

Abstract
Application of basic fibroblast growth factor (FGF-2) to the optic nerve after axotomy promotes the survival of retinal ganglion cells (RGCs) in the frog, Rana pipiens, and results in a rapid up-regulation of BDNF and TrkB synthesis by the RGCs. Here we investigate whether this upregulation is maintained in the long term, and whether it is required for FGF-2's survival effect. At 6 weeks after axotomy and FGF-2 treatment we found more RGCs immunopositive for BDNF protein and higher intensity of BDNF and TrkB immunostaining, accompanied by increases in BDNF and TrkB mRNA in RGCs. Application of fluorescently-labeled siRNA targeted against BDNF to the cut RGC axons showed that it was transported to the cell bodies. Axonal siRNA treatment eliminated the increases in BDNF immunostaining and mRNA that were induced by FGF-2, and had no effect on TrkB mRNA. This reduction in BDNF synthesis by siRNA greatly reduced the long term survival effect of FGF-2 on RGCs. This, taken together with previous results, suggests that, while FGF-2 may initially activate survival pathways via ERK signaling, its main long-term survival effects are mediated via its upregulation of BDNF synthesis by the RGCs.
Keywords: growth factor, retinal ganglion cells, axotomy, frog, neurotrophin
Introduction
The survival of mammalian central neurons depends upon neurotrophins produced by their synaptic targets. After axotomy the majority of retinal ganglion cells (RGCs) degenerate and die because they are unable to reacquire this trophic support (Lewin and Barde 1996; Snider 1994; Thoenen 1995; von Bartheld 1998), although a few survive for longer periods (Germain et al. 2007). Frog retinal ganglion cells are a good model to study the neuronal reaction to axotomy because some of them also undergo cell death, but, unlike mammalian RGCs, some survive and are able to regenerate (Beazley et al. 1986; Blanco et al. 2000; Scalia et al. 1985).
We have found that in frog, many RGCs are rescued from axotomy-induced cell death when fibroblast growth factor-2 (FGF-2) is applied to the optic nerve at the time of axotomy, followed by a second application midway through the regeneration period (Blanco et al. 2000). The same is true in rat as well, although the mechanisms involved have not been investigated (Sievers et al. 1987). Recently we reported that FGF-2 application to the injured frog optic nerve activates the ERK signaling pathway, which in turn modulates the expression of Bcl-2 family proteins in the RGCs, thus contributing to the long term survival of the regenerating cells (Ríos-Muñoz et al. 2005).
However, in the visual system it is brain-derived neurotrophic factor (BDNF), not FGF-2, that is considered to be the most important neurotrophin for RGCs (von Bartheld 1998). In the adult retina of several species, including the frog, both BDNF and its receptor, TrkB, are present in the majority of the RGCs, however, these adult cells still depend upon target-derived BDNF for survival (Duprey-Diaz et al. 2002; Pease et al. 2000; Quigley et al. 2000; von Bartheld 1998). Optic nerve axotomy in the rat transiently increases BDNF mRNA in the RGCs, however this expression rapidly declines and most of the cells die in only two weeks (Gao et al. 1997; Hirsch et al. 2000). In the frog we have shown that BDNF is also lost from RGCs after axotomy, with a partial recovery only after the surviving cells reconnect to the tectum (Duprey-Diaz et al. 2002), suggesting that loss of BDNF may contribute to cell death.
We recently obtained evidence that FGF-2 treatment can stimulate BDNF production. A single application of FGF-2 to the transected frog optic nerve rapidly stimulated the upregulation of BDNF and TrkB mRNA in RGCs, via activation of the FGF receptor-1 and by the ERK and PKA signaling pathways (Soto et al. 2006). However, we do not know if this upregulation of BDNF in the short term can promote the long-term survival of the RGCs.
The objectives of the present study are to bring all these results together and determine whether the FGF-2 treatment that induces long-term survival does indeed increase the amounts of BDNF in RGCs, and then, most importantly, to test whether inhibiting this BDNF synthesis reduces long term RGC survival. We find that FGF-2 treatment of the cut optic nerve does partially prevent the loss of BDNF staining in RGCs for up to 6 weeks after axotomy. As predicted, inhibiting synthesis of BDNF with BDNF siRNA applied selectively to the RGC axons at the time of FGF-2 treatment greatly inhibits the long term survival effect of FGF-2.
Materials and Methods
Surgical technique for optic nerve axotomy and bFGF application
Under 0.3% tricaine anesthesia the right eyeball of a series of frogs (Rana pipiens) was approached from the palate and an incision made; the extraocular muscles were teased aside and the intraorbital section of the optic nerve was exposed. While avoiding large blood vessels, the nerve, with its meningeal sheath, was transected, allowing complete separation of both stumps. The incision was sutured and the animal allowed to recuperate, feeding on crickets. All protocols conformed to NIH guidelines and were approved by the institutional animal care and use committee.
For FGF-2 treatment (Roche Diagnostics, Indianapolis, IN), the growth factor was dissolved at a concentration of 25 μg/ml in phosphate buffered saline (0.01M PBS) pH 7.4. Immediately after being cut, the nerve stump was lifted and placed on a strip of Parafilm and 5 μl of FGF-2 was applied directly to the cut end with a Hamilton micro-syringe. Control applications consisted of 5 μl PBS. For long term experiments, a second application of FGF-2 or PBS was made to the regenerating nerve at 3 weeks after axotomy.
SiRNA optic nerve application
Annealed siRNA oligonucleotides for Rana pipiens BDNF were designed and synthesized by Cenix BioScience (Silencer pre-designed siRNA, Ambion). The siRNA sequences used for this study were the anti-BDNF-antisense 5’-AAU AAC CUG CUC AAA GGU Atc-3’ and anti-BDNF-sense 5’-UAC CUU UGA GCA GGU UAU Utt-3’. Negative control siRNA #1 (Ambion cat. 4635) was used to control for possible siRNA toxicity. For Cy-3 labeled annealed siRNA oligonucleotides the Silencer siRNA labeling kit (Ambion) was used. To inhibit BDNF expression specifically in the RGCs of FGF-2 treated and untreated animals, 5 μl siRNA (500 ng/μl) were applied mixed with the FGF-2 or PBS directly onto the proximal optic nerve stump immediately after it was cut. In one group of animals used for long-term experiments (6 w), a second treatment of BDNF siRNA was applied to the optic nerve at three weeks after axotomy. To confirm that BDNF siRNA was retrogradely transported to the axotomized RGCs, Cy-3 labeled BDNF siRNA was applied to the cut optic nerve of another group of animals.
Real-Time Quantitative RT-PCR
Control and experimental RNA extractions (N = 9 animals) were carried out for each time point. Retinas from 3 animals were pooled and RNA was extracted using the Nucleospin Nucleic Acid RNA II Purification Kit (BD Bioscience, Palo Alto, CA). RNA quality was assessed using denaturing gel electrophoresis and concentrations were determined with a spectrophotometer. First strand cDNA templates were prepared from 5 μg of RNA using the First strand Superscript II kit (Invitrogen, Eugene, OR). Real-time quantitative RT-PCR was performed in a Smartcycler (Cepheid, Sunnyvale, CA) using SYBR-green (Quantitec Master Mix, Qiagen, Valencia, CA) and specific primers for BDNF (Forward 5’-GAA CCG CAC GTA AGA CTG AGT-3’; Reverse 5’-GTG ACT GCA GCC GAC AAG AAA), TrkB (Forward 5’-ATG GCA GAA GGG AAC CGC-3’; Reverse 5’-CGA TGG ACG AAG TGC TGT GCT GTG AGG-3’) and the internal standard cyclophilin (Forward 5’-CAT GAT TCA GGG TGG TGA TTT CAC-3’; Reverse 5’ GGA GGT CTT TAC TGT GGT CAT GAA-3’). Reactions were performed in a 25 μl volume with 0.3−0.5 μM primers and 1X SYBR-green master mix. For each of the conditions studied, three assays were run for each pair of primers. Starting quantity for each sample was determined by the interpolation of the threshold cycle to a standard curve derived from serial dilutions of the isolated template. To verify that a single PCR product was obtained in each run, the melting temperature of each product was measured and the PCR product was run on a 10% polyacrylamide gel. The BDNF and TrkB levels were normalized to cyclophilin or glyceraldehyde-3-phosphate dehydrogenase (GAPDH), both giving similar results. Finally, each of normalized BDNF and TrkB values were divided by the normalized control retinal BDNF and TrkB value to generate the relative expression levels of BDNF and TrkB mRNA. The statistical significance was determined using ANOVA and post-hoc Tukey or Tukey-Kramer tests. In the results, one asterisk indicates p<0.05, two asterisks p<0.01 and 3 asterisks, p<0.001.
In situ hybridization
In situ hybridization was performed essentially as described by the Genedetect protocol for DIG-labeled oligonucleotide probes. The Genedetect Company (Aukland, New Zealand) designed 48mer digoxigenin-labeled oligonucleotide probes that recognize our partial Rana pipiens BDNF (239 bp) and TrkB sequences (452 bp).
The eyes from control and experimental animals (48 hours, 1 week and 6 weeks after axotomy and FGF-2 application) were dissected and fixed in 4% paraformaldehyde for 60 min. 16 μm cryostat sections were cut, air dried, and stored at −80°C. Just before use, tissue sections were washed in PBS, dehydrated and air-dried. Then the sections were hybridized overnight at 37°C with 200 ng/ml of digoxigenin-labeled oligonucleotide probes in the hybridization solution (10% dextran sulfate, 4 X SSC (1X=150 mM NaCl, 15 mM sodium citrate, pH 7.2), 50% formamide, 10 mM DTT, 0.25 mg/ml Poly A, 0.25mg/ml of denatured and sheared salmon sperm DNA and 1X Denhardts buffer). After hybridization, sections were quickly washed once with 1 X SSC (with 10mM DTT) at RT, followed by two 15 min washes with 1 X SSC (10 mM DTT) at 55°C, and finally two more 15 min washes with 0.5 X SSC (10 mM DTT) at 55°C. The slides were then washed three times with TBS and incubated 30 min in a blocking solution (TBS + 0.3% Triton X-100 + 2% normal sheep serum). The oligonucleotide probes were detected using a Cy3-labeled mouse anti-DIG antibody (Jackson ImmunoLabs) in a 1:1000 dilution in the blocking solution. After 4 h of incubation, the slides were washed with TBS six times (10 min each) and mounted in an anti-fading agent (Vectashield, Vector labs, Burlingame, CA).
The staining intensity was quantitatively analysed in sections from different animals by comparing the intensities of the inner nuclear layer (INL) and the ganglion cell layer (GCL). This was done by measuring mean pixel intensity in areas of interest using ImageJ. The result was expressed as the GCL/INL intensity ratio so as to account for any effects of possible variations in tissue preparation. INL staining does increase after axotomy but is not affected by the different treatments of the axons in the optic nerve.
Immunohistochemistry
After dissection, three eye cups were fixed per condition from control and experimental frogs (1 week and 6 weeks after operation and treatment) with buffered 4% paraformaldehyde solution for 1 h. After PBS washing, the tissues were placed in 30% sucrose for cryoprotection at 4°C overnight and, after being frozen, cryostat sections of 16 μm were cut. The sections were washed two times (5 min each) in PBS containing 0.3% Triton X-100 and 1% BSA and incubated 30 min in the same buffer containing 10% normal donkey serum (NDS). They were then incubated overnight with polyclonal antisera against human BDNF (1:200, Promega, cat. G1541) or mouse full-length TrkB (1:200, Promega, cat. G1561) diluted in 0.1M PBS + 0.3% Triton X-100 + 1% BSA. The BDNF antibody has been shown to be highly specific (Goodman et al. 1996). In addition, Western blot analysis of the BDNF antibody showed a single band in retina (data not shown). The TrkB antibodies detect the intracellular domain of the full-length TrkB receptor, which shows 95% conservation of amino acid sequence between mouse and Xenopus. Western blots showed a strong band at the appropriate size for the full-length fragment (data not shown). After several washes with 0.1M PBS + 0.3% Triton X-100 + 0.5% BSA the sections were incubated with donkey anti-chicken IgG-Cy3 for 1hr at room temperature. The sections were rinsed in PBS 6 times, 10 min each, and mounted in Polymount.
Images were taken from inferior and temporal areas of retinas of controls, and animals at 6 weeks after axotomy. Staining intensity measurements were collected from each cell in the GCL using ImageJ (Wayne Rasband, NIH). Each cell was outlined using the TDA stain as a guide, and the mean intensity of antibody staining within the outline was calculated, converting pixel values to scale of 0 to 100. Mean intensity measurements were also taken from the lowest staining region of the ONL and from the IPL; the lower of these two values was subsequently defined as the background staining intensity. A neuron was defined as being significantly stained if its mean intensity was more than 2.33 standard deviations greater than the background mean (99% probability level). The number of stained RGCs was expressed as a percentage of the total number of TDA-labeled ganglion cells.
TDA labeling of retinal ganglion cells
A 1mm square piece of cellulose acetate membrane filter (0.8 μm pore size, Schleicher and Schuell, Keene, NH) was infused with a saturated solution of tetramethylrhodamine dextran amine (TDA, 3000 MW, Molecular Probes) in ethanol and left to air-dry so that particles of TDA were embedded within the filter. The optic nerve of control or experimental animals was exposed and cut close to the back of the eyeball. The piece of filter was inserted into the stump and the palate was sutured. After 48 − 60 hours, the frog was anesthetized, fixed and processed for immunohistochemistry as described above. With these parameters, TDA consistently labeled 80 − 84% of the cells in the GCL of control retinas. The percentage of displaced amacrine cells in Rana pipiens retina has been previously calculated as 16% (Scalia et al. 1985), so we appear to be labeling most of the RGCs. The percentage of total cells in the GCL labeled with TDA decreased with time after axotomy; this was not due to a reduced efficacy of the retrograde labeling procedure but corresponded to the expected decline in RGC numbers at these times.
Results
FGF-2 increases BDNF and TrkB immunoreactivity in RGCs
We do not know how axonal FGF-2 treatment enables the survival of more RGCs in the long term. In the present study, the first objective was to determine whether FGF-2 treatment alters the distribution of BDNF in RGCs for long periods (up to 6 weeks), in a way that could promote long term survival.
Western blot analysis of BDNF protein levels was not used for this long-term study because, as we have previously observed (Duprey-Diaz et al. 2002), axotomy alone produces a large increase in BDNF in amacrine cells of the inner nuclear layer at one month after axotomy, which masks the signal from the thin layer of RGCs. Instead, we quantitatively analyzed BDNF antibody staining intensity, using previously validated methods (Duprey-Diaz et al. 2002) (see Methods). The total number of stained neurons (those with a staining intensity of more than 2.33 standard deviations above background levels) was expressed as a percentage of the total number of RGCs that were retrogradely labeled with TDA. Thus the final results are not affected by changes in the number of surviving RGCs (Blanco et al. 2000).
In control animals, in which the nerve was cut and only PBS applied to the nerve, BDNF immunostaining was present in 63.9% ± 6.9% of 349 RGCs at 6 weeks after axotomy (Fig 1 A, B, E). The mean RGC staining intensity was 19.8 ± 1.8 (on a scale of 0 − 100, N = 3 animals).
Figure 1.

FGF-2 treatment increases the proportion of RGCs that contain BDNF immunostaining. A. Retrograde TDA labeling of RGCs at 6 weeks after axotomy and PBS treatment. B. BDNF immunostaining of the same section as in A, showing approximately 60% of RGCs containing BDNF immunoreactivity (arrows). The inner nuclear layer (INL), inner plexiform layer (IPL) and ganglion cell layer (GCL) are indicated. Other TDA-labeled RGCs do not contain BDNF (asterisks). C. Retrograde TDA labeling of RGCs at 6 weeks after axotomy and FGF-2 treatment. D. BDNF immunostaining of the same section as in C; almost all RGCs contain BDNF. E. Percentage of RGCs that contain significant BDNF immunoreactivity, with a significant increase after FGF-2 treatment. F. The percentage of RGCs that contain significant TrkB immunoreactivity shows no change with FGF-2 treatment. G. The intensity of BDNF immunoreactivity in stained RGCs significantly increases after FGF-2 treatment. H. The intensity of TrkB immunoreactivity in stained RGCs significantly increases after FGF-2 treatment. Scale bar in A: 30 μm.
In experimental animals, the optic nerve was axotomized and treated with FGF-2, with a second FGF-2 application to the regenerating nerve at 3 weeks. Examination of these retinas showed numerous RGCs remaining at 6 weeks, with an apparent increase over those in controls (Fig. 1C). This 25−35% increase in RGC survival with FGF-2 treatment has been quantified in an early study (Blanco et al. 2000). Here, in an analysis of 713 FGF-2-treated RGCs, we found that a significantly higher proportion of them contained BDNF staining (Fig 1D, E), with 92.9% ± 0.8% of RGCs being BDNF immunopositive (P<0.05, N = 4 animals, Aspin-Welch t-test for unequal variances). In addition, the mean staining intensity within these neurons was significantly greater (26.9 ± 1.44; P<0.05, N = 4 animals, Student's t-test for equal variances) (Fig. 1G).
The same retinas were also used for immunostaining with TrkB antibody. Unlike BDNF, the percentage of TrkB-immunoreactive RGCs was not significantly altered by FGF-2 treatment (Fig.1 F), with 89−97% being stained in both cases (P = 0.17, Aspin-Welch t-test). However, the mean intensity within these neurons did significantly increase (Fig. 1H), from 20.9 ± 2.1 (350 RGCs, N = 3 animals) to 35.5 ± 1.4 (511 RGCs, N = 4 animals; P < 0.01, Student's t-test).
The above results show that FGF-2 treatment leads to proportionately more RGCs containing BDNF, and that the amounts of BDNF and TrkB within the neurons also increase. However, the possibility remains that the RGCs could acquire BDNF and TrkB protein from other sources within the retina, such as presynaptic amacrine neurons. To determine whether BDNF and TrkB synthesis was increased in RGCs (as opposed to other retinal regions) we carried out in-situ hybridization on retinal sections, using specifically designed oligonucleotide probes for Rana pipiens BDNF and TrkB sequences. This showed that mRNA for these proteins was localized in the ganglion cell layer at 6 weeks after axotomy (Fig. 2). A quantitative analysis comparing the relative staining intensities of the GCL and INL within individual sections from different animals showed that the GCL/INL intensity ratio for BDNF mRNA increased from 0.80 ± 0.02 with PBS treatment to 1.29 ± 0.04 with FGF-2 treatment (Fig. 2B; N = 4, P<0.001, Student's t-test). The GCL/INL intensity ratio for TrkB mRNA also increased from 1.16 ± 0.03 with PBS treatment to 1.47 ± 0.07 with FGF-2 treatment (Fig. 2D; N = 4, P<0.05, Aspin-Welch t-test). These results indicate that FGF-2 treatment caused an increase in both BDNF and TrkB mRNA labeling.
Figure 2.
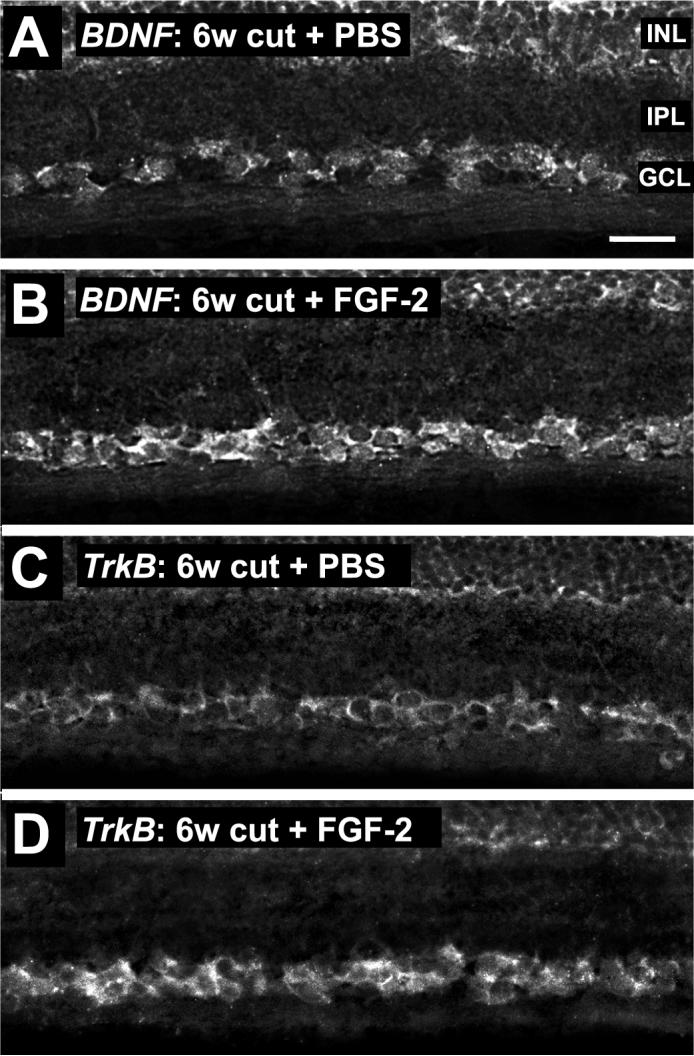
BDNF and TrkB mRNA in RGCs at 6 weeks after axotomy. A. A PBS-treated retina shows some BDNF mRNA in the ganglion cell layer (GCL) and inner nuclear layer (INL). The inner plexiform layer (IPL) is also indicated. B. 6 weeks after axotomy and FGF-2 treatment, more BDNF mRNA is present in the GCL. C. At 6 weeks after axotomy and PBS treatment, low levels of TrkB mRNA are present in the GCL. D. After axotomy and FGF-2 treatment, more TrkB mRNA is present in the GCL. Scale bar in A: 30 μm.
Axonally-applied siRNA is transported to the RGC cell bodies
In order to selectively inhibit BDNF synthesis in RGCs only, we applied small interfering double-stranded RNA (siRNA) oligonucleotides that had been designed specifically to target BDNF mRNA, to the ends of the cut axons at the time of axotomy. To determine whether these siRNAs had only a local effect, or were transported retrogradely to the cell body, as in the rat (Lingor et al. 2005), we also applied siRNA that had been labeled with Cy3. Flat mounts of retinas 3d later showed that most RGC cell bodies contained punctate deposits of labeled siRNA (Fig. 3A).
Figure 3.

Cy3-labeled BDNF siRNA applied to the cut axons is transported retrogradely to the RGC cell bodies. A. At 3 days after axotomy, the GCL of a flat-mounted retina shows punctate accumulations of Cy3-labeled siRNA in RGCs. B. The GCL of a control retina with PBS application only shows only background fluorescence. Scale bar in B: 50 μm.
BDNF siRNA inhibits the increase in BDNF elicited by FGF-2 treatment
To ascertain the efficacy of the axonal siRNA treatment, we carried out real-time PCR of BDNF mRNA from retinas. As shown before (Soto et al. 2006), axotomy alone had no effect on BDNF mRNA levels at 48 h, although later, at 1 week, there was a 2-fold increase (Fig. 4A). FGF-2 treatment brought about an early increase in BDNF mRNA and augmented the later increase. Axonal treatment with siRNA reduced BDNF mRNA levels to control levels at 48h, and reduced both the 1 week increase in BDNF mRNA due to axotomy and the further increase due to FGF-2 (Fig. 4A). To control for any possible nonspecific effects, we applied a control siRNA, designed to not be homologous to any known mRNA, to the axons. 48 h later there was no significant difference in BDNF mRNA levels compared to PBS-treated axons.
Figure 4.

BDNF siRNA inhibits the increase in BDNF mRNA elicited by FGF-2. A. Real-time PCR of retinal mRNA shows that axotomy alone has no effect on BDNF mRNA levels at 48 h after axotomy but induces a significant increase by 1 week. FGF-2 treatment increases BDNF mRNA at both 48 h and 1 week. These FGF-2-induced increases are reduced by siRNA treatment, as is the increase due to axotomy at 1 week. Negative control (NC) siRNA has no effect on mRNA levels at 48 h. B. In situ hybridization shows BDNF mRNA in RGCs at 1 week after axotomy and FGF-2 treatment. The inner nuclear layer (INL), inner plexiform layer (IPL) and ganglion cell layer (GCL) are indicated. C. SiRNA application reduces BDNF mRNA in RGCs. Scale bar in B: 30 μm.
To confirm that these retina-wide changes in mRNA levels could be localized to the ganglion cell layer, we carried out in-situ hybridization experiments. These showed that the increase in BDNF mRNA produced by FGF-2 treatment was localized to the GCL (Fig. 4B), as was the decrease in mRNA after siRNA treatment (Fig. 4C). The GCL/INL intensity ratio decreased from 1.26 ± 0.02 to 0.54 ± 0.06 (N = 3, P<0.01, Aspin-Welch t-test).
Having established that siRNA treatment affects the levels and distribution of BDNF mRNA, we then investigated whether distribution of the protein was affected. Controls showed normal distribution of BDNF protein, with strong immunoreactivity in the majority of RGCs (Fig. 5A), as reported before (Duprey-Diaz et al. 2002). The GCL/INL intensity ratio was 1.68 ± 0.18 (N = 4). One week after axotomy most cells in the GCL were stained, but few with high intensity (Fig. 5B), and there was an increase in immunoreactivity in the amacrine cells of the INL. This resulted in a significant decrease in the GCL/INL intensity ratio to 1.10 ± 0.07 (N = 4, P<0.05, Tukey test). These results suggest that part of the increase in retinal BDNF mRNA after 1 week axotomy is due to upregulation by amacrine cells. In addition, part of the apparent loss of BDNF protein in RGCs may be due to loss of target-derived BDNF.
Figure 5.
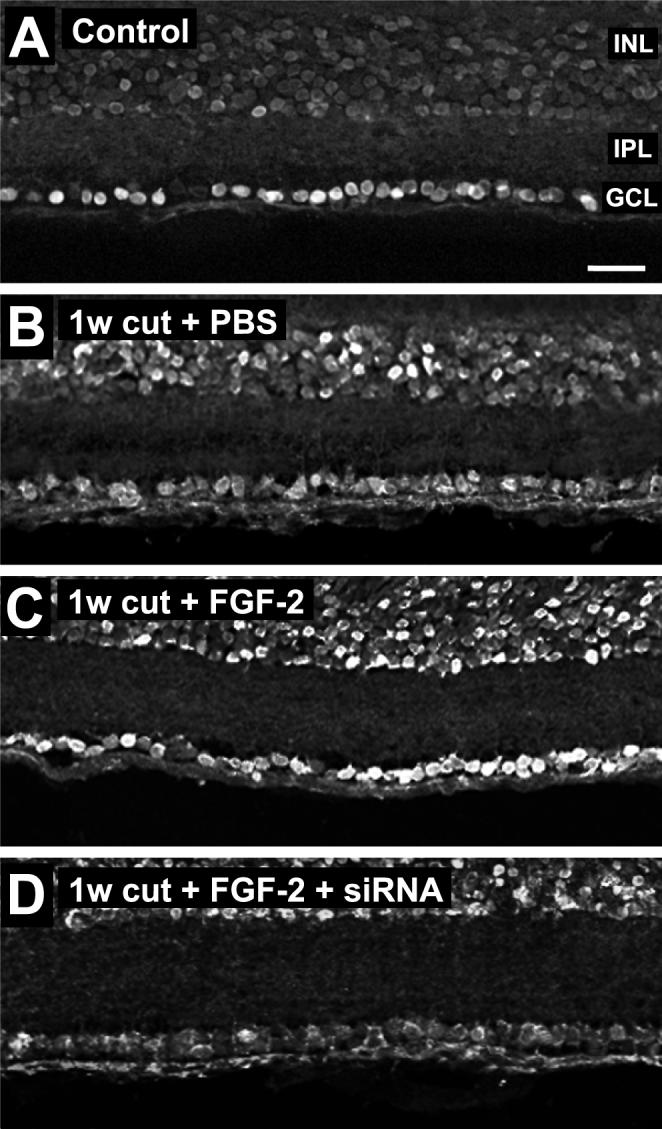
Application of BDNF siRNA to cut axons reduces the increase in BDNF staining elicited by FGF-2. A. Control retina shows strong BDNF immunostaining in RGCs. The inner nuclear layer (INL), inner plexiform layer (IPL) and ganglion cell layer (GCL) are indicated. B. At 1 week after axotomy, BDNF immunostaining is reduced in RGCs. C. FGF-2 treatment increases BDNF staining in RGCs. D. FGF-2 coupled with siRNA reduces BDNF staining. Scale bar in A: 30 μm.
Application of FGF-2 to the cut axons resulted in more intense BDNF staining in the GCL (Fig. 5C), with the GCL/INL intensity ratio increasing to 1.61 ± 0.08 (N = 4, P<0.05, Tukey test). Application of siRNA concomitantly with the FGF-2 prevented the increase in BDNF staining, reducing it to levels at or below those elicited by axotomy alone (Fig. 5D). The GCL/INL intensity ratio was significantly decreased to 0.70 ± 0.03 (N = 4, P<0.001, Tukey test). Thus the changes in BDNF protein distribution in the GCL appear to parallel the changes in BDNF mRNA levels.
As a further, positive control for the specificity of action of the BDNF siRNA, we also assayed TrkB mRNA levels in the retina following BDNF siRNA application. We found that, as previously described (Soto et al. 2006), axotomy alone causes a doubling in TrkB mRNA levels (Fig. 6A) and that, at 48 h (but not one week), FGF-2 treatment results in an additional two-fold increase. Neither treatment with BDNF siRNA nor with control siRNA has a significant effect on TrkB mRNA levels at 48 h, with or without concomitant FGF-2 treatment. These results are confirmed by in situ hybridization of TrkB mRNA (Fig. 6B).
Figure 6.

Application of BDNF siRNA to cut axons has no effect on TrkB mRNA. A. Real-time PCR of retinas shows that axotomy and PBS treatment increase TrkB mRNA levels at 48 h and 1 week after axotomy. FGF-2 treatment of the cut axons doubles this increase, at 48 h only. Neither BDNF nor negative control siRNA has any significant effect upon the increases in TrkB mRNA induced by axotomy or by FGF-2. B. In situ hybridization shows TrkB mRNA in RGCs at 48 h after axotomy and FGF-2 treatment. C. SiRNA application has no effect on TrkB mRNA in RGCs. Scale bar in B: 30 μm.
BDNF siRNA prevents the increase in long-term RGC survival elicited by FGF-2 treatment
The results so far support the idea that FGF-2 application to cut axons, followed by a second application during regeneration, upregulates BDNF synthesis by RGCs over the long term, consistent with the hypothesis that this is the mechanism by which FGF-2 prolongs the survival of RGCs. If this hypothesis were correct, we would expect that preventing the upregulation of BDNF mRNA and protein synthesis by application of BDNF siRNA would block the long-term survival effect of FGF-2 treatment. We therefore assayed the number of RGCs in whole mounts of retinas that had been retrogradely labeled with TDA, after different long-term treatments.
Control retinas reliably showed TDA labeling of the majority of RGCs (Fig. 7B), confirming that the retrograde labeling technique was an effective assay of survival. Six weeks after axotomy and two applications of PBS (one immediately after axotomy, the second at 3 weeks), there was a significant 41% drop in the numbers of labeled RGCs (Fig. 7A, C). These numbers closely parallel those found previously, in assays that used both Nissl staining of all GCL cells and retrograde labeling of RGCs only (Blanco et al. 2000). Two applications of FGF-2 to the optic nerve caused a significant 34% increase in the survival rate of the RGCs compared to PBS-treated, a result that, again, parallels the previous study (Fig. 7A, D). Application of siRNA to the axons concomitantly with both FGF-2 or PBS applications significantly reduced RGC survival rates. BDNF siRNA reduced the survival rate of RGCs treated only with PBS by a further 34% compared to PBS alone (Fig. 7A, E), while it reduced the survival of those treated with FGF-2 by a further 42% compared to FGF-2 alone (Fig. 7A, F), thus inhibiting FGF-2's survival effect. The first application of siRNA alone was just as effective as double application (results not shown), suggesting that its main point of entry is the cut axons.
Figure 7.

Application of BDNF siRNA to cut axons reduces the long term survival effect on RGCs elicited by FGF-2. A. Quantification of numbers of retrogradely-labeled RGCs at 6 weeks after axotomy. Axotomy alone leads to a 40% decrease in RGCs. Cell numbers decrease further with BDNF siRNA application to the cut axons. Negative control siRNA is not different from PBS alone. FGF-2 application to the cut axons rescues RGCs. Application of BDNF siRNA along with FGF-2 abolishes this survival effect. B-F. Representative flat mounts of retinas showing RGCs retrogradely labeled with axonally applied TDA with the different treatments. Scale bar in E: 50 μm.
Discussion
We showed previously that application of FGF-2 to the cut frog optic nerve can rescue many RGCs that would otherwise die (Blanco et al. 2000), in part by modulation of pro- and anti-apopotic proteins (Ríos-Muñoz et al. 2005). We have also shown that FGF-2 also upregulates the expression of BDNF and its receptor TrkB in the RGCs in the short term (Soto et al. 2006), but the physiological relevance of this to the long term survival of the neurons is not clear. Here we show that FGF-2 treatment causes long-term upregulation of BDNF in RGCs, and that preventing this upregulation blocks the FGF-2 survival effect.
FGF-2 upregulates BDNF and TrkB in the long term
We show here that FGF-2 treatment of the optic nerve, carried out at axotomy and repeated after 3 weeks, has a long term upregulatory effect on BDNF and TrkB in RGCs mediated, at least in part, through the increased synthesis of these proteins. In this study we used histological methods to localize these increases to retrogradely labeled RGCs, rather than measuring the overall changes in the retina. In this way we avoided any masking effects of the large-scale upregulation of BDNF expression that takes place after axotomy in the amacrine cells of the inner nuclear layer (Duprey-Diaz et al. 2002). With this histological analysis we determined that FGF-2 treatment to the cut optic nerve increased the number of RGCs containing BDNF, and also increased the amount of BDNF and TrkB in the RGCs. That this was due to increased synthesis, and not to acquisition of the proteins from other retinal sources, was indicated by the increase in mRNA in the GCL. These findings, coupled with those of the previous, short-term study (Soto et al. 2006), suggest that one possible mechanism by which FGF-2 rescues injured RGCs in the longer term is the endogenous upregulation of BDNF and TrkB. One way to test this is to knock down BDNF upregulation and determine whether this decreases long term survival.
Application of siRNA prevents BDNF upregulation
We used axonally-delivered BDNF siRNA to prevent the FGF-2-induced upregulation of BDNF synthesis in RGCs. This method, which, in contrast to intraocular injection, allows targeted delivery of the siRNA to the RGCs alone, was used previously to inhibit apoptotic proteins in rat RGCs (Lingor et al. 2005). At 48 h siRNA applied to FGF-2-treated axons apparently reduces total retinal BDNF mRNA levels to control levels. It should be pointed out that the mRNA total includes an unknown contribution from retinal amacrine cells, so the inhibition of BDNF synthesis in RGCs could be almost complete. At one week, BDNF mRNA has increased greatly in both PBS- and FGF-2-treated retinas, and siRNA treatment gives a correspondingly large reduction, although not down to control levels. While this could be due to a reduction in siRNA efficacy, it could simply be due to amacrine cells contributing more BDNF mRNA to the total as they begin to upregulate its synthesis.
There have been reports of off-target toxic effects of siRNAs (Fedorov et al. 2006). Although our siRNA sequences do not contain the UGGC motif that appears to be responsible for this effect, we also assayed the effects of applying control siRNAs that are not homologous to any coding sequences. These had no effect on BDNF or TrkB synthesis, suggesting that the siRNAs we use are non-toxic. In addition, we detected no effects of BDNF siRNA on TrkB mRNA levels, indicating that its effects were specific for BDNF.
BDNF upregulation is required for the survival effect of FGF-2
Having shown that FGF-2 does induce a long-term increase of BDNF staining in RGCs, and that BDNF synthesis can be blocked by applying siRNA to their axons, we then determined to what extent this would block the survival effect of FGF-2. The results show that the long-term survival effect of FGF-2 is greatly reduced by inhibiting BDNF synthesis. In fact, BDNF siRNA also reduces the survival rate after axotomy alone, showing that the BDNF that is synthesized by axotomized RGCs does in fact contribute to their long-term survival.
BDNF and its receptor, TrkB, have long been known to be co-expressed in CNS and retinal neurons (Kokaia et al. 1993; Vecino et al. 2002), with BDNF acting in autocrine or paracrine loops to prevent cell death during development (Acheson et al. 1995; Davies and Wright 1995). More recently, autocrine and paracrine actions of BDNF were shown to augment dendritic branching in cortical neurons (Horch 2004; Horch and Katz 2002; Horch et al. 1999). BDNF has different effects on Xenopus RGC dendrites depending on its source; the complexity of their branching is increased by BDNF supplied to the tectum but decreased by BDNF added directly to the eye (Lom et al. 2002; Lom and Cohen-Cory 1999). Recently, however, it was shown that dendritic stratification of mouse RGCs requires retinal BDNF, which is increased by environmental enrichment (Landi et al. 2007).
In developing amphibian and chick RGCs, there is also indirect evidence for autocrine/paracrine actions of BDNF (Cohen-Cory et al. 1996; Herzog et al. 1994). In axotomized adult rat RGCs there is upregulation of BDNF synthesis, suggesting that endogenous BDNF could potentially have a survival-promoting effect (Gao et al. 1997; Hirsch et al. 2000). Adenovirally-mediated BDNF overexpression in rat RGCs showed it to be neuroprotective during experimental glaucoma suggesting autocrine actions of this neurotrophin (Martin et al. 2003). However, to date, there has been no direct demonstration of the importance of endogenous BDNF synthesis on RGC survival after axotomy, which is what we show here.
Until recently, there were few studies indicating that there are any links between FGF-2 and BDNF and TrkB synthesis. Upregulation of TrkB mRNA by FGF-2 treatment was reported in the developing cochleovestibular ganglion (Brumwell et al. 2000), and more recently, it was shown that mice lacking endogenous FGF-2 failed to induce BDNF and TrkB mRNA expression in hippocampal cells after brain damage, suggesting that both are normally target genes for FGF-2 (Kiprianova et al. 2004). The neuroprotective effect of brimonidine on rat RGCs appears to be correlated with its upregulation of FGF-2 and also of BDNF synthesis (Gao et al. 2002; Lai et al. 2002), leaving open the possibility of a link between the two. We recently showed that FGF-2 upregulates the synthesis of both BDNF and TrkB in the short-term (Soto et al. 2006), and here we have extended those findings to show the relevance of this upregulation to long-term survival; up to 6 weeks after axotomy.
Upregulation of BDNF is clearly important for the survival of the RGCs, but only if the receptor, TrkB is still present. Rat RGCs, in fact, show a rapid, albeit transient, upregulation of BDNF expression after axotomy, but lose TrkB expression (Cheng et al. 2002; Gao et al. 1997). We have shown previously that frog RGCs show little long-term loss of TrkB after axotomy, although BDNF levels decline somewhat (Duprey-Diaz et al. 2002). The way in which axonally-applied FGF-2 promotes both BDNF and TrkB synthesis initially, therefore, makes it doubly effective in its neuroprotective effect on axotomized RGCs, boosting the response of these cells to the increased levels of BDNF in the retina. Similarly, longer survival effects and activation of the ERK signaling pathway are found in axotomized RGCs that are transfected with the TrkB gene and treated with exogenous BDNF in the rat retina (Cheng et al. 2002). We have shown previously that FGF-2 treatment to the optic nerve promotes the rapid activation of the ERK signaling pathway (Ríos-Muñoz et al. 2005), which upregulates BDNF synthesis (Soto et al. 2006), and that activity of the ERK pathway is also partially required for long term survival of RGCs. Taken together with our new data, we can suggest that sustained synthesis of BDNF by RGCs after axotomy is responsible for keeping many of them alive. FGF-2 treatment drives the early activation of the ERK pathway and affects the ratio of pro-and anti-apoptotic proteins directly, but in the long term it is FGF-2's stimulatory effects on BDNF synthesis that appear to be most critical for long-term survival. Further experiments are required to understand the signaling pathways involved in BDNF-TrkB mediated long-term survival of axotomized RGCs.
Acknowledgements
The authors would like to thank Clarissa del Cueto for expert technical assistance. REB and JMB are supported by NIH-SCORE SO6 GM08224, and in part by a Research Centers in Minority Institutions award, G12RR-03051, from the National Center for Research Resources, NIH. The confocal microscope was acquired with the support of NSF MRI 0115825 to the Institute of Neurobiology.
Grant Information: REB and JMB are supported by NIH-SCORE SO6 GM08224, and in part by a Research Centers in Minority Institutions award, G12RR-03051, from the National Center for Research Resources, NIH. The confocal microscope was acquired with the support of NSF MRI 0115825 to the Institute of Neurobiology.
References
- Acheson A, Conover JC, Fandl JP, DeChiara TM, Russell M, Thadani A, Squinto SP, Yancopoulos GD, Lindsay RM. A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature. 1995;374(6521):450–453. doi: 10.1038/374450a0. [DOI] [PubMed] [Google Scholar]
- Beazley LD, Darby JE, Perry VH. Cell death in the retinal ganglion cell layer during optic nerve regeneration for the frog Rana pipiens. Vision Res. 1986;26(4):543–556. doi: 10.1016/0042-6989(86)90003-9. [DOI] [PubMed] [Google Scholar]
- Blanco RE, Lopez-Roca A, Soto J, Blagburn JM. Basic fibroblast growth factor applied to the optic nerve after injury increases long-term cell survival in the frog retina. J Comp Neurol. 2000;423(4):646–658. [PubMed] [Google Scholar]
- Brumwell CL, Hossain WA, Morest DK, Bernd P. Role for basic fibroblast growth factor (FGF-2) in tyrosine kinase (TrkB) expression in the early development and innervation of the auditory receptor: in vitro and in situ studies. Exp Neurol. 2000;162(1):121–145. doi: 10.1006/exnr.2000.7317. [DOI] [PubMed] [Google Scholar]
- Cheng L, Sapieha P, Kittlerova P, Hauswirth WW, Di Polo A. TrkB gene transfer protects retinal ganglion cells from axotomy-induced death in vivo. J Neurosci. 2002;22(10):3977–3986. doi: 10.1523/JNEUROSCI.22-10-03977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Cory S, Escandon E, Fraser SE. The cellular patterns of BDNF and trkB expression suggest multiple roles for BDNF during Xenopus visual system development. Dev Biol. 1996;179(1):102–115. doi: 10.1006/dbio.1996.0244. [DOI] [PubMed] [Google Scholar]
- Davies AM, Wright EM. Neurotrophic factors. Neurotrophin autocrine loops. Curr Biol. 1995;5(7):723–726. doi: 10.1016/s0960-9822(95)00144-8. [DOI] [PubMed] [Google Scholar]
- Duprey-Diaz MV, Soto I, Blagburn JM, Blanco RE. Changes in brain-derived neurotrophic factor and trkB receptor in the adult Rana pipiens retina and optic tectum after optic nerve injury. J Comp Neurol. 2002;454(4):456–469. doi: 10.1002/cne.10451. [DOI] [PubMed] [Google Scholar]
- Fedorov Y, Anderson EM, Birmingham A, Reynolds A, Karpilow J, Robinson K, Leake D, Marshall WS, Khvorova A. Off-target effects by siRNA can induce toxic phenotype. Rna. 2006;12(7):1188–1196. doi: 10.1261/rna.28106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Qiao X, Cantor LB, WuDunn D. Up-regulation of brain-derived neurotrophic factor expression by brimonidine in rat retinal ganglion cells. Arch Ophthalmol. 2002;120(6):797–803. doi: 10.1001/archopht.120.6.797. [DOI] [PubMed] [Google Scholar]
- Gao H, Qiao X, Hefti F, Hollyfield JG, Knusel B. Elevated mRNA expression of brain-derived neurotrophic factor in retinal ganglion cell layer after optic nerve injury. Invest Ophthalmol Vis Sci. 1997;38(9):1840–1847. [PubMed] [Google Scholar]
- Germain F, Fernandez E, de la Villa P. Morphological signs of apoptosis in axotomized ganglion cells of the rabbit retina. Neuroscience. 2007;144(3):898–910. doi: 10.1016/j.neuroscience.2006.10.039. [DOI] [PubMed] [Google Scholar]
- Goodman LJ, Valverde J, Lim F, Geschwind MD, Federoff HJ, Geller AI, Hefti F. Regulated release and polarized localization of brain-derived neurotrophic factor in hippocampal neurons. Mol Cell Neurosci. 1996;7(3):222–238. doi: 10.1006/mcne.1996.0017. [DOI] [PubMed] [Google Scholar]
- Herzog KH, Bailey K, Barde YA. Expression of the BDNF gene in the developing visual system of the chick. Development. 1994;120(6):1643–1649. doi: 10.1242/dev.120.6.1643. [DOI] [PubMed] [Google Scholar]
- Hirsch S, Labes M, Bahr M. Changes in BDNF and neurotrophin receptor expression in degenerating and regenerating rat retinal ganglion cells. Restor Neurol Neurosci. 2000;17(2−3):125–134. [PubMed] [Google Scholar]
- Horch HW. Local effects of BDNF on dendritic growth. Rev Neurosci. 2004;15(2):117–129. doi: 10.1515/revneuro.2004.15.2.117. [DOI] [PubMed] [Google Scholar]
- Horch HW, Katz LC. BDNF release from single cells elicits local dendritic growth in nearby neurons. Nat Neurosci. 2002;5(11):1177–1184. doi: 10.1038/nn927. [DOI] [PubMed] [Google Scholar]
- Horch HW, Kruttgen A, Portbury SD, Katz LC. Destabilization of cortical dendrites and spines by BDNF. Neuron. 1999;23(2):353–364. doi: 10.1016/s0896-6273(00)80785-0. [DOI] [PubMed] [Google Scholar]
- Kiprianova I, Schindowski K, von Bohlen und Halbach O, Krause S, Dono R, Schwaninger M, Unsicker K. Enlarged infarct volume and loss of BDNF mRNA induction following brain ischemia in mice lacking FGF-2. Exp Neurol. 2004;189(2):252–260. doi: 10.1016/j.expneurol.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Kokaia Z, Bengzon J, Metsis M, Kokaia M, Persson H, Lindvall O. Coexpression of neurotrophins and their receptors in neurons of the central nervous system. Proc Natl Acad Sci U S A. 1993;90(14):6711–6715. doi: 10.1073/pnas.90.14.6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai RK, Chun T, Hasson D, Lee S, Mehrbod F, Wheeler L. Alpha-2 adrenoceptor agonist protects retinal function after acute retinal ischemic injury in the rat. Vis Neurosci. 2002;19(2):175–185. doi: 10.1017/s0952523802191152. [DOI] [PubMed] [Google Scholar]
- Landi S, Cenni MC, Maffei L, Berardi N. Environmental enrichment effects on development of retinal ganglion cell dendritic stratification require retinal BDNF. PLoS ONE. 2007;2:e346. doi: 10.1371/journal.pone.0000346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin GR, Barde YA. Physiology of the neurotrophins. Annu Rev Neurosci. 1996;19:289–317. doi: 10.1146/annurev.ne.19.030196.001445. [DOI] [PubMed] [Google Scholar]
- Lingor P, Koeberle P, Kugler S, Bahr M. Down-regulation of apoptosis mediators by RNAi inhibits axotomy-induced retinal ganglion cell death in vivo. Brain. 2005;128(Pt 3):550–558. doi: 10.1093/brain/awh382. [DOI] [PubMed] [Google Scholar]
- Lom B, Cogen J, Sanchez AL, Vu T, Cohen-Cory S. Local and target-derived brain-derived neurotrophic factor exert opposing effects on the dendritic arborization of retinal ganglion cells in vivo. J Neurosci. 2002;22(17):7639–7649. doi: 10.1523/JNEUROSCI.22-17-07639.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lom B, Cohen-Cory S. Brain-derived neurotrophic factor differentially regulates retinal ganglion cell dendritic and axonal arborization in vivo. J Neurosci. 1999;19(22):9928–9938. doi: 10.1523/JNEUROSCI.19-22-09928.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KR, Quigley HA, Zack DJ, Levkovitch-Verbin H, Kielczewski J, Valenta D, Baumrind L, Pease ME, Klein RL, Hauswirth WW. Gene therapy with brain-derived neurotrophic factor as a protection: retinal ganglion cells in a rat glaucoma model. Invest Ophthalmol Vis Sci. 2003;44(10):4357–4365. doi: 10.1167/iovs.02-1332. [DOI] [PubMed] [Google Scholar]
- Pease ME, McKinnon SJ, Quigley HA, Kerrigan-Baumrind LA, Zack DJ. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Invest Ophthalmol Vis Sci. 2000;41(3):764–774. [PubMed] [Google Scholar]
- Quigley HA, McKinnon SJ, Zack DJ, Pease ME, Kerrigan-Baumrind LA, Kerrigan DF, Mitchell RS. Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. Invest Ophthalmol Vis Sci. 2000;41(11):3460–3466. [PubMed] [Google Scholar]
- Ríos-Muñoz W, Soto I, Duprey-Díaz MV, Blagburn JM, Blanco RE. Fibroblast growth factor 2 applied to the optic nerve after axotomy increases Bcl-2 and decreases Bax in ganglion cells by activating the ERK signaling pathway. Journal of Neurochemistry. 2005;93:1422–1433. doi: 10.1111/j.1471-4159.2005.03129.x. [DOI] [PubMed] [Google Scholar]
- Scalia F, Arango V, Singman EL. Loss and displacement of ganglion cells after optic nerve regeneration in adult Rana pipiens. Brain Res. 1985;344(2):267–280. doi: 10.1016/0006-8993(85)90804-2. [DOI] [PubMed] [Google Scholar]
- Sievers J, Hausmann B, Unsicker K, Berry M. Fibroblast growth factors promote the survival of adult rat retinal ganglion cells after transection of the optic nerve. Neurosci Lett. 1987;76(2):157–162. doi: 10.1016/0304-3940(87)90708-7. [DOI] [PubMed] [Google Scholar]
- Snider WD. Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell. 1994;77(5):627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- Soto I, Rosenthal JJ, Blagburn JM, Blanco RE. Fibroblast growth factor 2 applied to the optic nerve after axotomy up-regulates BDNF and TrkB in ganglion cells by activating the ERK and PKA signaling pathways. J Neurochem. 2006;96(1):82–96. doi: 10.1111/j.1471-4159.2005.03510.x. [DOI] [PubMed] [Google Scholar]
- Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270(5236):593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- Vecino E, Garcia-Grespo D, Garcia M, Martinez-Millan L, Sharma SC, Carrascal E. Rat retinal ganglion cells co-express brain derived neurotrophic factor (BDNF) and its receptor TrkB. Vision Res. 2002;42(2):151–157. doi: 10.1016/s0042-6989(01)00251-6. [DOI] [PubMed] [Google Scholar]
- von Bartheld CS. Neurotrophins in the developing and regenerating visual system. Histol Histopathol. 1998;13(2):437–459. doi: 10.14670/HH-13.437. [DOI] [PubMed] [Google Scholar]