

Abstract
Protein kinase D1 (PKD1) is expressed ubiquitously and regulate diverse cellular processes such as oxidative stress, gene expression, cell survival, and vesicle trafficking. However, the presence and function of PKD1 in monocytic cells are currently unknown. Here we provide evidence that PKD1 is involved in Toll-like receptor (TLR) 9 signaling in macrophages. Class B type CpG DNA (CpG-B DNA) induced activation of PKD1 via a pathway that is dependent on endosomal pH, TLR9, MyD88, and IRAK1 in macrophages. Upon CpG-B DNA stimulation, PKD1 interacted with the TLR9/MyD88/IRAK/TRAF6 complex. Knockdown of PKD1 revealed that PKD1 is required for activation of NF-κB and MAPKs, and subsequent expression of cytokines in response to CpG-B DNA. Our findings identify PKD1 as a key signaling modulator in TLR9-mediated macrophage activation.
Keywords: Macrophages, Cytokines, Kinases, Inflammation, Toll-like receptor
INTRODUCTION
The success of host defense against microbial infection depends on the capacity of innate immune cells to detect invading pathogens. Innate immune cells express groups of structurally related proteins called pattern recognition receptors (PRR) such as Toll-like receptor (TLR)-family proteins, NACHT-leucine-rich repeat (NLR)-family proteins, and helicase domain-containing antiviral proteins (1–4). Recognition of the evolutionally conserved structures in microorganisms (pathogen-associated molecular pattern; PAMP), including lipopolysaccharide and microbial nucleic acids, by PRRs in innate immune cells initiates the complex signaling cascades leading to secretion of pro-inflammatory cytokines and mediators that control growth and dissemination of invading pathogens.
DNA released by bacterial and viral pathogens is recognized by host cells through at least two different mechanisms: TLR9 pathway and cytosolic DNA pathway (5–7). In the cytosolic DNA pathway, the yet to be discovered cytosolic DNA receptor recognizes double-stranded DNA with a native phosphodiester backbone in a DNA sequence-independent manner in cytoplasm. It induces production of type I interferon (IFN) and other immune responses by interacting with and activating Tank binding kinase (TBK) 1 and IFN regulatory factor (IRF) 3 (6, 8, 9). In contrast, TLR9 recognizes both single- and double-stranded DNA with synthetic phosphorothioate backbones as well as native phosphodiester backbones via an endosomal pH-sensitive and DNA sequence-specific (unmethylated CpG motif) manner in endosomal compartments, and its signal transduction requires an adaptor molecule, myeloid differentiation factor (MyD) 88 (10). Most, if not all, known biologic effects of the unmethylated CpG motifs containing bacterial DNA or synthetic DNA are mediated through a TLR9/MyD88 signaling pathway. Ligand (CpG motif containing DNA)-bound TLR9 recruits MyD88 and initiates a signaling pathway that sequentially involves IL-1R-associated kinase (IRAK) 4, IRAK1, tumor necrosis factor-α receptor-associated factor (TRAF) 6 and TRAF3 (2, 11). Activation of TRAF6 and TRAF3 initiates signaling cascades that lead to activation of NF-κB, mitogen-activated protein kinases (MAPKs) and IRFs, and subsequent expression of various proinflammatory cytokines, chemokines, and oncogenes. In addition to this well defined TLR9/MyD88 signaling pathway, CpG motif containing DNA has also been shown to induce generation of reactive oxygen species (ROS) and activation of Ras and Vav1 (12–15). The mechanisms by which CpG motif containing DNA activates these additional signaling modulators are yet to be revealed.
The protein kinase D family is a group of three structurally related serine/threonine kinases; PKD1/PKCμ, PKD2 and PKD3/PKCν (16). PKDs are expressed ubiquitously, including in B and T lymphocytes (17, 18). PKDs can be activated by upstream signaling modulators such as phosphoinositide 3-kinase (PI3K), phospholipase Cγ (PLCγ), diacylglycerol (DAG), and classical/novel protein kinase Cs (PKCs) that are activated through signals transduced by G-protein coupled-receptor agonists, ROS, epidermal growth factor receptor, FcγRII, B-cell antigen receptor (BCR), and T-cell receptor (TCR) (18–24). Depending on the cell context and the stimulation conditions, PKDs display dynamic changes in intracellular locations. They can be located in cytosol, the plasma membrane, the mitochondria, the Golgi apparatus, or the nucleus (20, 25–30). Cellular location of PKDs has been shown to dictate the biological role of the protein. PKDs have been demonstrated to be implicated in the regulation of a variety of cellular and subcellular processes such as vesicle transportation, ROS generation, activation of MAPKs and NF-κB, and regulation of cell shape, motility, adhesion, and gene expression (18, 20, 29–33). There have been a couple of studies that indicate a possibility that PKD family members may be involved in TLR signaling in non-immune cells. LPS-mediated p38 activation and TNF-α secretion in microglial cells is suppressed by a PKC/PKD inhibitor, Gö6976(34). PKD1 has been shown to bind and phosphorylate TLR5, and Gö6976 suppresses flagellin-mediated p38 activation and IL-8 production in epithelial cells (35). However, to date there is no evidence that pathogenic DNA induces activation of PKD family members, and it is not known whether innate immune cells like macrophages express PKD family proteins and whether PKD family members play an active role in macrophage activation induced by pathogenic DNA. In this study, we demonstrate that macrophages express all three PKD family members and that bacterial DNA and class B type CpG DNA (CpG-B DNA) induce activation of PKD1. In addition, we have identified a part of the biochemical mechanisms by which CpG-B DNA activates PKD1 and the biologic functions of PKD1 in macrophages.
MATERIALS AND METHODS
Mice
C57BL/6 and BALB/c mice at 4–5 weeks of age were obtained from The Frederick Cancer Research and Development Center, National Cancer Institute (Frederick, MD) and were used within 3 weeks. TLR9 gene-deficient (TLR9−/−) and MyD88−/− mice were provided by Dr. S. Akira (Osaka University, Osaka, Japan). All animal care and housing requirements set forth by the National Institutes of Health Committee on the Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources were followed, and animal protocols were reviewed and approved by the University of Tennessee Animal Care and Use Committee.
Isolation of primary murine macrophages, dendritic cells (DCs), and B cells, human peripheral blood mononuclear cells (PBMCs), cell lines, and culture conditions
Peritoneal cells were obtained by lavage of the peritoneal cavity of mice with 8 ml of cold sterile saline and washed three times with ice-cold PBS. Spleen cells were obtained by crushing spleen with a syringe plunger in 10 ml of cold sterile saline. After removing red blood cells (RBC) using RBC lysis buffer, peritoneal cell suspensions and splenic cell suspensions were subjected to Fico/Lite-LM (Atlanta Biologicals, Atlanta, GA) density gradient centrifugation. T-cells in peritoneal cell suspensions and splenic cell suspensions were depleted using Dynabeads® Mouse pan T (Thy1.2) according to the manufacturer’s protocol (Invitrogen, Carlsbad, CA). The resulting untouched peritoneal cells and splenic cells were suspended in complete medium and incubated at 37°C for 1hr in plastic petri dishes. Suspended cells were recovered, washed 3 times with PBS, and used as B lymphocytes. Plastic adherent cells were washed 3 times with pre-warmed PBS and then collected by scraping. The resulting plastic adherent cells were used as macrophages. Kupffer cells (KCs) were isolated as previously described (36). The purity of macrophage preparations was assessed by flow cytometry after staining with biotin-conjugated anti-mouse CD11b (Mac-1) Ab or anti-mouse F4/80 and streptavidin-PE (BD Biosciences, San Diego, CA). To isolate splenic conventional dendritic cells (cDCs) and plasmacytoid dendritic cells (pDCs), CD11c+ cells were enriched from splenic cell suspensions using a CD11c positive selection kit according to the manufacturer’s protocol (Miltenyi Biotech Inc., Auburn, CA). After enrichment, CD11c+ cells were stained with PE-conjugated anti-mouse CD11c and FITC-conjugated anti-mouse B220 (BD Biosciences, San Diego, CA) and then specific populations of cDC (CD11c+B220−) and pDC (CD11c+B220+) cells were separated by BD FACSAria II Cell Sorter (BD Biosciences, San Diego, CA). The purity of cDC and pDC was assessed by flow cytometry after sorting. Human peripheral blood was purchased from Mid-South Regional Blood Center (Memphis, TN) and PBMCs were isolated by Ficoll-paque (Roche Diagnostics, Mannheim, Germany) density gradient centrifugation. Primary macrophages, cDCs, pDCs, human PBMCs, and RAW264.7 (ATCC, Manassas, VA) were cultured in D-MEM supplemented with 10% (v/v) heat-inactivated fetal calf serum, 1.5 mM of L-glutamine, 100 units/ml of penicillin, and 100 μg/ml of streptomycin at 37 °C in a 5% CO2-humidified incubator. Splenic B cells were cultured in RPMI-1640 supplemented with 10% (v/v) heat-inactivated fetal calf serum, 50 μM of 2-ME, -glutamine, 100 units/ml of penicillin, 1.5 mM of Land 100 μg/ml of streptomycin at 37 °C in a purchased from Invitrogen and Sigma 5% CO2-humidified incubator. All culture reagents were(St. Louis, MO).
Oligodeoxynucleotides and reagents
Nuclease-resistant phosphorothioate oligodeoxynucleotides (S-ODN) 1585 (class A type CpG DNA for mouse), 1826 (class B type CpG DNA for mouse), 1982 (non-CpG DNA for mouse), 2088 (iCpG DNA for mouse and human), 2006 (class B type CpG DNA for human), and 2137 (non-CpG DNA for human) were purchased from Operon (Alameda, CA) and Coley Pharmaceutical Group (Kanata, Ontario, Canada) and further purified by ethanol precipitation. S-ODN had no detectable endotoxins by Limulus assay. Sequences of S-ODNs were previously reported (37–40). E. coli DNA (EC DNA), Calf thymus DNA (CT DNA), and EGTA were purchased from Sigma. PP1 was purchased from Invitrogen. SU6656, LY294002, U73122, Gö6976, Gö6983 and Gö6850 were purchased from Calbiochem (La Jolla, CA).
Plasmids, generation of FLAG-tagged PKD-expressing macrophages and gene-specific knockdown macrophages, transient transfection, reporter gene assay, and RT-PCR
To make the FLAG-tagged PKD1, PKD2, and PKD3 constructs, the coding regions of PKD1, PKD2, and PKD3 were amplified by PCR and then subcloned into pIRES-hrGFPII vector (Stratagene, La Jolla, CA). DNA sequences of all cloned genes were confirmed by DNA sequencing analysis and were identical with the previously reported sequences. To make the shRNA-expressing constructs, forward and reverse synthetic 60 nt ODNs containing the target sequences for C. elegans luciferase, PKD1, or IRAK1 were annealed and then inserted into the BglII/HindIII sites of pSuper.Retro.neo.GFP vectors, following the manufacturer’s instructions (Oligoengine, Seattle, WA). To generate stable transfectants that constitutively express FLAG-tagged PKD1, FLAG-tagged PKD2, FLAG-tagged PKD3, luciferase-shRNA, PKD1-shRNA, or IRAK1-shRNA, RAW264.7 cells were transfected with pIRES-hrGFPII, pIRES-hrGFPII-PKD1, pIRES-hrGFPII-PKD2, pIRES-hrGFPII-PKD3, pSuper.Retro.neo.GFP-luciferase-shRNA, pSuper.Retro.neo.GFP-PKD1-shRNA, or pSuper.Retro.neo.GFP-IRAK1-shRNA using FuGENE 6 transfection reagent (Roche Diagnostics, Mannheim, Germany) according to the manufacturer’s instructions. Stable transfectants were selected and maintained in selection medium containing G418 (1 mg/ml). Transient transfections, reporter gene assays, and RT-PCR were done as previously described (37, 41). Luciferase activity was normalized using pRL-TK-luciferase activity (Renilla) in each sample. Actin or GAPDH was used as a loading control for all RT-PCR. All primers for cloning, gene-specific shRNA targeting, and RT-PCR were purchased from Integrated DNA Technologies, INC (Coralville, IA) and sequences of primers are listed in Table 1.
Table 1.
Sequences of primers
| Gene | Forward/sense primer | Reverse/anti-sense primer | |
|---|---|---|---|
| Cloning | PKD1 | ATGAGCGTCCCTCCGCTGCT | GAGGATGCTGACACGCTCAC |
| PKD2 | ATGGCCGCCGCCCCCTCCCAT | GAGGATGCTGATGCGCTCAG | |
| PKD3 | ATGTCTGCAAATAATTCCCCTC | AGGATCCTCCTCCATGTCGT | |
|
| |||
| shRNA | Luciferase | GACAGTGTGGCATTTATTA | TAATAAATGCCACACTGTC |
| PKD1 802 | CTCCTGATGTCTAAGGTGA | TCACCTTAGACATCAGGAG | |
| PKD1 2453 | CCATTGATCTTATCAATAA | TTATTGATAAGATCAATGG | |
| IRAK1 845 | GCTTATACTGCCTTGTTTA | TAAACAAGGCAGTATAAGC | |
|
| |||
| RT- PCR | TNF-α | CGATGGGTTGTACCTTGTCTACTC | TGGAAGACTCCTCCCAGGTATATG |
| IL-6 | AAGTCCGGAGAGGAGACTTCACAG | GTCTTGGTCCTTAGCCACTCCTTC | |
| IL-10 | TATGCTGCCTGCTCTTACTGACTG | TTCATGGCCTTGTAGACACCTTGG | |
| IL-12 p40 | AGACCCTGACCATCACTGTCAAAG | GAAGAAGCTGGTGCTGTAGTTCTC | |
| * IFN-α | ATGGCTAGRCTCTGTGCTTTCCT | AGGGCTCTCCAGAYTTCTGCTCTG | |
| * IFN-β | CATCAACTATAAGCAGCTCCA | TTCAAGTGGAGAGCAGTTGAG | |
| GAPDH | GTCTTCACCACCATGGAGAAGGC | GGCATGGACTGTGGTCATGA | |
| PKD1 | CGGTACTGTCCGCACAGCAT | ATTCTGCACCAGGGCTAAGTAAT | |
| PKD2 | ATTCTGTGCGCCTTGGCAG | GCTTCCTCCATCGGCACG | |
| PKD3 | GCCAGGACCTGGCCTCTC | GGGCATGTCTGCATCAGTC | |
| PKCα | AAATCGCCGACTTTGGGATG | TTGGGCTTGAATGGTGGTTG | |
| PKCε | GGCTCGGAAACACCCTTATC | CAAGTCGTCCTCGTTGTCAG | |
| PKCζ | CTGTCATGCCTTCCCAAGAG | CTGGAAGCAGGAGTGTAAGC | |
| TLR9 | AGCTGAACATGAACGGCATCT | AAAACTGAAATTGTGGCCTA | |
| MyD88 | CCCAACGATATCGAGTTTGT | TTCTTCATCGCCTTGTATTT | |
| IRAK1 | ATGGCAGCCTCTAGTAGTGA | CTATCCAAGACCCCTTCTTC | |
| IRAK4 | CAAAATATCTGACTTTGGGC | TAGTTGAGGTTCACGGTTTT | |
| TRAF6 | AGGCAGGTTTCTTGTGTAAA | ATGTGCAACTGGGTATTCTC | |
| Actin | AAGGCCAACCGTGAAAAGATGACC | ACCGCTCGTTGCCAATAGTGATGA | |
| hIL-6 | ACTCACCTCTTCAGAACGAA | CTCAAACTCCAAAAGACCAG | |
| hIL-8 | CACCCCAAATTTATCAAAGA | TCAAAAACTTCTCCACAACC | |
| hActin | GTGGGGCGCCCCAGGCACCA | CTCCTTAATGTCACGCACGATTTC | |
: Primer sequences of IFN-α and -β were described previo/usly (58)
Enzyme-linked immunosorbent assay (ELISA), Western blot assay and electrophoretic mobility shift assay (EMSA)
Concentrations of the selected cytokines in culture supernatants, levels or phosphorylation status of specific proteins in whole cell extracts, and nuclear DNA binding activities of NF-κB and AP-1 were analyzed by ELISA, Western blot assay, and EMSA, respectively, as described previously (15, 37, 42). Actin was used as a loading control for all Western blot assays. Specificity of the NF-κB and AP-1 bands was confirmed by cold competition studies and super-shift assay (data not shown). All recombinant murine cytokines and antibodies (Abs) specific for murine cytokines were purchased from BD Biosciences (San Diego, CA). IFN-α ELISA kit was purchased from R&D Systems (Minneapolis, MN). Abs specific for actin, PKD, IκBα and IκBβ were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). All phospho-specific Abs were purchased from Cell Signaling (Beverly, MA).
In vitro kinase assays
RAW264.7 cells stably expressing empty vector, FLAG-tagged PKD1, FLAG-tagged PKD2, or FLAG-tagged PKD3 were stimulated with medium or CpG-B DNA. Lysates were prepared and each FLAG-tagged PKD protein was immunoprecipitated with anti-FLAG Ab. The resulting immune complexes were subjected to in vitro kinase assay using Syntide-2 (Sigma) as a PKD substrate, as previously described (43).
Analysis of protein-protein interaction
RAW264.7 cells stably expressing empty vector, FLAG-tagged PKD1, FLAG-tagged PKD2, or FLAG-tagged PKD3 were stimulated with medium or CpG-B DNA for 30 min. Each FLAG-tagged PKD family protein or endogenous protein in TLR9 signaling (TLR9, MyD88, IRAK1, IRAK4, TRAF6, or TRAF3) in the cell lysates was immunoprecipitated with EZview Red ANTI-FLAG M2 Affinity Gel (Sigma) or Abs specific for each individual protein. The presence of PKDs and proteins in the TLR9-signaling pathway in the resulting immunoprecipitates was detected by Western blot using Abs specific for FLAG (Sigma), TLR9 (Imgenex, San Diego, CA), IRAK1 (Millipore, Billerica, MA), IRAK4 (Abgent, San Diego, CA), MyD88 (Santa Cruz), TRAF6 (Santa Cruz), or TRAF3 (Santa Cruz).
RESULTS
Class B type CpG DNA activates PKD1 in macrophages
PKD family proteins are expressed in T- and B-lymphocytes and play a regulatory role in TCR- and BCR-mediated lymphocyte activation (17, 18). However, the presence and function of PKD proteins in macrophages are currently unknown. The results obtained from PKD gene-specific RT-PCR revealed that all three PKD family members were expressed in murine primary macrophages and a murine macrophage cell line RAW264.7 cells (Fig. 1A). Abundant expression of PKD family proteins in macrophages suggests a possibility that PKDs may be involved in innate immune responses. Therefore, we have investigated whether pathogenic DNA activates one or more PKD family members in macrophages. As demonstrated in Figure 1B–1E, CpG-B DNA that transduces signal through TLR9 induced phosphorylation of PKD family members in a time-, dose-, and sequence-dependent manner in RAW264.7 cells and primary murine macrophages (in vivo and in vitro). However, liposome combined transfection of poly dA/dT that induces IFN-β expression through yet to be identified cytosolic DNA receptor (44) did not induce activation of PKD family proteins in macrophages (Fig. 1F). Our results indicate that certain pathogenic DNA, such as CpG motif containing DNA, induces activation of one or more PKD family members in macrophages.
Figure 1. CpG-B DNA induces phosphorylation of PKD family members in macrophages.
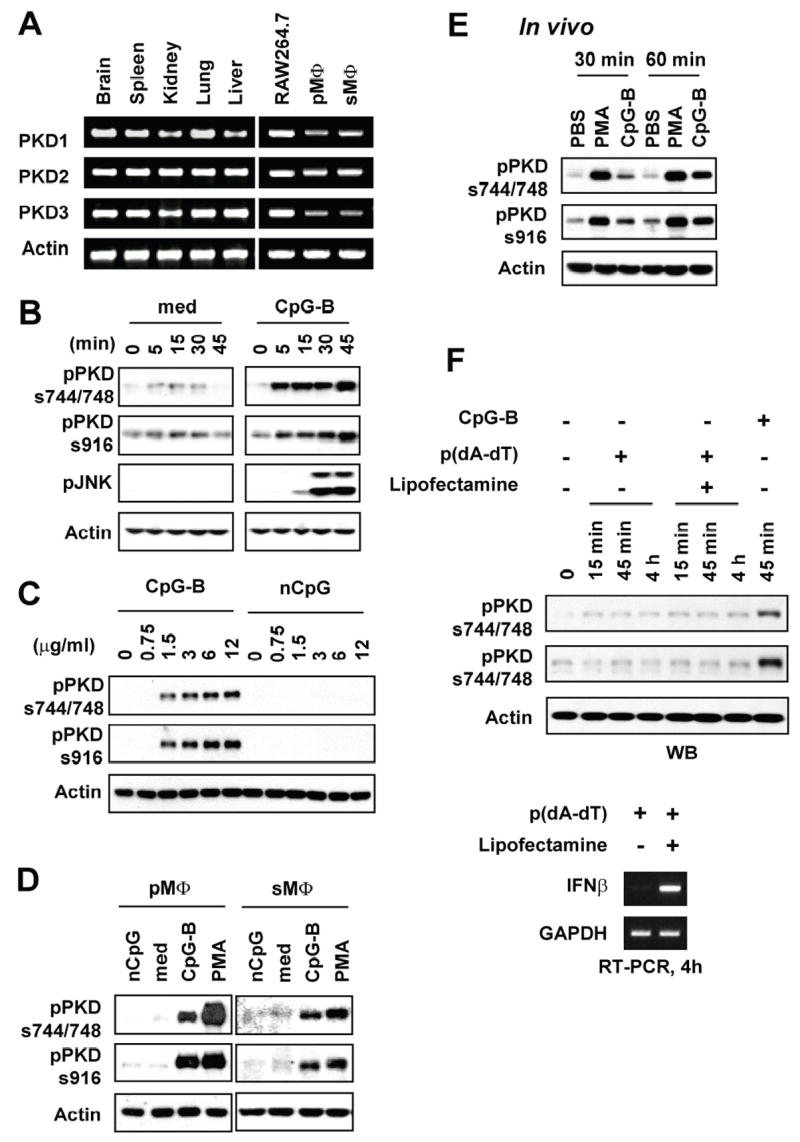
A, Levels of the indicated mRNA in various tissues, RAW264.7 cells and primary macrophages isolated from C57BL/6 mice were detected by RT-PCR analysis. PMΦ: peritoneal macrophages, sMΦ: splenic macrophages. B–D, RAW264.7 cells (Panels b and c), peritoneal (pMΦ) or splenic (sMΦ) macrophages isolated from C57BL/6 mice (Panel d) were stimulated with medium (med), CpG-B DNA (CpG-B), non-CpG DNA (nCpG), or PMA. Concentrations of DNA used are either indicated or 12 μg/ml. PMA was used at 10 ng/ml. Periods of DNA stimulation are either indicated or 45 min. Activation status of PKDs was detected by phospho-specific Western blot assay using Abs specific for the phosphorylated forms of PKD (pPKDs744/748, pPKDs916). E, BALB/c mice were injected intraperitoneally with PBS, CpG-B DNA (30 μg/mouse), or PMA (30 ng/mouse). At the indicated times later, peritoneal cells were isolated. Activation status of PKDs was detected by phospho-specific Western blot assay. F, RAW264.7 cells were stimulated with 6 μg of poly(dA-dT) in the presence or absence of lipofectamine or CpG-B DNA (6 μg) up to 4 h. Activation status of PKDs was detected by phospho-specific Western blot assay. Expression of IFN-β as a stimulation indicator was detected by RT-PCR.
Because, in addition to macrophages, other types of cells, including B cells and DCs, also express TLR9 (the CpG motif containing DNA receptor) and respond to CpG motif containing DNA, we further investigated whether CpG motif containing DNA can induce activation of PKD family members in B cells, pDCs, cDCs, and human PBMCs. As shown in Figure 2, CpG-B DNA induced phosphorylation of PKD family members in murine primary splenic B cells, pDCs, and cDCs. In addition, CpG-B DNA and EC DNA induced activation of PKD family proteins in human PBMCs. In contrast, CpG-A DNA, and controls CT DNA, non-CpG DNA, and DNase I-treated EC DNA did not induce phosphorylation of PKDs in these cells. Taken together, our results suggest that type B CpG motif containing DNA induces activation of one or more PKD family members in murine macrophages, B cells, pDCs, and cDCs, and human PBMCs.
Figure 2. CpG-B DNA, not CpG-A DNA, induces phosphorylation of PKD proteins in murine splenic B cells, cDCs, and pDCs, and human PBMCs.
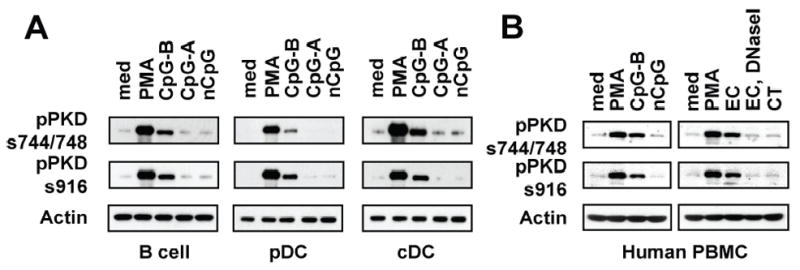
A, Splenic B cells, pDCs and cDCs isolated from BALB/c mice were stimulated with medium (med), CpG-A DNA, CpG-B DNA, non-CpG DNA, or PMA for 45 min. B, Human PBMCs were stimulated with medium, PMA, CpG-B DNA, nCpG, E. coli DNA (EC), EC plus DNase I, or calf thymus DNA (CT) for 45 min. Activation status of PKDs was detected by phospho-specific Western blot assay.
As mentioned previously, three PKD family proteins have been identified up to date. Due to high homologies between three PKD family members, pPKDs744/748-specific antibody detects phosphorylation of all PKD family proteins and pPKDs916-specific antibody detects phosphorylation of PKD1 and PKD2 (data not shown). Therefore, it was necessary for us to further identify PKD family members activated by CpG-B DNA. RAW264.7 cells stably expressing each FLAG-tagged PKD family member were stimulated with CpG-B DNA. Phospho-specific Western blot and in vitro kinase assay following immunoprecipitation of each FLAG-tagged PKD family member showed that CpG-B DNA induced kinase activity and phosphorylation of PKD1 in RAW264.7 cells (Fig. 3). However, we could not detect kinase activity and phosphorylation of PKD2 and PKD3 in RAW264.7 cells stimulated with CpG-B DNA under our experimental conditions. Of note, PMA induced kinase activity and phosphorylation of three PKD family proteins (data not shown). Taken together, our results provided evidence that macrophages express all three PKD family members and CpG-B DNA activates PKD1.
Figure 3. CpG-B DNA induces activation of PKD1 but not PKD2 and PKD3.
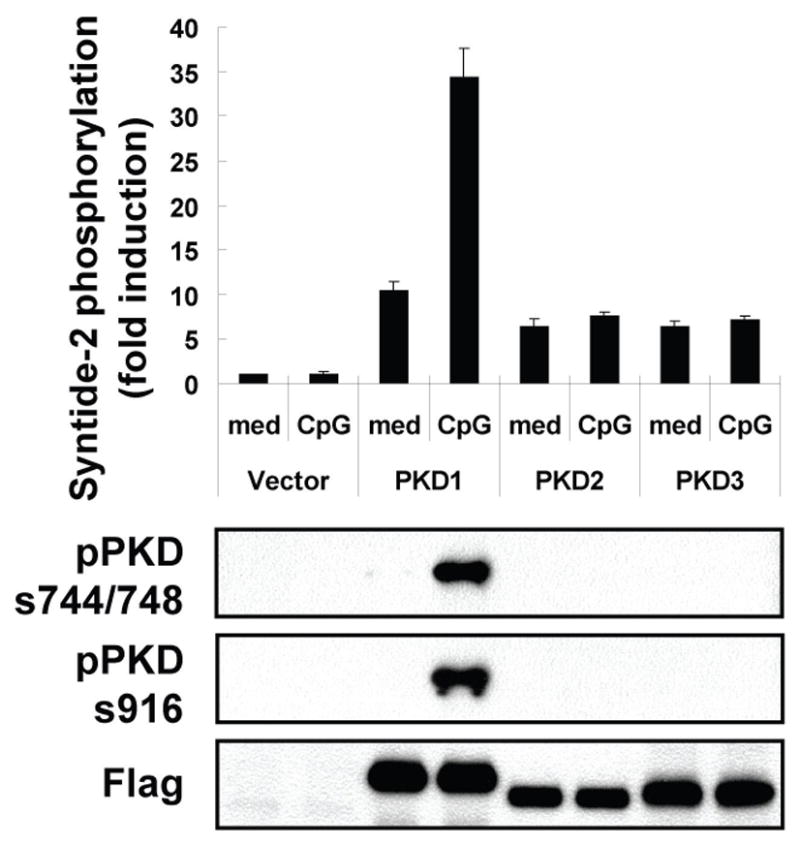
RAW264.7 cells stably expressing empty vector, FLAG-tagged PKD1, FLAG-tagged PKD2, or FLAG-tagged PKD3 were stimulated with medium, CpG-B DNA (12 μg/ml) for 45 min. Each PKD family protein in whole cell lysates was immunoprecipitated with anti-FLAG Ab. Kinase activity of PKDs was analyzed by in vitro kinase assay using syntide-2 as a PKD substrate (top). Expression and phosphorylation status of each PKD was analyzed by immunoblotting with anti-FLAG and anti-phospho-PKD Abs, respectively (bottom).
CpG-B DNA-mediated cytokine production is ablated in PKD1-knockdown macrophages
Pharmacological inhibitors that inhibit function or activation of specific signaling molecules can be useful tools to uncover the biological roles of the signaling molecule as well as effective therapeutic agents if the signaling molecule is involved in the pathogenesis of certain diseases. Although the pharmacological inhibitor that specifically inhibits only PKD has yet to be developed, careful use of several pharmacological inhibitors can provide insight to the biologic role of PKD. To investigate the biologic role of CpG-B DNA-activated PKD1, we employed three pharmacological inhibitors: Gö6850 selectively inhibits PKCα (IC50 = 8 nM) and βI (IC50 = 18 nM); Gö6976 selectively inhibits PKCα (IC50 = 2.3 nM), βI (IC50 = 6.2 nM), and PKD (IC50 = 20 nM) but does not inhibit PKCδ, ε, and ζ; Gö6983 selectively inhibits PKCα (IC50 = 7 nM), β (IC50 = 7 nM), γ (IC50 = 6 nM), δ (IC50 = 10 nM), and ζ (IC50 = 60 nM) but does not inhibit PKD, and is used together with Gö6976 to differentiate PKD from other PKC isozymes (45, 46). Murine macrophage RAW264.7 cells, murine splenic B cells, murine pDCs, and human PBMCs were pre-treated with DMSO, PKD inhibitor Gö6976 or PKC inhibitors Gö6983 or Gö6850. As shown in Figure 4A–4D, PKD inhibitor Gö6976 inhibited CpG-B DNA-mediated expression of cytokines (TNF-α, IL-6, IL-8, IL-10, IL-12) in RAW264.7 cells, pDCs, splenic B cells, and human PBMCs. In contrast, PKC inhibitors Gö6983 and Gö6850 (that do not inhibit PKD) failed to inhibit CpG-B DNA-mediated expression of these cytokines. As expected, neither Gö6976 nor Gö6983 affected the ability of CpG-A DNA (which does not induce activation of PKD1) to induce IFN-α production in pDCs (Fig. 4B). To further investigate whether PKD1 is also critical in CpG-B DNA-mediated expression of cytokines in vivo, BALB/c mice were stimulated with CpG-B DNA in the presence or absence of PKD inhibitor Gö6976 or PKC inhibitor Gö6983 for indicated time periods and protein and mRNA levels of the selected cytokines (TNF-α, IL-6, IL-10, and IL-12) in serum, peritoneal macrophages, spleen, and liver residential macrophage KCs were analyzed. As demonstrated in Figure 4E, Gö6976, but not Gö6983, selectively inhibited CpG-B DNA-mediated cytokine mRNA expression in peritoneal macrophages, KCs, and spleen in mice in vivo. In addition, serum levels of cytokines TNF-α, IL-6, IL-10, and IL-12 in response to CpG-B DNA were dramatically suppressed in the mice pretreated with Gö6976, but not in the mice pretreated with Gö6983 (Fig. 4F). Taken together, these results suggested that PKD1 plays a pivotal role in CpG-B DNA-mediated cytokine expression in CpG-B DNA-responding cells, including macrophages, B cells and pDCs, in vitro and in vivo.
Figure 4. Effect of PKC/PKD inhibitors on CpG-B DNA-mediated cytokine expression.
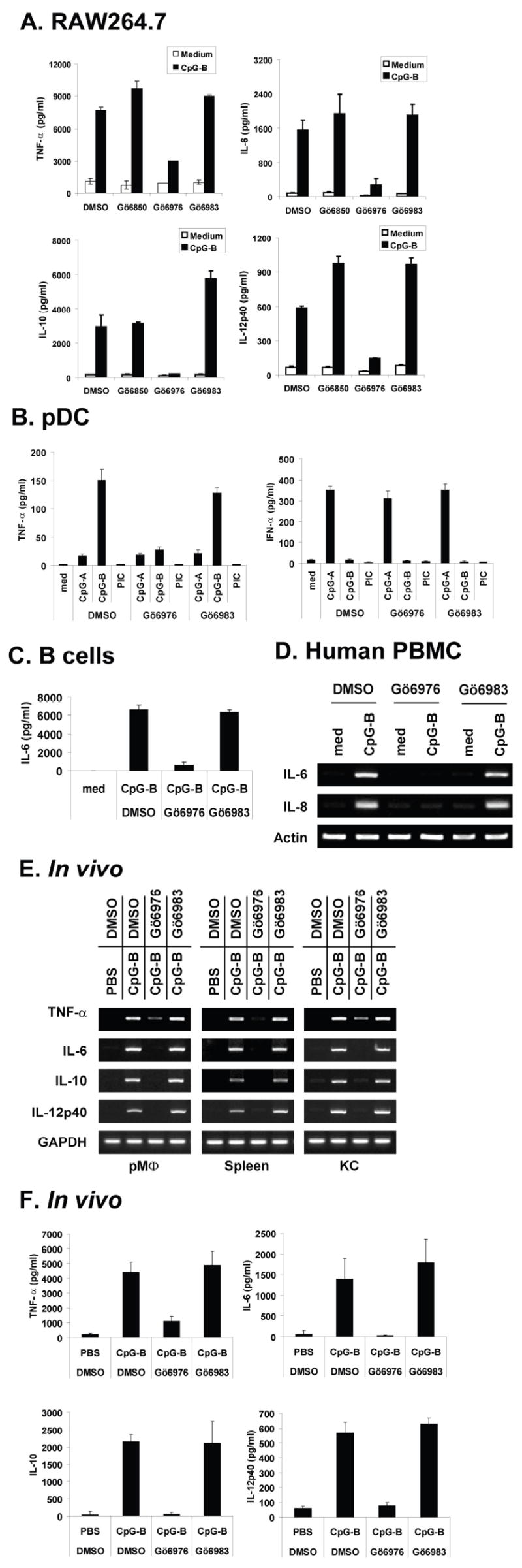
A–C, RAW264.7 cells (A), splenic pDCs (B), and splenic B cells (C) were pre-treated with DMSO, Gö6850 (500 ng/ml), Gö6976 (250 ng/ml), or Gö6983 (250 ng/ml) for 1 h and then stimulated with medium, CpG-B DNA, CpG-A DNA, or polyI:C (PIC) for 24 h. Concentrations of the selected cytokines (TNF-α, IL-6, IL-10, IL-12p40, IFN-α) in culture supernatants were analyzed by cytokine-specific ELISAs. Data represent the mean concentration (pg/ml) ± SD of triplicates. D, Human PBMCs were pre-treated with DMSO, Gö6976 (250 ng/ml) or Gö6983 (250 ng/ml) for 1 h and then stimulated with medium or CpG-B DNA (12 μg/ml) for 4 h. Levels of IL-6 and IL-8 mRNA were analyzed by RT-PCR. E and F, BALB/c mice (n=3) were injected intraperitoneally with DMSO, Gö6976 (2.5 mg/kg body weight), or Gö6983 (2.5 mg/kg body weight) and then 1 hr later injected intraperitoneally with PBS or CpG-B DNA (30 μg/mouse). Two hr later, mice were bled to obtain serum and then euthanized. Peritoneal cells, spleen, and Kupffer cells were isolated. Levels of TNF-α, IL-6, IL-10, and IL-12p40 mRNA in peritoneal macrophages (pMØ), spleen, Kupffer cells (KC) were analyzed by RT-PCR (E). Cytokine levels in serum were analyzed by cytokine-specific ELISAs. Data represent the mean concentration (pg/ml) ± SD of triplicates (F).
To further confirm the finding that PKD1 is required for CpG-B DNA-mediated innate immune cell activation, we generated PKD1-knockdown macrophages by stably transfecting RAW264.7 cells with vectors that express PKD1-specific shRNAs. Expression of PKD1 mRNA was almost completely inhibited in PKD1-knockdown macrophages (Fig. 5A). In contrast, mRNA levels of other PKC family members, TLR9 and TLR9-downstream signaling modulators in PKD1-knockdown macrophages were comparable to those in control luciferase-knockdown macrophages. Protein levels of PKD were also dramatically reduced in PKD1-knockdown macrophages (Fig. 5B). Of note, the anti-PKD antibody we used to detect PKD mainly recognizes PKD1 but it also reacts weakly with PKD2, but not to PKD3 (data not shown). Therefore, the remaining PKD protein in PKD1-knockdown cells could be remaining PKD1 and intact PKD2. In addition, CpG-B DNA failed to induce phosphorylation of PKDs in PKD1-knockdown macrophages, further supporting our finding that CpG-B DNA activates PKD1, but not PKD2 or PKD3, in macrophages (Fig. 5C). These results demonstrate that PKD1-shRNA specifically and effectively silenced PKD1 expression in RAW264.7 cells and that these PKD1-knockdown macrophages can be a useful model for studying the biologic role of PKD1 in macrophages. Using the PKD1-knockdown macrophages, we further investigated whether PKD1 plays a role in CpG-B DNA-mediated cytokine expression in macrophages. Compared to those in the control luciferase-knockdown macrophages, expression of cytokines TNF-α, IL-6, IL-10 and IL-12p40 at both mRNA and protein levels in response to CpG-B DNA was almost completely, if not completely, inhibited in PKD1-knockdown macrophages (Fig. 5D and 5E). The degree of inhibition of CpG-B DNA-mediated cytokine expression was correlated with the level of PKD1 expression in these cells. Of note, TNF-α mRNA expression and protein production in response to IFN-γ were not altered in PKD1-knockdown macrophages, indicating that the absence of PKD1 specifically affects the CpG-B DNA-signaling pathway but not the general biology of the cell. These results demonstrate that PKD1 plays an indispensable role in CpG-B DNA-mediated cytokine expression.
Figure 5. PKD1 plays an indispensable role in cytokine production in macrophages in response to CpG-B DNA.
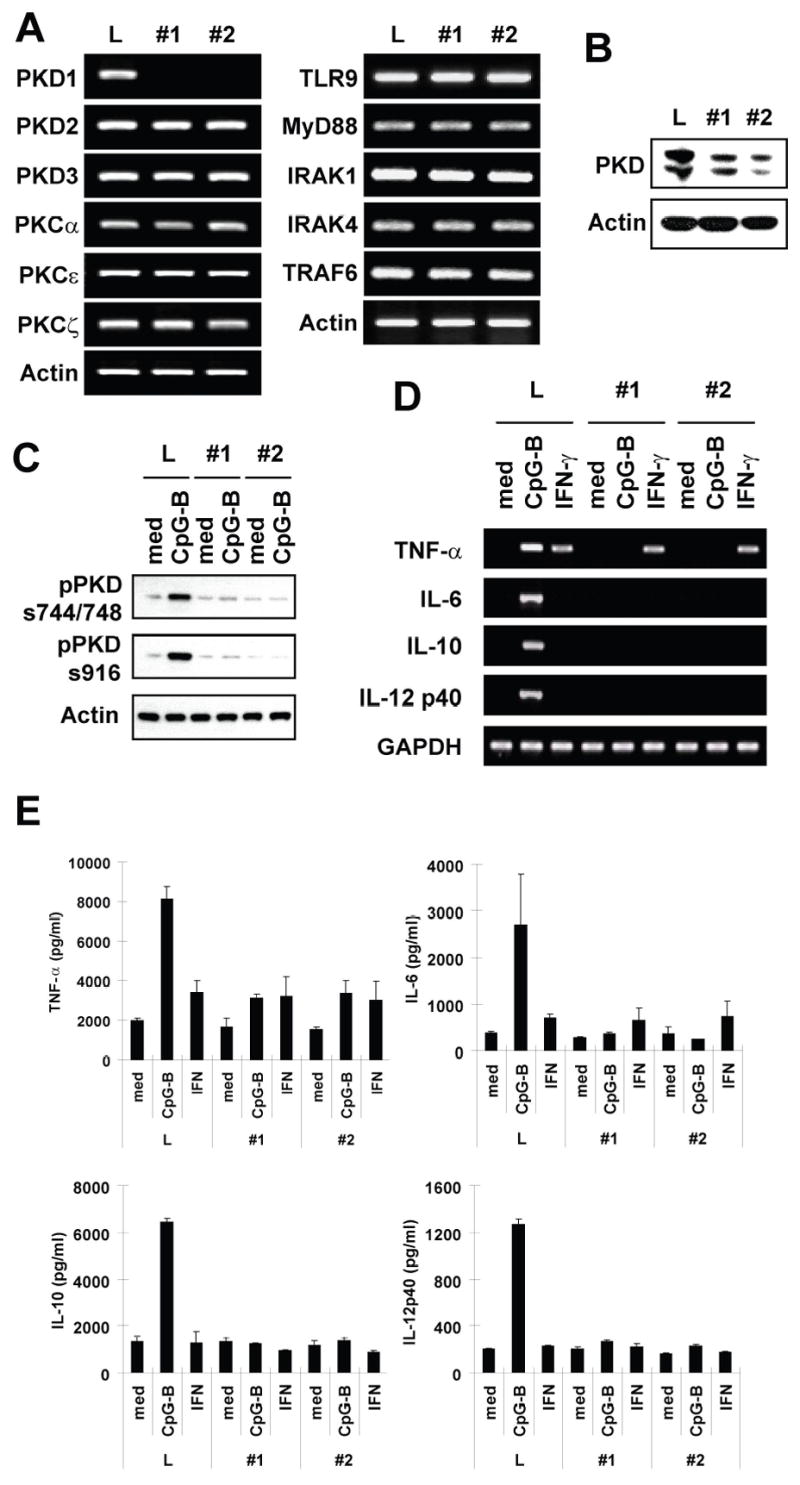
A–C, RAW264.7 cells were stably transfected with vectors expressing control luciferase-shRNA (L) or PKD1shRNA (clone #1 and #2) under control of the H1 promoter. Positive transfectants were selected and maintained in selection medium containing G418 (1 mg/ml). Messenger RNA levels of selected genes in PKD family members, PKC family members, and TLR9 and its down-stream signaling modulators were analyzed by RT-PCR (A). PKD protein levels in gene-specific knockdown RAW264.7 cells were examined using Western blot analysis (B). Phosphorylation of PKD in gene-specific knockdown RAW264.7 cells after CpG-B DNA stimulation (12 μg/ml for 45 min) was detected by Western blot (C). D and E, Control luciferase-knockdown (L) or PKD1-knockdown (#1 and #2) RAW264.7 cells were stimulated with medium, CpG-B DNA (6 μg/ml) or IFN-γ (10 ng/ml) for 4 h (D) or 24 h (E). Levels of cytokine mRNA (D) and protein (E) were analyzed by RT-PCR and cytokine-specific ELISA, respectively. ELISA data represent the mean cytokine concentration (pg/ml) ± SD of triplicates.
CpG-B DNA-mediated activation of transcription factors and MAPKs is inhibited in PKD1-knockdown macrophages
Our results (Fig. 5D and 5E) indicate that PKD1 regulates CpG-B DNA-mediated cytokine expression at or before the gene transcription level rather than at the level of protein secretion. CpG-B DNA-mediated expression of various cytokines is regulated by MAPKs and several transcription factors, including NF-κB, AP-1, CREB, and STAT1 (47–49). Therefore, we investigated whether PKD1 is involved in the CpG-B DNA-mediated activation of MAPKs and various transcription factors. As shown in Figure 6, CpG-B DNA-mediated activation of all three MAPKs (JNK, ERK, and p38), NF-κB (judged by degradation of IκBα and IκBβ, increased nuclear DNA binding activity and luciferase activity), AP-1 (judged by increased nuclear DNA binding activity and luciferase activity), CREB (judged by increased luciferase activity and phosphorylation at serine 133 residue), and STAT1 (judged by increased phosphorylation) was dramatically, if not completely, inhibited in PKD1-knockdown macrophages. The degree of inhibition of CpG-B DNA-mediated activation of MAPKs and transcription factors was correlated with the level of PKD1 expression in PKD1-knockdown macrophages. However, IFN-γ-mediated NF-κB activation and STAT1 phosphorylation were not suppressed in PKD1-knockdown macrophages compared to those in control luciferase-knockdown macrophages. Collectively, these results demonstrated that PKD1 is required for CpG-B DNA-mediated activation of MAPKs and transcription factors, including NF-κB, AP-1, CREB, and STAT1. Our results also suggest that PKD1 activation might take place at the proximal step in CpG-B DNA-signaling, which can profoundly influence all downstream biologic outcomes.
Figure 6. PKD1 is required for CpG-B DNA-mediated activation of MAPKs and transcription factors.
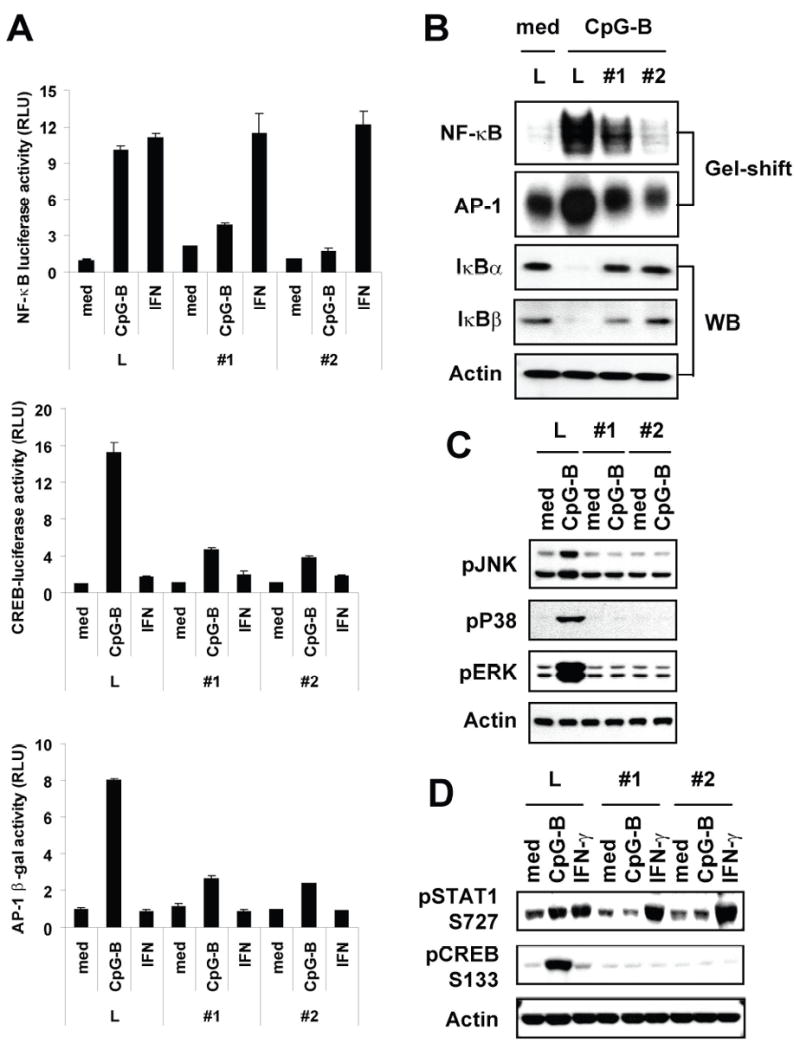
A, Control luciferase-knockdown (L) or PKD1-knockdown (#1 and #2) RAW264.7 cells were transiently transfected with NF-κB-luciferase (left), AP-1-β-galactosidase (middle), or CREB-luciferase (right) reporter genes and then stimulated with medium (med), CpG-B DNA (6 μg/ml) or IFN-γ (10 ng/ml) for 12 h. Luciferase (NF-κB or CREB) or β-galactosidase (AP-1) activities in cell extracts were analyzed. Data represent the mean relative luciferase unit (RLU) ± SD of triplicates. B, Control luciferase-knockdown (L) or PKD1-knockdown (#1 and #2) RAW264.7 cells were stimulated with medium or CpG-B DNA for 1 h (NF-κB) or 4 h (AP-1). Cytoplasmic extracts and nuclear extracts were prepared. DNA-binding activities of transcription factor, NF-κB or AP-1, in equal amounts of nuclear extracts (3 μg/lane) were analyzed by EMSA (gel-shift) and degradation of IκBα and IκBβ in cytosolic extracts was detected by Western blot analysis (WB). C and D, Control luciferase-knockdown (L) or PKD1-knockdown (#1 and #2) RAW264.7 cells were stimulated with medium, CpG-B DNA or IFN-γ for 45 min. Phosphorylation status of MAPKs (JNK, p38, ERK), STAT1, and CREB was detected by Western blot analysis.
PKD1 activation by CpG-B DNA is independent of Src kinases and conventional PKCs
Previous studies have demonstrated that PKDs can be activated by various upstream signaling modulators such as conventional PKCs, Src family protein tyrosine kinases (PTKs), PI3K, and PLCγ (23, 50). Src PTKs and PI3K have been shown to be implicated in CpG DNA-mediated macrophage activation (13, 51, 52). Therefore, we investigated whether these known PKD upstream modulators are involved in CpG-B DNA-mediated PKD1 activation. As shown in Figure 7A and 7B, inhibition of Src PTKs by either pharmacological inhibitors (PP1 and SU6656) or over-expression of the membrane-targeted form of C-terminal Src kinase (mCsk; inhibits the activity of Src PTKs) (53) did not result in suppression of CpG-B DNA-mediated PKD1 phosphorylation. In addition, CpG-B DNA-mediated PKD1 phosphorylation was not altered in RAW264.7 cells treated with LY294002 (PI3K inhibitor) or U73122 (PLC-γ inhibitor) (Fig. 7C). In contrast, CpG-B DNA-induced phosphorylation of PTKs was completely inhibited in the presence of Src kinase inhibitor (Fig. 7A–7B). These results indicated that CpG-B DNA induces activation of PKD1 through a pathway that is independent of Src PTKs, PI3K, or PLCγ.
Figure 7. Src PTKs, PI3K, PLC-γ and PKCs are not required for CpG-B DNA-mediated PKD1 activation.
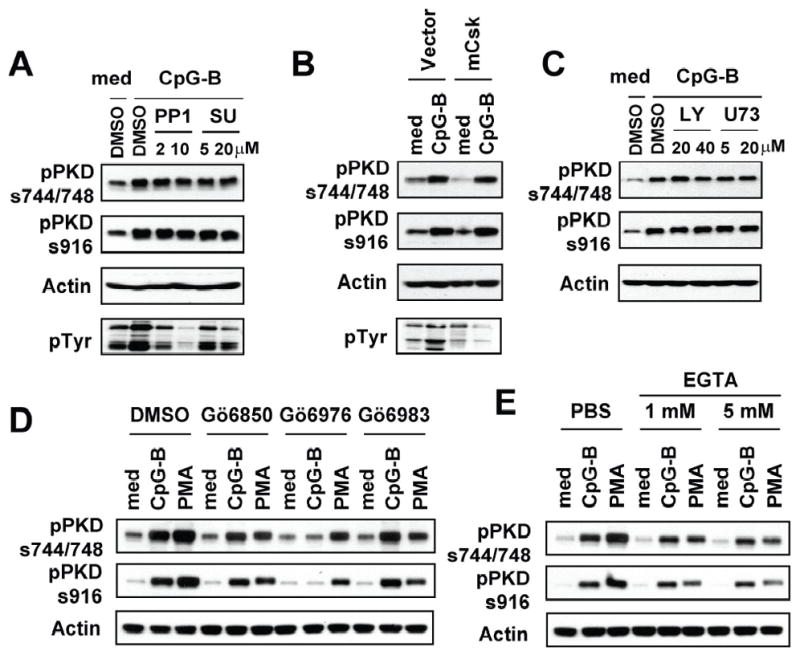
A and B, RAW264.7 cells pretreated with DMSO or Src PTK inhibitors (PP1 or SU6656) for 30 min (A), or that constitutively over-express empty vector or mCsk (B) were stimulated with medium or CpG-B DNA (12 μg/ml) for 45 min. C–E, RAW264.7 cells were pretreated with DMSO, PI3K inhibitor (LY294002), PLC-γ inhibitor (U73122), PKC inhibitors (Gö6850, Gö6976 or Gö6983 at 125 ng/ml), or EGTA for 30 min and then stimulated with medium, CpG-B DNA, or PMA for 45 min. Phosphorylation status of PKD1 and PTKs was detected by Western blot using a phospho-PKD specific antibody and phospho-tyrosine specific antibody (4G10), respectively. med: medium, SU: SU6656, LY: LY294002, U73: U73122.
Activation of PKDs by most stimuli has been shown to be dependent on conventional PKCs (50). More recently, several studies provided evidence that PKDs can be activated through PKC-independent pathways (54). CpG-B DNA-mediated cytokine production in CpG-B DNA-responding cells was insensitive to inhibitors (Gö6850 and Gö6983) that inhibit conventional PKCs (Fig. 4), suggesting a possibility that CpG-B DNA activates PKD1 via a conventional PKC-independent manner. As demonstrated in Figure 7D–7E, CpG-B DNA-mediated PKD1 activation was not suppressed in RAW264.7 cells pre-treated with Gö6850 (inhibits PKCα and βI), Gö6983 (inhibits PKCα, β, γ, δ, and ζ) or EGTA (inhibits activation of Ca++-dependent conventional PKCs), indicating that CpG-B DNA-mediated PKD1 activation is independent of Ca++-dependent conventional PKCs and PKCδ and ζ. In contrast, CpG-B DNA failed to activate PKD1 in the presence of Gö6976. PMA-mediated activation of PKDs in RAW264.7 cells was substantially inhibited in the presence of Gö6976, Gö6983 or EGTA. Collectively, these results suggested that CpG-B DNA activates PKD1 through a mechanism that is independent of Src PTKs, PI3K, PLCγ, conventional PKCs, PKCδ, and PKCζ.
CpG-B DNA activates PKD1 via a TLR9/MyD88/IRAK1-dependent pathway
Most biologic effects of CpG motif containing DNA, including bacterial DNA, are mediated through TLR9, present in endosomal compartments, and an adaptor molecule, MyD88. However, more recent studies have revealed that the TLR9-dependent pathway is not the sole signaling pathway utilized by bacterial DNA (6, 55). Therefore, it was necessary for us to investigate whether CpG-B DNA activates PKD1 through an endosomal pH-sensitive TLR9/MyD88-dependent signaling pathway. CpG-B DNA-mediated PKD1 activation was completely ablated by chloroquine (an endosomal acidification inhibitor) (14) or inhibitory CpG DNA (iCpG DNA; a TLR9 antagonist) (56) in RAW264.7 cells, murine primary macrophages, and human PBMCs (Fig. 8A and data not shown). In addition, CpG-B DNA failed to activate PKD1 in TLR9−/− macrophages and MyD88−/− macrophages (Fig. 8B). Furthermore, CpG-B DNA-mediated PKD1 activation was completely ablated in IRAK1-knockdown macrophages (Fig. 8C). Of note, PMA-mediated PKD activation was affected neither by the presence of chloroquine or iCpG DNA nor by the absence of TLR9, MyD88, or IRAK1. These results provide direct evidence that CpG-B DNA induces PKD1 activation through an endosomal pH-sensitive TLR9/MyD88/IRAK1-dependent signaling pathway.
Figure 8. CpG-B DNA induces activation of PKD1 through an endosomal pH-sensitive TLR9/MyD88/IRAK1-dependent signaling pathway.
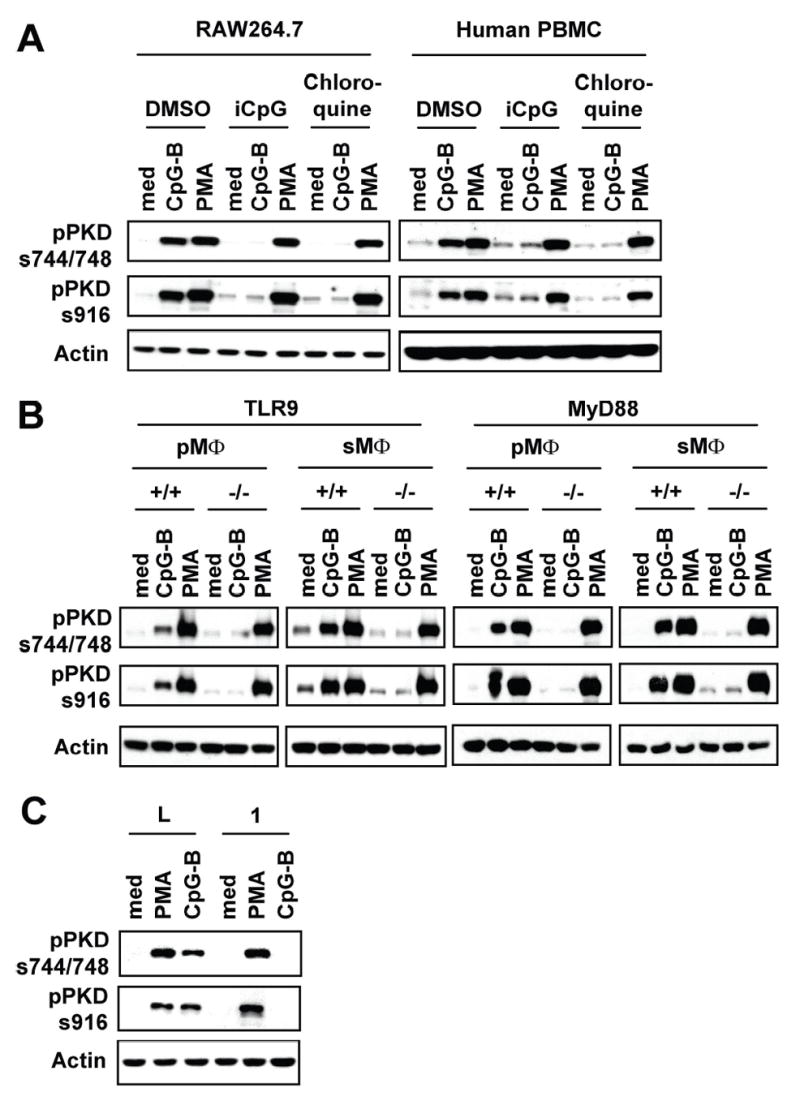
A, RAW264.7 cells and human PBMCs were pretreated with chloroquine (2.5 μg/ml) or iCpG DNA (12 μg/ml) for 30 min and then stimulated with medium (med), CpG-B DNA (12 μg/ml) or PMA (10 ng/ml) for 45 min. B, Peritoneal (pMΦ) or splenic (sMΦ) macrophages isolated from TLR9+/+ (BALB/c), TLR9−/−, MyD88+/+ (wild-type littermates), or MyD88−/− were stimulated with medium, CpG-B DNA (12 μg/ml) or PMA (10 ng/ml) for 45 min. Activation status of PKD1 was detected by phospho-specific Western blot assay. C, Control luciferase-knockdown (L) or IRAK1-knockdown (1) RAW264.7 cells were stimulated with medium, PMA, or CpG-B DNA for 45 min. Phosphorylation status of PKD1 was detected by Western blot analysis. Lack of IRAK1 expression in IRAK1-knockdown macrophages was confirmed (36).
CpG-B DNA induces recruitment of PKD1 to the TLR9/MyD88 signaling complex
Since our results (Figs. 5, 6, and 8) indicate that CpG-B DNA activates PKD1 through an endosomal pH-sensitive TLR9/MyD88/IRAK1-dependent signaling pathway, and that PKD1 activation might take place at the step that can regulate all known downstream biologic effects of CpG-B DNA, we hypothesized that PKD1 is recruited to the TLR9/MyD88 receptor signaling complex upon CpG-B DNA stimulation. To test this hypothesis, we examined formation of the protein complex between PKD1 and the proteins involved in TLR9 signaling. Initial studies using HEK293T cells transiently co-transfected with vectors expressing FLAG-tagged PKD1 and epitope-tagged proteins in the TLR9 signaling pathway revealed that PKD1, but not PKD2 and PKD3, binds to TLR9, MyD88, IRAK1, IRAK4, and TRAF6 upon CpG DNA stimulation (data not shown). Since transient overexpression system can give false positive results, we further investigated whether PKD1 can be recruited to the endogenous TLR9/MyD88 receptor signaling complex in RAW264.7 cells stably expressing FLAG-tagged PKD family proteins. As demonstrated in Figure 9, PKD1 was not associated with TLR9 or any known proximal downstream signaling modulators in the TLR9 signaling pathway in unstimulated cells. However, PKD1 co-precipitated with TLR9, MyD88, IRAK1, IRAK4, and TRAF6 after CpG-B DNA stimulation, indicating that PKD1 interacts, either directly or indirectly, with TLR9, MyD88, IRAK1, IRAK4, and TRAF6 upon CpG-B DNA stimulation in macrophages. In contrast, association of PKD1 with TRAF3 was not detected. No association between any of the tested TLR9 signaling molecules and PKD2 or PKD3 was detected under our experimental conditions. Taken together, these results demonstrate that PKD1 is recruited to the TLR9/MyD88/IRAK/TRAF6 complex upon CpG-B DNA stimulation.
Figure 9. PKD1 interacts with TLR9 signaling complex in response to CpG-B DNA.
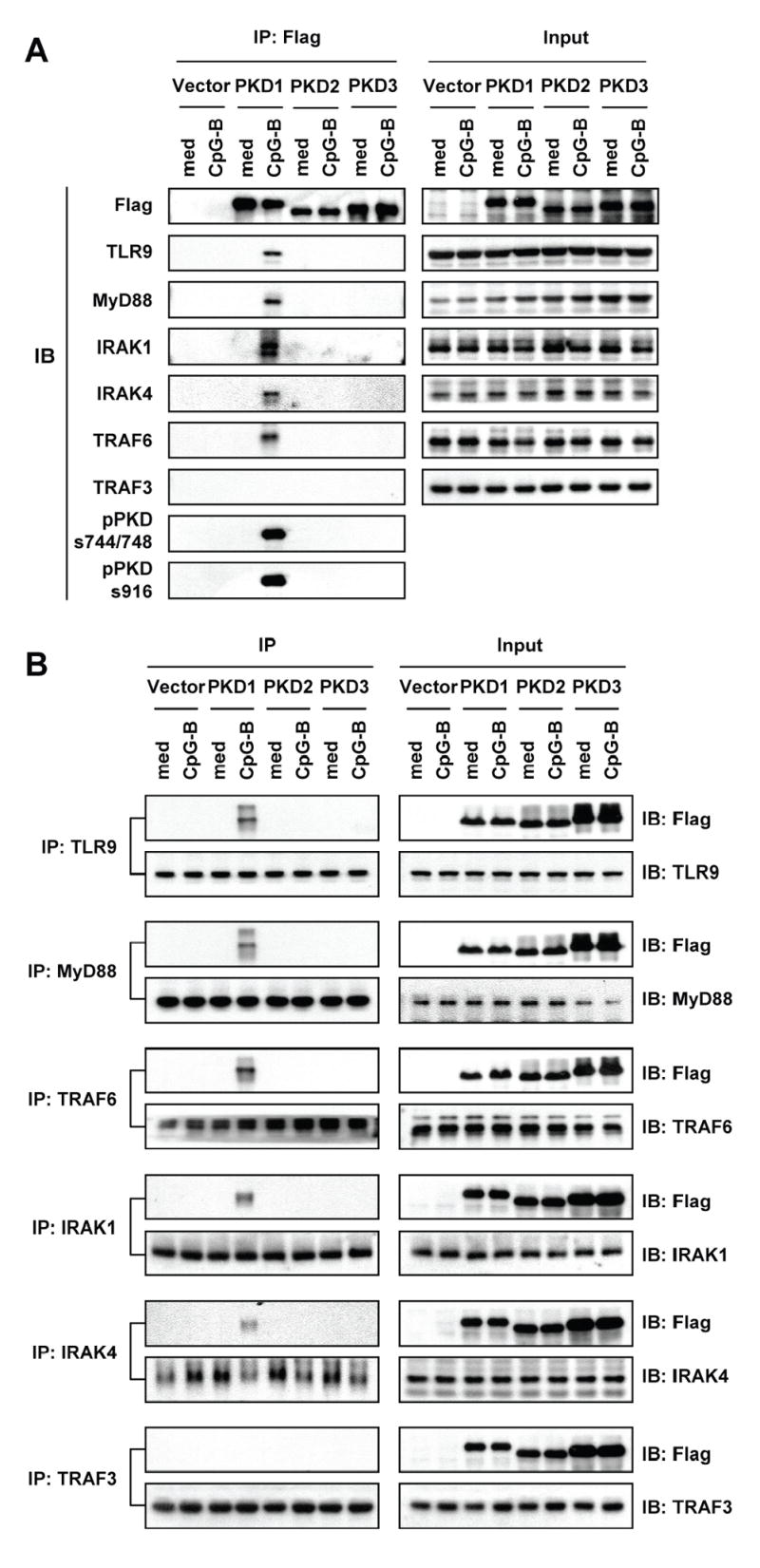
RAW264.7 cells stably expressing empty vector, FLAG-tagged PKD1, FLAG-tagged PKD2, or FLAG-tagged PKD3 were stimulated with medium (med) or CpG-B DNA for 30 min. A, FLAG-tagged PKD family proteins in the cell lysates were immunoprecipitated. The presence of proteins in the TLR9-signaling pathway and phosphorylated forms of PKD in the resulting immunoprecipitates were detected by Western blot. B, The indicated endogenous proteins in the TLR9-signaling pathway in the cell lysates were immunoprecipitated. The presence of FLAG-tagged PKD family proteins and proteins in the TLR9-signaling pathway in the resulting immunoprecipitates were detected by Western blot.
DISCUSSION
The innate immune system is responsible for detecting microbial pathogens and combating infection. Discovery of TLRs has dramatically enhanced our understanding of innate immune responses. Although large amounts of accumulated studies have provided the big picture for how our innate immune system recognizes and responds to pathogenic DNA originated from invading pathogens and dying self cells, understanding of the biochemical mechanisms of pathogenic DNA-mediated innate immune response is far from complete. In the present study, we identified PKD1, whose presence and function in monocytic cells is currently unknown, as a new key component of the TLR9 signaling complex. Upon type B CpG motif containing DNA stimulation, PKD1 is recruited to the TLR9/MyD88/IRAK/TRAF6 complex. Activation of PKD1 by CpG-B DNA is dependent on endosomal pH, TLR9, MyD88, and IRAK1 and is required for activation of NF-κB and MAPKs and subsequent production of proinflammatory cytokines in macrophages.
Serine/threonine kinase PKD1 is involved in dynamic cellular processes, such as Golgi function and organization, vesicle trafficking, activation of transcription factors and signaling modulators, and detoxification of mitochondrial ROS, in various types of cells, including T and B lymphocytes (18, 20, 21, 23, 25, 27). However, it is yet to be revealed whether any PKD family protein is involved in innate immune responses. Using various PKC inhibitors of different specificity and sensitivity, several recent studies indicate a possibility that PKD family members may be involved in TLR4 and TLR5 signaling in non-immune cells. PKCα, PKCβI, and PKD inhibitor Gö6976 inhibits LPS-mediated p38 activation and TNF-α secretion in microglial cells (34). Steiner and colleagues (35) showed that human TLR5 has a putative consensus PKD phosphorylation motif in the TIR domain, and PKD1 phosphorylates the TLR5-derived target peptide in vitro. This study also showed that PKD1 interacts with TLR5 in HEK293T cells upon flagellin stimulation, and that flagellin-mediated p38 activation and IL-8 production, but not NF-κB nuclear translocation, are inhibited by Gö6976 but not by Gö6983, suggesting that PKD1 may be an upstream molecule of TLR5 that directly phosphorylates TLR5 in epithelial cells. However, it is not known how PKD1 is being activated and whether flagellin induces activation of PKD1 in epithelial cells. Unlike human TLR5, mouse TLR5 and mouse TLR9 do not have this putative consensus PKD phosphorylation motif in the TIR domain (data not shown). In addition, we could not detect serine phosphorylation of TLR9 by PKD1 upon CpG-B DNA stimulation under our experimental conditions (data not shown). Similar to LPS and flagellin responses in neuron cells and epithelial cells, CpG-B DNA-mediated cytokine expression in murine macrophages, B cells, and pDCs, in mice in vivo, and in human PBMCs was ablated by Gö6976, but insensitive to Gö6850 or Gö6983, indicating a possible role of PKD family proteins in CpG-B DNA-induced cytokine expression. This possibility was confirmed in a set of more carefully controlled experiments. CpG-B DNA stimulation induced increased phosphorylation and kinase activity of PKD1, but not PKD2 and PKD3. In addition, PKD1 knockdown studies demonstrated that PKD1 is required for CpG-B DNA-mediated activation of MAPKs and transcription factors and subsequent production of various cytokines in macrophages. Taken together, our results provide direct evidence that CpG-B DNA activates PKD1 in macrophages, and that in spite of possible compensatory effects among three PKD family members, PKD1 plays an indispensable role in CpG-B DNA-mediated macrophage activation.
PKD can be activated via PKC-dependent or -independent pathways (50, 54). In the case of the PKC-dependent pathway, the phosphorylation of serine residues within an activation loop (S744/748 in mouse PKD1) of PKD is required (50). In contrast, phosphorylation of these serine residues is not required in the PKC-independent activation of PKD (54). Although CpG-B DNA induces the phosphorylation of serine residues within the activation loop of PKD1 in macrophages, CpG-B DNA-mediated PKD1 activation appears to be conventional PKCs- and PKCδ and ζ-independent. Activation of PKD1 by CpG-B DNA was not inhibited by conventional PKC inhibitors Gö6850, Gö6983, or EGTA. In addition, we did not find a role for upstream modulators, such as Src, PI3K, or PLCγ, in CpG-B DNA-mediated PKD1 activation in macrophages under our experimental conditions. Of note, LPS has been shown to induce activation of PKD in a PKC- and IL-1β-dependent manner in spinal neurons (57). It is unknown at this point whether other PKC isoforms that are not inhibited by Gö6983 or Gö6850 are involved in CpG-B DNA-mediated PKD1 activation, and it is a question that warrants further investigation.
Pathogenic DNA can be sensed by host cells via a TLR9-dependent or a TLR9-independent manner (5–7). Plasmid DNA spontaneously internalized by B lymphocytes induces expression of CD40 and CD86 costimulatory molecules through both TLR9-dependent and TLR9-independent mechanisms (55). Induction of type I IFN by cytosolic DNA released from intracellular pathogens or dying self cells is TLR9/MyD88/RIP2-independent and requires IRF3 and TBK1 without activation of NF-κB and MAPKs (6, 7, 44). Bacterial DNA and CpG-B DNA-mediated PKD1 activation appears to be mediated solely through a TLR9 rather than a yet to be identified cytosolic DNA receptor. Indeed, liposome combined transfection of poly dA/dT that activates cytosolic DNA receptor pathway did not induce activation of PKD1. In contrast, PKD1 activation by DNA was B type unmethylated CpG motif-specific, endosomal pH-sensitive, and TLR9-, MyD88-, and IRAK1-dependent. Moreover, PKD1 physically interacted with TLR9, MyD88, IRAK4, IRAK1, and TRAF6 upon CpG-B DNA stimulation, indicating that PKD1 is recruited to the TLR9 receptor signaling complex during signal transduction.
Several previous studies indicate that the TLR9/MyD88 signal diverges by the level of TRAF6. While CpG DNA-mediated proinflammatory cytokine production is dependent on TRAF6, CpG DNA-mediated production of IL-10 and type I IFN is dependent on both TRAF6 and TRAF3 (11). In addition, CpG DNA-mediated activation of ERK is dependent on Ras but independent of TRAF6 (37). We found that CpG-B DNA activates PKD1 in an IRAK1-dependent manner and induces interaction between PKD1 and IRAK1, IRAK4 and TRAF6, but not TRAF3. However, PKD1 was required for CpG-B DNA-mediated activation of ERK as well as all other MAPKs and transcription factors, indicating a possibility that PKD1 may be required for both TRAF6-dependent and -independent pathways and that TRAF6 may bind to PKD1 not as the upstream activator of PKD1, but perhaps instead as a PKD1 downstream effector or a signaling partner involved in the transmission of signals downstream of PKD1. It is yet to be revealed whether TRAF6 and TAK1 play a role in CpG-B DNA-mediated PKD1 activation, or whether, conversely, PKD1 plays a role in activation of TRAF6 and its downstream signaling molecules. Also, it should be addressed in future studies whether PKD1 binds to the TLR9 receptor signaling complex directly or indirectly through a yet-to-be identified modulator, whether IRAK1 and/or IRAK4 are direct upstream kinase(s) that phosphorylate PKD1, and what the downstream substrate(s) of PKD1 are in TLR9 signaling.
In summary, the present study provides the first direct evidence that CpG-B DNA induces recruitment of PKD1 to the TLR9/MyD88/IRAK/TRAF6 receptor complex and activation of PKD1 via an endosomal pH-sensitive, TLR9/MyD88/IRAK1-dependent pathway that is required for activation of MAPKs and transcription factors and subsequent production of cytokines. Our findings also raise the possibility that pharmacological inhibitors specifically targeting PKD1 can be useful therapeutics for acute and chronic inflammatory diseases and autoimmune diseases where type B CpG motif containing DNA/TLR9 contributes to the pathogenesis.
Acknowledgments
We thank Drs. S. Akira (Osaka Univ., Osaka, Japan) and Z.-I. Honda (Univ. of Tokyo, Tokyo, Japan) for kindly providing TLR9 and MyD88 gene-deficient mice and mCsk construct, respectively. We also thank Mrs. Andrea Patters for her assistance with preparation of this manuscript. A.-K. Y. was supported by the Children’s Foundation Research Center at Le Bonheur Children’s Medical Center, and grants from NIH (AI053137, AR47757) and the Children’s Foundation of Memphis. J.-E. P. was supported by a grant from Le Bonheur Children’s Medical Center. Y.-I. K. was supported by a postdoctoral fellowship from the Korea Research Foundation (KRF-2005-214-E00045) and a grant from Le Bonheur Children’s Medical Center. Animal experiments were supported in part by transgenic mice program grant from the Children’s Foundation of Memphis.
References
- 1.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 2.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 3.Fritz JH, Ferrero RL, Philpott DJ, Girardin SE. Nod-like proteins in immunity, inflammation and disease. Nat Immunol. 2006;7:1250–1257. doi: 10.1038/ni1412. [DOI] [PubMed] [Google Scholar]
- 4.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 5.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 6.Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 7.Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, Kawai T, Uematsu S, Takeuchi O, Takeshita F, Coban C, Akira S. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature. 2008;451:725–729. doi: 10.1038/nature06537. [DOI] [PubMed] [Google Scholar]
- 8.Okabe Y, Kawane K, Akira S, Taniguchi T, Nagata S. Toll-like receptor-independent gene induction program activated by mammalian DNA escaped from apoptotic DNA degradation. J Exp Med. 2005;202:1333–1339. doi: 10.1084/jem.20051654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yasuda K, Yu P, Kirschning CJ, Schlatter B, Schmitz F, Heit A, Bauer S, Hochrein H, Wagner H. Endosomal translocation of vertebrate DNA activates dendritic cells via TLR9-dependent and -independent pathways. J Immunol. 2005;174:6129–6136. doi: 10.4049/jimmunol.174.10.6129. [DOI] [PubMed] [Google Scholar]
- 10.Krieg AM. CpG motifs: the active ingredient in bacterial extracts? Nat Med. 2003;9:831–835. doi: 10.1038/nm0703-831. [DOI] [PubMed] [Google Scholar]
- 11.Hacker H, Redecke V, Blagoev B, Kratchmarova I, Hsu LC, Wang GG, Kamps MP, Raz E, Wagner H, Hacker G, Mann M, Karin M. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature. 2006;439:204–207. doi: 10.1038/nature04369. [DOI] [PubMed] [Google Scholar]
- 12.Xu H, An H, Yu Y, Zhang M, Qi R, Cao X. Ras participates in CpG oligodeoxynucleotide signaling through association with toll-like receptor 9 and promotion of interleukin-1 receptor-associated kinase/tumor necrosis factor receptor-associated factor 6 complex formation in macrophages. J Biol Chem. 2003;278:36334–36340. doi: 10.1074/jbc.M305698200. [DOI] [PubMed] [Google Scholar]
- 13.Stovall SH, Yi AK, Meals EA, Talati AJ, Godambe SA, English BK. Role of vav1- and src-related tyrosine kinases in macrophage activation by CpG DNA. J Biol Chem. 2004;279:13809–13816. doi: 10.1074/jbc.M311434200. [DOI] [PubMed] [Google Scholar]
- 14.Yi AK, Tuetken R, Redford T, Waldschmidt M, Kirsch J, Krieg AM. CpG motifs in bacterial DNA activate leukocytes through the pH-dependent generation of reactive oxygen species. J Immunol. 1998;160:4755–4761. [PubMed] [Google Scholar]
- 15.Yi AK, Klinman DM, Martin TL, Matson S, Krieg AM. Rapid immune activation by CpG motifs in bacterial DNA. Systemic induction of IL-6 transcription through an antioxidant-sensitive pathway. J Immunol. 1996;157:5394–5402. [PubMed] [Google Scholar]
- 16.Rozengurt E, Rey O, Waldron RT. Protein kinase D signaling. J Biol Chem. 2005;280:13205–13208. doi: 10.1074/jbc.R500002200. [DOI] [PubMed] [Google Scholar]
- 17.Matthews SA, Rozengurt E, Cantrell D. Characterization of serine 916 as an in vivo autophosphorylation site for protein kinase D/Protein kinase Cmu. J Biol Chem. 1999;274:26543–26549. doi: 10.1074/jbc.274.37.26543. [DOI] [PubMed] [Google Scholar]
- 18.Arnold R, I, Patzak M, Neuhaus B, Vancauwenbergh S, Veillette A, Van Lint J, Kiefer F. Activation of hematopoietic progenitor kinase 1 involves relocation, autophosphorylation, and transphosphorylation by protein kinase D1. Mol Cell Biol. 2005;25:2364–2383. doi: 10.1128/MCB.25.6.2364-2383.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zugaza JL, Waldron RT, Sinnett-Smith J, Rozengurt E. Bombesin, vasopressin, endothelin, bradykinin, and platelet-derived growth factor rapidly activate protein kinase D through a protein kinase C-dependent signal transduction pathway. J Biol Chem. 1997;272:23952–23960. doi: 10.1074/jbc.272.38.23952. [DOI] [PubMed] [Google Scholar]
- 20.Storz P, Doppler H, Toker A. Protein kinase D mediates mitochondrion-to-nucleus signaling and detoxification from mitochondrial reactive oxygen species. Mol Cell Biol. 2005;25:8520–8530. doi: 10.1128/MCB.25.19.8520-8530.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bagowski CP, Stein-Gerlach M, Choidas A, Ullrich A. Cell-type specific phosphorylation of threonines T654 and T669 by PKD defines the signal capacity of the EGF receptor. Embo J. 1999;18:5567–5576. doi: 10.1093/emboj/18.20.5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matthews SA, Dayalu R, Thompson LJ, Scharenberg AM. Regulation of protein kinase Cnu by the B-cell antigen receptor. J Biol Chem. 2003;278:9086–9091. doi: 10.1074/jbc.M211295200. [DOI] [PubMed] [Google Scholar]
- 23.Vigorito E, Kovesdi D, Turner M. Synergistic activation of PKD by the B cell antigen receptor and CD19 requires PI3K, Vav1 and PLCgamma. Cell Signal. 2006;18:1455–1460. doi: 10.1016/j.cellsig.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 24.Davidson-Moncada JK, Lopez-Lluch G, Segal AW, Dekker LV. Involvement of protein kinase D in Fc gamma-receptor activation of the NADPH oxidase in neutrophils. Biochem J. 2002;363:95–103. doi: 10.1042/0264-6021:3630095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maeda Y, Beznoussenko GV, Van Lint J, Mironov AA, Malhotra V. Recruitment of protein kinase D to the trans-Golgi network via the first cysteine-rich domain. Embo J. 2001;20:5982–5990. doi: 10.1093/emboj/20.21.5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prestle J, Pfizenmaier K, Brenner J, Johannes FJ. Protein kinase C mu is located at the Golgi compartment. J Cell Biol. 1996;134:1401–1410. doi: 10.1083/jcb.134.6.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matthews SA, Iglesias T, Rozengurt E, Cantrell D. Spatial and temporal regulation of protein kinase D (PKD) Embo J. 2000;19:2935–2945. doi: 10.1093/emboj/19.12.2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spitaler M, Emslie E, Wood CD, Cantrell D. Diacylglycerol and protein kinase D localization during T lymphocyte activation. Immunity. 2006;24:535–546. doi: 10.1016/j.immuni.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 29.Dequiedt F, Van Lint J, Lecomte E, Van Duppen V, Seufferlein T, Vandenheede JR, Wattiez R, Kettmann R. Phosphorylation of histone deacetylase 7 by protein kinase D mediates T cell receptor-induced Nur77 expression and apoptosis. J Exp Med. 2005;201:793–804. doi: 10.1084/jem.20042034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matthews SA, Liu P, Spitaler M, Olson EN, McKinsey TA, Cantrell DA, Scharenberg AM. Essential role for protein kinase D family kinases in the regulation of class II histone deacetylases in B lymphocytes. Mol Cell Biol. 2006;26:1569–1577. doi: 10.1128/MCB.26.4.1569-1577.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kennett SB, Roberts JD, Olden K. Requirement of protein kinase C micro activation and calpain-mediated proteolysis for arachidonic acid-stimulated adhesion of MDA-MB-435 human mammary carcinoma cells to collagen type IV. J Biol Chem. 2004;279:3300–3307. doi: 10.1074/jbc.M305734200. [DOI] [PubMed] [Google Scholar]
- 32.Eiseler T, Schmid MA, Topbas F, Pfizenmaier K, Hausser A. PKD is recruited to sites of actin remodelling at the leading edge and negatively regulates cell migration. FEBS Lett. 2007;581:4279–4287. doi: 10.1016/j.febslet.2007.07.079. [DOI] [PubMed] [Google Scholar]
- 33.Hausser A, Storz P, Martens S, Link G, Toker A, Pfizenmaier K. Protein kinase D regulates vesicular transport by phosphorylating and activating phosphatidylinositol-4 kinase IIIbeta at the Golgi complex. Nat Cell Biol. 2005;7:880–886. doi: 10.1038/ncb1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jeohn GH, Cooper CL, Jang KJ, Liu B, Lee DS, Kim HC, Hong JS. Go6976 inhibits LPS-induced microglial TNFalpha release by suppressing p38 MAP kinase activation. Neuroscience. 2002;114:689–697. doi: 10.1016/s0306-4522(02)00356-1. [DOI] [PubMed] [Google Scholar]
- 35.Ivison SM, Graham NR, Bernales CQ, Kifayet A, Ng N, Shobab LA, Steiner TS. Protein Kinase D Interaction with TLR5 Is Required for Inflammatory Signaling in Response to Bacterial Flagellin. J Immunol. 2007;178:5735–5743. doi: 10.4049/jimmunol.178.9.5735. [DOI] [PubMed] [Google Scholar]
- 36.Kim YI, Park JE, Martinez-Hernandez A, Yi AK. CpG DNA prevents liver injury and shock-mediated death by modulating expression of IL-1R-associated kinases. J Biol Chem. 2008 doi: 10.1074/jbc.M709549200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yeo SJ, Yoon JG, Yi AK. Myeloid differentiation factor 88-dependent post-transcriptional regulation of cyclooxygenase-2 expression by CpG DNA: tumor necrosis factor-alpha receptor-associated factor 6, a diverging point in the Toll-like receptor 9-signaling. J Biol Chem. 2003;278:40590–40600. doi: 10.1074/jbc.M306280200. [DOI] [PubMed] [Google Scholar]
- 38.Hartmann G, Weeratna RD, Ballas ZK, Payette P, Blackwell S, Suparto I, Rasmussen WL, Waldschmidt M, Sajuthi D, Purcell RH, Davis HL, Krieg AM. Delineation of a CpG phosphorothioate oligodeoxynucleotide for activating primate immune responses in vitro and in vivo. J Immunol. 2000;164:1617–1624. doi: 10.4049/jimmunol.164.3.1617. [DOI] [PubMed] [Google Scholar]
- 39.Rothenfusser S, Hornung V, Ayyoub M, Britsch S, Towarowski A, Krug A, Sarris A, Lubenow N, Speiser D, Endres S, Hartmann G. CpG-A and CpG-B oligonucleotides differentially enhance human peptide-specific primary and memory CD8+ T-cell responses in vitro. Blood. 2004;103:2162–2169. doi: 10.1182/blood-2003-04-1091. [DOI] [PubMed] [Google Scholar]
- 40.Krug A, Rothenfusser S, Hornung V, Jahrsdorfer B, Blackwell S, Ballas ZK, Endres S, Krieg AM, Hartmann G. Identification of CpG oligonucleotide sequences with high induction of IFN-alpha/beta in plasmacytoid dendritic cells. Eur J Immunol. 2001;31:2154–2163. doi: 10.1002/1521-4141(200107)31:7<2154::aid-immu2154>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 41.Yi AK, Yoon H, Park JE, Kim BS, Kim HJ, Martinez-Hernandez A. CpG DNA-mediated induction of acute liver injury in D-galactosamine-sensitized mice: the mitochondrial apoptotic pathway-dependent death of hepatocytes. J Biol Chem. 2006;281:15001–15012. doi: 10.1074/jbc.M601337200. [DOI] [PubMed] [Google Scholar]
- 42.Yi AK, Chace JH, Cowdery JS, Krieg AM. IFN-gamma promotes IL-6 and IgM secretion in response to CpG motifs in bacterial DNA and oligodeoxynucleotides. J Immunol. 1996;156:558–564. [PubMed] [Google Scholar]
- 43.Waldron RT, Rozengurt E. Protein kinase C phosphorylates protein kinase D activation loop Ser744 and Ser748 and releases autoinhibition by the pleckstrin homology domain. J Biol Chem. 2003;278:154–163. doi: 10.1074/jbc.M208075200. [DOI] [PubMed] [Google Scholar]
- 44.Ishii KJ, Coban C, Kato H, Takahashi K, Torii Y, Takeshita F, Ludwig H, Sutter G, Suzuki K, Hemmi H, Sato S, Yamamoto M, Uematsu S, Kawai T, Takeuchi O, Akira S. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat Immunol. 2006;7:40–48. doi: 10.1038/ni1282. [DOI] [PubMed] [Google Scholar]
- 45.Gschwendt M, Dieterich S, Rennecke J, Kittstein W, Mueller HJ, Johannes FJ. Inhibition of protein kinase C mu by various inhibitors. Differentiation from protein kinase c isoenzymes. FEBS Lett. 1996;392:77–80. doi: 10.1016/0014-5793(96)00785-5. [DOI] [PubMed] [Google Scholar]
- 46.Stempka L, Girod A, Muller HJ, Rincke G, Marks F, Gschwendt M, Bossemeyer D. Phosphorylation of protein kinase Cdelta (PKCdelta) at threonine 505 is not a prerequisite for enzymatic activity. Expression of rat PKCdelta and an alanine 505 mutant in bacteria in a functional form. J Biol Chem. 1997;272:6805–6811. doi: 10.1074/jbc.272.10.6805. [DOI] [PubMed] [Google Scholar]
- 47.Yi AK, Yoon JG, Yeo SJ, Hong SC, English BK, Krieg AM. Role of mitogen-activated protein kinases in CpG DNA-mediated IL-10 and IL-12 production: central role of extracellular signal-regulated kinase in the negative feedback loop of the CpG DNA-mediated Th1 response. J Immunol. 2002;168:4711–4720. doi: 10.4049/jimmunol.168.9.4711. [DOI] [PubMed] [Google Scholar]
- 48.Yi AK, Yoon JG, Krieg AM. Convergence of CpG DNA- and BCR-mediated signals at the c-Jun N-terminal kinase and NF-kappaB activation pathways: regulation by mitogen-activated protein kinases. Int Immunol. 2003;15:577–591. doi: 10.1093/intimm/dxg058. [DOI] [PubMed] [Google Scholar]
- 49.Yeo SJ, Yoon JG, Hong SC, Yi AK. CpG DNA induces self and cross-hyporesponsiveness of RAW264.7 cells in response to CpG DNA and lipopolysaccharide: alterations in IL-1 receptor-associated kinase expression. J Immunol. 2003;170:1052–1061. doi: 10.4049/jimmunol.170.2.1052. [DOI] [PubMed] [Google Scholar]
- 50.Cabrera-Poch N, Sanchez-Ruiloba L, Rodriguez-Martinez M, Iglesias T. Lipid raft disruption triggers protein kinase C and Src-dependent protein kinase D activation and Kidins220 phosphorylation in neuronal cells. J Biol Chem. 2004;279:28592–28602. doi: 10.1074/jbc.M312242200. [DOI] [PubMed] [Google Scholar]
- 51.Park Y, Lee SW, Sung YC. Cutting Edge: CpG DNA inhibits dendritic cell apoptosis by up-regulating cellular inhibitor of apoptosis proteins through the phosphatidylinositide-3′-OH kinase pathway. J Immunol. 2002;168:5–8. doi: 10.4049/jimmunol.168.1.5. [DOI] [PubMed] [Google Scholar]
- 52.Gelman AE, LaRosa DF, Zhang J, Walsh PT, Choi Y, Sunyer JO, Turka LA. The adaptor molecule MyD88 activates PI-3 kinase signaling in CD4+ T cells and enables CpG oligodeoxynucleotide-mediated costimulation. Immunity. 2006;25:783–793. doi: 10.1016/j.immuni.2006.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Honda Z, Suzuki T, Hirose N, Aihara M, Shimizu T, Nada S, Okada M, Ra C, Morita Y, Ito K. Roles of C-terminal Src kinase in the initiation and the termination of the high affinity IgE receptor-mediated signaling. J Biol Chem. 1997;272:25753–25760. doi: 10.1074/jbc.272.41.25753. [DOI] [PubMed] [Google Scholar]
- 54.Lemonnier J, Ghayor C, Guicheux J, Caverzasio J. Protein kinase C-independent activation of protein kinase D is involved in BMP-2-induced activation of stress mitogen-activated protein kinases JNK and p38 and osteoblastic cell differentiation. J Biol Chem. 2004;279:259–264. doi: 10.1074/jbc.M308665200. [DOI] [PubMed] [Google Scholar]
- 55.Cortez-Gonzalez X, Pellicciotta I, Gerloni M, Wheeler MC, Castiglioni P, Lenert P, Zanetti M. TLR9-independent activation of B lymphocytes by bacterial DNA. DNA Cell Biol. 2006;25:253–261. doi: 10.1089/dna.2006.25.253. [DOI] [PubMed] [Google Scholar]
- 56.Stunz LL, Lenert P, Peckham D, Yi AK, Haxhinasto S, Chang M, Krieg AM, Ashman RF. Inhibitory oligonucleotides specifically block effects of stimulatory CpG oligonucleotides in B cells. Eur J Immunol. 2002;32:1212–1222. doi: 10.1002/1521-4141(200205)32:5<1212::AID-IMMU1212>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 57.Song MJ, Wang YQ, Wu GC. Lipopolysaccharide-induced protein kinase D activation mediated by interleukin-1beta and protein kinase C. Brain Res. 2007;1145:19–27. doi: 10.1016/j.brainres.2007.01.128. [DOI] [PubMed] [Google Scholar]
- 58.Marie I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. Embo J. 1998;17:6660–6669. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]