

Abstract
In an effort to overcome previous problems with the preparation of Co(II)-substituted metallo-β-lactamase L1, two strategies were undertaken. Attempts to prepare Co(II)-substituted L1 using biological incorporation resulted in an enzyme that contained only one equivalent of cobalt and exhibited no catalytic activity. Co(II)-substituted L1 could be prepared by refolding metal-free L1 in the presence of Co(II), and the resulting enzyme contained 1.8 equivalents of cobalt, yielded a UV-Vis spectrum consistent with five-coordinate Co(II), and exhibited a kcat of 63 s−1 and Km of 20 µM when using nitrocefin as the substrate. Pre-steady state fluorescence and UV-Vis studies demonstrated that refolded, Co(II)-substituted L1 utilizes the same kinetic mechanism as Zn(II)-containing L1 in which a reaction intermediate is formed when using nitrocefin as substrate. The described refolding strategy can be used to prepare other Co(II)-substituted, Zn(II)-metalloenzymes, particularly those that contain a solvent-exposable disulfide, which often causes oxidation of Co(II) to Co(III).
Keywords: Co(II)-substitution, Zn(II)-metalloenzyme, metallo-β-lactamase, refolding
Introductory Statement
Bacterial resistance to β-lactam-containing antibiotics is most often accomplished by the production of β-lactamases, which cleave the C-N bond of the β-lactam ring and render these antibiotics ineffective as antimicrobial agents [1–4]. There are nearly 500 β-lactamases that have been identified, and these enzymes have been categorized into 4 distinct groups [5, 6]. Group A, C, and D β-lactamases utilize an active site serine for nucleophilic attack on the β-lactam carbonyl [7, 8]. The group B enzymes require the presence of Zn(II) and are called metallo-β-lactamases (mbl’s). There are currently over 30 mbl’s, and these enzymes have been categorized into 3 distinct subgroups based on sequence homology and metal content of the fully-active enzyme [1, 5, 6]. Subgroup B1 enzymes require 2 Zn(II) ions for full catalytic activity and share two conserved Zn(II) sites: Zn1 is coordinated by 3 His and a bridging solvent molecule and Zn2 is coordinated by one histidine, one aspartate, one cysteine, the bridging solvent molecule, and a terminally-bound solvent molecule. The B1 enzymes are represented by β-lactamase II from B. cereus [9], IMP-1 from Pseudomonas aeruginosa [10], Bla2 from B. anthracis [11], and CcrA from Bacteroides fragilis [12]. Subgroup B2 enzymes require 1 Zn(II), coordinated by one aspartate, one histidine, one cysteine, and one solvent molecule, for full activity, prefer carbapenems as substrates, share 11% amino acid sequence identity with the subgroup B1 enzymes, and are represented by CphA from A. hydrophila and ImiS from A. sobria [13, 14]. Subgroup B3 enzymes require 2 Zn(II) ions for full activity, contain a Zn1 site similar to that observed in the B1 enzymes, contain a Zn2 site with 2 histidines, 1 aspartate, the bridging solvent molecule, and a terminally-bound solvent molecule, prefer penicillins as substrates, share only 9 conserved residues with the B1 enzymes, and are represented by L1 and FEZ (Figure 1) [15, 16]. The first metallo-β-lactamase was discovered in 1966 [17], and this enzyme was considered to be an oddity. However as the use of β-lactam-containing antibiotics increased, more strains of emerging pathogens that harbor a mbl appeared in the clinic. For example, mbl’s have been found in strains such as B. anthracis, P. aeruginosa, and Acinetobacter spp. [4, 11]. Importantly, there are no known clinical inhibitors of any mbl.
Figure 1.
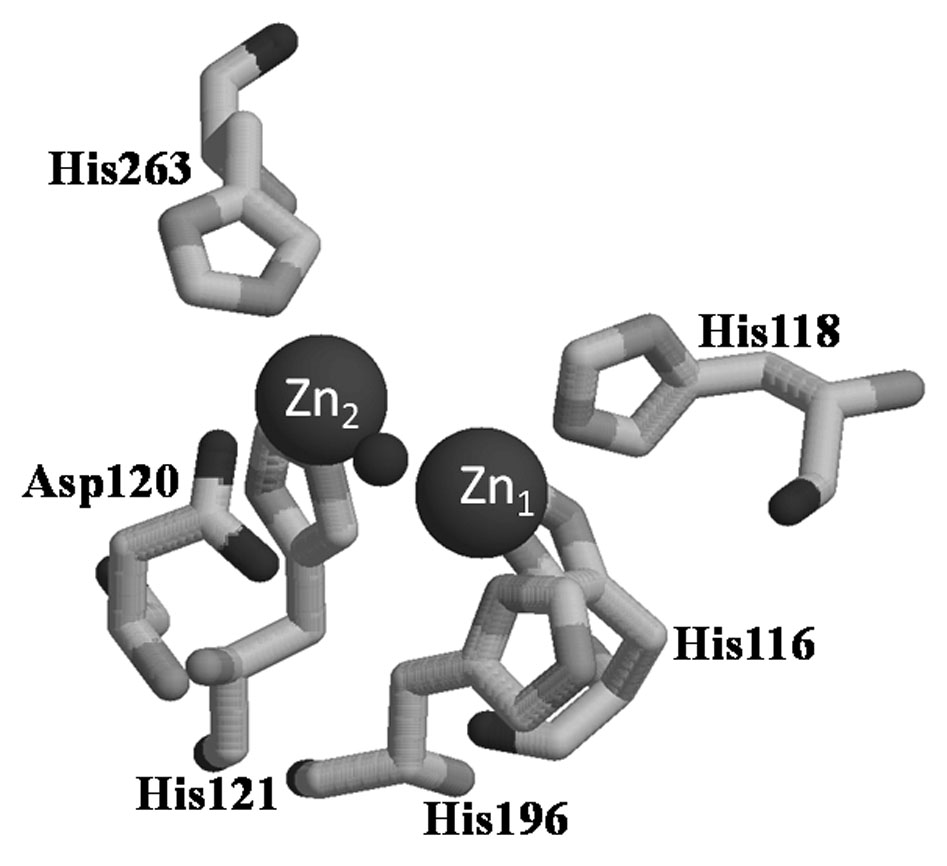
Active site of L1. Zn1 site has His116, His118, His196, and a bridging hydroxide as ligands. Zn2 site has Asp120, His121, His263, bridging hydroxide, and a terminally-bound water (not shown) as ligands.
In an effort to discover novel inhibitors of the mbl’s, we have been characterizing an enzyme from each of the distinct mbl subgroups in hopes of uncovering a common structural or mechanistic aspect towards which an inhibitor can be designed. Unfortunately due its electron configuration of [Ar]3d10, Zn(II) is silent to all common spectroscopic techniques except EXAFS spectroscopy. However in most Zn(II)-metalloenzymes, the Zn(II) can be replaced by high-spin Co(II) (electron configuration of [Ar]3d7 and a S = 3/2) to yield a structurally-similar and catalytically-active analog [18–23]. The two most common ways to prepare Co(II)-substituted enzymes is (1) direct addition of Co(II) to metal-free, Zn(II)-metalloenzyme, which was prepared by the use of chelators and exhaustive dialysis steps, and (2) biological incorporation of Co(II) into a recombinant enzyme during over-expression [18].
Our previous studies on mbl L1 demonstrated that a solvent-exposable disulfide can complicate the preparation of active, Co(II)-substituted enzyme [24]. Nonetheless, we were able to prepare L1 containing Co(II) if the disulfide was reduced with TCEP (tris(2-carboxyethyl)phosphine) before addition of Co(II). The resulting enzyme was active and exhibited UV-Vis, 1H NMR, and EPR spectra consistent with the presence of Co(II). However, the protein bound 2.5 equivalents cobalt even after extensive dialyses. The 0.5 equivalents of excess Co(II) was predicted to bind the reduced cysteines, and although the cysteine-containing site is remote from the active site, the presence of extra Co(II) complicated the interpretation of subsequent spectroscopic studies.
In an effort to overcome the problem with preparing Co(II)-substituted L1 using the direct addition method, we sought to test other methods. First, we attempted to prepare Co(II)-containing L1 using a biological incorporation strategy. Unfortunately, the resulting enzyme was isolated containing only 1 equivalent of cobalt and was not catalytically-active. Therefore, we attempted to prepare Co(II)-substituted L1 by unfolding apo-L1 and then refolding the enzyme in the presence of Co(II). By using this technique, we were able to prepare catalytically-active, Co(II)-substituted L1.
Material and Methods
Over-expression and purification of L1
A 50 mL overnight preculture of BL21(DE3)pLysS E. coli cells containing the pET26b-based plasmid that encodes for L1 was used to innoculate 4 × 1L flasks of Luria-Bertani (LB) medium, and L1 was over-expressed and purified as described previously [25]. In an effort to prepare Co(II)-substituted L1 using a biological incorporation method, the innoculum was grown in minimal medium at 37 °C with shaking until the culture reached an optical density at 600 nm of 0.6–0.8. The culture was then cooled to 15 °C for 30 minutes, was made 0.5 mM in IPTG and 100 µM in CoCl2, and shaken overnight at 15 °C for roughly 16 hours. The culture was centrifuged for 15 minutes (8200 × g), and the resulting cell pellet was resuspended in 30–40 mL of 30 mM Tris, pH 8.5 (buffer A). The cells were lysed by passing the resuspended cells through a French Press three times at a pressure of 1000–1500 psi. After removal of insoluble components by centrifugation (25 minutes at 23,400 × g), the supernatant was dialyzed versus 2L buffer A overnight. The dialyzed protein solution was centrifuged (25 min at 23,400 × g) to remove any precipitated proteins and subjected to FPLC as previously reported [25]. The FPLC fractions were analyzed by SDS-PAGE gels, and the fractions containing > 90% pure protein were pooled and concentrated by using an Amicon equipped with a YM-10 membrane.
Preparation of metal-free (apo) L1
A concentrated solution (< 10 mL) of L1 (~ 0.3 mM) was dialyzed against 4 × 1 L of 50 mM Hepes, pH 7.0, containing 10 mM 1,10-phenanthroline and then dialyzed against 6 × 1 L of 50 mM Hepes, pH 7.0. The metal content of the resulting sample was ascertained by ICP-AES, as previously reported [25]. The sample was stored at −80 °C.
Preparation of Co(II)-substituted L1 with TCEP
Apo-L1 was incubated with 1 mM tris-(carboxyethyl)phosphine (TCEP) on ice for 30 min. This enzyme was titrated with Co(II), and the UV-Vis spectrum of each sample was obtained using an Agilent 8453 UV-Vis spectrophotometer.
In vitro unfolding and refolding of L1
Apo-L1 (2 ml, 100 µM) was diluted with 18 ml of 6 M guanidinium chloride (Gdn-HCl). The sample was incubated on ice for 1 hour and then dialyzed versus 1 L of 50 mM Hepes, pH 7.0, containing no added metal or 50 µM Zn(II) or Co(II). Refolded L1 was further dialyzed versus 5 × 1L of Chelex-treated, 50 mM HEPES, pH 7.0, to remove any unbound metals. The resulting solution was centrifuged (25 min at 23,400 × g) to remove any precipitated protein.
Metal analyses
The metal content of protein samples was determined by using a Perkin-Elmer Inductively Coupled Plasma Spectrometer with Atomic Emission detection (ICP-AES), as previously described [25].
Steady-st1ate kinetics
Steady-state kinetic studies were performed on an Agilent 8453A UV-Vis diode array spectrophotometer at 25 °C using nitrocefin as the substrate and 50 mM cacodylate, pH 7.0, as the buffer.
Stopped-flow kinetic studies
Stopped-flow kinetic experiments were performed on an Applied Photophysics SX18MV apparatus equipped with a constant temperature circulating water bath. The pathlength of the observation cell was 0.2 cm for the fluorescence measurements, and both excitation and emission slits were maintained at 4 mm. Fluorescence data were collected using an excitation wavelength of 295 nm and a WG320 nm cut-on filter on the emission photomultiplier. The photomultiplier input was adjusted for each protein concentration to maintain a total signal change of 1V between protein in the absence of substrate and dark current readings. Data were recorded as photomultiplier output in volts. For absorbance experiments, data were recorded as absorbance units, with photomultiplier input first adjusted to give a reading of 0 for buffer only at each wavelength used. All experiments were performed in Chelexed-treated, 50 mM cacodylate, pH 7.0, at 10 °C using nitrocefin as substrate.
Fluorescence spectra
A Perkin-Elmer LS55 Luminescence spectrometer, tuned to an excitation wavelength of 295 nm and emission wavelength of 340 nm with a slit width of 5 nm, was used to monitor fluorescence emission intensities of the proteins. A 4 mm quartz cuvette was used, and the protein concentrations were 1 µM. Chelex-treated 50 mM HEPES, pH 7.0, was used as a buffer blank.
Results
Adding Co(II) to TCEP-treated apo-L1
We previously reported that adding Co(II) to apo-L1 resulted in a rapid oxidation of Co(II) to Co(III) [24]. We reasoned that oxidation was due to the presence of a solvent-accessible disulfide in L1. The addition of TCEP (tris(2-carboxyethyl)phosphine) before adding Co(II) resulted in a pink colored protein that was stable for several weeks. In an effort to further probe Co(II) binding, the UV-Vis difference (spectrum of Co-L1 minus spectrum of apo-L1) spectra of TCEP-treated, apo-L1 upon titration with CoCl2 were obtained [24] (Figure 2 top). Two distinct sets of absorption peaks were observed: (1) a broad band that is positioned at 325 nm when 1 equivalent of Co(II) is present but shifts to 360 nm when higher concentrations of Co(II) are present and (2) a broad peak between 450 and 700 nm comprised of at least 4 distinct features. The former peak that shifts from 325 to 360 nm exhibits an extinction coefficient of ε340 = 86 M−1cm−1 (per Co(II)), and the position of this peak is consistent with it being due to a cysteine S → Co(II) ligand to metal charge transfer (LMCT) transition, which has been observed in electronic spectra of other Co(II)-substituted metallo-β-lactamases [26–29]. However, the peak at 340 nm in the spectrum of Co(II)-substituted L1 is much broader than those in other metallo-β-lactamases [26–29]. It is possible that there may be additional peaks (for example at 400 nm) under the putative LMCT. The intensities of S → Co(II) LMCT’s in the other metallo-β-lactamases are typically equal to ε320nm = 1,000 M−1 cm−1, suggesting that cysteine-containing Co(II) binding site in L1 is minimally-occupied. The only two cysteines in L1 are Cys218 and Cys246 that form a disulfide bond [16]. The addition of TCEP presumably reduces this disulfide bond and allows for Co(II) binding at substoichiometric levels. As a control, the UV-Vis spectra of 1 mM TCEP and 1 mM TCEP + 1 mM Co(II) were obtained, and TCEP exhibits an absorbance at 310 nm (ε = 430 M−1cm−1). There are no additional peaks in the spectrum of TCEP + Co(II) (data not shown). The absorbance due to TCEP was subtracted in the difference spectra shown in Figure 2 (top). As another control, a sample of 1 mM 2Zn-L1 (sample of L1 containing 2 equivalents of Zn(II)) was made 1 mM in TCEP, and 1 equivalent of Co(II) was added to this sample. A peak at 320 nm appeared that had an ε = 460 M−1cm−1, which we have assigned to a cysteinyl S to Co(II) LMCT (data not shown).
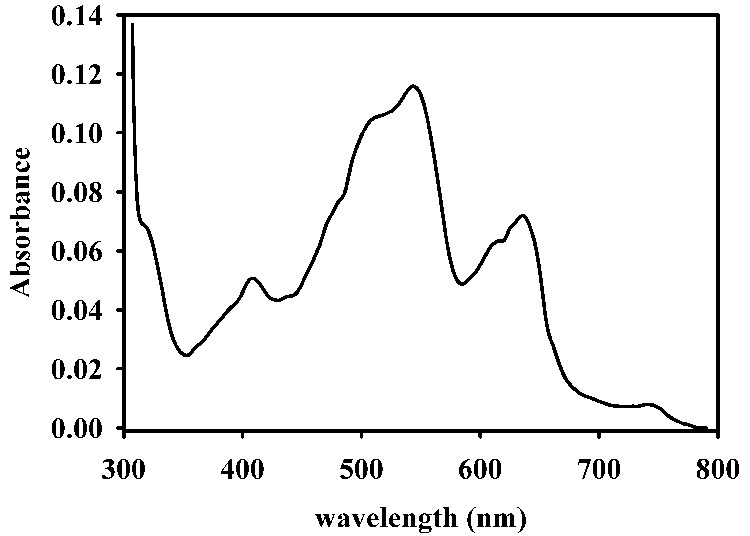
Figure 2.
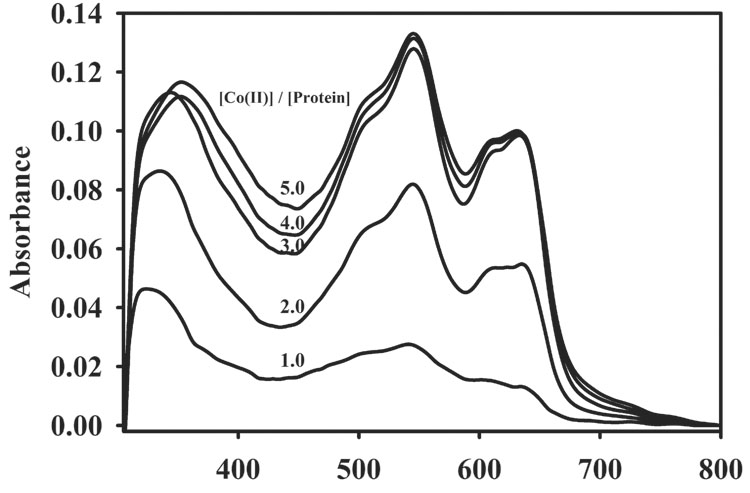
UV-Vis difference spectra (spectrum of Co-L1 minus spectrum of apo-L1) of cobalt-containing analogs of L1: (top) cobalt-containing L1 prepared by biological incorporation and (bottom) refolded, cobalt-containing L1. The enzyme concentration in all samples was 550 µM, and the buffer was 50 mM cacocylate, pH 7.0.
The other features from 450 and 700 nm are ligand field transitions of high-spin Co(II) [22, 30]. The absorbance of these transitions increased until 3 equivalents of Co(II) were added to the enzyme, suggesting that the metal binding sites in TCEP-treated L1 are saturated only after 3 equivalents of Co(II) are added. Since the extinction coefficient of Co(II) ligand field transitions is dependent on the coordination number of Co(II) [22, 30], we determined the extinction coefficients of these transitions during the Co(II) titration. The extinction coefficient of the peak at 545 nm increased from 26, 75, 120 M−1cm−1 as the Co(II)/protein stoichiometry increased from 1.0, 2.0, to 3.0 equivalents. Since our previous EXAFS studies demonstrate sequential binding of Zn(II) [31], this result suggests that the first equivalent of Co(II) is 6-coordinate and the second and third equivalents are 5-coordinate [22, 30]. However, we cannot rule out the possibility that Co(II) binds differently than Zn(II) to L1, and recent work on BcII suggests a very complicated pathway for Co(II) binding to this enzyme [32]. If Co(II) does not bind sequentially to L1, the coordination numbers of the first and second equivalents of Co(II) cannot be determined.
The catalytic properties of TCEP-reduced L1 containing 1, 2, and 3 equivalents of Co(II) were ascertained (Table 1) in assays using nitrocefin as the substrate. TCEP-reduced L1 containing 1 equivalent of Co(II) (1 CoL1) exhibited a kcat of 14 s−1 and a Km of 10 ± 3 µM. A second Co(II) resulted in an enzyme (2 CoL1) with a similar Km value (8 ± 1µM) but a kcat that is roughly twice that of enzyme containing 1 equivalent of Co(II) (kcat = 27 ± 1 s−1). The third equivalent of Co(II) resulted in an enzyme (3 CoL1) that exhibits a Km of 71 ± 9 s−1; however, this enzyme also had a larger Km value of 30 ± 7µM.
Table 1.
Characterization of Co(II)-substituted L1 analogs.
| Species | Cobalt content (eq) | kcat (s−1) | Km (µM) |
|---|---|---|---|
| Biologicallyincorporated L1 | 1.0 ± 0.1 | 2.8 ± 0.1 | 13 ± 2 |
| 1 CoL1 | 1 | 14 ± 1 | 10 ± 3 |
| 2 CoL1 | 2 | 27± 1 | 8 ± 1 |
| 3 CoL1 | 3 | 71± 9 | 30± 7 |
| L1 refolded w Co(II) | 1.8 ± 0.1 | 63 ± 3 | 20 ± 1 |
Steady state kinetic studies were conducted in Chelex-treated 50 mM cacodylate buffer, pH 7.0, using nitrocefin as the substrate.
Preparation of Co(II)-substituted L1 by biological incorporation
Metallo-β-lactamase L1 was over-expressed in minimal medium (pH 6.8) containing 100 µM CoCl2 according to previously published procedures [25]. After FPLC, the Q-Sepharose fractions containing L1, as identified by SDS-PAGE gels, were not pink in color, as expected for Co(II)-substituted L1 [24, 33]. Metal analyses revealed that the purified enzyme bound only 1.0 ± 0.1 equivalent of cobalt (Table 1) and showed that the sample contained less than 0.1 equivalents of Zn(II), Fe, or Mn. Steady-state kinetics revealed that the enzyme exhibited a kcat of 2.8 ± 0.1 s−1 and a Km of 13 ± 2 µM (Table 1) when using nitrocefin as the substrate. Steady-state kinetics were repeated using buffers containing 100 µM CoCl2 in an attempt to further load the enzyme with its full complement of metal. However, steady-state kinetic constants were not greatly changed. The UV-Vis difference (spectrum of Co-L1 minus the spectrum of apo-L1) spectrum of the isolated enzyme showed a broad, small feature at 340 nm and no resolved peaks between 500 – 600 nm, which are due to Co(II) ligand field transitions. These characteristics suggest that most of the cobalt in the sample is Co(III) [24]. In an effort to increase the cobalt content in L1 via biological incorporation, L1 was over-expressed in minimal medium containing 500 µM or 1 mM CoCl2. However, L1 over-expressed in the presence of 500 µM CoCl2 contained only 1 equivalent of cobalt, and E. coli cells cultured in medium containing 1 mM CoCl2 lysed.
In vitro refolding of L1 in the absence of metal and in the presence of Zn(II) or Co(II)
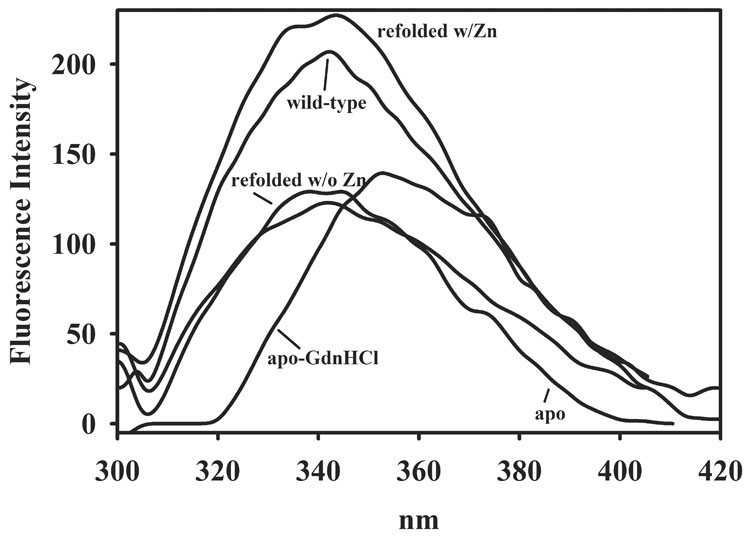
Since the methods of biological incorporation and direct addition of Co(II) did not result in the preparation of Co(II)-substituted L1 that could be used for future spectroscopic/mechanistic studies, we attempted to prepare Co(II)-substituted L1 by refolding L1 in the presence of Co(II). Our initial efforts to unfold and refold L1 involved using L1 that contained 2 equivalents of Zn(II) (2Zn-L1); however, our data suggested that 2Zn-L1 did not unfold completely even in the presence of 6 M guanidinum hydrochloride (data not shown), and the resulting refolded enzyme contained significant amounts of Zn(II). Therefore, we utilized metal-free (apo) L1 in the unfolding/refolding experiments. As previously reported [34], the fluorescence emission spectrum of apo-L1 is less intense than that of wild-type L1 (Figure 3). The addition of 6 M guandinium hydrochloride resulted in a red shift of the fluorescence emission spectrum from 335 nm to 345 nm, suggesting that apo-L1 is unfolded [35]. A similar shift occurred when RNase T1 was denatured in guanidinium hydrochloride, and the shift was attributed to a buried trytophan residue that was fully exposed to water upon unfolding [35, 36].
Figure 3.
Fluorescence emission spectra of L1 samples. The concentration of L1 in the samples was 1 µM, and the buffer was 50 mM Hepes, pH 7.0. An excitation wavelength of 295 nm was used.
The resulting unfolded L1 was refolded by dialyzing the denaturant out of the sample in the (a) absence of metal ions, (b) in the presence of 100 µM Zn(II), or (c) in the presence of 100 µM Co(II). The fluorescence emission spectrum of L1 refolded in all conditions returned back to the position of apo- or wild-type L1 before Gdn-HCl was added; however, the intensities of L1 refolded in the absence of metal or in the presence of Co(II) were lower than that of L1 refolded in the presence of Zn(II) (data not shown). L1 refolded in the absence of metal was shown to bind < 0.03 equivalents of Zn(II) and exhibit a kcat of < 0.1 s−1. This protein did apparently fold correctly since the addition of 2 equivalents of Zn(II) to L1 refolded in the absence of metal resulted in a protein that exhibited a kcat of 31 ± 1 s−1 and a Km of 3.6 ± 0.5 µM, when using nitrocefin as the substrate (Table 2).
Table 2.
Metal content and steady-state kinetic constants of Zn(II)-containing L1 analogs.
| Species | Zn(II) content (eq) | kcat(s−1) | Km (µM) |
|---|---|---|---|
| Wild-type L1a | 1.9 ± 0.1 | 27 ± 1 | 3.8 ± 0.5 |
| Wild-type L1 | 1.9 ± 0.1 | 41 ± 1b | 4 ± 1b |
| L1 refolded w/ Zn(II) | 2.0 ± 0.1 | 41 ± 1 | 4.8 ± 0.3 |
| L1 refolded w/o Zn(II) | < 0.03 | < 0.1 | ND |
| L1 refolded w/o Zn(II)c | 2 | 31±1 | 3.6 ± 0.5 |
Steady state kinetic studies were carried out in Chelex-treated 50 mM cacodylate, pH 7.0, using nitrocefin as the substrate.
L1 over-expressed in LB medium according to previously published protocol [25].
Kinetic reactions conducted in Chelex-treated 50 mM cacodylate, pH 7.0, containing 100 µM Zn(II).
Two equivalents of Zn(II) were added to this sample before the steady state kinetic studies were conducted.
On the other hand, L1 refolded in the presence of 100 µM Zn(II) (and after extensive dialysis versus Chelex-treated buffer) was found to bind 2.0 ± 0.1 equivalents of Zn(II) and exhibit steady-state kinetic constants of kcat = 41 ± 1 s−1 and Km of 4.8 ± 1.0 μM. This kcat value is almost 50% larger than that of wild-type (as-isolated) L1 and similar to that of wild-type L1 that is assayed in the presence of 100 µM Zn(II) ([25] and Table 2). This protein exhibited a fluorescence emission spectrum similar to that of as-isolated L1 (Figure 3). Unfolded L1 was also refolded in the presence of 100 µM Co(II). After dialysis to remove Gdn-HCl and excess Co(II), the resulting refolded protein was purple and bound 1.8 ± 0.1 equivalents of cobalt and <0.1 equivalent of Zn(II). The refolded, Co(II)-substituted L1 exhibited a kcat of 63 ± 3 s−1 and a Km of 20 ± 1 µM when using nitrocefin as the substrate. These steady-state kinetic constants are similar to those of TCEP-reduced L1 containing 3 equivalents of Co(II) (Table 1 and Table 2). The UV-Vis spectrum of refolded, Co(II)-substituted L1 showed several peaks, and there is a small, relatively sharp shoulder at 310 nm (Figure 2 (bottom)). The small peak at 400 nm (ε < 40 M−1cm−1) is attributed to small amounts of Co(III) in the sample. The remaining peaks are attributed to d-d bands of high-spin Co(II), and the extinction coefficient for the peak at 550 nm is 180 M−1cm−1, which is consistent with 5-coordinate Co(II) [22, 30].
To ascertain whether the refolded, Co(II)-substituted L1 exhibits similar kinetic properties as 2Zn-L1, we conducted stopped-flow fluorescence and UV-Vis studies using nitrocefin as the substrate. The stopped-flow fluorescence time course of 50 µM refolded 2Co-L1 with 50 µM nitrocefin showed a rapid decrease in fluorescence over the first 20 milliseconds of the reaction followed by a rapid return of fluorescence over the subsequent 100–150 milliseconds. The shape of the stopped-flow fluorescence trace and rates of fluorescence change are very similar to those previously reported for 2Zn-L1 (data not shown) [37]. The reaction of 50 µM refolded 2Co-L1 with 50 µM nitrocefin was also monitored with stopped-flow UV-Vis studies. The absorbance at 390 nm, which corresponds to substrate [38, 39], decreased over the first 40 milliseconds (Figure 4B). The increase in absorbance at 50 ms has been observed previously by Spencer et al. and was attributed to the greater overlap of the absorbance of substrate with product as compared to that with intermediate [37]. The relatively larger dip in the absorbance of the 390 nm peak in the stopped-flow data of 2Co-L1 (Figure 4B), as compared to that of 2Zn-L1 (Figure 4C), suggests that the overlap in absorbance of substrate and intermediate is greater in the case of 2Co-L1, and previous stopped-flow studies on Co-L1 demonstrated this scenario [33]. The absorbance at 485 nm, which corresponds to the concentration of product [38, 39], rapidly increased over 100 milliseconds for both 2Co-L1 and 2Zn-L1 (Figure 4B and 4C). The absorbance at 665 nm, which is attributable to reaction intermediate [38, 39], rapidly increased during the first 20 milliseconds of the reaction and decreased over the next 100 milliseconds for both enzymes. It appears that the decay of the intermediate in the reaction with 2Co-L1 (Figure 4B) is slightly slower than that in the reaction with 2Zn-L1 (Figure 4C). In addition, more intermediate (1.6-fold) is observed in the traces with 2Zn-L1 (absorbance of 1.8) as compared to 2Co-L1 (absorbance of 1.1). Nonetheless, the 2Co-L1 analog exhibits very similar kinetic properties as 2Zn-L1.
Figure 4.
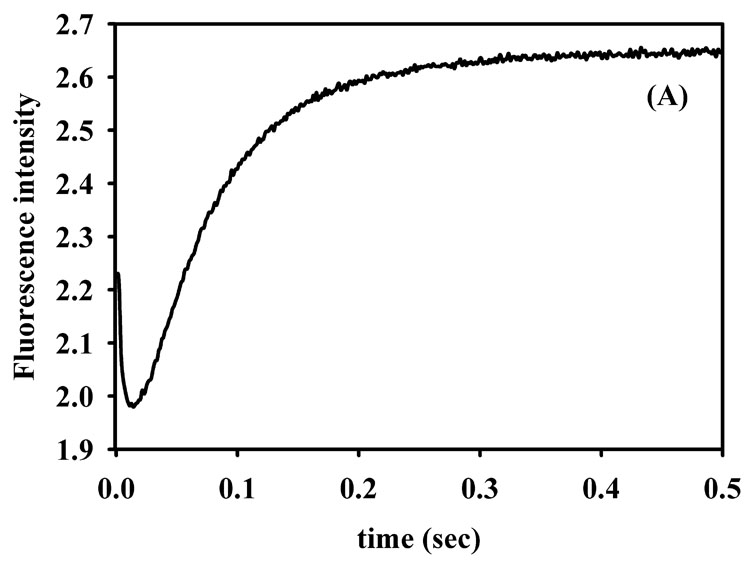
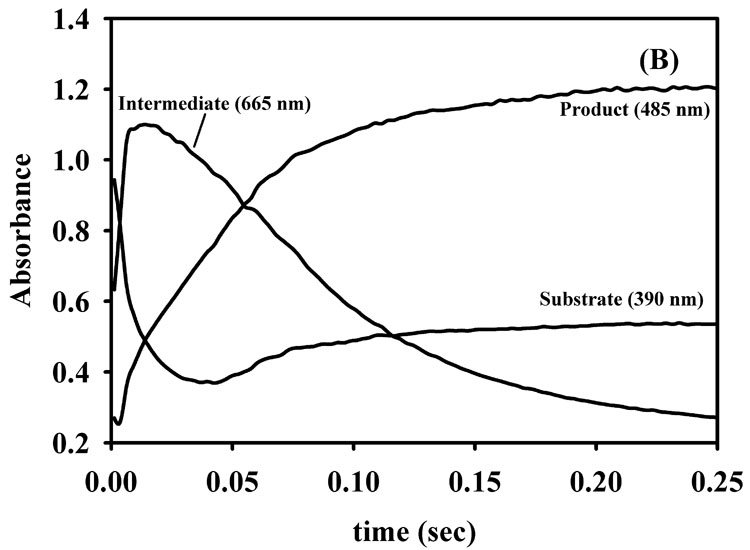
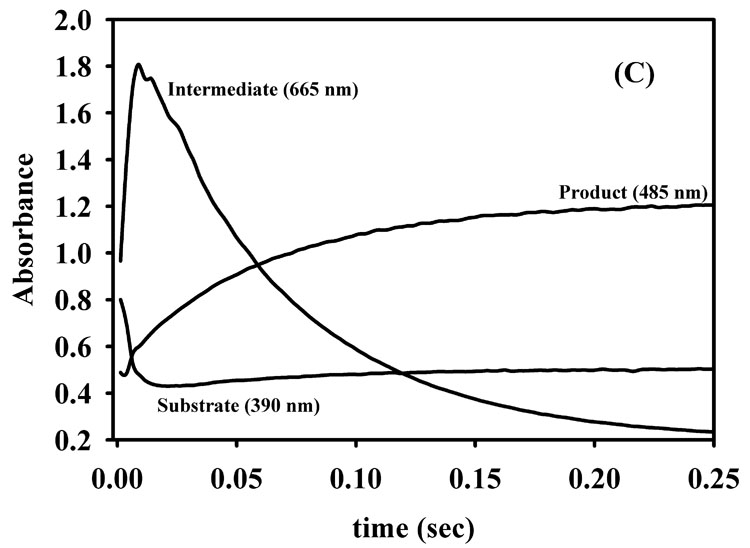
Stopped-flow fluorescence (A) progress curve for reaction of refolded 2Co-L1 and nitrocefin. UV-Vis stopped-flow progress curves on (B) refolded 2Co-L1 and (C) 2Zn-L1 reacted with nitrocefin. The reactions were carried out in Chelex-treated, 50 mM cacodylate, pH 7.0, at 10 °C, and the enzyme and substrate concentrations were 50 µM.
Discussion
Zn(II) has been predicted to be a required cofactor in one third of all proteins, and Zn(II) is a cofactor in enzymes from all six major classes of enzymes [40, 41]. Zn(II) metalloproteins play vital biological roles in all organisms and are targets for a number of drugs and drug candidates [42]. However, the physicochemical properties (diamagnetism, color) of Zn(II) limit the number of physical techniques that can be used to structurally characterize the metal binding sites in Zn(II) metalloproteins, and only EXAFS spectroscopy and X-ray crystallography are commonly used to directly probe these metal binding sites. Therefore, Zn(II) is often replaced with other metal ions to afford spectroscopically-active analogs, and Co(II) is the most commonly used metal ion for this purpose. Co(II) has a similar ionic radius to Zn(II) and can accommodate the binding geometry and ligation of Zn(II) in biological molecules [18, 43]. Moreover, almost all Co(II)-substituted enzymes are catalytically-active and allow for the use of UV-Vis, EPR, and paramagnetic 1H NMR spectroscopies to be used to characterize the metal binding sites. The coupling of rapid-freeze quench techniques with EPR spectroscopy allows for detailed structural characterization of reaction intermediates [33, 44, 45].
In an effort to probe the metal binding sites to offer information that could lead to inhibitors, we and others have prepared Co(II)-substituted mbl’s and performed spectroscopic studies on the resulting enzymes [26, 29, 33, 44, 45]. However, the preparation of Co(II)-substituted mbl L1 from Stenotrophomonas maltophilia (subgroup 3C) has been hampered by the presence of a solvent-accessible disulfide in the enzyme [24]. The direct addition of Co(II) to metal-free L1 resulted in oxidation of Co(II) to Co(III) and an inactive L1 analog [24]. To circumvent this problem, we reported a strategy in which the disulfide bond in metal-free L1 is reduced with TCEP before addition of Co(II). A catalytically-active analog of L1 was obtained; however, the resulting enzyme was shown to bind 2.5 equivalents of Co(II), which complicated the interpretation of subsequent spectroscopic studies. In fact, this present study shows that it takes 3 equivalents of Co(II) to saturate the metal binding sites in L1 (Figure 2). Two of the Co(II) ions are probably bound to the active site. Based on the extinction coefficient of the S to Co(II) LMCT at ~320 nm, the reduced Cys site is minimally occupied. We believe that there is some Co(III) in the sample (peak at ~400 nm), and it is possible that Co(II) could also bind to the site identified in the recent crystal structure of Cu(II)-L1 [46].
To prepare Co(II)-substituted L1, we attempted a biological incorporation strategy in which we over-expressed L1 in minimal medium containing various concentrations of Co(II). We reasoned that the disulfide bond would be reduced during folding and that the folding of L1 in the presence of Co(II) would result in a Co(II)-substituted enzyme. We were careful to make sure that the pH of the growth medium was under 7.0, since air oxidation of Co(II) is more favored at basic pH values [43]. Unfortunately, the purified protein only bound 1 equivalent of cobalt regardless of the concentration of Co(II) in the growth medium, and UV-Vis spectra strongly suggest that the cobalt is Co(III).
Since both traditional techniques to prepare Co(II)-substituted L1 were unsuccessful, we developed a new strategy that involved in vitro folding of L1 in the presence of metal ions. Our first test of this strategy involved the refolding of L1 in the absence of added metal ions. Apo-L1 was unfolded in the presence of 6M Gdn-HCl, and fluorescence emission spectroscopy was used to verify that the enzyme unfolded. The removal of Gdn-HCl by dialysis resulted in an enzyme that contained 0.03 equivalents of Zn(II) and exhibited very little activity. However, the addition of 2 equivalents of Zn(II) to this protein resulted in an enzyme that exhibited steady-state kinetic constants and fluorescence emission spectra very similar to those of as-isolated L1. This result was surprising since Periyannan et al. previously suggested that the proper in vivo folding of L1 requires the presence of Zn(II) [34]. We also refolded apo-L1 in the presence of 100 µM Zn(II), and the resulting enzyme was shown to bind 2 equivalents of Zn(II) and exhibit a kcat value that was 30% higher than that exhibited by as-isolated L1 (Table 2). This result suggests that some of the as-isolated L1 may not be folded correctly and that the in vitro folding procedure results in a higher percentage of correctly folded, catalytically-active enzyme. The refolding of apo-L1 in the presence of 100 µM Co(II) resulted in a pink protein that contained 1.8 equivalents of cobalt, and a UV-Vis spectrum is consistent with the presence of Co(II). The extinction coefficient of the ligand field bands suggests that the Co(II) ions are 5/6 coordinate. Interestingly, Co(II)- substituted L1 exhibits a larger kcat value, as compared to that of 2Zn-L1, and this result is similar to the results on L1 containing 3 equivalents of cobalt.
The results presented in this work demonstrate a new strategy for preparing Co(II)-substituted analogs of the large number of Zn(II)-containing proteins that also have solvent-accessible disulfide bonds. For example, mung bean nuclease [47], the anti-sigma factor RsrA from S. coelicolor [48], the peroxide regulon repressor PerR [49], and E. coli primase [50] are Zn(II)-binding proteins that also contain a disulfide bond. The resulting Co(II)-substituted proteins could be structurally-characterized using UV-Vis, EPR, and 1H NMR spectroscopies and mechanistically-characterized using rapid-freeze quench EPR/EXAFS spectroscopies or stopped-flow UV-Vis studies. This technique may also be useful for generating Co(II)-substituted, Zn(II)-proteins that have a Cys as a metal binding ligand. The successful preparation of 2Co(II)-L1 now positions us to conduct rapid-freeze quench EPR and EXAFS studies to probe for intermediates in the reaction of the enzyme. These studies are currently underway.
Acknowledgments
The authors would like to thank Karen Anderson for assistance in purifying the enzyme and the National Institutes of Health (GM40052) for funding this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bush K. Metallo-β-lactamases: A Class Apart. Clinical Infectious Diseases. 1998;27 Supplement 1:S-48–S-53. doi: 10.1086/514922. [DOI] [PubMed] [Google Scholar]
- 2.Crowder MW, Spencer J, Vila AJ. Metallo-β-lactamases: novel weaponry for antibiotic resistance in bacteria. Acc. Chem. Res. 2006;39:721–728. doi: 10.1021/ar0400241. [DOI] [PubMed] [Google Scholar]
- 3.Toney JH, Moloughney JG. Metallo-β-lactamase inhibitors: promise for the future? Curr. Opin. Invest. Drugs. 2004;5:823–826. [PubMed] [Google Scholar]
- 4.Walsh TR, Toleman MA, Poirel L, Nordmann P. Metallo-β-lactamases: the quiet before the storm? Clin. Microbiol. Rev. 2005;18:306–325. doi: 10.1128/CMR.18.2.306-325.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rasmussen BA, Bush K. Carbapenem-hydrolyzing β-lactamases. Antimicro. Agents Chemo. 1997;41:223–232. doi: 10.1128/aac.41.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galleni M, Lamotte-Brasseur J, Rossolini GM, Spencer J, Dideberg O, Frere JM. Standard numbering scheme for class B β-lactamases. Antimicro. Agents Chemo. 2001;45:660–663. doi: 10.1128/AAC.45.3.660-663.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frere JM, Dubus A, Galleni M, Matagne A, Amicosante G. Mechanistic diversity of β-lactamases. Biochem. Soc. Trans. 1999;27:58–63. doi: 10.1042/bst0270058. [DOI] [PubMed] [Google Scholar]
- 8.Page MI, Laws AP. The mechanism of catalysis and the inhibition of β- lactamases. J. Chem. Soc. Chem. Commun. 1998:1609–1617. [Google Scholar]
- 9.Fabiane SM, Sohi MK, Wan T, Payne DJ, Bateson JH, Mitchell T, Sutton BJ. Crystal structure of the zinc-dependent β-lactamase from Bacillus cereus at 1.9 Å resolution: Binuclear active site with features of a mononuclear enzyme. Biochemistry. 1998;37:12404–12411. doi: 10.1021/bi980506i. [DOI] [PubMed] [Google Scholar]
- 10.Oelschlaeger P, Schmid RD, Pleiss J. Insight into the mechanism of the IMP-1 metallo-β-lactamase by molecular dynamics simulations. Protein Engineering. 2003;16:341–350. doi: 10.1093/protein/gzg049. [DOI] [PubMed] [Google Scholar]
- 11.Materon IC, Queenan AM, Koehler TM, Bush K, Palzkill T. Biochemical characterization of β-lactamases Bla1 and Bla2 from Bacillus anthrasis. Antimicro. Agents Chemo. 2003;47:2040–2042. doi: 10.1128/AAC.47.6.2040-2042.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Concha NO, Rasmussen BA, Bush K, Herzberg O. Crystal structure of the wide-spectrum binuclear zinc β-lactamase from Bacteroides fragilis. Structure. 1996;4:823–836. doi: 10.1016/s0969-2126(96)00089-5. [DOI] [PubMed] [Google Scholar]
- 13.Crawford PA, Sharma N, Chandrasekar S, Sigdel T, Walsh TR, Spencer J, Crowder MW. Over-expression, purification, and characterization of metallo-β-lactamase ImiS from Aeromonas veronii bv. sobria. Prot. Express. Purif. 2004;36:272–279. doi: 10.1016/j.pep.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 14.Garau G, Bebrone C, Anne C, Galleni M, Frere JM, Dideberg O. A metallo-β-lactamase enzyme in action: crystal structure of the monozinc carbapenemase CphA and its complex with biapenem. J. Mol. Biol. 2005;345:785–795. doi: 10.1016/j.jmb.2004.10.070. [DOI] [PubMed] [Google Scholar]
- 15.Garcia-Saez I, Mercuri PS, Papamicael C, Kahn R, Frere JM, Galleni M, Rossolini GM, Dideberg O. Three-dimensional structure of FEZ-1, a monomeric subclass B3 metallo-β-lactamase from Fluoribacter gormanii, in native form and in complex with D-captopril. J. Mol. Biol. 2003;325:651–660. doi: 10.1016/s0022-2836(02)01271-8. [DOI] [PubMed] [Google Scholar]
- 16.Ullah JH, Walsh TR, Taylor IA, Emery DC, Verma CS, Gamblin SJ, Spencer J. The crystal structure of the L1 metallo-β-lactamase from Stenotrophomonas maltophilia at 1.7 angstrom resolution. J. Mol. Biol. 1998;284:125–136. doi: 10.1006/jmbi.1998.2148. [DOI] [PubMed] [Google Scholar]
- 17.Sabath LD, Abraham EP. Zinc as a cofactor for cephalosporinase from Bacillus cereus 569. Biochem. J. 1966;98:11c–13c. doi: 10.1042/bj0980011c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bennett B. EPR of Co(II) as a structural and mechanistic probe of metalloprotein active sites: Characterization of an aminopeptidase. Curr. Topics Biophys. 2002;26:49–57. [Google Scholar]
- 19.Bennett B, Holz RC. EPR studies on the mono- and dicobalt(II)-substituted forms of the aminopeptidase from Aeromonas proteolytica. Insight into the catalytic mechanism of dinuclear hydrolases. J. Am. Chem. Soc. 1997;119:1923–1933. [Google Scholar]
- 20.Breece RM, Costello A, Bennett B, Sigdel TK, Matthews ML, Tierney DL, Crowder MW. A five-coordinate metal center in Co(II)-substituted VanX. J. Biol. Chem. 2005;280:11074–11081. doi: 10.1074/jbc.M412582200. [DOI] [PubMed] [Google Scholar]
- 21.Gilson HSR, Krauss M. Structure and spectroscopy of metallo-lactamase active sites. J. Am. Chem.Soc. 1999;121:6984–6989. [Google Scholar]
- 22.Lever ABP. Co(II) d7, Inorganic Electronic Spectroscopy. 2nd ed. 1984. [Google Scholar]
- 23.Maret W, Vallee BL. Meth. Enzymol. Vol. 226. New York: Academic Press; 1993. pp. 52–71. [Google Scholar]
- 24.Crowder MW, Yang KW, Carenbauer AL, Periyannan G, Seifert MA, Rude NE, Walsh TR. The problem of a solvent exposable disulfide when preparing Co(II)-substituted metallo-β-lactamase L1 from Stenotrophomonas maltophilia. J. Biol. Inorg. Chem. 2001;6:91–99. doi: 10.1007/s007750000173. [DOI] [PubMed] [Google Scholar]
- 25.Crowder MW, Walsh TR, Banovic L, Pettit M, Spencer J. Overexpression, purification, and characterization of the cloned metallo-β-lactamase L1 from Stenotrophomonas maltophilia. Antimicro. Agents Chemo. 1998;42:921–926. doi: 10.1128/aac.42.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Seny D, Heinz U, Wommer S, Kiefer M, Meyer-Klaucke W, Galleni M, Frere JM, Bauer R, Adolph HW. Metal ion binding and coordination geometry for wild type and mutants of metallo-β-lactamase from Bacillus cereus 569/H/9 (BcII) - A combined thermodynamic, kinetic, and spectroscopic approach. J. Biol. Chem. 2001;276:45065–45078. doi: 10.1074/jbc.M106447200. [DOI] [PubMed] [Google Scholar]
- 27.Davies AM, Rasia RM, Vila AJ, Sutton BJ, Fabiane SM. Effect of pH on the active site of an Arg121Cys mutant of the metallo-β-lactamase from Bacillus cereus Implications for the enzyme mechanism. Biochemistry. 2005;44:4841–4849. doi: 10.1021/bi047709t. [DOI] [PubMed] [Google Scholar]
- 28.Orellano EG, Girardini JE, Cricco JA, Ceccarelli EA, Vila AJ. Spectroscopic characterization of a binuclear metal site in Bacillus cereusβ-lactamase II. Biochemistry. 1998;37:10173–10180. doi: 10.1021/bi980309j. [DOI] [PubMed] [Google Scholar]
- 29.Wang Z, Benkovic SJ. Purification, characterization, and kinetic studies of soluble Bacteroides fragilis metallo-β-lactamase. J. Biol. Chem. 1998;273:22402–22408. doi: 10.1074/jbc.273.35.22402. [DOI] [PubMed] [Google Scholar]
- 30.Garmer DR, Krauss M. Ab initio quantum chemical study of the cobalt d-d spectroscopy of several substituted zinc enzymes. J. Am. Chem. Soc. 1993;115:10247–10257. [Google Scholar]
- 31.Costello A, Periyannan G, Yang KW, Crowder MW, Tierney DL. Site-selective binding of Zn(II) to metallo-β-lactamase L1 from Stenotrophomonas maltophilia. J. Biol. Inorg. Chem. 2006;11:351–358. doi: 10.1007/s00775-006-0083-z. [DOI] [PubMed] [Google Scholar]
- 32.Llarrull LI, Tioni MF, Kowalski J, Bennett B, Vila AJ. Evidence for a dinuclear active site in the metallo-β-lactamase BcII with substoichiometric Co(II): A new model for metal uptake. J. Biol. Chem. 2007;282:30586–30595. doi: 10.1074/jbc.M704613200. [DOI] [PubMed] [Google Scholar]
- 33.Garrity JD, Bennett B, Crowder MW. Direct evidence that reaction intermediate in metallo-β-lactamase is metal bound. Biochemistry. 2005;44:1078–1087. doi: 10.1021/bi048385b. [DOI] [PubMed] [Google Scholar]
- 34.Periyannan G, Shaw PJ, Sigdel T, Crowder MW. In vivo folding of recombinant metallo-β-lactamase L1 requires the presence of Zn(II) Prot. Sci. 2004;13:2236–2243. doi: 10.1110/ps.04742704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lakowicz JR. Principles of fluorescence spectroscopy Kluwer Academic Plenum. New York: 1999. [Google Scholar]
- 36.Wilson CJ, Apiyo D, Wittung-Stafshede P. Role of cofactors in metalloprotein folding. Q. Rev. Biophys. 2004;37:285–314. doi: 10.1017/S003358350500404X. [DOI] [PubMed] [Google Scholar]
- 37.Spencer J, Clark AR, Walsh TR. Novel mechanism of hydrolysis of therapeutic β-lactams by Stenotrophomonas maltophilia L1 metallo-β-lactamase. J. Biol. Chem. 2001;276:33638–33644. doi: 10.1074/jbc.M105550200. [DOI] [PubMed] [Google Scholar]
- 38.McMannus-Munoz S, Crowder MW. Kinetic mechanism of metallo-β-lactamase L1 from Stenotrophomonas maltophilia. Biochemistry. 1999;38:1547–1553. doi: 10.1021/bi9826512. [DOI] [PubMed] [Google Scholar]
- 39.Garrity JD, Carenbauer AL, Herron LR, Crowder MW. Metal binding Asp-120 in Metallo-β-lactamase L1 from Stenotrophomonas maltophilia plays a crucial role in catalysis. J. Biol. Chem. 2004;279:920–927. doi: 10.1074/jbc.M309852200. [DOI] [PubMed] [Google Scholar]
- 40.Auld DS. Zinc coordination sphere in biochemical zinc sites. Biometals. 2001;14:271–313. doi: 10.1023/a:1012976615056. [DOI] [PubMed] [Google Scholar]
- 41.Auld DS. In: Handbook of Metalloproteins. Messerschmidt A, editor. Vol. 3. New York: John Wiley & Sons; 2004. pp. 416–431. [Google Scholar]
- 42.Holz R, Bzymek K, Swierczek S. Co-catalytic metallopeptidases as pharmaceutical targets. Curr. Opin. Chem. Biol. 2003;7:197–206. doi: 10.1016/s1367-5931(03)00033-4. [DOI] [PubMed] [Google Scholar]
- 43.Bertini I, Luchinat C. High-spin cobalt(II) as a probe for the investigation of metalloproteins. Adv. Inorg. Biochem. 1984;6:72–111. [PubMed] [Google Scholar]
- 44.Sharma NP, Hajdin C, Chandrasekar S, Bennett B, Yang KW, Crowder MW. Mechanistic studies on the mononuclear Zn(II)-containing metallo-β-lactamase ImiS from Aeromonas sobria. Biochemistry. 2006;45:10729–10738. doi: 10.1021/bi060893t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matthews ML, Periyannan G, Hajdin C, Sidgel TK, Bennett B, Crowder MW. Probing the reaction mechanism of the D-ala-D-ala dipeptidase, VanX, by using stopped-flow kinetic and rapid-freeze quench EPR studies on the Co(II)-substituted enzyme. J. Am. Chem. Soc. 2006;128:13050–13051. doi: 10.1021/ja0627343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nauton L, Kahn R, Garau G, Hernandez JF, Dideberg O. Structural insights into the design of inhibitors for the L1 metallo-β-lactamase from Stenotrophomonas maltophilia. J. Mol. Biol. 2008;375:257–269. doi: 10.1016/j.jmb.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 47.McCutchan T, Hansen J, Dame J, Mullins J. Mung bean nuclease cleaves Plasmodium genomic DNA at sites before and after genes. Science. 1984;225:625–628. doi: 10.1126/science.6330899. [DOI] [PubMed] [Google Scholar]
- 48.Bae J-B, Park J-H, Hahn M-Y, Kim M-S, Roe J-H. Redox-dependent changes in RsrA, an anti-sigma factor in Streptomyces coelicolor: Zinc release and disulfide bond formation. J. Mol. Biol. 2004;335:425–435. doi: 10.1016/j.jmb.2003.10.065. [DOI] [PubMed] [Google Scholar]
- 49.Herbig AF, Helmann JD. Roles of metal ions and hydrogen peroxide in modulating the interaction of the Bacillus subtilis PerR peroxide regulon repressor with operator DNA. Mol. Microbiol. 2001;41:849–859. doi: 10.1046/j.1365-2958.2001.02543.x. [DOI] [PubMed] [Google Scholar]
- 50.Griep MA, Lokey ER. The role of zinc and the reactivity of cysteines in Escherichia coli primase. Biochemistry. 1996;35:8260–8267. doi: 10.1021/bi952948p. [DOI] [PubMed] [Google Scholar]