

Abstract
The dentate gyrus (DG) normally functions as a filter, preventing propagation of synchronized activity into the seizure-prone hippocampus. This filter or ‘gatekeeper’ attribute of the DG is compromised in various pathological states, including temporal lobe epilepsy (TLE). This study examines the role that altered inhibition may play in the deterioration of this crucial DG function. Using the pilocarpine animal model of TLE, we demonstrate that inhibitory synaptic function is altered in principal cells of the DG. Spontaneous miniature inhibitory postsynaptic currents (mIPSCs) recorded in dentate granule cells (DGCs) from epileptic animals were larger, more sensitive to blockade by zinc and less sensitive to augmentation by the benzodiazepine type site 1 modulator zolpidem. Furthermore, mIPSCs examined during a quiescent period following injury but preceding onset of epilepsy were significantly smaller than those present either in control or in TLE DGCs, and had already acquired sensitivity to blockade by zinc prior to the onset of spontaneous seizures. Rapid agonist application experiments demonstrated that prolonged (>35 ms) exposure to zinc is required to block GABAA receptors (GABAARs) in patches pulled from epileptic DGCs. Therefore, zinc must be tonically present to block DGC GABAARs and alter DG function. This would occur only during repetitive activation of mossy fibres. Thus, in the pilocarpine animal model of TLE, an early, de novo, expression of zinc-sensitive GABAARs is coupled with delayed, epilepsy-induced development of a zinc delivery system provided by aberrant sprouting of zinc-containing mossy fibre recurrent collaterals. The temporal and spatial juxtaposition of these pathophysiological alterations may compromise normal ‘gatekeeper’ function of the DG through dynamic zinc-induced failure of inhibition, predisposing the hippocampal circuit to generate seizures.
Keywords: dentate gyrus, electrophysiology, pilocarpine, zinc, zolpidem
Introduction
In temporal lobe epilepsy (TLE), the hippocampus becomes involved in the generation of seizures. The factors responsible for this altered hippocampal function is an area of active investigation. In TLE, mossy fibres (MFs), the axons of dentate granule cells (DGCs), frequently sprout and aberrantly innervate the somatic and inner molecular layers of the dentate gyrus (Tauck & Nadler, 1985; Cronin & Dudek, 1988; Sutula et al., 1989; Babb et al., 1991; Okazaki et al., 1995). In addition to glutamate, MF terminals also contain high concentrations of zinc, which is colocalized in synaptic vesicles and coreleased with glutamate. Stimulation of MFs can produce extracellular zinc concentrations as high as 100–300 μM in the control hippocampus (Assaf & Chung, 1984; Howell et al., 1984).
In addition to circuit rearrangements, there are alterations in expression and function of neurotransmitter receptors in the epileptic dentate gyrus (DG), including prominent changes in function of DGC GABAA receptors (GABAARs). Both GABA efficacy and the amplitude of miniature inhibitory postsynaptic currents (mIPSCs) increase in DGCs from epileptic animals, reflecting an overall increase in the numbers of both total membrane and subsynaptic GABAARs (Gibbs et al., 1997; Nusser et al., 1998a; Otis et al., 1994). Accompanying this increase in GABAARs are significant changes in their pharmacology. Most notably, DGC GABAARs in epileptic hippocampus become sensitive to blockade by zinc, with an IC50 of 30 μM (Buhl et al., 1996; Gibbs et al., 1997). In contrast, GABAARs in DGCs from control animals are virtually zinc-insensitive. This altered zinc pharmacology of DGC GABAARs is due to a transcriptionally mediated alpha subunit switch, from receptors containing predominantly alpha1 and alpha2 subunits to receptors containing much higher levels of alpha4 subunit (Brooks-Kayal et al., 1998; reviewed in Coulter, 2000).
This apposition of a zinc delivery system (sprouted MFs) together with zinc-sensitive DGC GABAARs in the epileptic hippocampus led to the formulation of a hypothesis that postulates that the combination of these two pathological factors could lead to a dynamic zinc-mediated collapse of inhibition in the epileptic DG. During seizure initiation, zinc, released from sprouted MFs, may diffuse to and block neighbouring GABAARs, and unmask excitatory recurrent collateral activity. This could compromise the normal ‘gatekeeper’ function of this area, and facilitate seizure generation (Heinemann et al., 1992; Lothman et al., 1992; Buhl et al., 1996; Gibbs et al., 1997; reviewed in Coulter, 2000).
In the present study, we examine several interrelated issues relating to the above hypothesis. TLE is induced by brain injury, following which there is a delay (or latent period) before spontaneous seizures emerge. This delay can be weeks or months in animals, and years in humans. Within 24 h following an injury which will go on to elicit TLE, zinc-sensitive GABAARs appear (Brooks-Kayal et al., 1998). Do these aberrant receptors reach the synapse? If so, when? Finally, what are the kinetics of zinc blockade of GABAARs in animals with TLE, and how might this relate to seizure initiation and/or propagation?
Materials and methods
Pilocarpine injections
All experimental procedures and protocols for animal studies were approved by the CHOP/UPENN Institutional Animal Care and Use Committee. Pilocarpine animals were produced using previously reported methods (Mello et al., 1993; Gibbs et al., 1997; Brooks-Kayal et al., 1998). Adult male Sprague-Dawley rats, ≈120 days postnatal, were pretreated with scopolamine methyl nitrate (1 mg/kg, i.p.) to antagonize peripheral effects induced by subsequent pilocarpine (350 mg/kg, i.p.) injection. Thirty minutes after scopolamine pretreatment, pilocarpine injection triggered status epilepticus (SE; i.e. sustained seizures lasting >30 min) within 10–30 min after injection. Rats that did not exhibit behavioural seizures within 1 h of pilocarpine injection received a booster injection of pilocarpine (175 mg/kg, i.p.) One hour after onset of SE, diazepam (4 mg/kg, i.p.) was administered in order to quell seizure activity. Additional doses of diazepam at 3 and 5 h after SE onset were administered as needed. Sham pilocarpine rats were treated in exactly the same manner as pilocarpine-injected rats, except that a subconvulsive dose of pilocarpine (35 mg/kg) was used. This animal model shares a natural history of development with human TLE in that it is characterized by a latent or silent period between the initial injury (here the initial precipitating event is pilocarpine-induced SE) and the onset of recurrent seizures (Engel, 1989; French et al., 1993; Spencer & Spencer, 1994). This seizure-free or silent period present in this model is a prominent feature of clinical epilepsy, and typically lasts 2–6 weeks in the pilocarpine model in our laboratory. An additional experimental group was termed latent-period animals, and was derived from animals killed 6–8 days following pilocarpine-induced SE, well before the onset of spontaneous seizures in >95% of animals. Rats were video-monitored beginning 10–14 days after pilocarpine injection to document at least two spontaneous seizures before being classified as epileptic. To minimize acute effects of seizures on GABAA receptor properties, epileptic rats were further monitored to ensure that no seizures had occurred 24 h before use.
Tissue preparation
Rats were divided into four groups: pilocarpine-treated, latent (6–8 days post-SE onset), sham latent and control. Animals in the sham latent group were treated identically to animals in the latent group except that they received a subconvulsive dose of pilocarpine. Naive rats (i.e. without pilocarpine injections) were also used as controls and results obtained from these animals were not significantly different from subconvulsive treated rats; therefore, results from sham latent, sham pilocarpine and naive animals were pooled. The total number of animals used were 14 controls (six naïve, eight sham), four latent, three sham latent and 15 pilocarpine-induced epileptic. Brain slices were prepared using previously reported methods (Rafiq et al., 1993; Cohen et al., 2000; Lin et al., 2001). In brief, rats were anaesthetized with halothane, decapitated, and the brain quickly removed and chilled for 2 min in a modified sucrose-based artificial cerebral spinal fluid (aCSF) composed of (in mM): sucrose, 201; KCl, 3.2; NaHPO4, 1.25; MgCl2, 2; CaCl2, 2; NaHCO3, 26; and glucose, 10 (equilibrated with 95%O2, 5% CO2 at 32.5 °C). The brain was then hemisected and each side was glued rostral-side up onto a 12° agar ramp with cyanoacrylate cement, and 225-μm hippocampal–entorhinal–cortical (HEC) brain slices were sectioned using a Vibratome (Lancer 1000, St Louis, MO, USA). HEC brain slices have been previously shown to keep the maximal number of intact dentate granule cell–CA3 axons and synapses, which enhances the frequency of spontaneous activity (Rafiq et al., 1993). Brain slices were subsequently transferred to a holding chamber and incubated in warm (35 °C) normal aCSF containing 126 mM NaCl substituted for sucrose, and allowed to equilibrate for at least 2 h before being transferred to the recording chamber.
Patch recording in slices
Whole-cell voltage-clamp recordings were conducted at room temperature from visually identified DGCs using either Hoffman modulation or Nomarski differential interference contrast video microscopy. DGCs were voltage clamped at −60 mVand signals were recorded and amplified with an Axopatch 1D (Axon Instruments, Foster City, CA, USA), filtered at 1 kHz, digitized and sampled at 44 kHz with a PCM digitizer (Neuro-Corder DR-890; Neurodata Instruments, NY, NY, USA) and stored on videotape for off-line analysis. Electrodes were fabricated from thick-walled borosilicate glass (World Precision Instruments, Sarasota, FL, USA) and pulled on a two-stage puller (Narishige PP-83, East Meadow, NY, USA) to a resistance between 2 and 6 MΩ when filled with an internal solution composed of (in mM): CsCl, 135; HEPES, 10; MgCl2, 2; NaATP, 4; pH 7.25 (CsOH).
Rapid agonist application
Fast application of agonists was performed as described by Jonas (1995). Theta glass was mounted on a piezoelectric transducer (Burleigh, Fishers, NY, USA). Waveform protocols were generated using Clampex 7.0 software (Axon Instruments). Agonists were applied at 10–20 s intervals, and traces shown in figures are averaged from at least five applications. Upon excision of an outside-out patch, the tip of the patch electrode was positioned in the control solution, ≈20 μm from the interface separating the control and drug streams, which was visualized by the addition of 25 mM sucrose to the drug solution. The patches yielded GABA-evoked currents between 100 and 1500 pA in amplitude. After rupturing the patch, the 20–80% exchange times of the liquid junction currents between control and a 90% control/10% distilled H2O solution was typically between 200 and 250 μs, and these ‘patch-less’ recordings are used as the stimulus protocol above the traces in Fig. 7 below.
Fig. 7.
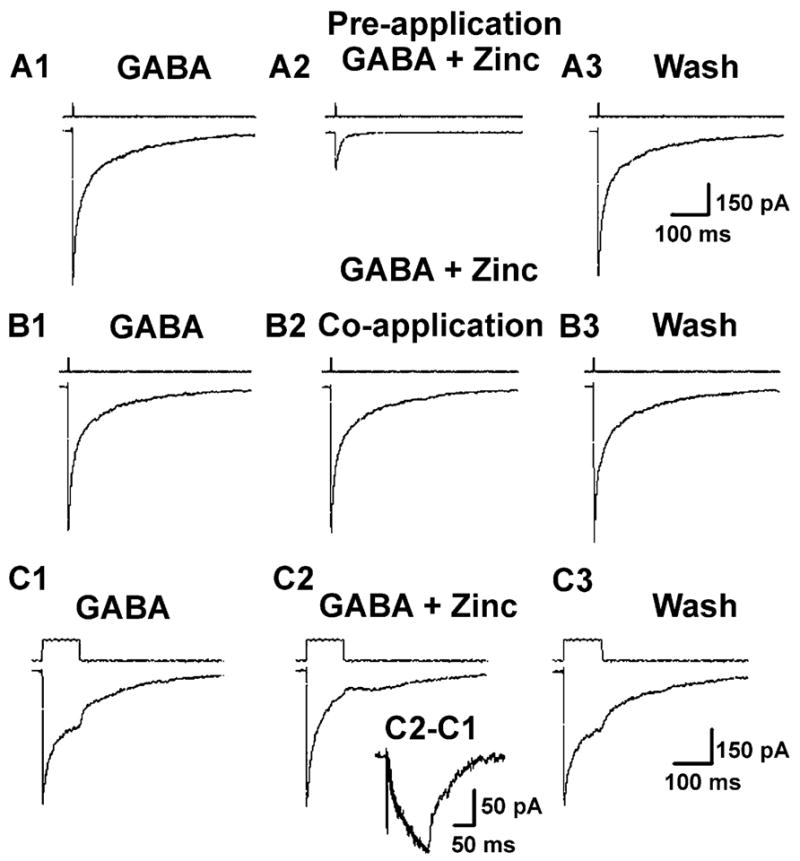
Zinc had to be tonically applied to block currents evoked by rapid GABA application. All records in A and B are from the same patch, pulled from an epileptic DGC. (A1) Currents elicited by 1 ms pulse of 1 mM GABA (IGABA = 712 pA). (A2) Currents elicited by 1 ms pulse of 1 mM GABA + 200 μM zinc after the patch had been pre-exposed to 200 μM zinc for 60 s (IGABA = 178 pA, a 75% decrease compared to A1). (A3) currents elicited by 1 ms pulse of 1 mM GABA alone to the same patch after zinc wash (IGABA = 672 pA). (B) The same patch re-exposed to (B1) GABA alone, (B2) zinc coapplied with GABA (note: no 60-s zinc pre-exposure), and (B3) GABA alone after zinc washout. Notice no blockade of IGABA by zinc when coapplied. The open-tip electrode solution switching responses are displayed above each current trace. (C1) Currents elicited by 100 ms pulse of 1 mM GABA. (C2) Currents elicited by 100 ms pulse of 1 mM GABA +200 μM zinc. (C3) currents elicited by 100 ms pulse 1 mM GABA alone after zinc washout. (Inset) Trace derived by subtracting GABA alone response (C1) from GABA +zinc (C2). Superimposed on the subtracted trace is a single exponential curve (smooth line) with a time constant (τ) = 42 ms. All traces are GABA-evoked currents from an epileptic DGC outside-out patch (VHOLD = −60 mV), and are averaged from five consecutive responses.
Analysis of miniature inhibitory postsynaptic currents
Recorded mIPSCs were reacquired using Dempster software (Strathclyde, Glasgow, UK), which collects events using a manually controlled threshold detector, and is capable of detecting events as small as 2–3× the baseline noise. After a stretch of 200–500 detected events was reacquired, individual events were re-examined and only mIPSCs with 10–90% rise times <1 ms were kept (usually >95% of events). In order to minimize cases of inadequate space clamp, neurons were used for analysis only when series resistance (Rs) was <20 MΩ and at least 80% series resistance compensation was achieved. Rs was checked frequently throughout experiments, and neurons in which Rs increased by >20% were discarded. In addition, event amplitudes were plotted against rise times and examined for a possible correlation, where a significant correlation (r2 >0.5) was assumed to signify inadequate space clamp. These neurons were also discarded (<4% of cells). mIPSC kinetics measures (conductance, 50% decay times) were analysed using cumulative probability plots. mIPSC frequency was determined using Mini Analysis program (Synaptosoft Inc., Leonia, NJ, USA).
Reagents and statistical tests
Reagents were purchased from the following vendors: all salts, zinc and zolpidem (ZOL) from Sigma (St Louis, MO, USA); D-2-amino-5-phosphonopentanoic acid (AP5) and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) from Research Biochemicals International (Natick, MA, USA); tetrodotoxin (TTX) from Calbiochem (La Jolla, CA, USA).
Statistical significance between cumulative probability distributions in control and drug conditions in individual neurons was assessed at the P <0.005 confidence level using the Kolmogorov–Smirnov nonparametric statistical test. Two-tailed unpaired Student’s t-tests were performed to determine statistical significance at the P <0.05 confidence level when comparing different treatment groups. All data are presented as group means ±SD.
Results
mIPSC properties in control and epileptic DGCs
In the presence of TTX (400 nM) and the excitatory amino acid antagonists D-AP5 (50 μM) and CNQX (6 μM), spontaneous inward currents with varying amplitudes were recorded (Fig. 1A and B; note larger amplitude events in Epileptic sweeps). The GABAergic identity of these events was confirmed by their blockade by the GABAA antagonist bicuculline methiodide (30 μM, n = 4, data not shown). Median mIPSC conductances [calculated by dividing the mean median mIPSC amplitude by the driving force (EHOLD − ECL = −60 mV)] were larger in epileptic neurons than in control DGC mIPSCs, while decay times were similar in both groups (see Fig. 1C and D, and cumulative probability histograms in Fig. 1E and F). On average, the median mIPSC conductance in epileptic neurons was significantly larger (60% enhancement; P <0.05, unpaired t-test; Fig. 4C). Mean mIPSC conductance was 0.69 ± 0.10; 1.06 ± 0.13 nS for control (n = 21) and epileptic (n = 24) neurons, respectively. These data are similar to the 75% increase recorded in DGCs in kindled animals (Otis et al., 1994), and support our previously reported results demonstrating a 78% increase in GABAAR current density in acutely dissociated DGCs from chronic epileptic animals (Gibbs et al., 1997). Interestingly, epileptic mIPSCs decayed with similar kinetics to controls (10.77 ± 1.7 and 12.07 ± 1.5 ms 50% decay time, epileptic and control, respectively; Fig. 1F). However, because the measurement of the 50% decay time is sensitive to noise in the recordings, we calculated the weighted decay τ-values for all groups. Weighted decay τ-values were calculated by dividing the area of the events by the peak amplitudes and the average values compared. The mean weighted τ-value was 17.43 ± 1.44 (n = 24) for the epileptic population and significantly slower (P <0.05) than the mean τ-value (14.89 ± 1.55, n = 11) for the control group. Furthermore, because mIPSC conductance and kinetic responses can be affected by clamp control problems we compared the series resistance in all groups. No significant differences were found between any of the groups (data not shown).
Fig. 1.
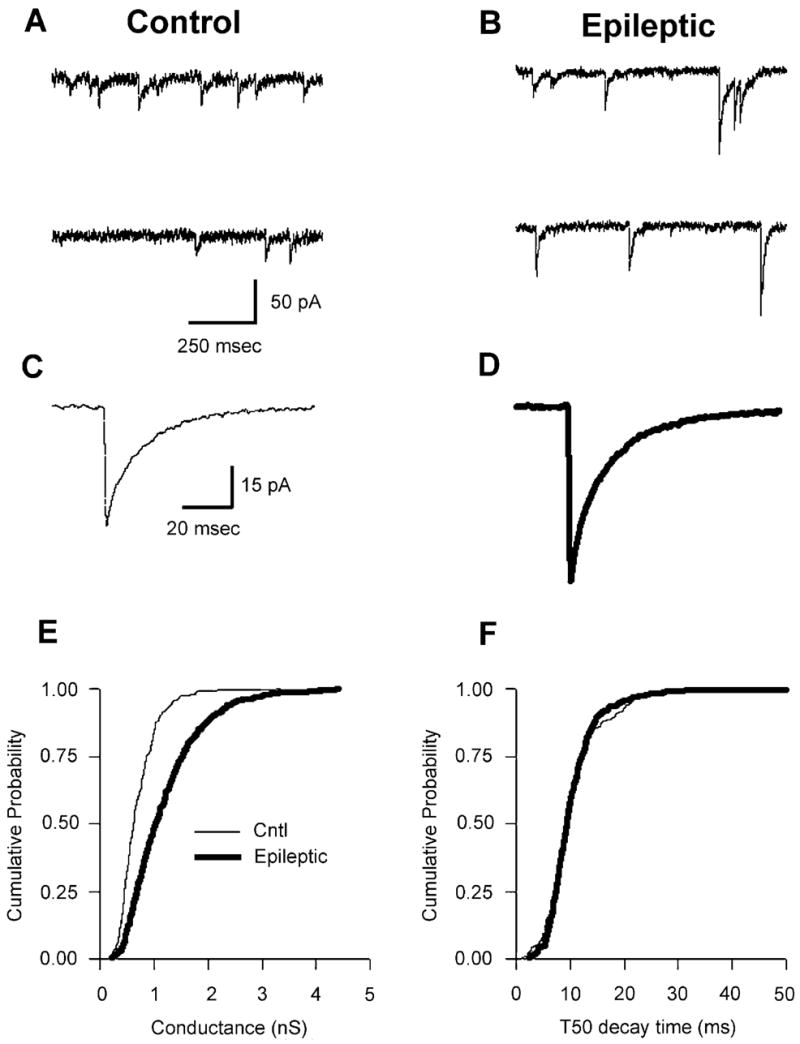
Comparison of mIPSCs in control and epileptic DGCs. (A and B) mIPSCs from (A) control and (B) epileptic DGCs. Data are continuous sweeps in control and epileptic neurons, respectively. (C and D) Averaged mIPSCs (from 50 random events) in the same cells as in A and B. Note larger average amplitude mIPSC in the epileptic neuron (D). (E) Cumulative probability–conductance plot demonstrating that mIPSCs recorded in epileptic DGCs (thick trace) were larger than those present in control DGCs. (F) Cumulative probability–50% decay time (T50) plot demonstrating that mIPSCs recorded in epileptic DGCs (thick trace) were identical to those recorded in control DGCs. However, the averaged weighted decay τ was significantly slower than averaged value in controls (see text). All data are from whole-cell patch-clamp recordings of DGCs in the presence of TTX (400 nM), CNQX (6 μM) and D-AP5 (50 μM). VHOLD = −60 mV in all experiments.
Fig. 4.
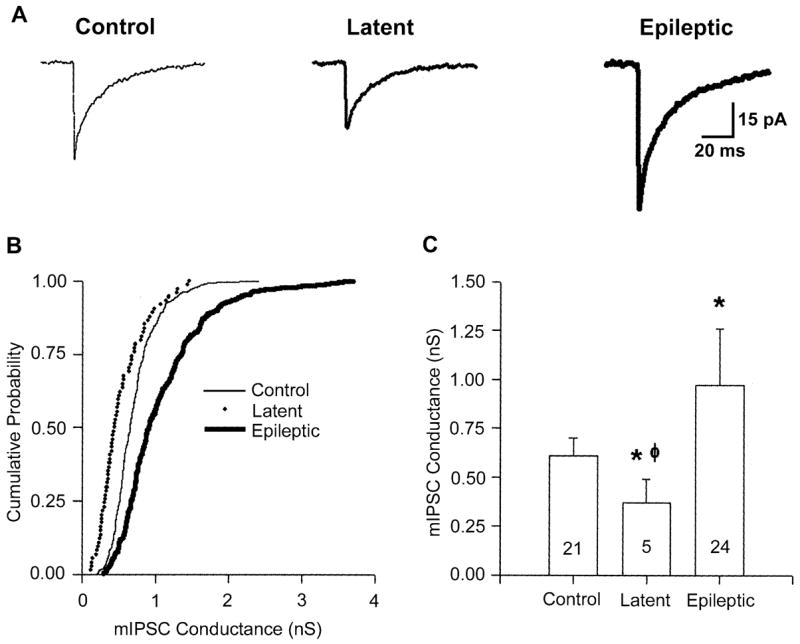
Reduced amplitude mIPSCs in DGCs during the latent period. (A) Averaged mIPSCs (50 random traces) recorded in control, latent and epileptic DGCs, demonstrating that mIPSCs recorded from DGCs in the latent period were smaller than those recorded in control and epileptic DGCs. (B) Cumulative probability–conductance plot for the neurons represented in A. Note the smaller conductance (and therefore smaller amplitude mIPSCs) in the entire population of events in the latent period DGC compared to both the control and epileptic DGC events. (C) Histogram of median mIPSC conductances for the entire population of recorded cells, demonstrating a significant decrease in latent-period mIPSC conductance compared to both control (*) and epileptic (φ) mIPSC conductances (*,φP ≤0.05, two-tailed Student’s t-test.
Although mIPSC conductances and weighted decay τ-values were significantly different between groups, mIPSC frequencies were similar. The mean mIPSC frequency was 2.8 ± 1.2 Hz in epileptic (n = 10) neurons, not significantly different than controls (3.5 ± 1.1; n = 10; data not shown). The frequency values in our controls are similar to those reported previously by Hajos et al. (2000), but are lower than those reported by Otis et al. (1994) and Buhl et al. (1996). However, this may be explained by the fact that both of the latter studies were performed at 34 °C, while the present study, like that of Hajos and colleagues, was carried out at room temperature.
mIPSCs in epileptic DGCs are zinc-sensitive
In addition to the increased DGC mIPSC conductance (Otis et al., 1994) and enhanced GABAAR current density (Gibbs et al., 1997), the pharmacological properties of GABAARs recorded in DGCs from epileptic animals was also altered. GABAARs are pentameric structures composed of subunits from several related subunit families. At present, eight different GABAAR subunit families have been cloned (with many families having multiple members), including alpha (1–6), beta (1–4), gamma (1–3), delta, rho (1–3), epsilon, pi and theta (reviewed in Macdonald & Olsen, 1994; Barnard et al., 1998). The subunit stoicheometry is the primary determinant of the pharmacology of the receptor. For example, GABAARs lacking a gamma2 subunit are significantly more sensitive to blockade by zinc (Verdoorn et al., 1990; Hosie et al., 2003). However, if a gamma2 subunit is present within the GABAAR pentamer, zinc sensitivity varies as a function of the specific alpha subtypes expressed, i.e. low alpha1 yields high zinc sensitivity (White & Gurley, 1995; Burgard et al., 1996; Fisher & Macdonald, 1998). We have previously shown that GABAARs in DGCs isolated from epileptic animals are zinc-sensitive (Gibbs et al., 1997; Brooks-Kayal et al., 1998). In kindled animals, DGC mIPSCs are also blocked by zinc, unlike controls (Buhl et al., 1996). In the present study, we examined whether synaptic GABAARs in DGCs from epileptic animals are also sensitive to blockade by zinc. Bath application of zinc (300 μM) significantly (P <0.05) decreased the conductance of mIPSCs recorded in DGCs from epileptic animals [Figs 2 (A2 and B2) and 5C). The mean conductance was significantly reduced from 1.06 ± 0.13 to 0.85 ± 0.12 nS (a 20% reduction; n = 5, P <0.05; see Fig. 5C, below). The median as well as the mean 50% mIPSC decay time was unaltered by zinc (Fig. 2, C2). These results contrast with zinc effects on mIPSCs recorded from control DGCs, where a 20-min bath application of zinc had no discernable effects on either mIPSC conductance or decay time [Figs 2 (A1–C1) and 5C).
Fig. 2.
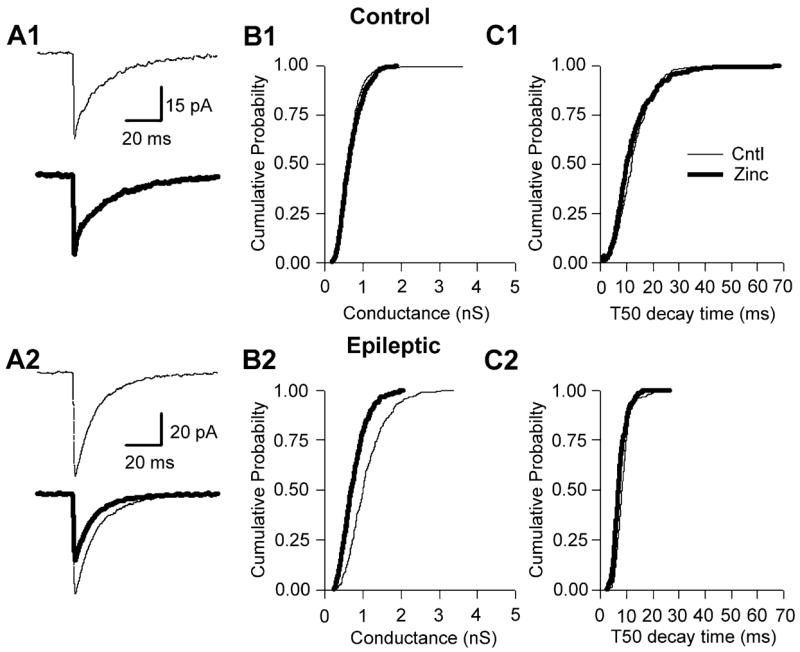
Zinc blocked mIPSCs in epileptic, but not control, DGCs. (A1) Averaged mIPSCs (50 events) taken before (thin line) and during (thick line) bath application of zinc (300 μM) to a control DGC. (B1) Cumulative probability–conductance and (C1) cumulative probability −50% decay time (T50) plots for the same cell as represented in A1, demonstrating that zinc has no effect on mIPSC conductance or T50 in control DGCs. (A2) Averaged mIPSCs taken before (thin line) and during (thick line) bath application of zinc to epileptic DGCs, demonstrating that zinc significantly reduced mIPSC amplitude (see A2 lower sweep; note difference in scale bar amplitude). (B2) Cumulative probability–conductance plot demonstrating a significant reduction in mIPSC conductance. (C2) However, the cumulative probability–decay plot for the same neuron as in A2 clearly demonstrates that zinc was without effect on epileptic mIPSC decay time.
Fig. 5.
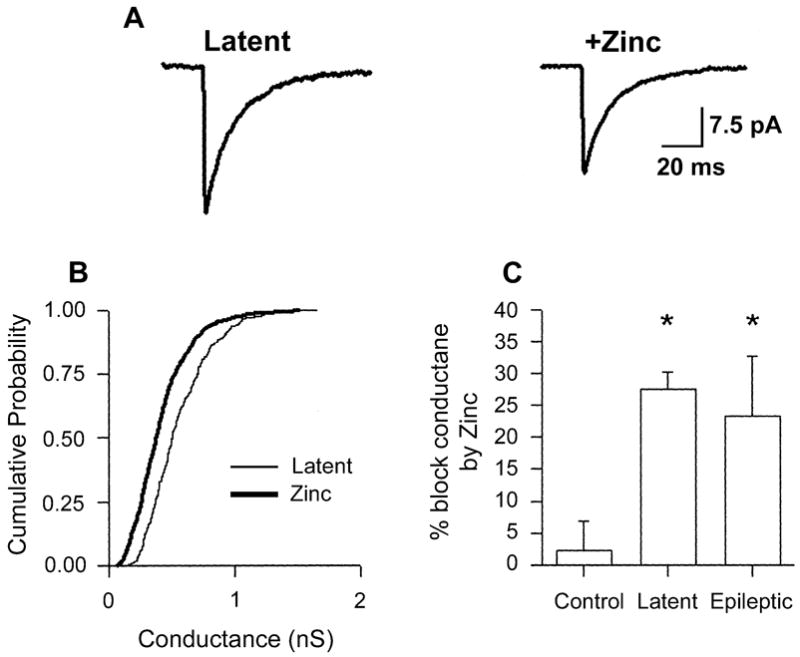
mIPSCs in DGCs during the latent period are zinc-sensitive. (A) Averaged mIPSCs (50 events) taken before and during bath application of zinc (300 μM) to a latent-period DGC demonstrating a decrease in mIPSC amplitude during zinc exposure. (B) Cumulative probability–conductance plots for the same cell as represented in A, demonstrating that zinc significantly reduced mIPSC conductance. (C) Histogram of percentage block of mIPSC conductance by zinc (300 μM) in control (n = 4), latent period (n = 4) and epileptic (n = 5) DGCs. Zinc had no effect in controls, compared to 25% block of mIPSC conductance in epileptic DGCs. *P ≤0.05 vs. control, two-tailed Student’s t-test.
We then conducted additional pharmacological studies to explore possible alterations in subunit composition of GABAARs which might explain the enhanced zinc sensitivity of subsynaptic inhibitory receptors in the DG of epileptic animals.
mIPSCs in epileptic DGC are zolpidem-insensitive
One mechanism which could confer zinc sensitivity onto GABAARs is a down-regulation of alpha1 subunits in DGCs from epileptic animals (reviewed in Coulter, 2001). This would also concomitantly decrease GABAAR sensitivity to benzodiazepine (BDZ) site 1 agonists, such as zolpidem (ZOL; Arbilla et al., 1986; reviewed in Macdonald & Olsen, 1994; Barnard et al., 1998). To examine this issue, ZOL was applied to control and TLE slices. Bath application of ZOL (200 nM) significantly augmented the decay of the control DGC mIPSCs (Fig. 3, A1 and C1). The mean 50% mIPSC decay time (T50) increased 214% (from 10.77 ± 1.75 to 23.12 ± 4.7 ms, n = 5). As expected, control DGC mIPSC conductances were unaffected by ZOL application (see Fig. 3, A1 and B1) similar to previous reports (Otis & Mody, 1992; Poncer et al., 1996; Cohen et al., 2000). These results are in marked contrast to ZOL effects on DGCs from epileptic animals. Bath application of ZOL to slices prepared from epileptic animals had virtually no effect on mIPSC amplitude and conductance (see Fig. 3, A2 and B2; n = 8) or decay (Fig. 3, A2 and C2), supporting our hypothesis that alpha1 subunit expression in synaptic DGC GABAARs is decreased.
Fig. 3.
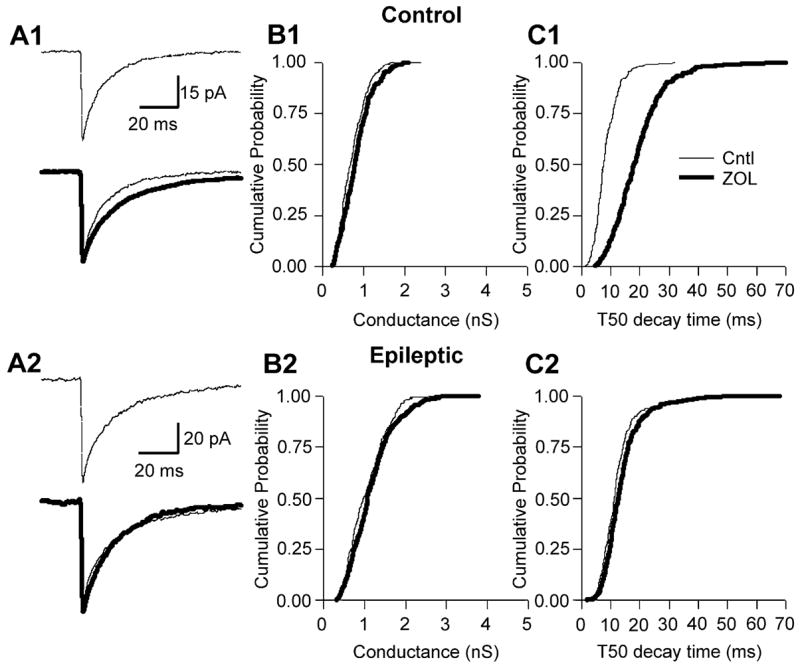
Zolpidem augmented mIPSCs in control, but not epileptic, DGCs. (A1) Averaged mIPSCs (50 events) taken before (thin line) and during (thick line) bath application of ZOL (200 nM) to a control DGC. (B1) Cumulative probability–conductance and (C1) cumulative probability –50% decay time (T50) plots for the same cell as represented in A1, demonstrating that ZOL significantly augmented mIPSC T50 in control DGCs. (A2) Averaged mIPSCs taken before (thin line) and during (thick line) bath application of ZOL to epileptic DGCs, demonstrating no effect on mIPSC amplitude or T50 (see A2 lower sweep; note difference in scale bar amplitude). (B2 and C2) Cumulative probability–conductance and –T50 plots, demonstrating no effect of ZOL on mIPSC conductance or T50.
We next examined when, during the time course of development of TLE, do the alterations in subsynaptic GABAARs emerge, specifically focusing on the question of whether these alterations precede the onset of spontaneous seizures in animals destined to become epileptic.
mIPSCs recorded in DGCs during the latent period are small and zinc-sensitive
Human TLE is characterized by a latent or silent period between the initial precipitating event and the onset of recurrent spontaneous seizures defining a chronic epileptic state (Avanzini et al., 1997). That is, following the initial injury there is a temporal window in which no overt seizures are detected. Animal models of TLE, including the pilocarpine model, also have a characteristic latent period (Mello et al., 1993). The length of the latent period is variable. In humans, this period can last for years or even decades. In the pilocarpine animal model, this period typically lasts 2–8 weeks. To examine possible changes in GABAAR properties during the latent period, we recorded mIPSCs in slices from animals 6–8 days post-SE. At this time point, <5% of our animals have recurrent spontaneous seizures. In 10 DGCs recorded from latent-period animals, the amplitude of mIPSCs was always significantly smaller when compared to either control or epileptic DGCs (Fig. 4A–C). The median conductance amplitude for mIPSCs in latent period DGCs was 0.53 ± 0.2 nS (n = 10), significantly (P <0.05) smaller than those present in control (by 30%) and TLE (by 58%) DGCs (Fig. 4C). Interestingly, in 3/13 neurons from these latent-period animals, no mIPSCs were evident; however, all three cells were recorded from slices generated from the same animal. Therefore, the absence of mIPSCs in these cells might be anomalous. This contrasts with slices from both control and epileptic animals where every DGC recorded exhibited robust mIPSC activity (45/45). Furthermore, in six sham-latent DGCs (recorded from animals receiving a subconvulsive pilocarpine dose and killed 6–8 days following treatment) the mean conductance was 0.68 ± 0.14 nS, not significantly different from mIPSC conductance present in controls (0.69 ± 0.10 nS). Despite the above finding that mIPSCs were significantly smaller during the latent period, the GABAAR pharmacological transition evident in neurons from chronically epileptic animals had already occurred, very early in the development of TLE. Bath application of zinc (300 μM) significantly (P <0.05) decreased the conductance of mIPSCs recorded from DGCs in latent-period animals (Fig. 5A–C). The median conductance was significantly reduced, from 0.53 ± 0.2 to 0.41 ± 0.06 nS (a 20% reduction; n = 4, P <0.05; Fig. 5C). The T50 as well as the weighted decay τ was unaltered by zinc (data not shown).
We then went on to further investigate the mechanism of zinc blockade of GABAARs in order to better understand the temporal requirements for zinc presence to block zinc-sensitive GABAARs present in the epileptic hippocampus, thereby altering circuit function in the DG.
Mechanism of action of GABAAR blockade by zinc
Recent evidence using cultured hippocampal neurons has demonstrated that zinc slows the microscopic GABAAR gating parameters responsible for binding rate (kon) and the transition rate from closed to open state (beta2), thereby slowing mIPSC rise times (Barberis et al., 2000). We compared mIPSC rise times in epileptic neurons in the presence and absence of zinc by measuring 10–90% rise times of 20 random mIPSCs in control and epileptic neurons in normal and zinc-containing aCSF (n = 4 for both control and epileptic populations) following reacquisition at 100 kHz and filtered at 5 kHz. Average epileptic mIPSC rise times were not significantly different from those measured in control DGCs (Fig. 6C); however, they were significantly faster (28%; Fig. 6A and C) than those measured from epileptic mIPSCs in the presence of zinc (Fig. 6B; 0.51 ± 0.02 and 0.71 ± 0.02 ms for control and epileptic, respectively, P <0.05). The difference in rise times in control and epileptic neurons is clearly evident when the epileptic trace is scaled to the control trace (Fig. 6B). Our data supports the idea that zinc slows the binding rate kon and the transition rate from closed to open state (beta2) (Barberis et al., 2000).
Fig. 6.
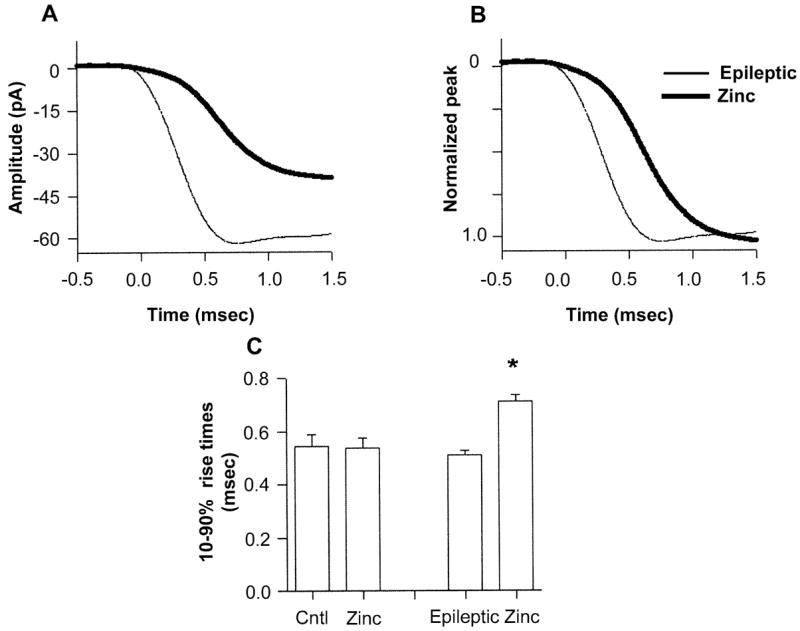
mIPSC kinetics were significantly altered during zinc exposure in epileptic neurons. (A) Averaged rise time (n = 20) from an epileptic DGC (thin line) in control aCSF (mean 0.47 ms), and in the presence of zinc (thick line, mean 0.76 ms). (B) Normalization of the zinc and control traces more clearly illustrates the slowed onset of mIPSCs during zinc exposure. The traces were lined up by aligning their 10% rise times. (C) Histogram of mean mIPSC 10–90% rise times in control and epileptic populations in normal and zinc-containing aCSF. *P ≤ 0.05, paired t-test, between epileptic in control and zinc-containing aCSF.
To further investigate the mechanism of GABAAR blockade by zinc, we conducted rapid agonist application experiments on perisomatic patches excised from epileptic DGCs. This allowed us to temporally mimic synaptic release of GABA and to precisely control the concentration and time of exposure of agonists and antagonists. Patches were first exposed to 1 mM GABA to determine the magnitude of the GABA-evoked transient. Patches were then either pre-exposed to zinc (200 μM) for 60 s and subsequently exposed to zinc coapplied with GABA (1 mM), or directly exposed to zinc coapplied with GABA. In addition, the duration of the GABA exposure pulse was varied, with either 1- or 100-ms exposure times. The peak of GABA-evoked transients were only blocked when zinc was preapplied to the patch, and was unaffected by the duration of the pulse. The mean zinc-induced block of GABA-evoked transients was 83.65 ± 7.03% (n = 8, Fig. 7A) and 86.08 ± 7.06% (n = 6, data not shown) for 1- and 100-ms pulse durations, respectively. When zinc was coapplied with GABA, no significant block of the peak GABA-evoked transient occurred (n = 6; see Fig. 7B). However, the falling (desensitization) phase of the GABA response was significantly accelerated during zinc exposure, presumably due to zinc slowly binding to and blocking the receptors. We were able to quantify the time course of zinc blockade of GABAARs by subtracting the zinc +GABA trace from the GABA trace (Fig. 7C and inset). We found that the mean time constant for zinc blockade was 35.25 ± 9.5 ms (n = 4).
Discussion
In the hippocampus, zinc is normally stored in synaptic vesicles present in MF terminals (Frederickson & Danscher, 1990; Slomianka, 1992), and is coreleased with glutamate (Assaf & Chung, 1984; Vogt et al., 2000). In TLE, MFs sprout and form monosynaptic feedback innervation onto DGCs present in the inner molecular layer (Babb et al., 1991; Okazaki et al., 1992). This serves to increase excitatory drive and form synaptic interconnections necessary for synchronization in this region (Buckmaster & Dudek, 1997; Patrylo & Dudek, 1998; Wuarin & Dudek, 2001). Recordings in slices obtained from epileptic animals exhibiting robust MF sprouting demonstrated that the dentate gyrus becomes epileptogenic, i.e. will generate and propagate seizure activity (Wuarin & Dudek, 1996). However, this occurred primarily under conditions of reduced inhibition and enhanced membrane excitability, suggesting that compensatory mechanisms may oppose synchronization mediated by these recurrent collaterals.
In the epileptic brain, the altered cellular and circuit players are set in place for seizure generation and propagation, i.e., disinhibition, recurrent excitatory connections and bursting conductances (Wong et al., 1986). Why, then, are seizures not more frequent? The answer may lie in the augmented tonic inhibition that we as well as others have documented. That is, enhanced excitatory drive by sprouted MFs might be kept in check by augmented inhibition and, under most conditions, the epileptic dentate gyrus will function normally. To address the possible mechanism(s) underlying the increased amplitude mIPSCs recorded in DGCs from kindled rats, Otis et al. (1994) conducted peak scaled nonstationary noise analysis. Their analysis led them to conclude that enhanced mIPSC amplitude was due to an increase in the number of activated postsynaptic channels (NPo) with no change in single-channel conductance. Nusser et al. (1998a) corroborated this data by demonstrating that immunogold labeling of GABAARs was significantly increased at kindled DG inhibitory synapses. Accompanying the increased number of subsynaptic GABAARs present at epileptic DG inhibitory synapses was a concomitant shift in receptor pharmacological sensitivity. That is, GABAARs were now sensitive to blockade by zinc (Figs 2 and 5C) and virtually insensitive to augmentation by ZOL (Fig. 3). This new pharmacological profile is virtually the antithesis of that present in the control condition, i.e., little zinc but robust ZOL mIPSC modulation.
This altered pharmacology of DGC GABAARs may have an integral role in the initiation and propagation of seizures in the epileptic hippocampus. In addition to releasing glutamate, sprouted MFs serve as a zinc delivery system in TLE. During sustained firing of these fibres, zinc coreleased with glutamate onto DGCs may diffuse sufficiently away from excitatory synapses to interact with neighbouring inhibitory synapses. Normally, this would be of little consequence, because (i) zinc-containing terminals are quite distant from DGC somata in control brain, and (ii) control DGC GABAARs are virtually zinc-insensitive. Both of these situations are altered in the epileptic hippocampus. Our rapid application experiments as well as synaptic recordings demonstrate that epileptic GABAARs are significantly more sensitive to blockade by zinc (Figs 2 and 5C), and that zinc blockade occurs very slowly (Fig. 7). This implies that only robust or sustained MF stimulation (and concomitantly maintained zinc release) might be sufficient to counteract TLE-enhanced tonic inhibition. Once this zinc accumulation occurs, a cataclysmic failure of inhibition could break down the DG gate, uncover the synchronizing effects of the sprouted recurrent collaterals and enhanced function of NmDA receptors (Mody & Heinemann, 1987; Kohr et al., 1993) and potentially throw the DG into a positive feedback loop, freely amplifying and propagating seizure activity.
To test whether zinc released by MF stimulation could diffuse to and block GABAARs present at nearby inhibitory terminals, an experiment examining hippocampal slices with pilocarpine-induced MF sprouting was recently conducted. This study demonstrated that robust MF stimulation had no discernable inhibitory effect on DGC responses to photoreleased caged GABA (Molnar & Nadler, 2001a). This suggests that synaptically released zinc may not be sufficiently mobile to block neighbouring GABAARs. However, this lack of effect could also be attributable to an inability to release MF zinc by electrical stimulation under in vitro conditions. Supporting evidence for this possibility has come from a study by Suh et al. (2000), which reported that vibratome sectioning of a brain to generate slices results in an irreversible loss of up to 50% of synaptic zinc, with the ability to release zinc further reduced by conducting experiments at low temperatures (<30 °C), depressing endogenous release of zinc by >75% compared to in vivo conditions (Suh et al., 2000). However, Nadler and colleagues did find that the NMDA component of the MF response in the DG was enhanced by the incorporation of a high affinity zinc chelator in the bathing media, suggesting that at least some zinc was released from sprouted MFs in their studies (Molnar & Nadler, 2001b). To definitively determine whether zinc released from sprouted MFs is capable of blocking GABAARs, experiments similar to those used by Molnar & Nadler (2001a) need to be replicated under conditions where synaptic zinc levels are maintained, i.e. in organotypic cultures (Gahwiler, 1984) or in vivo.
In order for the altered DGC GABAARs described in the present set of studies to play a primary contributory role in seizure initiation and/or propagation in the epileptic hippocampus, they must appear prior to or simultaneously with the onset of the spontaneous seizures defining TLE. TLE in both humans and animals is usually triggered by some injurious brain insult which, following a delay of weeks to months in animals, and years in humans, triggers the subsequent development of recurrent spontaneous seizures. If DGC GABAAR alterations only appear following the onset of these seizures then they must result from the seizures and not from the primary underlying disease process. Instead, we found that the altered GABAARs actually develop far in advance of the onset of seizures (Fig. 5), and therefore are temporally poised to play a role as one fundamental epileptogenic mechanism. What then could explain the protracted latent period, because GABAAR changes develop so early? Because we are hypothesizing that aberrant GABAARs may interact with zinc released from sprouted MFs, both would need to be present for this to occur. Functional assays have demonstrated that robust MF sprouting takes weeks or months to develop in animal models of TLE (Lynch & Sutula, 2000; Wuarin & Dudek, 2001), and so this may explain the pronounced delay in onset of seizures following hippocampal injury.
GABAAR pharmacological sensitivity is primarily determined by receptor subunit composition. Zinc and ZOL sensitivity is governed to a large degree by the specific alpha subunit present within the receptor. GABAARs containing alpha1 subunits have high ZOL and low zinc sensitivity. Conversely, receptors containing other alpha subunits have high zinc and low ZOL sensitivity (reviewed in Macdonald & Olsen, 1994; Barnard et al., 1998). Interestingly, this altered pharmacological profile correlates nicely with our previous data derived from single cell antisense RNA amplified from acutely dissociated epileptic DGCs. This demonstrated decreased alpha1 together with increased alpha4 and delta subunit mRNA expression relative to controls (Brooks-Kayal et al., 1998). Past research using immunoprecipitation methodology has further demonstrated that thalamic delta subunits coassemble exclusively with alpha4-containing receptors whereas alpha4 subunits will coassemble with both delta and gamma2 subunits (Sur et al., 1999). However, delta-containing receptors are thought to be located at extrasynaptic sites (Nusser et al., 1998b) because they lack a gamma2 subunit which is critical in tethering the receptor to subsynaptic sites (Essrich et al., 1998; Wang et al., 1999). It remains undetermined whether TLE-induced up-regulation in delta and alpha4 subunits leads to the assembly of putatively delta/alpha4-containing extrasynaptic receptors that now impinge on the synapse or alternatively to the assembly of gamma2/alpha4-containing synaptic receptors (Coulter, 2001). Nonetheless, either one of these two hypothesized receptor configurations, individually or in concert, could readily explain the altered pharmacology exhibited by epileptic DGCs.
In conclusion, there are several chronic alterations in inhibitory neurotransmission that develop during the latent period and lead to altered GABAAergic function in temporal lobe epilepsy. Although inhibitory neurotransmission is enhanced in chronic epileptic dentate gyrus, the appearance of heightened zinc sensitivity of these receptors may be pivotal in generating spontaneous recurrent seizures that are the hallmark of TLE. Heightened zinc sensitivity of GABAARs and the emergence of a de novo zinc delivery system via aberrant sprouted MFs may synergistically interact to trigger an intermittent catastrophic failure of inhibition in the epileptic hippocampus. This may compromise the gatekeeper function of the dentate gyrus and facilitate seizure induction and propagation.
Acknowledgments
This work was supported by an Epilepsy Foundation postdoctoral fellowship to A.S.C. and NIH-NINDS RO1 grants (NS-32403 and NS-38572) to D.A.C. We are grateful to Drs M. A. Dichter, D. E. Pleasure, M. B. Robinson, G. Carlson and K. V. Sharma for constructive suggestions on the manuscript, M. A. Sarda and H. Ahmed for technical assistance, and E. Johnson for administrative support.
Abbreviations
- aCSF
artificial cerebral spinal fluid
- AP5
D-2-amino-5-phosphonopentanoic acid
- BDZ
benzodiazepine
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- DG
dentate gyrus
- DGC
dentate granule cell
- GABAARs
GABAA receptors
- MF
mossy fibre
- mIPSC
miniature inhibitory postsynaptic current
- Rs
series resistance
- SE
status epilepticus
- T50
50% mIPSC decay time
- TLE
temporal lobe epilepsy
- TTX
tetrodotoxin
- ZOL
zolpidem
References
- Arbilla S, Allen J, Wick A, Langer SZ. High affinity [3H]zolpidem binding in the rat brain: an imidazopyridine with agonist properties at central benzodiazepine receptors. Eur J Pharmacol. 1986;130:257–263. doi: 10.1016/0014-2999(86)90276-1. [DOI] [PubMed] [Google Scholar]
- Assaf T, Chung SH. Release of endogenous Zn2+ from brain tissue during activity. Nature. 1984;308:736–738. doi: 10.1038/308734a0. [DOI] [PubMed] [Google Scholar]
- Avanzini G, Moshe SL, Schwarzkroin PA, Engel J. Animal models of localization-related epilepsy. In: Engel J Jr, Pedley TA, editors. Epilepsy: A comprehensive textbook. Chapter 37. Lippencott-Raven; Philadelphia: 1997. [Google Scholar]
- Babb TL, Kupfer WR, Pretorius JK, Crandall PH, Levesque MF. Synaptic reorganization by mossy fibers in human epileptic fascia dentata. Neuroscience. 1991;42:351–363. doi: 10.1016/0306-4522(91)90380-7. [DOI] [PubMed] [Google Scholar]
- Barberis A, Cherubini E, Mozrzymas JW. Zinc inhibits miniature GABAergic currents by allosteric modulation of GABAA receptor gating. J Neurosci. 2000;20:8681–8627. doi: 10.1523/JNEUROSCI.20-23-08618.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard EA, Skolnick P, Olsen RW, Mohler H, Sieghart W, Biggio G, Braestrup C, Bateson AN, Langer SZ. International Union of Pharmacology. XV. Subtypes of gamma-aminobutyric acidA receptors: classification on the basis of subunit structure and receptor function. Pharmacol Rev. 1998;50:291–313. [PubMed] [Google Scholar]
- Brooks-Kayal AR, Shumate MD, Jin H, Rikhter TR, Coulter DA. Selective changes in single cell GABA (A) receptor subunit expression and function in temporal lobe epilepsy. Nature Med. 1998;4:1166–1172. doi: 10.1038/2661. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Dudek FE. Network properties of the dentate gyrus in epileptic rats with hilar neuron loss and granule cell axon reorganization. J Neurophysiol. 1997;77:2685–2696. doi: 10.1152/jn.1997.77.5.2685. [DOI] [PubMed] [Google Scholar]
- Buhl EH, Otis TS, Mody I. Zinc-induced collapse of augmented inhibition by GABA in a temporal lobe epilepsy model. Science. 1996;271:369–373. doi: 10.1126/science.271.5247.369. [DOI] [PubMed] [Google Scholar]
- Burgard E, Tietz E, Neelands T, Macdonald RL. Properties of recombinant gamma-aminobutyric acid A receptor isoforms containing the alpha 5 subunit subtype. Mol Pharmacol. 1996;50:119–127. [PubMed] [Google Scholar]
- Cohen AS, Lin DD, Coulter DA. Protracted postnatal development of inhibitory synaptic transmission in rat hippocampal area CA1 neurons. J Neurophysiol. 2000;84:2465–2476. doi: 10.1152/jn.2000.84.5.2465. [DOI] [PubMed] [Google Scholar]
- Coulter DA. Mossy fiber zinc and temporal lobe epilepsy: Pathological association with altered ‘epileptic’ gamma-aminobutyric acid A receptors in dentate granule cells. Epilepsia. 2000;41:S96–S99. doi: 10.1111/j.1528-1157.2000.tb01565.x. [DOI] [PubMed] [Google Scholar]
- Coulter DA. Epilepsy-associated plasticity in gamma-aminobutyric acid receptor expression, function, and inhibitory synaptic properties. Int Rev Neurobiol. 2001;45:237–252. doi: 10.1016/s0074-7742(01)45013-6. [DOI] [PubMed] [Google Scholar]
- Cronin J, Dudek FE. Chronic seizures and collateral sprouting of dentate mossy fibers after kaininc acid treatment in rats. Nature. 1988;474:181–184. doi: 10.1016/0006-8993(88)90681-6. [DOI] [PubMed] [Google Scholar]
- Engel J. Seizures and Epilepsy. FA Davis; Philadelphia: 1989. [Google Scholar]
- Essrich C, Lorez M, Benson JA, Fritschy JM, Luscher B. Postsynaptic clustering of major GABAA receptor subtypes requires the gamma 2 subunit and gephyrin. Nature Neurosci. 1998;1:563–571. doi: 10.1038/2798. [DOI] [PubMed] [Google Scholar]
- Fisher JL, Macdonald RL. The role of an alpha subtype M2–M3 His in regulating inhibition of GABAA receptor current by zinc and other divalent cations. J Neurosci. 1998;18:2944–2953. doi: 10.1523/JNEUROSCI.18-08-02944.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederickson CJ, Danscher G. Zinc-containing neurons in hippocampus and related CNS structures. Prog Brain Res. 1990;83:71–84. doi: 10.1016/s0079-6123(08)61242-x. [DOI] [PubMed] [Google Scholar]
- French JA, Williamson PD, Thadani VM, Darcey TM, Marson RH, Spencer SS, Spencer DD. Characteristics of medial temporal lobe epilepsy. I. Results of history and physiology examination. Ann Neurol. 1993;34:774–780. doi: 10.1002/ana.410340604. [DOI] [PubMed] [Google Scholar]
- Gahwiler BH. Development of the hippocampus in vitro: cell types, synapses and receptors. Neuroscience. 1984;4:751–760. doi: 10.1016/0306-4522(84)90192-1. [DOI] [PubMed] [Google Scholar]
- Gibbs JW, Shumate MD, Coulter DA. Differential epilepsy-associated alterations in postsynaptic GABA (A) receptor function in dentate granule and CA1 neurons. J Neurophysiol. 1997;77:1924–1938. doi: 10.1152/jn.1997.77.4.1924. [DOI] [PubMed] [Google Scholar]
- Hajos N, Nusser Z, Rancz EA, Freund TF, Mody I. Cell type-and synapse-specific variability in synaptic GABAA receptor occupancy. Eur J Neurosci. 2000;12:810–818. doi: 10.1046/j.1460-9568.2000.00964.x. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Beck H, Dreier JP, Ficker E, Stabel J, Zhang CL. The dentate gyrus as a regulated gate for the propagation of epileptiform activity. Epilepsy Res Suppl. 1992;7:273–280. [PubMed] [Google Scholar]
- Hosie AM, Dunne EI, Harvey RJ, Smart TG. Zinc-mediated inhibition of GABAA receptors: discrete binding sites underlie subtype specificity. Nature Neurosci. 2003;6:362–369. doi: 10.1038/nn1030. [DOI] [PubMed] [Google Scholar]
- Howell GA, Welch MG, Frederickson CJ. Stimulation-induced uptake and release of zinc in hippocampal slices. Nature. 1984;308:736–738. doi: 10.1038/308736a0. [DOI] [PubMed] [Google Scholar]
- Jonas P. Fast application of agonists to isolated membrane patches. In: Sakmann B, Neher E, editors. Single-Channel Recording. Plenum Press; New York: 1995. pp. 231–243. [Google Scholar]
- Kohr G, De Koninck Y, Mody I. Properties of NMDA receptor channels in neurons acutely isolated from epileptic (kindled) rats. J Neurosci. 1993;13:3612–3627. doi: 10.1523/JNEUROSCI.13-08-03612.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DD, Cohen AS, Coulter DA. Zinc-induced augmentation of synaptic currents and glutamate receptor responses in hippocampal CA3 neurons. J Neurophysiol. 2001;85:1185–1196. doi: 10.1152/jn.2001.85.3.1185. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Stringer JL, Bertram EH. The dentate gyrus as a control point for seizures in the hippocampus and beyond. Epilepsy Res. 1992;(Supplement 7):301–313. [PubMed] [Google Scholar]
- Lynch M, Sutula T. Recurrent excitatory connectivity in the dentate gyrus of kindled and kainic acid-treated rats. J Neurophysiol. 2000;83:693–704. doi: 10.1152/jn.2000.83.2.693. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Olsen RW. GABAA receptor channels. Annu Rev Neurosci. 1994;17:569–602. doi: 10.1146/annurev.ne.17.030194.003033. [DOI] [PubMed] [Google Scholar]
- Mello LEAM, Cavalheiro EA, Tan AM, Kupfer WR, Pretorious JK, Babb TL, Finch DM. Circuit mechanisms of seizures in the pilocarpine model of chronic epilepsy: cell loss and mossy fiber sprouting. Epilepsia. 1993;34:985–995. doi: 10.1111/j.1528-1157.1993.tb02123.x. [DOI] [PubMed] [Google Scholar]
- Mody I, Heinemann U. NMDA receptors of dentate gyrus granule cells participate in synaptic transmission following kindling. Nature. 1987;326:701–704. doi: 10.1038/326701a0. [DOI] [PubMed] [Google Scholar]
- Molnar P, Nadler JV. Lack of effect of mossy fiber-released zinc on granule cell GABAA receptors in the pilocarpine model of epilepsy. J Neurophysiol. 2001a;85:1932–1940. doi: 10.1152/jn.2001.85.5.1932. [DOI] [PubMed] [Google Scholar]
- Molnar P, Nadler JV. Synaptically-released zinc inhibits N-methyl-D-aspartate receptor activation at recurrent mossy fiber synapses. Brain Res. 2001b;910:205–207. doi: 10.1016/s0006-8993(01)02720-2. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Hajos N, Somogyi P, Mody I. Increased number of synaptic GABAA receptors underlies potentiation at hippocampal inhibitory synapses. Nature. 1998a;395:172–177. doi: 10.1038/25999. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Sieghart W, Somogyi P. Segregation of different GABAA receptors to synaptic and extrasynaptic membranes of cerebellar granule cells. J Neurosci. 1998b;18:1693–1703. doi: 10.1523/JNEUROSCI.18-05-01693.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki MM, Evenson DA, Nadler JW. Hippocampal mossy fiber sprouting and synapse formation after status epilepticus in rats: visualization after retrograde transport of biocytin. J Comp Neurol. 1995;352:515–534. doi: 10.1002/cne.903520404. [DOI] [PubMed] [Google Scholar]
- Otis TS, De Koninck Y, Mody I. Lasting potentiation of inhibition is associated with an increased number of gamma-aminobutyric acid type A receptors activated during miniature inhibitory postsynaptic currents. Proc Natl Acad Sci USA. 1994;91:7698–7702. doi: 10.1073/pnas.91.16.7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis TS, Mody I. Modulation of decay kinetics and frequency of GABAA receptor-mediated spontaneous inhibitory postsynaptic currents in hippocampal neurons. Neuroscience. 1992;49:13–32. doi: 10.1016/0306-4522(92)90073-b. [DOI] [PubMed] [Google Scholar]
- Patrylo PR, Dudek FE. Physiological unmasking of new glutamatergic pathways in the dentate gyrus of hippocampal slices from kainite-induced epileptic rats. J Neurophysiol. 1998;79:418–429. doi: 10.1152/jn.1998.79.1.418. [DOI] [PubMed] [Google Scholar]
- Poncer JC, Durr R, Gahwiler BH, Thompson SM. Modulation of synaptic GABAA receptor function by benzodiazepines in area CA3 of rat hippocampal slice cultures. Neuropharmacology. 1996;35:1169–1179. doi: 10.1016/s0028-3908(96)00055-x. [DOI] [PubMed] [Google Scholar]
- Rafiq A, DeLorenzo RJ, Coulter DA. Generation and propagation of epileptiform discharges in a combined entorhinal cortex/hippocampal slice. J Neurophysiol. 1993;70:1962–1974. doi: 10.1152/jn.1993.70.5.1962. [DOI] [PubMed] [Google Scholar]
- Slomianka L. Neurons of origin of zinc-containing pathways and the distribution of zinc-containing boutons in the hippocampal region of the rat. Neuroscience. 1992;48:325–352. doi: 10.1016/0306-4522(92)90494-m. [DOI] [PubMed] [Google Scholar]
- Spencer DD, Spencer SS. Hippocampal resections and the use of human tissue in defining temporal lobe epilepsy syndromes. Hippocampus. 1994;4:243–249. doi: 10.1002/hipo.450040303. [DOI] [PubMed] [Google Scholar]
- Suh SW, Danscher G, Jensen MS, Thompon R, Motamedi M, Frederickson CJ. Release of synaptic zinc is substantially depressed by conventional brain slice preparations. Brain Res. 2000;879:7–12. doi: 10.1016/s0006-8993(00)02675-5. [DOI] [PubMed] [Google Scholar]
- Sur C, Farrar SJ, Kerby J, Whiting PJ, Atack JR, McKernan RM. Preferential coassembly of alpha4 and delta subunits of the gamma-aminobutyric acidA receptor in rat thalamus. Mol Pharmacol. 1999;56:110–115. doi: 10.1124/mol.56.1.110. [DOI] [PubMed] [Google Scholar]
- Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol. 1989;26:321–330. doi: 10.1002/ana.410260303. [DOI] [PubMed] [Google Scholar]
- Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci. 1985;5:1016–1022. doi: 10.1523/JNEUROSCI.05-04-01016.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdoorn TA, Draguhn A, Ymer S, Seeburg PH, Sakmann B. Functional properties of recombinant GABAA receptors depend upon subunit composition. Neuron. 1990;4:919–928. doi: 10.1016/0896-6273(90)90145-6. [DOI] [PubMed] [Google Scholar]
- Vogt K, Mellor J, Tong G, Nicoll R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron. 2000;26:187–196. doi: 10.1016/s0896-6273(00)81149-6. [DOI] [PubMed] [Google Scholar]
- Wang H, Bedford FK, Brandon NJ, Moss SJ, Olsen RW. GABAA-receptor-associated protein links GABAA receptors and the cytoskeleton. Nature. 1999;397:69–72. doi: 10.1038/16264. [DOI] [PubMed] [Google Scholar]
- White G, Gurley D. Benzodiazepine site inverse agonists can selectively inhibit subtypes of the GABAA receptor. Neuroreport. 1995;6:1313–1316. doi: 10.1097/00001756-199506090-00021. [DOI] [PubMed] [Google Scholar]
- Wong RKS, Traub RD, Miles R. Cellular basis of neuronal synchrony in epilepsy. Adv Neurol. 1986;44:583–592. [PubMed] [Google Scholar]
- Wuarin JP, Dudek FE. Electrographic seizures and new recurrent excitatory circuits in the dentate gyrus of hippocampal slices from kainate-treated epileptic rats. J Neurosci. 1996;16:4438–4448. doi: 10.1523/JNEUROSCI.16-14-04438.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuarin JP, Dudek FE. Excitatory synaptic input to granule cells increases with time after kainate treatment. J Neurophysiol. 2001;85:1067–1077. doi: 10.1152/jn.2001.85.3.1067. [DOI] [PubMed] [Google Scholar]