mice lacking PPARδ in the macrophage compartment were at an increased risk of developing metabolic syndrome. To conduct these experiments, we adoptively transferred wild type and PPARδ-deficient bone marrow into lethally irradiated wild type animals (Weisberg et al., 2003). After reconstitution for 4 weeks, age-matched cohorts were placed on a high fat diet (HFD) for 22 weeks to promote maximal infiltration of macrophages into WAT (Xu et al., 2003). Q-PCR analysis of genomic DNA confirmed greater than 99% replacement of wild type marrow by PPARδ null cells (Figure S6A). Importantly, in this time frame, even the relatively radio-resistant Kupffer cells get replaced with new cells from the bone marrow (Figure S6B, D). Moreover, isolation of F4/80-positive macrophages from WAT and liver confirmed that majority of tissues macrophages were derived from PPARδ null hematopoietic cells (Figure S6C, D). Remarkably, reconstitution of wild type mice with PPARδ −/− bone marrow (PPARδ −/− BMT) led to development of glucose intolerance on the HFD (Figure 4A). In addition, compared to mice transplanted with wild type bone marrow (WT BMT), PPARδ −/− BMT mice were more resistant to glucose lowering by exogenous insulin (Figure 4B). Consistent with this decrease in insulin sensitivity, fasting insulin levels were ~2-fold higher in PPARδ −/− BMT mice (Figure 4C). However, fasting levels of circulating lipids were not significantly different between the two cohorts of transplanted mice (Table S1). To identify the sites of insulin resistance, WT BMT and PPARδ −/− BMT mice were injected with insulin via their inferior vena cava, and tissues were quickly harvested for biochemical analyses of insulin signaling (Arkan et al., 2005; Cai et al., 2005). As shown in Figures 4D–F, phosphorylation of Akt was markedly reduced in liver, quadriceps and WAT of PPARδ −/− BMT mice, indicating onset of global insulin resistance. Since reduction in oxidative phosphorylation (OXPHOS) is causally linked to development of insulin resistance in mice and humans (Mootha et al., 2003; Patti et al., 2003; Petersen et al., 2003; Vianna et al., 2006), we looked for evidence of mitochondrial dysfunction in all three tissues. Notably, RT-qPCR analyses revealed a profound decrease (30–50%) in expression of enzymes for fatty acid oxidation (Acox1, Acadm, Adcadl) and oxidative phosphorylation (Ndufs1, Sdhb, Uqcrc1, Cox4i1 and Atp5j) in livers of PPARδ −/− BMT mice (Figure 4G). Similarly, genes important in oxidative metabolism (Acox1, Cpt1b) and electron transport (Ndusf1, Uqcrc1, Cox4i1) are suppressed by 60–90% in quadriceps of PPARδ −/− BMT animals (Figure 4H). Importantly, in both tissues, expression of transcriptional regulators controlling mitochondrial oxidative metabolism (Tfam, Nrf1, Pgc1a and Pgc1b) is dramatically reduced (30–80%). However, metabolic programs for β-oxidation and oxidative phosphorylation were largely intact in WAT of PPARδ −/− BMT mice (Figure S7C), suggesting that hematopoietic deficiency of PPARδ primarily suppresses oxidative metabolism in myocytes and hepatocytes.
Figure 4. Hematopoietic deficiency of PPARδ exacerbates insulin resistance and impairs glucose tolerance.
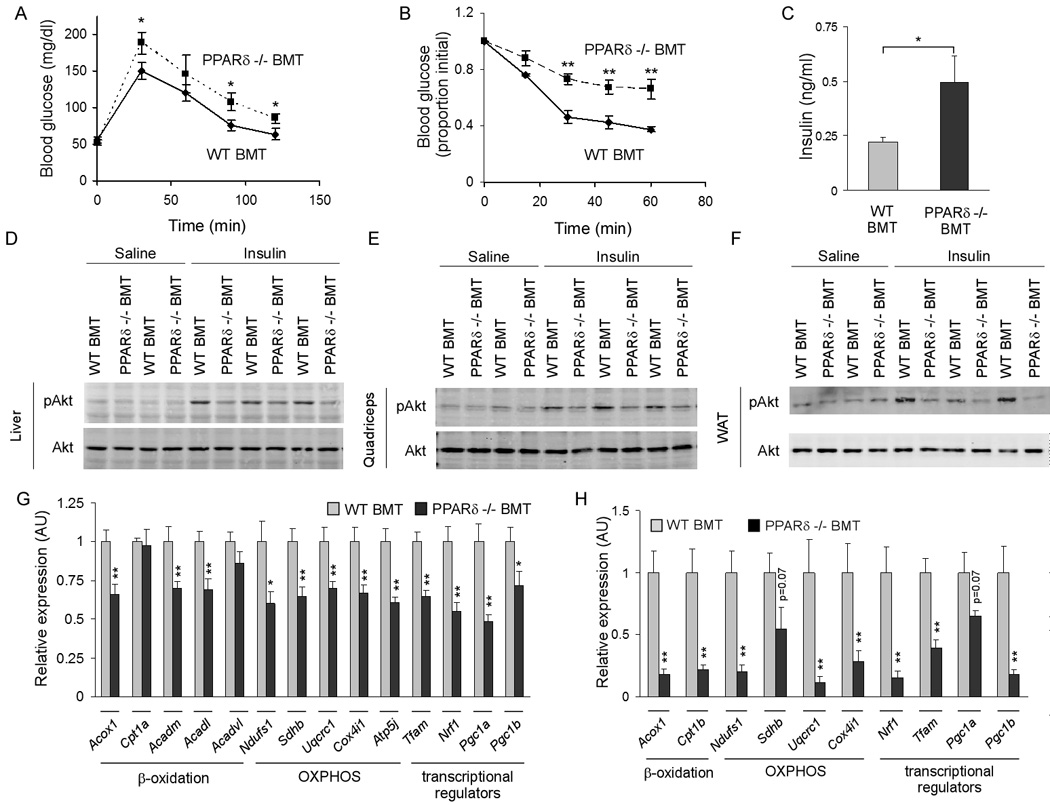
(A) Oral glucose tolerance test (1g/kg) were carried out in male WT BMT and PPARδ −/− BMT mice after 18 weeks of high fat diet (n=5–7 per cohort). (B) Insulin tolerance test (0.65U/kg) were performed in obese mice after a 4 hour fast (n=5–7 per cohort). Similar results were obtained in two other cohorts of transplanted mice (n=7 per group). (C) Fasting serum levels of insulin in WT BMT and PPARδ −/− BMT mice after a 5 hr fast. (D–F) Impairment in insulin action in PPARδ −/− BMT mice. Total cell lysates were immunoblotted for pAkt or total Akt in liver (D), quadriceps muscle (E) and epididymal WAT (F). (G, H) Mitochondrial dysfunction in peripheral tissues of PPARδ −/− BMT mice. Relative transcript levels for genes encoding key enzymes in β-oxidation, oxidative phosphorylation (OXPHOS), and of transcriptional regulators controlling these pathways in liver (G) and quadriceps (H). Data presented as mean ± s.e.m. *P < 0.05, **P < 0.01.
Hematopoietic PPARδ protects against diet-induced obesity
We recently demonstrated that alternatively activated ATMs protect against diet-induced obesity (Odegaard et al., 2007a), prompting us to evaluate changes in adiposity in PPARδ null chimeric animals. At 18 weeks of HFD, dual energy x-ray absorptiometry indicated that weight-matched PPARδ −/− BMT mice had significantly higher fat content (43.4 ± 0.39% vs. 37.8 ± 1.81%, P < 0.01) and slightly lower lean body mass (18.2 ± 0.47g vs. 19.9 ± 0.52g, P < 0.05) than WT BMT controls (Figure 5A). Furthermore, gross post-mortem analyses show marked enlargement of adipose depots (Figure 5B), paralleling the ~50% increase in epididymal fat pad mass, ~7-fold increase in serum leptin and 20% reduction in serum adiponectin (Figure 5C and Table S1). However, in contrast to adipocyte cell size in Mac-PPARγ null mice (Odegaard et al., 2007a), adipocytes were significantly larger in PPARδ −/− BMT mice (Figure 5D and Figure S7A), suggesting adipocyte hypertrophy likely accounts for the increase in total adiposity. Because adipocyte morphology is markedly different in these two models, we next quantified ATM content and activation. In striking contrast to Mac-PPARδ null mice, ATM content trended toward being higher in PPARδ −/− BMT mice without significant change in the signature of alternatively activated macrophages (Figures 5 E, F and Figure S7B). Moreover, unlike WAT of Mac-PPARγ null mice, expression analysis of WAT showed only modest decrease in adipocyte programs of lipid accretion, storage, and utilization (Figure S7C), findings that are consistent with the minor increase in inflammatory response in this tissue (Figure 5F) (Weisberg et al., 2003). Direct co-culture of adipocytes with macrophages further affirmed that PPARδ null macrophages do not significantly impact adipocyte biology, as assessed by insulin-stimulated glucose uptake (Figure S7D) and expression of cytokines and adipokines in these cells (Figure S7E). Together, these data suggest that nutritional status of the animal is a key determinant of whether PPARδ signaling is required for alternative activation of ATMs. While PPARδ signaling is necessary for alternative activation of ATMs in lean mice (Figure S5A), it is largely dispensable for maintenance of this macrophage population in WAT of obese mice (Figure 5F). Future experiments utilizing the recently generated macrophage-specific PPARδ/γ double knockout mice will allow us to determine whether induction of PPARγ compensates for loss of PPARδ signaling in ATMs of obese animals. Nonetheless, the lack of significant macrophage phenotype in WAT of PPARδ −/− BMT mice suggests potential involvement of alternatively activated macrophages in other metabolic tissues.
Figure 5. Increased adiposity in PPARδ −/− BMT mice.
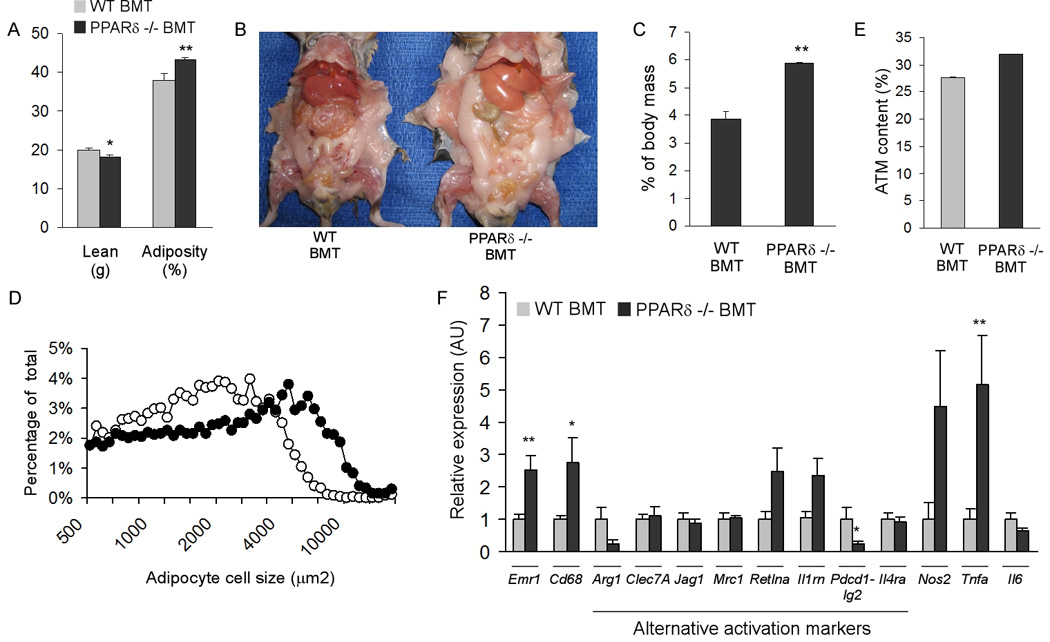
(A) Body composition was quantified by dual-energy X-ray absorptiometry in weight matched transplanted mice (n=7 per cohort). (B) Representative images of necropsied WT BMT and PPARδ null BMT mice after 22 weeks of high fat diet. (C) Increased epididymal fat pad mass in PPARδ null BMT mice (n=5 per cohort). (D) Increased adipocyte cell size in PPARδ null BMT mice. Adipocyte cell size was measured using dark field images. (E, F) Macrophage content and activation in white adipose tissue. ATM content was determined by immunostaining for the macrophage antigen F4/80, paired T-test P value = 0.16; n=4 per cohort (E). (F) Interrogation of ATM activation state by qRT-PCR. Emr1, F4/80; Cd68, macrosialin; Arg1, arginase I; Clec7A, dectin-1; Jag1, jagged 1; Mrc1, mannose receptor; Retlna, resistin-like alpha; Il1rn, IL-1 receptor antagonist; Pdcd1lg2, programmed cell death 1 ligand 2; Il4ra, IL-4 receptor alpha; Nos2, inducible nitric oxide synthase; Il6, interleukin-6; Tnfa, tumor necrosis factor alpha. Data presented as mean ± s.e.m. *P < 0.05, **P < 0.01.
Alternative activation of Kupffer cells preserves hepatic function
Kupffer cells in lean PPARδ null mice exhibited diminished capacity for alternative activation (Figure S5B), prompting us to examine the number and activation state of hepatic macrophages in obese animals. Although HFD feeding led to a modest increase (~20%) in hepatic macrophage content in mice reconstituted with PPARδ −/− bone marrow (Figure S8A, B), the activation state of resident macrophages was dramatically different between WT BMT and PPARδ −/− BMT mice. Notably, the signature of alternatively activated Kupffer cells was greatly reduced in livers of obese PPARδ −/− BMT mice (Figure 6A). Furthermore, liver arginase activity was also reduced by ~41% in PPARδ −/− BMT mice (Figure 6B), confirming that resident macrophages were gradually being replaced by cells derived from the bone marrow (Figure S6C, E). To ascertain whether PPARδ is required for IL-4 mediated alternative activation of Kupffer cells, wild type and PPARδ null mice were challenged with IL-4 and activation state of Kupffer cells was interrogated by qRT-PCR. Remarkably, injection of IL-4 induced the expression of alternative activation signature genes Arg1, Clec7a, Chi3l3, and Tgfb1 in a PPARδ-dependent manner (Figure 6C), thus proving an absolute requirement for PPARδ in the IL-4 driven program of Kupffer cell alternative activation.
Figure 6. Impaired alternative activation of Kupffer cells and hepatic dysfunction in PPARδ−/− BMT mice.
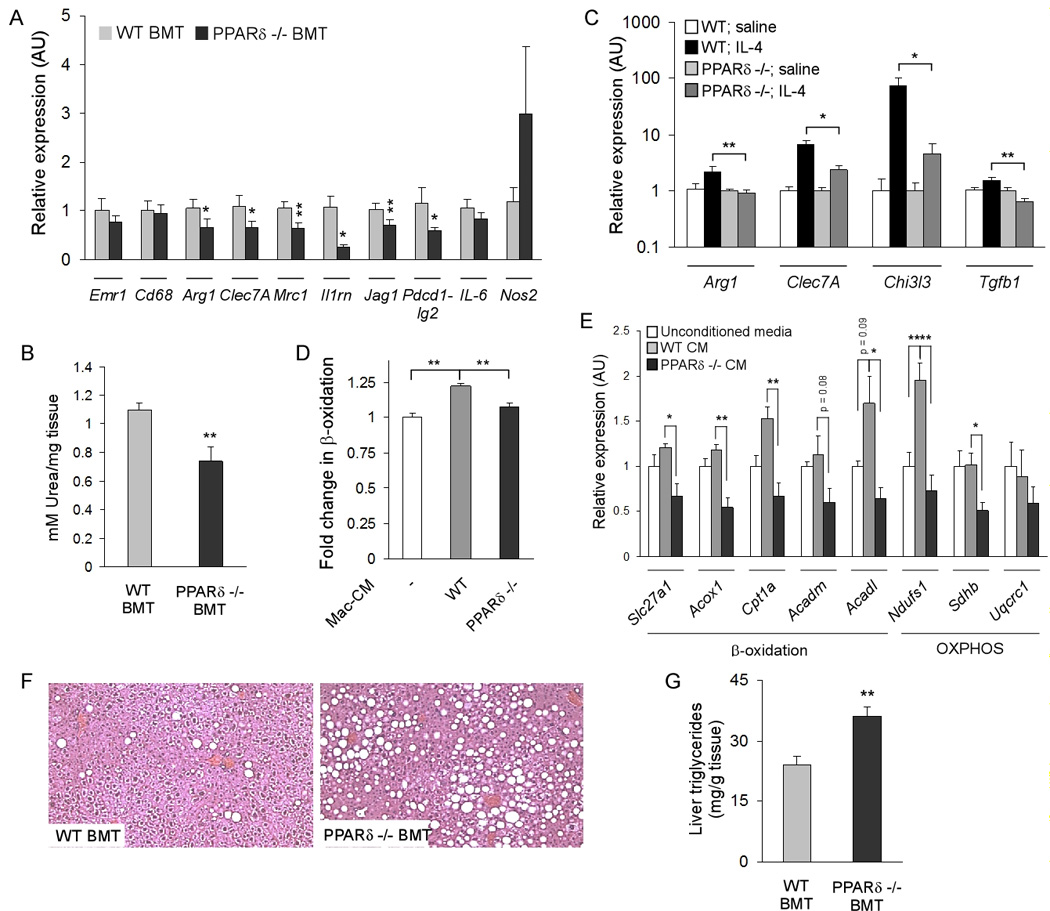
(A) Decreased expression of signature genes for alternative activation macrophages in PPARδ −/− BMT livers (n=5–7 per cohort). (B) Reduction in liver arginase activity in PPARδ−/− BMT mice (n=5–7 per cohort). (C) IL-4 is unable to induce alternative activation of Kupffer cells in PPARδ −/− mice (n=4 per cohort). Arg1, arginase I; Clec7a, dectin-1; Chi3l3, chitinase 3-like 3; Tgfb1, (transforming growth factor β1). (D, E) Treatment of primary hepatocytes with macrophage conditioned media alters their oxidative metabolism. Conditioned media from PPARδ null macrophages suppresses oxidative metabolism, as monitored by β-oxidation of fatty acids (D) and expression of fatty acid oxidation and OXPHOS genes (E). (F, G) Histologic (F) and biochemical (G) evidence of increased triglyceride accumulation in livers of PPARδ −/− BMT mice. Data presented as mean ± s.e.m. *P < 0.05, **P < 0.01.
We have previously shown that impairment in alternative macrophage activation is linked to development of mitochondrial dysfunction in metabolic tissues (Odegaard et al., 2007a). Because PPARδ −/− chimeric mice have markedly reduced expression of β-oxidation and OXPHOS genes in liver (Figures 4G), it suggests that alternatively activated Kupffer cells might directly modulate hepatic metabolism. To test this hypothesis, β-oxidation of fatty acids was analyzed in wild type primary hepatocytes that had been co-cultured with wild type or PPARδ null BMDMs for 72 hours. Strikingly, hepatocytes co-cultured with PPARδ null macrophages exhibit ~25% decrease in the rate of fatty acid oxidation compared to those co-cultured with wild type macrophages (Figure S8C). Furthermore, qRT-PCR analyses of co-cultured primary hepatocytes demonstrate potent suppression of β-oxidation and OXPHOS pathways by PPARδ null macrophages (Figure S8D). Since similar results were obtained when primary hepatocytes were treated with conditioned media from wild type or PPARδ null macrophages (Figure 6D, E), it suggests that factors secreted by macrophages can directly modulate oxidative metabolism in parenchymal cells. Finally, confirming a causal relationship between PPARδ-deficient Kupffer cells and dysregulation of hepatic metabolism, livers of PPARδ −/− BMT mice exhibited gross and histological evidence of hepatic steatosis (Figure 5B and Figure 6F), and had ~50% increase in extractable liver triglycerides (Figure 6G).
DISCUSSION
The infiltration and activation of macrophages in metabolic tissues is a key event in the pathogenesis of diet-induced obesity and insulin resistance (Qatanani and Lazar, 2007). However, most metabolic tissues, including adipose tissue and liver, contain a resident population of less inflammatory alternatively activated macrophages (Gordon, 2003), whose functions in obesity-induced metabolic disease remain poorly understood. To address this critical question, we have taken a systematic approach to identify transcriptional regulators that control the maturation of alternatively activated macrophages in tissues. Previously, we demonstrated that PPARγ orchestrates the program of oxidative metabolism in alternatively activated macrophages, a requisite for alternative maturation of ATMs (Odegaard et al., 2007a). In contrast, we now show that PPARδ coordinates the immune phenotype of alternatively activated macrophages, both in vitro and in vivo. Interestingly, hematopoietic deficiency of PPARδ selectively impairs alternative activation of Kupffer cells in obese mice, leading to reduction in oxidative metabolism and insulin sensitivity. Furthermore, direct co-culture of primary hepatocytes with PPARδ null macrophages recapitulates the liver phenotypes of PPARδ −/− BMT mice. Taken together, our findings suggest that PPARδ and PPARγ control distinct aspects of alternative macrophage activation to ameliorate obesity-induced insulin resistance (Figure 7).
Figure 7.
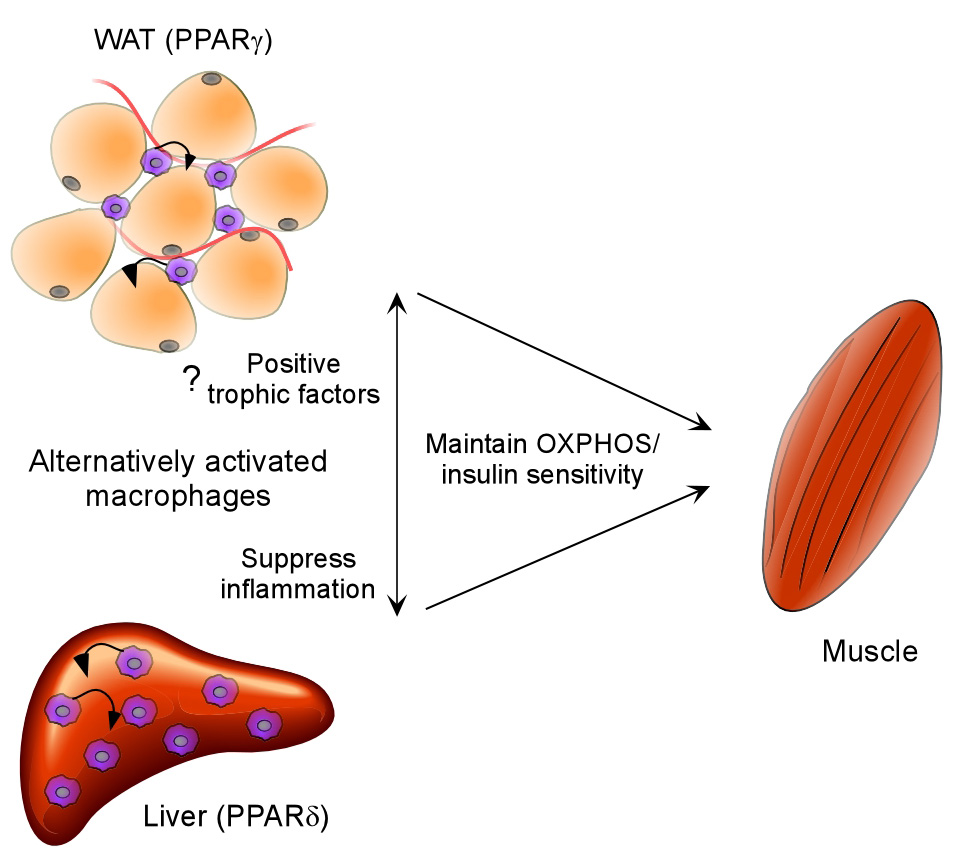
Model highlighting the metabolic functions of alternatively activated macrophages. PPARγ transcriptional signaling is required for maturation of these cells in adipose tissue, whereas PPARδ controls the expression of alternative phenotype in Kupffer cells of obese mice. Two potential mechanisms by which alternatively activated macrophages improve insulin action in obese mice are: paracrine inhibition of inflammation and secretion of trophic factors that can directly modulate oxidative metabolism in parenchymal cells (small arrows).
While inflammatory activation of Kupffer cells by bacterial by-products induces hepatic insulin resistance (Arkan et al., 2005; Li and Diehl, 2003), the role of alternatively activated Kupffer cells in liver metabolism has remained enigmatic. Unexpectedly, we discovered that the PPARδ regulated program of Kupffer cell alternative activation mediates beneficial effects on metabolic disease, an observation that is supported by three independent lines of evidence. First, in lean animals, genetic deletion of PPARδ reduces the expression of genes that constitute the signature of alternatively activated Kupffer cells. Second, transplantation of PPARδ null bone marrow into wild type mice diminishes alternative activation of Kupffer cells, leading to mitochondrial dysfunction and insulin resistance in hepatocytes. Third, direct co-culture of PPARδ null macrophages with primary hepatocytes leads to dramatic suppression of oxidative metabolism in the parenchymal cells. Lastly, since Kupffer cell replacement in the PPARδ −/− BMT mice is incomplete (Figure S6D), our studies likely underestimate the metabolic benefits of the alternatively activated Kupffer cells.
Extensive metabolic characterization of PPARδ −/− BMT and Mac-PPARγ null mice revealed that the activation state of resident macrophages plays an integral role in regulation of peripheral metabolism (Figure 7). For instance, in Mac-PPARγ null mice, loss of alternatively activated ATMs causes impairment in adipocyte function, resulting in reduction in oxidative metabolism and insulin sensitivity (Hevener et al., 2007; Odegaard et al., 2007a). Similarly, Kupffer cells in PPARδ −/− BMT mice have diminished capacity to undergo alternative activation, leading to a dramatic reduction in oxidative phosphorylation and insulin sensitivity. Hence, these findings suggest that, irrespective of their sites of residence, alternatively activated macrophages have a profound influence on oxidative metabolism and lipid homeostasis in peripheral tissues. Interestingly, the maturation of alternatively activated macrophages is itself regulated by fatty acids and their cognate receptors, PPARγ and PPARδ, thereby positioning these cells to be the cellular integrators of lipid metabolism. In support of this idea, saturated fatty acids (the “bad” fats) bind and activate TLR4 signaling in macrophages, thereby increasing adipose tissue inflammation and insulin resistance (Kim et al., 2007; Shi et al., 2006). In contrast, monounsaturated fatty acids (the “good” fats) activate PPARδ to bias tissue macrophages to the alternative state, thus improving insulin sensitivity. However, the precise molecular mechanisms by which alternatively activated macrophages modulate insulin action and peripheral metabolism remain unclear. On the one hand, attenuation of tissue inflammation by paracrine actions of these cells might lead to improvement in insulin signaling (Bouhlel et al., 2007). On the other hand, alternatively activated macrophages might secrete trophic factors that act in a paracrine or endocrine manner to enhance oxidative metabolism of peripheral tissues (Figure 7). Experimental support for either hypothesis will require further investigations.
While our findings clearly demonstrate a role for alternatively activated ATMs and Kupffer cells in lipid homeostasis, the stimuli driving this program of macrophage activation and the regulation thereof remain unclear. In addition to the Th2 cytokines IL-4 and IL-13, the microenvironment of tissue macrophages might contain other stimuli that support alternative activation (Rauh et al., 2005). In this study, we provide strong evidence that one such signal is likely to be the composition of fatty acids to which the macrophage is exposed. For instance, monounsaturated fatty acids activate PPARδ to enhance maturation of alternatively activated macrophages in tissues, suggesting an increase in cytosolic concentration of free fatty acids might promote alternative macrophage activation and improve insulin action. In support of this notion, deletion or inhibition of cytosolic fatty acid binding proteins improves insulin sensitivity (Furuhashi et al., 2007; Hotamisligil et al., 1996); however, the potential involvement of fatty acid binding proteins in alternative macrophage activation will require further investigation.
Although our findings suggest subtype specificity for PPARs in acquisition of the metabolic and immune phenotypes of alternatively activated macrophages, it remains unknown whether pharmacologic activation of either receptor can rescue the observed defects in single receptor knockout macrophages. For instance, it will be important to determine whether treatment of PPARδ null macrophages with synthetic activators of PPARγ can compensate for absence of PPARδ signaling. Thus, these types of gain-of-function experiments will be carried out using macrophages deficient in PPARγ, PPARδ or both PPARγ/δ.
In summary, we have demonstrated for a critical role for alternatively activated Kupffer cells in metabolic syndrome. In absence of PPARδ, Kupffer cells are unable to maintain the alternative phenotype, leading to suppression of oxidative metabolism and worsening of insulin resistance in peripheral tissues. Since our findings indicate that macrophage activation plays a critical role when caloric intake exceeds expenditure, cellular targeting of Kupffer cells to undergo alternative activation might be an effective strategy for treating obesity and insulin resistance.
EXPERIMENTAL PROCEDURES
In vivo metabolic analyses
PPARδ null mice used in these studies were generated by the Evans lab, and backcrossed onto the 129/SvJ strain for 8 generations (Barak et al., 2002). 129/SvJ mice were used as wild type controls for these experiments. All in vivo studies were initiated at 8–12 weeks of age. For bone marrow transplant studies, whole bone marrow was prepared from wild type and PPARδ null mice, and injected intravenously (5 × 106 cells/recipient) into lethally irradiated (850rad) wild type recipients. After 2–4 weeks rest for bone marrow reconstitution, mice were fed a high fat diet (Bio-Serv F3283) for 18–25 weeks to promote obesity. Dual energy x-ray absorptiometry was performed using a Piximus II (GE Healthcare). Oral glucose tolerance tests (1 g/kg) were done after 14 hour fast. For insulin tolerance tests, mice were challenged with an intraperitoneal injection of human insulin (0.65 U/kg) after a 4hr fast. For biochemical analysis of insulin signaling, human insulin (5 U/kg) was injected into the inferior vena cava after a 4hr fast. Liver and quadriceps were isolated, and snap-frozen in liquid nitrogen at 2 and 5min, respectively. Tissue homogenates were immunoblotted for total Akt and S-473 phospho-Akt (Cell Signaling). Adipocyte cell size analysis and ATM quantification were performed as previously described (Odegaard et al., 2007a). Serum levels of lipids (total cholesterol and triglycerides), cytokines (TNFα, IL-6, and IL-1β), adiopokines (leptin, resistin, and adiponectin), and insulin in fasted mice were quantified using commercially available kits. For liver triglyceride quantification, samples were finely minced and extracted overnight in acetone at 4°C. Extractable lipid content was measured using a commercially available kit (StanBio) (Qu et al., 2006). To activate Kupffer cells with IL-4, mice were injected with IL-4 complexed with anti-IL-4 antibody, a procedure that has been shown to extend the half-life of this cytokine in vivo (Finkelman et al., 1993). Briefly, soluble complexes of IL-4 (2 µg) with anti-IL-4 antibody (BVD4-11, 10 µg) were injected intraperitoneally into recipient mice on day 1 and 4, and livers were harvested on day 5 for qRT-PCR analysis
Quantitative RT-PCR analyses
Gene expression analyses were performed as previously described (Odegaard et al., 2007a; Odegaard et al., 2007b). Briefly, total RNA was extracted from homogenized samples using Trizol reagent (Invitrogen), and used as template for cDNA synthesis (Marligen). Real time quantitative PCR assays were carried in triplicate using DNA Engine Opticon 2. Relative expression level of mRNAs was calculated by the comparative threshold cycle method using L32 as an internal control. All primer sequences were verified by BLASTing against NCBI mouse genome sequence database to ensure specificity.
In vitro macrophage activation analyses
Bone marrow-derived macrophages were cultured from wild-type and PPARδ null 129/SvJ as previously described (Odegaard et al., 2007a). Cells were stimulated with recombinant mIL-4 (10ng/ml) or mIL-13 (10–40 ng/ml) for 48–72hr in low-glucose media (1 g/l). Arginase activity was measured by monitoring colorimetrically the evolution of urea from arginine (Rutschman et al., 2001). To assess cytokine production, cells were pretreated with IL-4 for 24–48hr with subsequent stimulation with LPS (5 ng/ml). Secreted cytokines were quantified by ELISA as per manufacturer’s protocols (BD Pharmingen). Cellular proliferation was assessed by measuring 3H-thymidine incorporation over a 16hr pulse. For the oleic acid experiments, low glucose media was supplemented with fatty acid-free BSA (0.35%, Roche) and filter sterilized. Oleic acid (100µM, Sigma) was added to the media and allowed to equilibrate for 4–5hr at 37°C with periodic mixing. Subsequently, differentiated BMDMs were switched to BSA containing media and simultaneously stimulated with IL-4 for 24–48 hours. All experiments were independently repeated at least three times.
Immunoblotting and flow cytometry
Total cellular proteins were subjected to electrophoresis and immunoblotted for arginase I (BD Biosciences) (Rauh et al., 2005). For flow cytometry, BMDMs were treated with vehicle or IL-4 (10 ng/ml) for 48–72 hours, harvested in PBS and resuspended in PBS containing 2% FCS and 0.2mM EDTA. Macrophages were blocked with 200µg/ml normal mIgG, stained with antibodies directed against dectin-1 or mannose receptor, and analyzed on FACSCalibur.
Transient transfections
Transient transfection experiments were carried out as previously described (Vats et al., 2006). Briefly, plasmid DNAs were introduced into RAW264.7 cells by electroporation (300V, 1000µF), and cells were allowed to recover for 2 hours in Macrophage Serum Free Media (Invitrogen). Luciferase activity was quantified 16 hours after stimulation with IL-4 (10 ng/ml) or GW501516 (100 nM) using the Dualluciferase reporter assay kit (Biotium). All experiments were performed in triplicate and repeated at least three times.
Macrophage-hepatocyte co-culture
Primary hepatocytes were isolated and cultured using Invitrogen liver media and reagents. Briefly, the livers of anesthetized 8–12wk wild type mice were sequentially perfused with liver perfusion and digest media via catheterization of the inferior vena cava. Digested livers were then resected and dissociated in liver wash buffer. Hepatocytes were purified over 50% percoll, and allowed to recover overnight on collagen-coated plates in Hepatozyme serum-free medium. Wild type and PPARδ null BMDMs were seeded onto hepatocyte cultures at a 1:10 ratio, and co-cultured for a further 72hr. Fatty acid oxidation assays were performed using a sodium hydroxide trap in a modified tissue culture flask (Vats et al., 2006). Parallel co-culture plates were assayed for gene expression by qRT-PCR. Similarly, fatty acid oxidation and hepatic gene expression was assessed in primary hepatocytes 72 hours after treatment with wild type or PPARδ null macrophage conditioned media. All experiments were repeated at least three independent times.
Macrophage-adipocyte co-culture
3T3-L1 cells were differentiated as previously described. On day 10, wild type and PPARδ null BMDMs were seeded onto adipocyte cultures to an approximate density of 1:10, and cultured in macrophage medium for 48hr. For glucose uptake, cultures were washed and stimulated with insulin (100nM) or medium alone; glucose uptake was subsequently measured as previously described (Odegaard et al., 2007a).
Isolation of tissue macrophages from transplanted mice
Kupffer cell-enriched and stromal vascular fractions were isolated from livers and adipose tissue of transplanted mice, as previously described (Weisberg et al., 2003). Macrophages present in these fractions were subsequently isolated using F4/80-coupled Dynabeads (Invitrogen), as per manufacturer’s protocol. qRT-PCR analyses were performed for F4/80 and region encompassing exon 4 of PPARδ, which is absent in PPARδ null cells.
PPARδ ligand studies
For in vivo analyses, wild type 129/SvJ mice were given intraperitoneal injections of vehicle or GW0742 (20mg/Kg/day) for 5 days (Graham et al., 2005). Twenty four hours after the first injection, 1 ml of Bio-gel beads, suspended in sterile PBS, was injected into the peritoneal cavity. Four days later, elicited macrophages were isolated by peritoneal lavage and subjected to qRT-PCR analyses. Similarly, the activation state of Kupffer cells and adipose tissue macrophages was determined in mice after treatment with vehicle or GW0742 for 5 days (n=3–4/treatment). For in vitro analyses, bone marrow cells from wild type 129/SvJ mice were treated with GW501516 (100 nM) for 7 days, with media changes every 2 days. To assess whether PPARδ ligands can synergize with IL-4 to promote alternative macrophage activation, BMDMs from 129/SvJ wild type mice were pre-stimulated with low-dose IL-4 (2 ng/ml) for 24 hours followed by treatment with GW501516 for an additional 24 hours.
Statistical analyses
Data are presented as mean ± s.e.m. All P values were calculated using two-tailed distribution, two-sample unequal variance Student’s t-test. Statistical significance is indicated by one asterisk (P < 0.05) or two asterisks (P< 0.01).
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Chawla lab and A. Loh for valuable comments, and Peter Murray for providing key reagents. This work was supported by grants made available to AC: NIH (DK076760 and HL076746), Rockefeller Brothers Fund (Goldman Philanthropic Partnerships), and American Diabetes Association. AC is a Charles E. Culpeper Medical Scholar. Support was provided by Stanford MSTP (JIO and ARE), AHA (JIO), HHMI Gilliam fellowship (ARE), NRSA AI066402 (RRRG). All animal care was in accordance with Stanford University’s A-PLAC committee guidelines.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- Barak Y, Liao D, He W, Ong ES, Nelson MC, Olefsky JM, Boland R, Evans RM. Effects of peroxisome proliferator-activated receptor delta on placentation, adiposity, and colorectal cancer. Proc Natl Acad Sci U S A. 2002;99:303–308. doi: 10.1073/pnas.012610299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhlel MA, Derudas B, Rigamonti E, Dievart R, Brozek J, Haulon S, Zawadzki C, Jude B, Torpier G, Marx N, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–143. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11:183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A, Lee C, Barak Y, He W, Rosenfeld J, Liao D, Han J, Kang H, Evans R. PPAR delta is a very low-density lipoprotein sensor in macrophages. Proc Natl Acad Sci U S A. 2003;100:1268–1273. doi: 10.1073/pnas.0337331100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science. 2001;294:1866–1870. doi: 10.1126/science.294.5548.1866. [DOI] [PubMed] [Google Scholar]
- Evans RM, Barish GD, Wang Y-X. PPARs and the complex journey to obesity. Nat Med. 2004;10:355–361. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- Finkelman FD, Madden KB, Morris SC, Holmes JM, Boiani N, Katona IM, Maliszewski CR. Anti-cytokine antibodies as carrier proteins. Prolongation of in vivo effects of exogenous cytokines by injection of cytokine-anti-cytokine antibody complexes. J Immunol. 1993;151:1235–1244. [PubMed] [Google Scholar]
- Furuhashi M, Tuncman G, Gorgun CZ, Makowski L, Atsumi G, Vaillancourt E, Kono K, Babaev VR, Fazio S, Linton MF, et al. Treatment of diabetes and therosclerosis by inhibiting fatty-acid-binding protein aP2. Nature. 2007;447:959–965. doi: 10.1038/nature05844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Graham TL, Mookherjee C, Suckling KE, A Palmer CN, Patel L. The PPAR[delta] agonist GW0742X reduces atherosclerosis in LDLR−/− mice. Atherosclerosis. 2005;181:29–37. doi: 10.1016/j.atherosclerosis.2004.12.028. [DOI] [PubMed] [Google Scholar]
- Herbert DR, Holscher C, Mohrs M, Arendse B, Schwegmann A, Radwanska M, Leeto M, Kirsch R, Hall P, Mossmann H, et al. Alternative macrophage activation is essential for survival during schistosomiasis and downmodulates T helper 1 responses and immunopathology. Immunity. 2004;20:623–635. doi: 10.1016/s1074-7613(04)00107-4. [DOI] [PubMed] [Google Scholar]
- Hevener AL, Olefsky JM, Reichart D, Nguyen MT, Bandyopadyhay G, Leung HY, Watt MJ, Benner C, Febbraio MA, Nguyen AK, et al. Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J Clin Invest. 2007;117:1658–1669. doi: 10.1172/JCI31561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Johnson RS, Distel RJ, Ellis R, Papaioannou VE, Spiegelman BM. Uncoupling of obesity from insulin resistance through a targeted mutation in aP2, the adipocyte fatty acid binding protein. Science. 1996;274:1377–1379. doi: 10.1126/science.274.5291.1377. [DOI] [PubMed] [Google Scholar]
- Kamei N, Tobe K, Suzuki R, Ohsugi M, Watanabe T, Kubota N, Ohtsuka-Kowatari N, Kumagai K, Sakamoto K, Kobayashi M, et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J Biol Chem. 2006;281:26602–26614. doi: 10.1074/jbc.M601284200. [DOI] [PubMed] [Google Scholar]
- Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K, Kasuga M. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy DW, Abkowitz JL. Kinetics of central nervous system microglial and macrophage engraftment: analysis using a transgenic bone marrow transplantation model. Blood. 1997;90:986–993. [PubMed] [Google Scholar]
- Kim F, Pham M, Luttrell I, Bannerman DD, Tupper J, Thaler J, Hawn TR, Raines EW, Schwartz MW. Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ Res. 2007;100:1589–1596. doi: 10.1161/CIRCRESAHA.106.142851. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Xu HE, Lambert MH, Willson TM. Peroxisome proliferator-activated receptors: from genes to physiology. Recent Prog Horm Res. 2001;56:239–263. doi: 10.1210/rp.56.1.239. [DOI] [PubMed] [Google Scholar]
- Li Z, Diehl AM. Innate immunity in the liver. Curr Opin Gastroenterol. 2003;19:565–571. doi: 10.1097/00001574-200311000-00009. [DOI] [PubMed] [Google Scholar]
- Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U S A. 2003;100:5336–5341. doi: 10.1073/pnas.0931259100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Munder M, Eichmann K, Moran JM, Centeno F, Soler G, Modolell M. Th1/Th2-regulated expression of arginase isoforms in murine macrophages and dendritic cells. J Immunol. 1999;163:3771–3777. [PubMed] [Google Scholar]
- Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Eagle AR, Vats D, Brombacher F, Ferrante AW, Chawla A. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007a;447:1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard JI, Vats D, Zhang L, Ricardo-Gonzalez R, Smith KL, Sykes DB, Kamps MP, Chawla A. Quantitative expansion of ES cell-derived myeloid progenitors capable of differentiating into macrophages. J Leukoc Biol. 2007b;81:711–719. doi: 10.1189/jlb.0906590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver WR, Jr, Shenk JL, Snaith MR, Russell CS, Plunket KD, Bodkin NL, Lewis MC, Winegar DA, Sznaidman ML, Lambert MH, et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc Natl Acad Sci U S A. 2001;98:5306–5311. doi: 10.1073/pnas.091021198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauleau AL, Rutschman R, Lang R, Pernis A, Watowich SS, Murray PJ. Enhancer-mediated control of macrophage-specific arginase I expression. J Immunol. 2004;172:7565–7573. doi: 10.4049/jimmunol.172.12.7565. [DOI] [PubMed] [Google Scholar]
- Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qatanani M, Lazar MA. Mechanisms of obesity-associated insulin resistance: many choices on the menu. Genes Dev. 2007;21:1443–1455. doi: 10.1101/gad.1550907. [DOI] [PubMed] [Google Scholar]
- Qu S, Altomonte J, Perdomo G, He J, Fan Y, Kamagate A, Meseck M, Dong HH. Aberrant Forkhead box O1 function is associated with impaired hepatic metabolism. Endocrinology. 2006;147:5641–5652. doi: 10.1210/en.2006-0541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raes G, De Baetselier P, Noel W, Beschin A, Brombacher F, Hassanzadeh Gh G. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. J Leukoc Biol. 2002;71:597–602. [PubMed] [Google Scholar]
- Rauh MJ, Ho V, Pereira C, Sham A, Sly LM, Lam V, Huxham L, Minchinton AI, Mui A, Krystal G. SHIP Represses the Generation of Alternatively Activated Macrophages. Immunity. 2005;23:361–374. doi: 10.1016/j.immuni.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Rutschman R, Lang R, Hesse M, Ihle JN, Wynn TA, Murray PJ. Cutting edge: Stat6-dependent substrate depletion regulates nitric oxide production. J Immunol. 2001;166:2173–2177. doi: 10.4049/jimmunol.166.4.2173. [DOI] [PubMed] [Google Scholar]
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Greaves DR, Murray PJ, Chawla A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006;4:13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vianna CR, Huntgeburth M, Coppari R, Choi CS, Lin J, Krauss S, Barbatelli G, Tzameli I, Kim YB, Cinti S, et al. Hypomorphic mutation of PGC-1beta causes mitochondrial dysfunction and liver insulin resistance. Cell Metab. 2006;4:453–464. doi: 10.1016/j.cmet.2006.11.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, Charo I, Leibel RL, Ferrante AW., Jr CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. 2006;116:115–124. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HE, Lambert MH, Montana VG, Parks DJ, Blanchard SG, Brown PJ, Sternbach DD, Lehmann JM, Wisely GB, Willson TM, et al. Molecular recognition of fatty acids by peroxisome proliferator- activated receptors. Mol Cell. 1999;3:397–403. doi: 10.1016/s1097-2765(00)80467-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.