

Abstract
We have reported that [methyl-11C](3R,5R)-5-(3-methoxy-phenyl)-3-((R)-1-phenyl-ethylamino)-1-(4-trifluoromethyl-phenyl)-pyrrolidin-2-one ([11C]8, [11C]MePPEP) binds with high selectivity to cannabinoid type-1 (CB1) receptors in monkey brain in vivo. We now describe the synthesis of 8 and four analogs, namely the 4-fluoro-phenyl (16, FMePPEP), 3-fluoromethoxy (20, FMPEP), 3-fluoromethoxy-d2 (21, FMPEP-d2) and 3-fluoro-ethoxy analogs (22, FEPEP), and report their activity in an ex vivo model designed to identify compounds suitable for use as PET ligands. These ligands showed high, selective potency at CB1 receptors in vitro (Kb < 1 nM). Each ligand (30 μg/kg, i.v.) was injected into rats under baseline and pretreatment conditions (3, rimonabant, 10 mg/kg, i.v.), and quantified at later times in frontal cortex ex vivo with LC-MS detection. Maximal ligand uptakes were high (22.6-48.0 ng/g). Under pretreatment, maximal brain uptakes were greatly reduced (6.5-17.3 ng/g). Since each ligand readily entered brain and bound with high selectivity to CB1 receptors, we then established and here describe methods to produce [11C]8, [11C]16 and [18F]20-22 in adequate activities for evaluation as candidate PET radioligands in vivo.
Introduction
The therapeutic potential of plant-derived phytocannabinoids from Cannabis sativa (marijuana) has been known for centuries and is arguably considered the world’s oldest therapeutic. Smoking or ingesting the resin from marijuana leaves or flowers provides medicinal properties such as analgesia, anti-emesis, anti-spasmodic, and appetite stimulation, but also less desirable side effects such as immunosupression, psychoactive “high”, drowsiness and memory impairment.1,2 Only over the last several decades have the biological mechanisms of phytocannabinoid exposure become better understood. A major advancement was the correct structural identification of (–)-(6aR,10aR)-6,6,9-trimethyl-3-pentyl-6a,7,8,10a-tetrahydro-6H-benzo[c]chromen-1-ol (1, Δ9-THC, Figure 1)3, which was identified as the major psychoactive phytocannabinoid. Subsequent attempts to improve on the potency of 1 through rational design led to the identification of 2-[(1S,2R,5S)-5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-5-(2-methyloctan-2-yl)phenol (2, CP-55,940) as a considerably higher affinity agonist.4 The use of [3H]2 (Figure 1) in vitro helped to provide direct evidence for cannabinoid receptors which are now known to belong to the superfamily of G protein-coupled receptor proteins.5 Two cannabinoid receptor family members have been cloned and sequenced (CB1 and CB26,7) but others subtypes may exist.8
Figure 1.

Structures of some CB1 receptor agonists. Asterisks mark positions of radiolabel.
CB1 receptors, which have two splice variants (CB1A and CB1B),9,10 and are localized in peripheral tissues (e.g. heart, lung, prostate, testis, bone, marrow, tonsils and spleen)8 and central tissues with highest densities in the brain’s globus pallidus, substania nigra, molecular layer of the cerebellum, cerebral cortex, striatum and hippocampus. The pons, thalamus and brain stem are almost devoid of CB1 receptor expression.11,12 CB2 receptors are located mainly in the peripheral immune system tissues such as macrophages of spleen, bone marrow, and pancreas, but are also localized at peripheral nerve terminals.7,13,14 Low levels of CB2 receptors have been found in brain in both neurons and glia though their physiological significance in the brain has not been fully elucidated.15,16
Abnormalities in regional brain CB1 receptor densities or function may contribute to an array of neuropsychiatric and/or neurodegenerative disorders. Non-invasive quantitative imaging of brain CB1 receptors with positron emission tomography (PET) could help to clarify the role of these receptors in many CNS disorders. However, an effective PET radioligand, labeled with either carbon-11 (t1/2 = 20.4 min) or fluorine-18 (t1/2 = 109.7 min), is required for such imaging. Such a radioligand would need to show suitable in vivo characteristics, including adequate maximal brain uptake, selectivity for binding to CB1 receptors, insignificant brain-penetrating radiometabolite(s) and low non-specific binding.17,18
Much progress has been made recently in the development of CB1 receptor PET radioligands. Past attempts at CB1 PET radioligand development have focused on modifying the substituents of the 1,5-diarylpyrazole core of rimonabant (3, Figure 2).19This approach led to the development of two promising radioligands, [11C]4 ([11C]JHU75528, Figure 2)20,21 and [11C]5 ([11C]JHU75575, Figure 2)21. Merck recently reported [18F]6 ([18F]MK-9470, Figure 2)22,23 as a suitable PET radioligand for in vivo quantification of brain CB1 receptors in humans. Further promising radioligands are [11C]7 ([11C]PipISB, Figure 2),24 [18F]7 ([18F]PipISB, Figure 2)24 and [11C]8 ([11C]MePPEP, Figure 2)25. [18F]7 is somewhat superior to [11C]7 and has a similar in vivo PET time-activity profile to [18F]6 but shows a greater specific signal, when expressed as receptor-specific to non-specific binding (9:1 vs. 5:1). In rhesus monkey, [11C]8 exhibits excellent maximal brain uptake and binds selectively to CB1 receptors.25 Furthermore, the binding of [11C]8 appears in some cases to reach equilibrium during the scan time (120 min after injection), which would allow for equilibrium-based pharmacokinetic analysis.25 However, the time of apparent equilibrium is variable and in some cases is not observed which then requires complex biomathematical modeling. In order to surmount this issue, analogs of 8 labeled with fluorine-18 are desirable to increase the allowable scan time.
Figure 2.

Structures of 3 and candidate CB1 receptor PET radioligands. Asterisks mark positions of radiolabel.
The structure and pharmacology of 8 suggested [18F]fluoro(m)ethoxy analogs to be plausible candidate radioligands. Direct incorporation of [18F]fluoride ion into the F3C-group of the N-1 aryl ring of 8 was not considered because of the risk of achieving only low specific radioactivity when high specific radioactivity would be required in an effective radioligand.18 The in vivo metabolism of [11C]8 in rhesus monkey is very rapid and can obfuscate accurate measurement of unchanged radioligand in plasma at late scan time. Such measurements are required for biomathematical compartmental modeling of acquired PET data.18,25 A more metabolically stable analog of 8 with similar potency and lipophilicity is desirable to allow for more accurate measurement of plasma radioligand. It has been reported that replacement of a fluoromethoxy group by a dideutero-fluoromethoxy group can lead to a decrease in the rate of defluorination.26,27 Hence, we considered a [18F]fluoromethoxy-d2 analog to take advantage of this kinetic isotope effect. Another potential metabolic route is aromatic hydroxylation of the (R)-1-phenyl-ethylamino moiety of 8. Incorporation of a fluorine atom at the para-position of an aromatic ring is known to increase the biological half-life by inhibiting metabolic aromatic hydroxylation.28 Hence, a 4-fluoro-phenyl analog of 8 was also considered.
The discovery of radioligands suitable for in vivo PET imaging is a challenging task. PET radioligands need to satisfy several criteria, such as high potency for the molecular target, facile penetration into the brain, and low non-specific binding.18 In many cases, the properties necessary for PET ligands are different from those found in therapeutic agents. While compound potency is relatively straightforward to measure and optimize, the evaluation of non-specific binding is particularly difficult and is generally done using radiolabeled compound. The necessity of preparing radiolabeled material has been an impediment to the rapid evaluation and discovery of new PET ligands. Consequently, investigators have used cLogP and cLogD7.4 as surrogate measures of non-specific binding; however, the correlation of these parameters with non-specific binding in in vivo experiments is often not satisfactory.29 The development of an ex vivo method to determine the brain penetration and non-specific binding of a non-radiolabeled compound would greatly facilitate the discovery of new PET ligands.
Highly sensitive LC-MS techniques have been developed for the quantification of drug levels in tissues.30 We reasoned that this methodology could be used to measure the concentration of candidate PET ligands in brain samples following administration of the compound at very low, tracer doses.30 Not only would this methodology provide a measure of brain penetration, it could also be used to determine non-specific binding. In the present case, brain penetration was determined by administration to rats of a tracer dose (30 μg/kg, i.v.) of the study compound. Non-specific binding was determined by pretreatment of rats with a high dose of 3 (10 mg/kg, i.v.) prior to administration of the tracer dose. Under these conditions, the vast majority of central CB1 receptors are occupied by 3 and the tissue levels of the tracer represent non-specifically bound compound. Compounds which displayed low brain levels of tracer in pretreated rats and high levels in non-pretreated rats were selected for radiolabeling with carbon-11 or fluorine-18 and evaluation in vivo.
The aims of this study were therefore: 1) to synthesize 8 and closely related analogs (16, 20-22); 2) determine their in vitro potencies at CB1 and CB2 receptors; 3) quantify 8, 16, 20 and 22 in CB1-rich frontal cerebral cortex of rat brains at times after injection under baseline and CB1 pre-blocked conditions, and 4) radiolabel the promising candidate radioligands with 11C or 18F, to make them available for detailed evaluation in vivo.
Results and Discussion
Chemistry
Synthesis of the potential PET ligands is outlined in Scheme 1. The pyrolidinone ring system was assembled in a single step using the four-component coupling method developed by Andreichikov, et al.31 Reaction of ethyl pyruvate with a solution of 4-aminobenzotrifluoride and 3-methoxybenzaldehyde in acetic acid led to the formation of pyrolidone 9 in 85% yield. Acid catalyzed hydrolysis of the enamine using a mixture of acetic acid and aqueous hydrochloric acid yielded ketolactam 10 in 80% yield. Condensation with R-α-methylbenzylamine provided a diastereomeric mixture of 11 and 12, which was readily separated by silica gel chromatography. Reduction of enamine 11 with sodium cyanoborohydride in acetic acid gave a 20:1 mixture of 8 and 15 that favored the desired diastereomer 8. The absolute stereochemical configuration of 8 was confirmed by single crystal X-ray crystallography (unpublished data). Demethylation of 8 in a pyridinium hydrochloride melt gave the desired phenol 18 in 60% yield accompanied by a small amount (12%) of the C-3 epimeric product. The 4-fluorophenyl analogue (16) and the corresponding phenol (19) were prepared in an analogous manner. The fluoroalkyl analogues 20, 21, and 22 were prepared by alkylation of phenol 18 with fluoromethyl tosylate, fluoromethyl tosylate-d2, and 1-bromo-2-fluoroethane in 53%, 43%, and 89% yields, respectively. No epimerization was observed during the course of these alkylations.
Scheme 1.

Synthesis of 8, 16 and 20–22). Reagents and conditions: a) 4-Aminobenzotrifluoride, ethyl pyruvate, AcOH; b) conc. HCl, AcOH; c) (R)-(+)-1-phenylethylamine, CH2Cl2; d) (R)-1-(4-fluoro-phenyl)-ethylamine, CH2Cl2; e) NaBH3CN, AcOH; f) pyridinium hydrochloride, heat; g) Cs2CO3, DMF, fluoromethyl tosylate; h) Cs2CO3, DMF, fluoromethyl tosylate-d2; i) Cs2CO3, DMF, F(CH2)2Br.
Lipophilicity calculation
Calculated lipophilicity (cLogD7.4) values can be useful for predicting the relative ability of ligands within the same structural class to pass the blood-brain barrier.18,29 Moderate lipophilicity, in the cLogP range of 2.5-3.5 is usually considered desirable for adequate brain entry without high non-specific binding to brain tissue.18,29 Many exceptions to this guideline are however known.29 Although [11C]8 has a high cLogD7.4 value (5.7) (Table 1), it passes the blood-brain barrier very readily, reaching almost 600% standardized uptake value (% SUV) within 10-20 min after injection in monkey.25 Candidate ligands 16, 20 and 22 have a cLogD value similar to or slightly above 8, suggesting they would have a similar or slightly less ability to penetrate the blood-brain barrier.
Table 1.
In vitro potency at CB1 and CB2 receptors, CB1 selectivity over CB2 receptors and cLogD7.4 data for 3, 8, 16, 20 and 22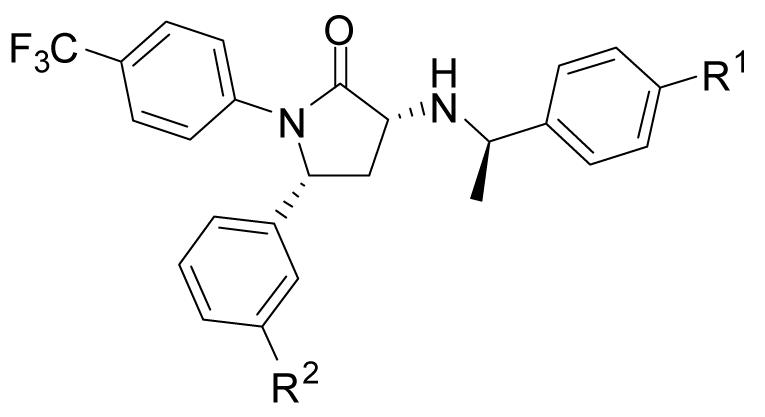
| Ligand | R1 | R2 | CB1Kb (nM)a | CB2Kb (nM)a | CB1 versus CB2 selectivity |
cLogD7.4b |
|---|---|---|---|---|---|---|
| 3 | 0.698 ± 0.200 | > 1,977 | >2,830 | 7.0 | ||
| 8 | H | OCH3 | 0.472 ± 0.160 | 363 ± 87 | 769 | 5.7 |
| 16 | F | OCH3 | 0.216 ± 0.004 | 466 ± 136 | 2,160 | 5.7 |
| 20 | H | OCH2F | 0.187 ± 0.018 | 669 ± 137 | 3,580 | 5.7 |
| 22 | H | OCH2CH2F | 0.424 ± 0.021 | >8260 | >19,500 | 5.8 |
All values represent mean ± SEM of at least three determinations.
cLogD7.4 data calculated by using Pallas 3.0 for windows.
Ligand potencies and selectivities at the CB1 and CB2 receptors
The in vitro potencies (Kb values) and selectivities of 8, 16, 20 and 22 for CB1 versus CB2 receptors compare well with other promising PET radioligands (Table 1). All the new ligands exhibited high potency for CB1 receptors in vitro and were > 769-fold selective for binding to CB1 over CB2 receptors. These data showed that all four ligands had adequate potency and selectivity to be considered for development as PET radioligands.
Ex vivo experiments
The coupling of single (LC-MS) or triple (LC-MS-MS) quad mass spectral detectors with liquid chromatography has produced analytical instrumentation with high enough sensitivity and selectivity to be a useful tool for quantifying the brain distribution of trace doses of new candidate drugs and candidate radioligands in rats ex vivo over time. This technique, although not yet widely applied, has been reported with rat brain receptor occupancy experiments targeting the dopamine-2 (D2), serotonin-2A (5-HT2A) and the neurokinin-1 (NK1) receptors.30,32 This approach to assessing the potential of new ligands for development as PET radioligands is attractive; useful definitive information on ligand brain entry and receptor-specific signal versus non-specific signal can be gained without first having to label the ligand with either a beta-emitter (e.g., 3H) or positron-emitter (e.g., 11C or 18F).
In this study, we applied the mass spectrometry technique to quantify levels of administered 8, 16, 20 and 22 (each 30 μg/kg, i.v.) in the frontal cerebral cortex of rat brains under baseline conditions and also under conditions in which the rats were pretreated with a selective CB1 receptor inverse agonist (3; 10 mg/kg, i.v.) at 15 min before injection of ligand (Figure 3, Panels A-D). Under baseline conditions, each ligand exhibited high brain tissue uptake, and reached concentrations of 39.0 ± 5.0 (at 0.25 h), 48.0 ± 2.8 (at 0.5 h), 39.3 ± 8.1 (at 0.5 h) and 22.0 ± 2.6 (at 0.25 h) ng/g for 8, 16, 20 and 22, respectively. The uptakes then reduced to 8.5 ± 0.6, 10.5 ± 0.0, 8.2 ± 0.5 and 0.7 ± 0.1 ng/g at 8 h after injection for 8, 16, 20 and 22 respectively. Ligand 16 exhibited the highest brain tissue uptake, which may be related to reduced metabolic hydroxylation caused by the introduction of the (R)-1-(4-fluoro-phenyl)-ethylamino moiety. 8 and 20 exhibited similar brain uptakes over time. 22 exhibited the lowest brain uptake over time, probably because its cLogD7.4 value is higher than for the other ligands. Under conditions in which the rats were pretreated with 3 (Figure 3, Panels A-D), maximal brain uptakes in CB1-rich frontal cortex were reduced to 11.5 ± 1.9, 17.3 ± 2.0, 9.7 ± 0.1 and 6.5 ± 1.1 ng/g at 0.25 h after injection. 8 and 20 were not quantifiable at 4 h after injection, 22 could not be quantified at 2 h after injection and 0.8 ± 0.0 ng/g of 16 remained at 8 h after injection. The reduced brain uptakes in CB1 rich cerebral cortex under pretreatment conditions demonstrates that the vast majority of each ligand in brain was specifically bound to CB1 receptors.
Figure 3.

Time course evaluation in the frontal cerebral cortex of male Sprague Dawley rats after administration (30 μg/kg, i.v.) of 8 (Panel A), 16 (Panel B), 20 (Panel C) and 22 (Panel D) under baseline and pretreated with 3 (10 mg/kg, i.v.) 15 min before ligand injection. Key: (░), baseline; (▓), pretreatment.
The excellent brain penetration of these ligands and also their high ratios of CB1 receptor-specific to non-specific (non-blockable) binding in rats, as measured ex vivo with mass spectrometry, strongly suggested that these ligands would be promising PET radioligands when labeled with a positron-emitter. We therefore set out to prepare such candidate PET radioligands for future more detailed evaluation with PET.
Labeling with carbon-11
[11C]8 and [11C]16 were prepared by treating the appropriate O-desmethyl precursor (18 or 19, respectively) with [11C]iodomethane under basic conditions in an Autoloop apparatus33 (Scheme 2). After reverse phase HPLC, the final formulated product ([11C]8 or [11C]16) was obtained in 2.50 ± 1.15%25 or 16.5% overall decay-corrected radiochemical yield (RCY) from cyclotron-produced [11C]carbon dioxide with high specific radioactivity (> 78.1 or > 261 GBq/μmol at EOS for [11C]8 or [11C]16, respectively) in a radiosynthesis time of 40 min. The radiochemical purities of [11C]8 and [11C]16 were > 95%. [11C]8 and [11C]16 were thus obtained in activities and purities adequate for further investigation in animal or human subjects.
Scheme 2.
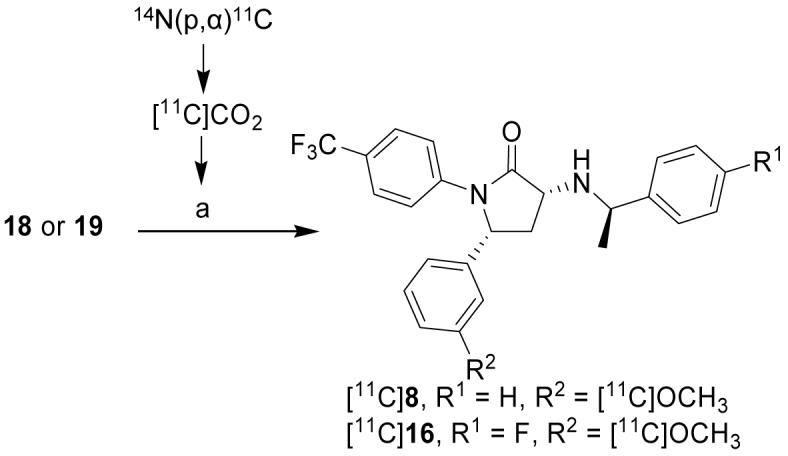
Radiosynthesis of [11C]8 and [11C]16. Reagents, conditions and yields: a) “Loop”, Precursor (18 or 19), DMF, 0.5M (n-Bu)4NOH in MeOH, [11C]MeI, rt, ∼ 3 min, RCY = 2.5 ± 1.1% (8, n = 57) and 16.5% ([11C]16, n = 2) decay-corrected from [11C]carbon dioxide.
Labeling with fluorine-18
[18F]20-22 were prepared by reaction of the O-desmethyl precursor (18) with the appropriate [18F]fluoroalkyl halide under basic conditions in a modified version of the automated TRACERlab FXF-N module.34 The RCY of an isolated labeling agent is an important factor affecting the overall RCY of a formulated radiopharmaceutical. We therefore compared radiochemical yields for two methods for preparing [18F]fluoromethyl bromide as a labeling agent in the radiosynthesis of [18F]20 (Scheme 4). The first method was based on nucleophilic displacement in dibromomethane with [18F]fluoride ion-K2.2.2-K+ complex in acetonitrile. This method is well established35 and has been shown to be useful for the routine production of radiopharmaceuticals, such as [18F]SPA-RQ34,36 and [18F]FMeNER-d2.27 However, there is one major disadvantage with this method, namely the difficult separation of the generated labeling agent (BrCH218F or BrCD218F) from the starting dibromomethane. Unseparated dibromomethane can act as a competing electrophile in the product labeling reaction, and also lead to difficult separation of labeled compound. To circumvent this problem gas chromatography has been used to isolate only the desired [18F]fluoromethyl bromide.37 Alternatively, the labeling agent can be purified by passage over a series of four silica plus SepPak columns, which can be troublesome.35 With the latter method we achieved a RCY of 27.5 ± 4.5% (n = 57) for isolated [18F]fluoromethyl bromide.34 We also explored the alternative use of bromomethyl tosylate as a more attractive precursor for generating the [18F]fluoromethyl bromide (Scheme 3). This non-volatile precursor obviates the need for GC or SepPak chromatography, as this precursor does not co-transfer during isolation of the labeling agent. After optimization of this method, we achieved a maximal RCY of 14.4% for isolated [18F]fluoromethyl bromide, a yield inferior to that from the first method. Therefore, we routinely produced [18F]fluoromethyl bromide from dibromomethane with Sep-pak isolation. Reaction of 18 with [18F]fluoromethyl bromide under basic conditions in DMF for 10 min at 110 °C gave [18F]20 in 5.92 ± 1.34% (n = 3) isolated (formulated) RCY with high radiochemical purity (> 95%) and high specific radioactivity (> 57 GBq/μmol at EOS). The radiosynthesis time was about 120 min.
Scheme 3.

Radiosynthesis of [18F]20–22. Reagents, conditions and yields: a) [18F]fluoride ion-K 2.2.2-K+ complex, solvent (MeCN for CH2Br2, CD2Br2 and o-DCB for bromo(m)ethyl tosylate), Δ; b) Precursor (18), Cs2CO3, DMF, Δ; c) Precursor (18), DMF, (n-Bu)4NOH in MeOH, decay-corrected RCY’s = 5.92 ± 1.34% ([18F]20; n = 3), 7.93 ± 2.48% ([18F]21; n = 6) and 7.92 ± 2.16% ([18F]22; n = 6) from [18F]fluoride ion.
[18F]21 was prepared in the same way as [18F]22, but with [18F]fluoromethyl bromide-d2 as the isolated labeling agent (Scheme 3), and was obtained in 7.93 ± 2.48% (n = 6) RCY, > 95% radiochemical purity and high specific radioactivity (> 68 GBq/μmol at EOS).
The radiosynthesis of [18F]22 used [18F]2-fluoroethyl bromide as the isolated labeling agent. [18F]2-Fluoroethyl bromide was itself generated from 2-bromoethyl tosylate as described previously (Scheme 3).38 Reaction of 18 with [18F]2-fluoromethyl bromide under basic conditions in DMF for 5 min at 110 °C gave formulated [18F]22 in 7.92 ± 2.16% (n = 6) RCY, in high radiochemical purity (> 95%) and with high specific radioactivity (> 86 GBq/μmol at EOS). Overall radiosynthesis time was about 120 min.
Conclusions
Ligands 8, 16 and 20-22 were synthesized efficiently. Each ligand exhibited high potency (Kb values) in vitro. Ex vivo experiments in rats using LC-MS and LC-MS-MS showed that 8, 16, 20 and 22 (and hence 21) adequately pass the blood-brain barrier after low dose administration and bind with high specificity to CB1 receptors in rat brain tissue. Radiosynthesis of [11C]8, [11C]16, [18F]20-22 gave adequate decay-corrected RCYs and purities to allow their further evaluation as promising radioligands with PET imaging. Our evaluation of [11C]8 in monkey has already been reported showing this to be a highly promising PET radioligand.25 Evaluations of the other radioligands will be reported elsewhere. Both [11C]8 and [18F]21 are being or will be studied in human subjects with PET under obtained exploratory INDs.
Experimental Section
Materials
All chemicals were purchased from commercial sources and used as received. The chemicals and solvents were of ACS or HPLC quality. 3 was synthesized at Eli Lilly and Company (Indianapolis, IN, USA). Fluoromethyl tosylate was prepared at Eli Lilly and Company as previously reported.39 Fluoromethyl tosylate-d2 was prepared at SYNCOM AB (Groningen, Netherlands) using the same procedure, but substituting dideutero-diiodomethane for diiodomethane. Bromo(m)ethyl toslylate was synthesized at the National Institute of Mental Health (Bethesda, MD, USA).40
Methods
Column chromatography was performed on silica gel columns (35-65 μm; Isco Inc., Lincoln, NE, USA). 1H NMR spectra (400 MHz) were measured on an INOVA spectrometer (Varian, Palo Alto, CA, USA). 1H NMR (500 MHz) and 13C NMR (125 MHz) were acquired on Varian System 500 equipped with a 5 mm direct cold probe and the sample temperature was maintained at 25 °C. Abbreviations s, d, dd, ddd, m and br denote singlet, doublet, double doublet, double double doublet, multiplet and broad respectively. Melting points (Mp) were determined on a Thomas Hoover capillary melting point apparatus and are uncorrected. Elemental analyses were acquired from Midwest Microlabs LLC (Indianapolis, IN, USA). Mass detection was performed with an 1100 Series LC-MSD single quadrupole spectrometer (Agilent; Santa Clara, CA, USA) with ESI interface. Samples were analyzed by methods 1 or 2. In method 1, LC-MS analyses, were performed with a heated (50 ± 10 °C) reverse phase column (Gemini; C18; 2.0 × 50 mm; 3.0 μm; Phenomenex., Torrence, CA, USA) eluted at 1 mL/min with a gradient of MeCN (A) and MeCN with 0.1% HCO2H (B), with A increased linearly from 5 to 100% v/v over 7 min and then held for 1 min. In method 2, LC-MS analyses, were performed with a heated (50 ± 10 °C) reverse phase column (Xterra C18; 2.1 × 50 mm; 3.5 μm; Waters Corp., Milford, MA, USA) eluted at 1 mL/min with a gradient of MeCN (A) and MeOH with 0.2% aq.-HCO2NH4 (B), with A increased linearly from 5 to 100% v/v over 7 min and then held for 1 min. After electrospray ionization of the eluted test sample, ions between m/z 150 and 750 were captured. High-resolution mass spectrometry (HRMS) was performed with a Thermo LTQFT (Thermo Electron Corporation, San Jose, CA, USA). Data was acquired in FTMS +ESI mode with a data range of 98-980 AMU. Sample (10 μL) was injected in loop mode. Solvent: Methanol with 0.1% formic acid.
γ-Radioactivity from 11C and 18F was measured with a dose calibrator (Atomlab 300™, Biodex Medical Systems) calibrated with Cs-137 and Co-57 sources. Low levels of radioactivity (< 40 kBq) were measured with a well type γ-counter (model 1080 Wizard, Perkin-Elmer) having an electronic window set between 360 and 1800 keV.
Specific radioactivities (GBq/μmol) were determined with analytical HPLC methods, described later, calibrated for absorbance (λ = 254 nm) response per mass of ligand. The radioactivity of the radioligand peak (decay-corrected) (GBq) was divided by the mass of the associated carrier peak (μmol).
5-(3-Methoxy-phenyl)-1-(4-trifluoromethyl-phenyl)-3-(4-trifluoromethyl-phenylamino)-1,5-dihydro-pyrrol-2-one (9)
4-Aminobenzotrifluoride (34.0 mL, 270.7 mmol) was added to a solution of 3-methoxybenzaldehyde (11.0 mL, 90.2 mmol) in glacial acetic acid (80 mL). Ethyl pyruvate (9.9 mL, 90.2 mmol) was added and the mixture stirred at ambient temperature for 18 h. The precipitate was filtered and washed with a mixture of 20% MTBE in heptane and dried under vacuum to afford 9 (44.4 g, 85%) as an off white powder. Mp 177-179 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.70 (s, 1H), 7.86 (d, 2H, J = 8.4 Hz), 7.68 (d, 2H, J = 8.8 Hz), 7.54 (d, 2H, J = 8.4 Hz), 7.44 (d, 2H, J = 8.8 Hz), 7.19 (dd, 1H, J = 7.8, 7.8 Hz), 6.88 (d, 1H, J = 1.8 Hz), 6.78 (dd, 2H, J = 7.9, 1.8 Hz), 6.59 (d, 1H, J = 2.6 Hz), 6.14 (d, 1H, J = 2.2 Hz), 3.67 (s, 3H). LC-MS ESI m/z: 491 (M-H)-, tR = 5.44 min, method 1.
5-(3-Methoxy-phenyl)-1-(4-trifluoromethyl-phenyl)-pyrrolidine-2,3-dione (10)
A slurry of 9 (89.7 g, 182 mmol), glacial acetic acid (400 mL) and c. HCl. (500 mL) was stirred at ambient temperature for 22 h. The heterogeneous mixture was heated to 60 °C for 1 h. The mixture was poured onto ice (1 L), stirred and stood for 1 h. The precipitate was filtered, washed with water and dried under vacuum to afford a solid. The solid still contained starting material. The solid was slurried with glacial acetic acid (500 mL) and c. HCl. (500 mL) and stirred at ambient temperature for 22 h. The mixture was poured onto ice and water (2 L), stirred and stood for 1 h. The solid was filtered, washed with wate, and dried under vacuum to afford 10 (50.8 g, 80%): 1H NMR showed the material to be a mixture of enol-keto tautomers. Mp 134-144 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.19 (s, 1H, enol), 7.80 (d, 2H, J = 8.8 Hz, enol), 7.75 (d, 2H, J = 8.7 Hz, keto), 7.70 (d, 2H, J = 8.7 Hz, keto), 7.63 (d, 2H, J = 8.8 Hz, enol), 7.19-7.14 (m, 1H enol, 1H keto), 7.02 (dd, 1H, J = 2.0, 2.0 Hz, keto), 6.93 (d, 1H, J = 7.9 Hz, keto), 6.81 (dd, 1H, J = 2.2, 2.2 Hz, enol), 6.77-6.71 (m, 2H enol, 1H keto), 5.97 (d, 1H, J = 2.6 Hz, enol), 5.91 (d, 1H, J = 2.6 Hz, enol), 5.73 (dd, 1H, J = 7.5, 4.0 Hz, keto), 3.66 (s, 3H, enol), 3.65 (s, 3H, keto), 3.35 (dd, 1H, J = 19.3, 7.5 Hz, keto), 2.59 (dd, 1H, J = 19.3, 4.0 Hz, keto). LC-MS ESI m/z: 349.8 (M+H)+, 347.8 (M-H)-, tR = 3.68 min, method 1.
(R)-5-(3-Methoxy-phenyl)-3-((R)-1-phenyl-ethylamino)-1-(4-trifluoromethyl-phenyl)-1,5-dihydro-pyrrol-2-one (11) and (S)-5-(3-methoxy-phenyl)-3-((R)-1-phenyl-ethylamino)-1-(4-trifluoromethyl-phenyl)-1,5-dihydro-pyrrol-2-one (12)
(R)-(+)-1-phenylethylamine (22.4 mL, 176 mmol) was added to a solution of 10 (30.8 g, 88.2 mmol) in CH2Cl2 (225 mL). The solution was stirred at ambient temperature for 18 h. The solution was then poured onto a silica gel column and the CH2Cl2 was evaporated off with a stream of nitrogen. The material was purified by silica gel chromatography (5–15% EtOAc-hexanes) to afford 12, the first eluting isomer, as a white foam (9.6 g, 24%) and 11, the second eluting isomer, as a yellow foam (6.7 g, 17%). A mixture of 11 and 12 (11.5 g, 29%) was also obtained. 11: 1H NMR (400 MHz, DMSO-d6): δ 7.77 (d, 2H, J = 8.4 Hz), 7.61 (d, 2H, J = 8.8 Hz), 7.33 (d, 2H, J = 7.0 Hz), 7.23 (dd, 2H, J = 7.6, 7.6 Hz), 7.15–7.10 (m, 1H), 7.03 (dd, 1H, J = 7.9, 7.9 Hz), 6.64 (ddd, 1H, J = 8.4, 2.6, 0.9 Hz), 6.57 (s, 1H), 6.53 (d, 1H, J = 7.9 Hz), 5.84–5.80 (m, 2H), 5.15 (d, 1H, J = 2.6 Hz), 4.34–4.25 (m, 1H), 3.56 (s, 3H), 1.42 (d, 3H, J = 6.6 Hz). LC—MS ESI m/z: 453 (M+H)+, tR = 6.06 min, method 2. 12: 1H NMR (400 MHz, DMSO-d6): δ 7.75 (d, 2H, J = 8.8 Hz), 7.61 (d, 2H, J = 8.8 Hz), 7.35 (d, 2H, J = 7.5 Hz), 7.28 (dd, 2H, J = 7.4, 7.4 Hz), 7.21–7.11 (m, 2H), 6.75–6.70 (m, 2H), 6.65 (d, 1H, J = 7.5 Hz), 5.92 (d, 1H, J = 7.5 Hz), 5.76 (d, 1H, J = 2.6 Hz), 5.15 (d, 1H, J = 2.6 Hz), 4.28–4.19 (m, 1H), 3.65 (s, 3H), 1.40 (d, 3H, J = 7.0 Hz). LC—MS ESI m/z: 453 (M+H)+, tR = 6.16 min, method 2.
(R)-3-[(R)-1-(4-Fluoro-phenyl)-ethylamino]-5-(3-methoxy-phenyl)-1-(4-trifluoromethyl-phenyl)-1,5-dihydro-pyrrol-2-one (13) and (S)-3-[(R)-1-(4-fluoro-phenyl)-ethylamino]-5-(3-methoxy-phenyl)-1-(4-trifluoromethyl-phenyl)-1,5-dihydro-pyrrol-2-one (14)
A slurry of 10 (6.6 g, 18.9 mmol) in CH2Cl2 (40 mL) was added to a solution of (R)-1-(4-fluoro-phenyl)-ethylamine (5.24 g, 37.6 mmol) in CH2Cl2(20 mL). The homogeneous solution was stirred at ambient temperature for 18 h. The solution was then poured onto a silica gel column and the CH2Cl2 was evaporated off with a stream of nitrogen. The material was purified by silica gel chromatography (5–20% EtOAc—hexanes) to afford 14, the first eluting isomer, as a white foam (3.34 g, 38%) and 13, the second eluting isomer, as a white foam (3.26 g, 37%). 13: 1H NMR (400 MHz, DMSO-d6): δ 7.74 (d, 2H, J = 8.4 Hz), 7.59 (d, 2H, J = 8.8 Hz), 7.36 (dd, 2H, J = 8.8, 5.7 Hz), 7.06–6.99 (m, 3H), 6.64 (dd, 1H, J = 8.1, 2.4 Hz), 6.55–6.53 (m, 1H), 6.51 (d, 1H, J = 7.9 Hz) 5.87 (d, 1H, J = 7.5 Hz), 5.80 (d, 1H, J = 2.6 Hz), 5.16 (d, 1H, J = 2.2 Hz), 4.34–4.25 (m, 1H), 3.56 (s, 3H), 1.40 (d, 3H, J = 6.6 Hz). LC—MS ESI m/z: 471 (M+H)+, tR = 6.53 min, method 2.14: 1H NMR (400 MHz, DMSO-d6): δ 7.74 (d, 2H, J = 8.4 Hz), 7.59 (d, 2H, J = 8.8 Hz), 7.38 (dd, 2H, J = 8.4, 5.7 Hz), 7.15–7.05 (m, 3H), 6.73–6.69 (m, 2H), 6.63 (d, 1H, J = 7.9 Hz), 5.95 (d, 1H, J = 7.5 Hz), 5.76 (d, 1H, J = 2.6 Hz), 5.16 (d, 1H, J = 2.6 Hz), 4.28–4.20 (m, 1H), 3.63 (s, 3H), 1.37 (d, 3H, J = 6.6 Hz). LC—MS ESI m/z: 471 (M+H)+, tR = 6.62 min, method 2.
(3R,5R)-5-(3-Methoxy-phenyl)-3-((R)-1-phenyl-ethylamino)-1-(4-trifluoromethyl-phenyl)-pyrrolidin-2-one (8) and (3S,5R)-5-(3-methoxy-phenyl)-3-((R)-1-phenyl-ethylamino)-1-(4-trifluoromethyl-phenyl)-pyrrolidin-2-one (15)
Sodium cyanoborohydride (780 mg, 12.4 mmol) was added to a solution of 11 (2.8 g, 6.19 mmol) in glacial acetic acid (31 mL). The reaction mixture was stirred at ambient temperature for 1 h and concentrated in vacuo. The residue was dissolved in EtOAc and washed with saturated NaHCO3 solution, water, and brine, dried (Na2SO4), and concentrated in vacuo. The material was purified by silica gel chromatography (10–30% EtOAc–hexanes) to afford 8, the first eluting isomer, as a white solid (2.26 g, 80%) and 15, the second eluting isomer, as a clear colorless oil (120 mg, 4.3%). 8: Mp 126–127.5 °C. 1H NMR (400 MHz, DMSO-d6): δ 7.57 (d, 2H, J = 8.8 Hz), 7.50 (d, 2H, J = 8.8 Hz), 7.34 (dd, 2H, J = 8.4, 1.3 Hz), 7.27 (dd, 2H, J = 7.5, 7.5 Hz), 7.20–7.15 (m, 1H), 7.12 (dd, 1H, J = 7.9, 7.9 Hz), 6.79 (dd, 1H, J = 2.0, 2.0 Hz), 6.76 (d, 1H, J = 7.9 Hz), 6.69 (ddd, 1H, J = 8.4, 2.6, 0.9 Hz), 5.16 (dd, 1H, J = 9.2, 6.6 Hz), 4.33–4.26 (m, 1H), 3.63 (s, 3H), 3.45–3.36 (m, 1H), 2.72–2.67 (m, 1H), 2.37 (ddd, 1H, J = 13.5, 6.9, 5.6 Hz), 1.49 (ddd, 1H, J = 16.3, 6.8, 5.5 Hz), 1.28 (d, 3H, J = 6.6 Hz). LC—MS ESI m/z: 455 (M+H)+, tR = 2.81 min, method 1. 13C NMR (125 MHz, DMSO-d6): δ 175.3, 159.3, 146.0, 142.5, 141.3, 129.8, 128.2 (2C), 126.8 (2C), 126.7, 125.3 (2C, q, J = 3.8 Hz), 124.5 (q, J = 32.0 Hz), 124.1 (q, J = 271.3 Hz), 122.9 (2C), 118.8, 112.7, 112.6, 58.6, 57.0, 56.1, 54.9, 38.7, 24.6. HRMS-FT (m/z): [M+H]+ calcd for C26H26F3N2O2, 455.1941; found 455.1936. Anal. (C26H25F3N2O2) C, H, N.
15: 1H NMR (400 MHz, DMSO-d6): δ 7.74 (d, 2H, J = 8.8 Hz), 7.64 (d, 2H, J = 8.8 Hz), 7.32 (d, 2H, J = 7.5 Hz), 7.27 (dd, 2H, J = 7.5, 7.5 Hz), 7.20–7.11 (m, 2H), 6.73 (dd, 1H, J = 8.2, 2.1 Hz), 6.69 (s, 1H), 6.63 (d, 1H, J = 7.9 Hz), 5.52 (d, 1H, J =7.5 Hz), 3.86–3.79 (m, 1H), 3.63 (s, 3H), 3.43–3.37 (m, 1H), 2.56 (br s, 1H), 2.43–2.34 (m, 1H), 2.19–2.11 (m, 1H), 1.27 (d, 3H, J = 7 .0 Hz). LC—MS ESI m/z: 455 (M+H)+, tR = 3.02 min, method 1.
(3R,5R)-3-[(R)-1-(4-Fluoro-phenyl)-ethylamino]-5-(3-methoxy-phenyl)-1-(4-trifluoromethyl-phenyl)-pyrrolidin-2-one (16)
Sodium cyanoborohydride (846 mg, 13.5 mmol) was added to a solution of 13 (3.16 g, 6.73 mmol) in glacial acetic acid (10 mL) and CH2Cl2 (10 mL). The reaction mixture was stirred at ambient temperature for 1 h and concentrated in vacuo. The residue was partitioned between EtOAc and 1N NaOH. The organic extract was washed with brine, dried (Na2SO4), and concentrated in vacuo. The material was purified by silica gel chromatography (20–30% EtOAc–hexanes) to afford 16 as a white solid (2.5 g, 79%): Mp 98–100 °C. 1H NMR (400 MHz, DMSO-d6): δ 7.56 (d, 2H, J = 8.8 Hz), 7.48 (d, 2H, J = 8.4 Hz), 7.38–7.33 (m, 2H), 7.13–7.04 (m, 3H), 6.79–6.73 (m, 2H), 6.70–6.66 (m, 1H), 5.15 (dd, 1H, J = 9.2, 6.6 Hz), 4.31 (q, 1H, J = 6.6 Hz), 3.61 (s, 3H), 3.37 (dd, 1H, J = 11.0, 8.4 Hz), 2.72 (br s, 1H), 2.37 (ddd, 1H, J = 13.6, 7.0, 5.3 Hz), 1.48 (ddd, 1H, J = 16.3, 6.8, 5.5 Hz), 1.25 (d, 3H, J = 6.6 Hz). 13C NMR (125 MHz, DMSO-d6): δ 175.3, 161.1 (d, J = 241.9 Hz), 159.3, 142.4, 142.1, 141.3, 129.8, 128.6 (2C, d, J = 7.9 Hz), 125.3 (2C, q, J = 3.9 Hz), 124.5 (q, J = 32.0 Hz), 124.1 (q, J = 271.4 Hz), 122.9 (2C), 118.8, 114.9 (2C, d, J = 21.1 Hz), 112.7, 112.6, 58.6, 56.8, 55.3, 54.9, 38.6, 24.6. LC–MS ESI m/z: 473 (M+H)+, tR = 5.82 min, method 2. HRMS-FT (m/z): [M+H]+ calcd for C26H25F4N2O2, 473.1847; found 473.1842. Anal. (C26H24F4N2O2) C, H, N.
(3R,5R)-5-(3-Hydroxy-phenyl)-3-((R)-1-phenyl-ethylamino)-1-(4-trifluoromethyl-phenyl)-pyrrolidin-2-one (18)
A mixture of 8 (1.03 g, 2.27 mmol) and pyridinium hydrochloride (20 g) was heated in a 185 °C oil bath for 2 h under a N2 atmosphere. The reaction mixture was cooled to ambient temperature. The reaction mixture was dissolved in water and the precipitate was filtered and the filtrate saved. The precipitate was dissolved in EtOAc and c. NH4OH. solution was added until the mixture was basic. The layers were separated and the organic portion was washed with water and brine, dried (Na2SO4), and concentrated in vacuo to gives an oil (660 mg). c. NH4OH solution was added to the above filtrate until basic. The basic solution was extracted with EtOAc. The organic extracts were washed with water and brine, dried (Na2SO4), and concentrated in vacuo give a foam (430 mg). The oil and the foam were combined and purified by silica gel chromatography (0–25% MTBE–CH2Cl2) to yield 18 (600 mg, 60%) as a white foam. 1H NMR (400 MHz, DMSO-d6): δ 9.31 (s, 1H), 7.57 (d, 2H, J = 8.8 Hz), 7.47 (d, 2H, J = 8.4 Hz), 7.32 (dd, 2H, J = 8.1, 1.5 Hz), 7.25 (dd, 2H, J = 7.4, 7.4 Hz), 7.18–7.13 (m, 1H), 6.98 (dd, 1H, J = 7.9, 7.9 Hz), 6.59 (d, 1H, J = 7.9 Hz), 6.54 (dd, 1H, J = 2.0, 2.0 Hz), 6.50 (ddd, 1H, J = 8.0, 2.5, 1.0 Hz), 5.08 (dd, 1H, J = 9.2, 6.6 Hz), 4.28–4.21 (m, 1H), 3.42–3.35 (m, 1H), 2.65 (br s, 1H), 2.30 (ddd, 1H, J =13.5, 7.1, 5.4 Hz), 1.42 (dd, 1H, J = 21.8, 10.8 Hz), 1.26 (d, 3H, J = 6.6 Hz). LC–MS ESI m/z: 441 (M+H)+, tR = 4.66 min, method 2.
(3R,5R)-3-[(R)-1-(4-Fluoro-phenyl)-ethylamino]-5-(3-hydroxy-phenyl)-1-(4-trifluoromethyl-phenyl)-pyrrolidin-2-one (19)
A mixture of 16 (1.22 g, 2.58 mmol) and pyridinium hydrochloride (24 g) was heated in a 185 °C oil bath for 2 h under a N2 atmosphere. The reaction mixture was cooled but while still warm was dissolved in water. c. NH4OH solution was added until the mixture was basic. The basic solution was extracted with EtOAc. The organic extracts were washed with water and brine, dried (Na2SO4), and concentrated in vacuo. The crude product was purified by silica gel chromatography (5–15% MTBE–CH2Cl2) to yield 19: The crude 19 was purified further by silica gel chromatography (0.5–1.5% MeOH-CH2Cl2) to give a white foam (790 mg, 67%): 1H NMR (400 MHz, DMSO-d6): δ 9.34 (s, 1H), 7.58 (d, 2H, J = 8.8 Hz), 7.49 (d, 2H, J = 8.4 Hz), 7.37 (dd, 2H, J = 8.4, 5.7 Hz), 7.09 (dd, 2H, J = 8.8, 8.8 Hz), 7.00 (dd, 1H, J = 7.8, 7.8 Hz), 6.62 (d, 1H, J = 7.5 Hz), 6.56 (s, 1H), 6.52 (dd, 1H, J = 7.9, 1.7 Hz), 5.10 (dd, 1H, J = 9.2, 6.6 Hz), 4.33–4.25 (m, 1H), 3.42–3.34 (m, 1H), 2.74–2.67 (m, 1H), 2.39–2.30 (m, 1H), 1.44 (dd, 1H, J = 22.0, 10.5 Hz), 1.26 (d, 3H, J = 6.6 Hz). LC–MS ESI m/z: 459 (M+H)+, 457 (M-H)-, tR = 2.50 min. method 1.
(3R,5R)-5-(3-Fluoromethoxy-phenyl)-3-((R)-1-phenyl-ethylamino)-1-(4- trifluoromethyl-phenyl)-pyrrolidin-2-one (20)
Cesium carbonate (1.19 g, 3.6 mmol) was added to a solution of 18 (267 mg, 0.61 mmol) in DMF (4 mL). A solution of fluoromethyl tosylate (149 mg, 0.73 mmol) in DMF (2 mL) was added and the reaction mixture was stirred at ambient temperature for 26 h. The mixture was diluted with water and extracted with EtOAc. The EtOAc extracts were washed with water and brine, dried (Na2SO4), and concentrated in vacuo. The crude material was purified by silica gel chromatography (0–25% EtOAc-hexanes) to afford the 20 as a white solid (153 mg, 53%): Mp 116–118 °C. 1H NMR (400 MHz, DMSO-d6): δ 7.56 (d, 2H, J = 8.8 Hz), 7.48 (d, 2H, J = 8.8 Hz), 7.32 (d, 2H, J = 7.0 Hz), 7.26 (dd, 2H, J = 7.5, 7.5 Hz), 7.21–7.14 (m, 2H), 6.97–6.91 (m, 2H), 6.85 (dd, 1H, J = 8.4, 2.2 Hz), 5.79 (dd, 1H, J = 16.9, 3.3 Hz), 5.66 (dd, 1H, J = 16.7, 3.4 Hz), 5.19 (dd, 1H, J = 9.2, 6.6 Hz), 4.33–4.25 (m, 1H), 3.43–3.35 (m, 1H), 2.69 (br s, 1H), 2.38 (ddd, 1H, J = 13.6, 6.8, 5.5 Hz), 1.48 (dd, 1H, J = 21.8, 10.8 Hz), 1.26 (d, 3H, J = 6.6 Hz). 13C NMR (125 MHz, DMSO-d6): δ 175.3, 156.1, 145.9, 143.0, 141.2, 130.1, 128.2 (2C), 126.8 (2C), 126.7, 125.4 (2C, q, J = 3.8 Hz), 124.6 (q, J = 31.7 Hz), 124.1 (q, J = 271.3 Hz), 123.0 (2C), 121.5, 114.9, 114.7, 100.2 (d, J = 215.4 Hz), 58.4, 56.9, 56.1, 38.6, 24.6. LC–MS ESI m/z: 473 (M+H)+, tR = 5.23 min, method 2. HRMS-FT (m/z): [M+H]+ calcd for C26H25F4N2O2, 473.1847; found 473.1842. Anal. (C26H24F4N2O2) C, H, N.
(3R,5R)-5-((3-Fluoromethoxy-d2)phenyl)-3-((R)-1-phenyl-ethylamino)-1-(4-trifluoromethyl-phenyl)-pyrrolidin-2-one (21)
Cesium carbonate (6.42 g, 19.70 mmol) was added to a solution of 18 (1.45 g, 3.29 mmol) in DMF (30 mL). A solution of fluoromethyl tosylate-d2 (1.02 g, 4.61 mmol) in DMF (10 mL) was added and the reaction mixture was stirred at ambient temperature for 26 h. The mixture was diluted with water and extracted with EtOAc. The EtOAc extracts were washed with water and brine, dried (Na2SO4), and concentrated in vacuo. The crude material was purified by silica gel chromatography (heptane–EtOAc (80: 20–50: 50 v/v)) to afford the 21 as a white solid (700 mg, 45%): Mp 116–118 °C. 1H NMR (500 MHz, DMSO-d6): δ 7.60 (d, 2H, J = 8.7 Hz), 7.52 (d, 2H, J = 8.6 Hz), 7.36 (d, 2H, J = 7.2 Hz), 7.30 (dd, 2H, J = 7.5, 7.5 Hz), 7.25–7.18 (m, 2H), 7.00 (s, 1H), 6.97 (d, 1H, J = 7.8 Hz), 6.89 (dd, 1H, J = 8.2, 2.3 Hz), 5.23 (dd, 1H, J = 9.3, 6.6 Hz), 4.36–4.30 (m, 1H), 3.47–3.40 (m, 1H), 2.76–2.70 (m, 1H), 2.42 (ddd, 1H, J = 13.6, 6.9, 5.6 Hz), 1.52 (dd, 1H, J = 21.6, 10.8 Hz), 1.30 (d, 3H, J = 6.6 Hz). 13C NMR (125 MHz, DMSO-d6): δ 175.3, 156.1, 145.9, 143.0, 141.2, 130.1, 128.2 (2C), 126.8 (2C), 126.7, 125.4 (2C, q, J = 3.8 Hz), 124.6 (q, J = 31.7 Hz), 124.1 (q, J = 271.3 Hz), 123.0 (2C), 121.5, 114.9, 114.7, 99.6 (d of pentets, J = 213.6 Hz, 27.5 Hz), 58.4, 56.9, 56.1, 38.6, 24.6. HRMS-FT (m/z): [M+H]+ calcd for C26H23D2F4N2O2, 475.1972; found 475.1966. Anal. (C26H24F4N2O2) C, H, N. (N.B., results are based on H because the instrumentation could not distinguish between H and D).
(3R,5R)-5-[3-(2-Fluoro-ethoxy)-phenyl]-3-((R)-1-phenyl-ethylamino)-1-(4-trifluoromethyl-phenyl)-pyrrolidin-2-one (22)
Cesium carbonate (426 mg, 1.3 mmol) and 1-bromo-2-fluoroethane (19 μL, 0.26 mmol) were added to a solution of 18 (96 mg, 0.22 mmol) in DMF (2 mL). The reaction mixture was stirred at ambient temperature for 22 h. The mixture was diluted with water and extracted with EtOAc. The EtOAc extracts were washed with water and brine, dried (Na2SO4), and concentrated in vacuo. The crude material was purified by silica gel chromatography (0–40% EtOAc–hexanes) to afford 22 as an oil (94 mg, 89%). 1H NMR (400 MHz, DMSO-d6): δ 7.56 (d, 2H, J = 8.8 Hz), 7.48 (d, 2H, J = 8.4 Hz), 7.32 (dd, 2H, J = 8.1, 1.5 Hz), 7.25 (dd, 2H, J = 7.6, 7.6 Hz), 7.18–7.14 (m, 1H), 7.11 (dd, 1H, J = 7.9, 7.9 Hz), 6.82 (dd, 1H, J = 2.0, 2.0 Hz), 6.78 (d, 1H, J = 7.5 Hz), 6.71 (dd, 1H, J = 8.3, 2.6 Hz), 5.14 (dd, 1H, J = 9.4, 6.4 Hz), 4.71–4.67 (m, 1H), 4.59–4.55 (m, 1H), 4.32–4.25 (m, 1H), 4.18–3.99 (m, 2H), 3.43–3.35 (m, 1H), 2.68 (br s, 1H), 2.35 (ddd, 1H, J = 13.5, 6.9, 5.6 Hz), 1.48 (dd, 1H, J = 21.7, 10.9 Hz), 1.26 (d, 3H, J = 6.6 Hz); LC–MS ESI m/z: 487 (M+H)+, tR = 5.16 min, method 2. 22 (94 mg, 0.19 mmol) was dissolved in EtOAc and HCl (0.97 mL, 0.97 mmol, 1M in ether) was added. The homogeneous solution was concentrated to give the hydrochloride salt (103 mg, 100%) as a yellow solid. Mp 137–144 °C. 13C NMR (125 MHz, DMSO-d6): δ 168.8, 158.3, 141.2, 140.2, 137.0, 129.9, 129.1, 129.0 (2C), 128.1 (2C), 125.6 (q, J = 32.3 Hz), 125.6 (2C, q, J = 3.5 Hz), 123.9 (q, J = 271.7 Hz), 123.6 (2C), 120.0, 113.8, 113.8, 82.0 (d, J = 166.6 Hz), 66.9 (d, J = 19.2 Hz), 59.2, 55.6, 54.0, 33.4, 18.7. LC-MS ESI m/z: 487 (M+H)+, tR = 5.17 min, method 2. HRMS-FT (m/z): [M+H]+ calcd for C27H27F4N2O2, 487.2003; found 487.1996. Anal. (C27H27ClF4N2O2) C, H, N.
Ex vivo experiments
The time course of CB1 receptor ligand uptake into rat brain was studied for 8, 16, 20 and 22. Ligand levels were measured in the rat frontal cerebral cortex, a structure which contains a high density of CB1 receptors. 3 (10 mg) was dissolved in 1 mL vehicle comprised of intralipid (Sigma-Aldrich Chemical Co. St. Louis, MO) containing 0.2%-AcOH. Ligand 8, 16, 20 or 22 (1 mg) was dissolved in 1 mL of intralipid containing 1%-AcOH, diluted with sterile water to a final concentration of 30 μg/mL, and administered intravenously to rats in a volume of 1 mL/kg.
Adult male Sprague-Dawley rats (225–250 g; Harlan Sprague-Dawley, Indianapolis, IN) were used. All studies were performed in accordance with National Institutes of Health guidelines under protocols approved by the Animal Care and Use Committee of Eli Lilly and Company. Rats were pretreated for 15 min with 3 (10 mg/kg, i.v.) or its vehicle administered by tail vein injection to groups of three rats per time point. After the 15 min pretreatment period, rats received 8, 16, 20 or 22 (30 μg/kg, i.v.). Rats were sacrificed at 0.25, 0.5, 1, 2, 4 or 8 h by cervical dislocation. A portion of each frontal cerebral cortex was dissected, weighed, and placed in a centrifuge tube on ice. Brain tissues were homogenized using a cell disrupter in four volumes (W/V) of MeCN containing 0.1% HCO2H. To extract the ligand (e.g., 8), tissues were centrifuged at 20,000 × g for 14 min. Aliquots of supernatant containing the tracer were diluted with water to an MeCN content less than that of the mobile phase and injected by auto sampler onto an HPLC employing a Zorbax SB C-18 narrow bore rapid resolution column (2.1 × 50 mm, Agilent Technologies, Wilmington, DE). Separation was achieved using isocratic conditions, in a mobile phase of 70% MeCN containing 1% HCO2H at a flow rate of 0.25 mL/min. Eluted 8, 16, 20 or 22 was measured using an Agilent model 1946 single quad mass spectrometer run in positive mode, fitted with an electrospray ion source and set to m/z 455, 473, 473 and 487 ion for each experiment, respectively. Clearly delineated chromatographic peaks with the retention time of authentic standards and expected molecular weight were seen after each injection of sample. Analytes were quantified based on peak area. Data for 8, 16, 20 and 22 were plotted with Graphpad Prism version 4.02 (Graphpad software; San Diego, CA) are presented as ng/g of tissue as mean ± SD (n = 3).
CB1 and CB2 GTPγ35S potency assay
GTPγ35S potency of 3, 8, 16, 20 and 22 in CHO (Lonza, Walkersville, MD) or Sf9 (PerkinElmer Life Sciences, Boston, MA) cell membranes that ectopically express human CB1 or CB2 receptors, respectively, was measured in a 96-well format with a modified antibody capture technique as described previously.25,41 The following exceptions applied to the CB1 CHO GTPγ35S assays: membranes (1 unit/well), compound, and GTPγ35S (500 pM) were incubated in GTP-binding assay buffer (20 mM HEPES, 100 mM NaCl, 5 mM MgCl2, 0.5% fatty acid free bovine serum albumin (Serologicals Corp, Norcross, GA), 1 μM GDP, pH 7.4) at room temperature for 30 min. Antagonist dose responses were performed in the presence of an 80% efficacious dose of full agonist (methanandamide). A mixture containing 0.2% Nonidet P40 detergent (Roche, Indianapolis, IN), anti-Gi antibody (final dilution of 1:362; Covance, Princeton, NJ), and 1.25 mg anti-rabbit antibody scintillation proximity assay beads (GE Healthcare, Piscataway, NJ) was added to all wells. Assay plates were sealed, vortexed, and incubated for an additional 2 h. Following incubation, plates were centrifuged at 700 × g for 10 min and counted for 1 min per well (Wallac MicroBeta TriLux scintillation counter; PerkinElmer, Boston, MA). Antagonist Kb values (potency) were calculated with a modification of the Cheng-Prusoff relationship as follows: Kb = IC50/[1 + [Agonist]/EC50], where IC50 is determined from a four parameter fit of displacement curves, [agonist] = EC50 of full agonist, and EC50 is determined from a four parameter fit of a full agonist concentration response curve.42 Mean Kb values were calculated as mean of at least three independent determinations ± standard error of the mean (SEM).
Computation of cLogD7.4
cLogD (at pH 7.4) values for 8, 16, 20 and 22 were computed with Pallas 3.0 for Windows (CompuDrug; San Francisco, CA).
Radionuclide production
No-carrier-added (NCA) [11C]carbon dioxide (∼ 52 GBq) was produced in a target of nitrogen gas (∼ 225 psi) containing oxygen (1%) via the 14N(p,α)11C reaction induced by irradiation with a 16 MeV proton beam (45 μA) for 20 min from a PETtrace cyclotron (GE; Milwaukee, WI). [11C]iodomethane was produced within a lead-shielded hot-cell from [11C]carbon dioxide via reduction to [11C]methane and iodination with a MeI MicroLab apparatus (GE; Milwaukee, WI).
NCA [18F]fluoride ion was produced with a PETtrace cyclotron to implement the 18O(p,n)18F reaction on 18O-enriched water (95 atom %; 1.8 mL). Typically, proton bombardments were with 18 MeV protons at 20 μA for 120 min. Portions of the irradiated water containing up to 18.5 GBq were used for individual experiments.
Radiosynthesis of [11C]8 and [11C]16
At about 3 min before the end of radionuclide production the O-desmethyl precursor 18 or 19 (∼ 1.5 mg, 3.4 μmol for 18 and 3.3 μmol for 19) dissolved in DMF (80 μL) was treated with tetra-n-butylammonium hydroxide in methanol (0.5 M; ∼ 6 μL). This solution was then loaded into a loop of stainless steel (internal volume, 2 mL) of a commercially available radiomethylation apparatus (Bioscan) housed within a lead-shielded hot-cell. The loop was conditioned by flushing with helium (12 mL/min) for about 3 min. [11C]Iodomethane was swept into the loop in a stream (12 mL/min) of helium until radioactivity in the loop had maximized (about 4 min after release of [11C]iodomethane from the MeI Microlab apparatus). Radiomethylation was allowed to proceed for about 3 min at room temperature. Crude [11C]8 or [11C]16 was flushed from loop the with HPLC mobile phase (2 mL, MeCN–10 mM HCO2NH4, 67:33 v/v) and purified with HPLC on a Luna C-18 column (10 μM, 10 × 250 mm, Phenomenex) eluted with mobile phase at 6 mL/min with eluate monitored for radioactivity and absorbance at 254 nm. [11C]8 (tR = 12 min) or [11C]16 (tR = 12 min) was collected and concentrated to dryness by rotary evaporation under reduced pressure and heat at 80 °C, formulated in sterile physiological saline (0.9% w/v; 10 mL) containing ethanol (5% v/v), Tween 80 (10 mg) and finally filtered through a sterile filter (0.2 μm pore size, Millex-GV, Millipore) into a sterile, pathogen free dose vial.
Specific radioactivity, chemical purity and radiochemical purity of [11C]8 and [11C]16 were assessed by HPLC of a 0.1-mL aliquot of formulated dose on a Luna C-18 column (4.6 × 250 mm, Phenomenex) eluted with MeCN–0.1%-TFA (45: 55 v/v) at 2 mL/min with eluate monitored for radioactivity and absorbance at 235 nM (tR’s of [11C]8 and [11C]16 = 5.12 and 6 min, respectively).
Synthesis of [18F]fluoro(m)ethyl bromide
Cyclotron-produced [18F]fluoride ion in [18O]water was delivered into a vial containing Kryptofix 2.2.2 (5.2 mg, 3.6 μmol; Aldrich Chem. Co.) and potassium carbonate (0.5 mg, 3.6 μmol) in MeCN-H2O (94: 4 v/v; 0.1 mL). The [18F]fluoride ion mixture was transferred to a modified version of the TRACERlab FXF-N module which was then followed by MeCN (1 mL). The mixture was evaporated to dryness at 90 °C under reduced pressure with nitrogen flow. MeCN (2 mL) was again added and then evaporated to dryness. The CH2Br2 (50 μL), CD2Br2(50 μL) or bromo(m)ethyl tosylate (30 μL) in solvent (1 mL; MeCN for CH2Br2 and CD2Br2 and o-dichlorobenzene for bromo(m)ethyl tosylate) was added to the dry [18F]fluoride ion-K2.2.2-K+ complex which was then heated to 110 °C for 15 min. The reaction vessel was then cooled to 35 °C. Nitrogen gas (30 mL/min) was used to transfer the volatile [18F]fluoro(m)ethyl bromide through a series of four silica gel cartridges (SepPak Plus) for CH2Br2, CD2Br2 and or one silica gel SepPak Plus for bromo(m)ethyl tosylate and then swept into a pre-cooled V-vial (volume 1-mL) with a crimp-seal silicone-Teflon septum cap.
Radiosynthesis of [18F]20, [18F]21 and [18F]22
A glass reaction vessel was loaded with 18 (∼ 0.5mg, 1.1 μmol), DMF (1 mL) and base [Cs2CO3 (0.5 mg, 1.5 μmol) and 18-crown-6 (5 mg, 19 μmol) for [18F]20 and [18F]21; (n-Bu)4NOH (0.176 M in MeOH; ∼ 8 μL) for [18F]22]. [18F]FMeBr, [18F]FMeBr-d2 or [18F]FEtBr was transferred to a solution under computer control from the TRACERlab FXF-N module. Radioactivity transfer was monitored by two external Bioscan detectors and was stopped when radioactivity in the vessel reached a maximum. The vessel was heated at 110 °C for 10 min ([18F]20, [18F]21) and 5 min for ([18F]22). The mixture containing [18F]20 or [18F]21 (tR = 34 min) was diluted with water (1 mL) and injected remotely onto a Luna C-18 column (10 × 250 mm; 10 μL) eluted with MeCN–1% aq.-TFA (35:65 v/v) over 40 min at 6 mL/min. The fraction containing [18F]22 (tR = 28 min) was diluted with water (1 mL) and injected remotely onto on a Luna C-18 column (10 × 250 mm; 10 μL) eluted with MeCN–10mM aq.-HCO2NH4 (55: 45 v/v) over 40 min at 6 mL/min. Eluate was monitored for absorbance at 254 nm and radioactivity. The purified [18F]20, [18F]21 or [18F]22 was collected, diluted in water (∼ 85 mL) and passed through a Sep-Pak Plus C-18 cartridge [preconditioned first with ethanol (10 mL) and then water (10 mL)]. Under TRACERlab FXF-N module control, the product ([18F]20, [18F]21 and [18F]22) was washed with sterile water (5 mL). The product ([18F]20, [18F]21 or [18F]22) was eluted from the cartridge with first with ethanol (USP; 9.5 mL) and then sodium chloride (0.9% USP; 9 mL), which were then combined in a separate vessel (20 mL). The mixture was pushed through a sterile filter (0.2 μm pore size, Millex-GV, Millipore for [18F]20 or [18F]21; 0.22 μm pore size, Anatop for [18F]22) into a sterile dose vial (10 mL).
Specific radioactivities, chemical purities and radiochemical purities of [18F]20 or [18F]27 (tR = 9.5 min) were assayed by HPLC of a 0.1-mL aliquot of formulated dose on a Luna C-18 column (4.6 × 250 mm) eluted with MeCN–%1 aq.-TFA (45: 55 v/v) at 2 mL/min with eluate monitored for radioactivity and absorbance at 254 nm. Specific radioactivity, chemical purity and radiochemical purity of [18F]22 (tR = 6.7 min) were assayed by HPLC of a 0.1-mL aliquot of formulated dose on a Luna C-18 column (4.6 × 250 mm) eluted with MeCN–10mM aq-HCO2NH4 (65: 35 v/v) at 2 mL/min with eluate monitored for radioactivity and absorbance at 254 nm.
Supplementary Material
Acknowledgements
This research was supported by the Intramural Research Program of the National Institutes of Health, specifically the National Institute of Mental Health, and also by a Cooperative Research and Development Agreement between Lilly Research Laboratories, NIMH and the Karolinska Institutet. We are grateful to Ms. Cheryl L. Morse and Mr. Jinsoo Hong for assistance in radioligand production. We thank the NIH PET Department for carbon-11 and fluorine-18 production.
Abbreviations
- CB1
cannabinoid subtype-1
- CB2
cannabinoid subtype-2
- CHO
chinese hamster ovary
- CNS
central nervous system
- D2
dopamine subtype-2
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- EOS
end of synthesis
- EtOAc
ethyl acetate
- GDP
guanosine diphosphate
- GTPγS
guanosine-5′-(γ-thio)-triphosphate
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HRMS
high-resolution mass spectrometry
- HPLC
high-performance liquid chromatography
- 5-HT2A
serotonin-2A
- IND
Investigational New Drug
- LC–MS
liquid chromatography–mass spectrometry
- Mp
melting point
- MTBE
methyl tert-butyl ether
- NK1
neurokinin-1
- PET
positron emission tomography
- RCY
decay-corrected radiochemical yield
- SUV
standardized uptake value
- TFA
trifluoroacetic acid
- THC
tetrahydrocannabinol
- ppm
parts per million
References
- (1).Mackie K. Cannabinoid receptors as therapeutic targets. Annu. Rev. Pharmacol. Toxicol. 2005;46:101–122. doi: 10.1146/annurev.pharmtox.46.120604.141254. [DOI] [PubMed] [Google Scholar]
- (2).Lambert DM, Fowler CJ. The endocannabinoid system: drug targets, lead compounds, and potential therapeutic applications. J. Med. Chem. 2005;48:5059–5087. doi: 10.1021/jm058183t. [DOI] [PubMed] [Google Scholar]
- (3).Mechoulam R, Gaoni Y. Absolute configuration of Δ1-tetrahydrocannabinol major active constituent of hashish. Tetrahedron Lett. 1967:1109–1111. doi: 10.1016/s0040-4039(00)90646-4. [DOI] [PubMed] [Google Scholar]
- (4).Johnson MR, Melvin LS. Cannabinoids as Therapeutic Agents. CRC Press; Boca Raton, FL: 1986. pp. 121–145. [Google Scholar]
- (5).Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- (6).Devane WA, Dysarz FA, Johnson MR, Melvin LS, Howlett AC. determination and characterization of a cannabinoid receptor in rat-brain. Mol. Pharmacol. 1988;34:605–613. [PubMed] [Google Scholar]
- (7).Munro S, Thomas KL, Abushaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- (8).Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, Mechoulam R, Pertwee RG. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- (9).Shire D, Carillon C, Kaghad M, Calandra B, Rinaldicarmona M, Lefur G, Caput D, Ferrara P. An amino-terminal variant of the central cannabinoid receptor resulting from alternative splicing. J. Biol. Chem. 1995;270:3726–3731. doi: 10.1074/jbc.270.8.3726. [DOI] [PubMed] [Google Scholar]
- (10).Ryberg E, Vu HK, Larsson N, Groblewski T, Hjorth S, Elebring T, Sjorgren S, Greasley PJ. Identification and characterisation of a novel splice variant of the human CB1 receptor. FEBS Lett. 2005;579:259–264. doi: 10.1016/j.febslet.2004.11.085. [DOI] [PubMed] [Google Scholar]
- (11).Herkenham M, Lynn AB, Johnson MR, Melvin LS, Decosta BR, Rice KC. Characterization and localization of cannabinoid receptors in rat-brain: a quantitative in vitro autoradiographic study. J. Neurosci. 1991;11:563–583. doi: 10.1523/JNEUROSCI.11-02-00563.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, Decosta BR, Rice KC. Cannabinoid receptor localization in brain. Proc. Natl. Acad. Sci. U.S.A. 1990;87:1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lynn AB, Herkenham M. Localization of cannabinoid receptors and nonsaturable high-density cannabinoid binding-sites in peripheral-tissues of the rat: implications for receptor-mediated immune modulation by cannabinoids. J. Pharmacol. Exp. Ther. 1994;268:1612–1623. [PubMed] [Google Scholar]
- (14).Griffin G, Fernando SR, Ross RA, McKay NG, Ashford MLJ, Shire D, Huffman JW, Yu S, Lainton JAH, Pertwee RG. Evidence for the presence of CB2-like cannabinoid receptors on peripheral nerve terminals. Eur. J. Pharmacol. 1997;339:53–61. doi: 10.1016/s0014-2999(97)01336-8. [DOI] [PubMed] [Google Scholar]
- (15).Gong JP, Onaivi ES, Ishiguro H, Liu QR, Tagliaferro PA, Brusco A, Uhl GR. Cannabinoid CB2 receptors: immunohistochemical localization in rat brain. Brain Res. 2006;1071:10–23. doi: 10.1016/j.brainres.2005.11.035. [DOI] [PubMed] [Google Scholar]
- (16).Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, Stella N, Makriyannis A, Piomelli D, Davison JS, Marnett LJ, Di Marzo V, Pittman QJ, Patel KD, Sharkey KA. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310:329–332. doi: 10.1126/science.1115740. [DOI] [PubMed] [Google Scholar]
- (17).Pike VW. Positron-emitting radioligands for studies in vivo: probes for human psychopharmacology. J. Psychopharmacology. 1993;7:139–158. doi: 10.1177/026988119300700202. [DOI] [PubMed] [Google Scholar]
- (18).Laruelle M, Slifstein M, Huang Y. Relationships between radiotracer properties and image quality in molecular imaging of the brain with positron emission tomography. Mol. Imaging Biol. 2003;5:363–375. doi: 10.1016/j.mibio.2003.09.009. [DOI] [PubMed] [Google Scholar]
- (19).Rinaldi-Carmona M, Barth F, Heaulme M, Shire D, Calandra B, Congy C, Martinez S, Maruani J, Neliat G, Caput D, Ferrara P, Soubrié P, Breliére J-C, Le Fur G. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994;350:240–244. doi: 10.1016/0014-5793(94)00773-x. [DOI] [PubMed] [Google Scholar]
- (20).Horti AG, Fan H, Kuwabara H, Hilton J, Ravert HT, Holt DP, Alexander M, Kumar A, Rahmim A, Scheffel U, Wong DF, Dannals RF. 11C-JHU75528: A radiotracer for PET imaging of CB1 cannabinoid receptors. J. Nucl. Med. 2006;47:1689–1696. [PubMed] [Google Scholar]
- (21).Fan H, Ravert HT, Holt DP, Dannals RF, Horti AG. Synthesis of 1- (2,4-dichlorophenyl)-4-cyano-5-(4-[11C]methoxyphenyl)-N-(piperidin-1-yl)-1H-pyrazole-3-carboxamide ([11C]JHU75528) and 1-(2-bromophenyl)-4-cyano-5-(4-[11C]methoxyphenyl)-N-(piperidin-1-yl)-1H-pyrazole-3-carboxamide ([11C]JHU75575) as potential radioligands for PET imaging of cerebral cannabinoid receptor. J. Label. Compd. Radiopharm. 2006;49:1021–1036. [Google Scholar]
- (22).Burns HD, Van Laere K, Sanabria-Bohorquez S, Hamill TG, Bormans G, Eng WS, Gibson R, Ryan C, Connolly B, Patel S, Krause S, Vanko A, Van Hecken A, Dupont P, De Lepeleire I, Rothenberg P, Stoch SA, Cote J, Hagmann WK, Jewell JP, Lin LS, Liu P, Goulet MT, Gottesdiener K, Wagner JA, de Hoon J, Mortelmans L, Fong TM, Hargreaves RJ. [18F]MK-9470, a positron emission tomography (PET) tracer for in vivo human PET brain imaging of the cannabinoid-1 receptor. Proc. Natl. Acad. Sci. U.S.A. 2007;104:9800–9805. doi: 10.1073/pnas.0703472104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Liu P, Lin LS, Hamill TG, Jewell JP, Lanza TJ, Gibson RE, Krause SM, Ryan C, Eng WS, Sanabria S, Tong XC, Wang JY, Levorse DA, Owens KA, Fong TM, Shen CP, Lao JL, Kumar S, Yin WJ, Payack JF, Springfield SA, Hargreaves R, Burns HD, Goulet MT, Hagmann WK. Discovery of N-{(1S,2S)-2-(3-cyanophenyl)-3-[4-(2-[18F]fluoroethoxy)phenyl]-1-methylpropyl}-2-methyl-2-[(5-methylpyridin-2-yl)oxy]propanamide, a cannabinoid-1 receptor positron emission tomography tracer suitable for clinical use. J. Med. Chem. 2007;50:3427–3430. doi: 10.1021/jm070131b. [DOI] [PubMed] [Google Scholar]
- (24).Donohue SR, Halldin C, Schou M, Hong J, Phebus LA, Chernet E, Hitchcock SA, Gardinier KM, Ruley KM, Krushinski JH, Schaus JM, Pike VW. Radiolabeling of a high potency cannabinoid subtype-1 receptor ligand, N-(4-fluoro-benzyl)-4-(3-(piperidin-1-yl)-indole-1-sulfonyl)benzamide (PipISB), with carbon-11 or fluorine-18. J. Label. Compd. Radiopharm. 2008;51:146–152. [Google Scholar]
- (25).Yasuno F, Brown AK, Zoghbi SS, Krushinski JH, Chernet E, Tauscher J, Schaus JM, Phebus LA, Chesterfield AK, Felder CC, Gladding RL, Hong J, Halldin C, Pike VW, Innis RB. The PET radioligand [11C]MePPEP binds reversibly and with high specific signal to cannabinoid CB1 receptors in nonhuman primate brain. Neuropsychopharmacology. 2008;33:259–269. doi: 10.1038/sj.npp.1301402. [DOI] [PubMed] [Google Scholar]
- (26).Hamill T, Burns H, Eng W, Ryan C, Krause S, Gibson R, Hargreaves R. An improved fluorine-18 labeled neurokinin-1 receptor ligand. Mol. Imaging Biol. 2002;4(Supp 1):S34. [Google Scholar]
- (27).Schou M, Halldin C, Sovago J, Pike VW, Hall H, Gulyas B, Mozley PD, Dobson D, Shchukin E, Innis RB, Farde L. PET evaluation of novel radiofluorinated reboxetine analogs as norepinephrine transporter probes in the monkey brain. Synapse. 2004;53:57–67. doi: 10.1002/syn.20031. [DOI] [PubMed] [Google Scholar]
- (28).Park BK, Kitteringham NR, O’Neill PM. Metabolism of fluorine-containing drugs. Ann. Rev. Pharmacol. Toxicol. 2001;41:443–470. doi: 10.1146/annurev.pharmtox.41.1.443. [DOI] [PubMed] [Google Scholar]
- (29).Waterhouse RN. Determination of lipophilicity and its use as a predictor of blood-brain barrier penetration of molecular imaging agents. Mol. Imaging Biol. 2003;5:376–389. doi: 10.1016/j.mibio.2003.09.014. [DOI] [PubMed] [Google Scholar]
- (30).Chernet E, Martin LJ, Li D, Need AB, Barth VN, Rash KS, Phebus LA. Use of LC/MS to assess brain tracer distribution in preclinical, in vivo receptor occupancy studies: dopamine D2, serotonin 2A and NK-1 receptors as examples. Life Sci. 2005;78:340–346. doi: 10.1016/j.lfs.2005.04.075. [DOI] [PubMed] [Google Scholar]
- (31).Andreichikov YS, Gein VL, Ivanenko OI, Maslivets AN. Synthesis and structure of 1,5-diaryltetrahydro-2,3-pyrrolediones. Zhurnal Organicheskoi Kimii. 1986;22:2208–2213. [Google Scholar]
- (32).Barth VN, Chernet E, Martin LJ, Need AB, Rash KS, Morin M, Phebus LA. Comparison of rat dopamine D2 receptor occupancy for a series of antipsychotic drugs measured using radiolabeled or nonlabeled raclopride tracer. Life Sci. 2006;78:3007–3012. doi: 10.1016/j.lfs.2005.11.031. [DOI] [PubMed] [Google Scholar]
- (33).Wilson AA, Garcia A, Jin L, Houle S. Radiotracer synthesis from [11C]-iodomethane: a remarkably simple captive solvent method. Nucl. Med. Biol. 2000;27:529–532. doi: 10.1016/s0969-8051(00)00132-3. [DOI] [PubMed] [Google Scholar]
- (34).Chin FT, Morse CL, Shetty HU, Pike VW. Automated radiosynthesis of [18F]SPA-RQ for imaging human brain NK1 receptors with PET. J. Label. Compd. Radiopharm. 2006;49:17–31. [Google Scholar]
- (35).Iwata R, Pascali C, Bogni A, Furumoto S, Terasaki K, Yanai K. [18F]Fluoromethyl triflate, a novel and reactive [18F]fluoromethylating agent: preparation and application to the on-column preparation of [18F]fluorocholine. Appl. Radiat. Isot. 2002;57:347–352. doi: 10.1016/s0969-8043(02)00123-9. [DOI] [PubMed] [Google Scholar]
- (36).Solin O, Eskola O, Hamill TG, Bergman J, Lehikoinen P, Gronroos T, Forsback S, Haaparanta M, Viljanen T, Ryan C, Gibson R, Kieczykowski G, Hietala J, Hargreaves R, Burns HD. Synthesis and characterization of a potent, selective, radiolabeled substance-P antagonist for NK1 receptor quantitation: ([18F]SPA-RQ) Mol. Imaging Biol. 2004;6:373–384. doi: 10.1016/j.mibio.2004.08.001. [DOI] [PubMed] [Google Scholar]
- (37).Bergman J, Eskola O, Lehikoinen P, Solin O. Automated synthesis and purification of [18F]bromofluoromethane at high specific radioactivity. Appl. Radiat. Isot. 2001;54:927–933. doi: 10.1016/s0969-8043(00)00358-4. [DOI] [PubMed] [Google Scholar]
- (38).Zhang MR, Tsuchiyama A, Haradahira T, Yoshida Y, Furutsuka K, Suzuki K. Development of an automated system for synthesizing 18F-labeled compounds using [18F]fluoroethyl bromide as a synthetic precursor. Appl. Radiat. Isot. 2002;57:335–342. doi: 10.1016/s0969-8043(02)00075-1. [DOI] [PubMed] [Google Scholar]
- (39).Iwata R, Furumoto S, Pascali C, Bogni A, Ishiwata K. Radiosynthesis of O-[11C]methyl-L-tyrosine and O-[18F]fluoromethyl-L-tyrosine as potential PET tracers for imaging amino acid transport. J. Label. Compd. Radiopharm. 2003;46:555–566. [Google Scholar]
- (40).Hahn RC, Tompkins J. Homogeneous Nucleophile Exchange. 2. Silver-free direct synthesis of primary alkyl sulfonates from alkyl-halides. J. Org. Chem. 1988;53:5783–5785. [Google Scholar]
- (41).DeLapp NW, McKinzie JH, Sawyer BD, Vandergriff A, Falcone J, McClure D, Felder CC. Determination of [35S]guanosine-5′-O-(3-thio)triphosphate binding mediated by cholinergic muscarinic receptors in membranes from Chinese hamster ovary cells and rat striatum using an anti-G protein scintillation proximity assay. J. Pharmacol. Exp. Ther. 1999;289:946–955. [PubMed] [Google Scholar]
- (42).Cheng Y, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.