

Abstract
A highly efficient total synthesis of (±)-Lycoricidine is described. The synthesis features the ready preparation of the lycoricidine skeleton by a Stille-IMDAF cycloaddition cascade. The resulting cycloadduct is then used for the stereocontrolled installation of the other functionality present in the C-ring of the target molecule.
Many of the lycorine-type Amaryllidaceae alkaloids display useful biological properties1 and as a consequence this family has captured the interest of a number of synthetic groups as targets for total synthesis.2 The history of the hydroxylated phenanthridones of the Amaryllidaceae group, their biological profiles and various syntheses have been reviewed on several occasions,3 most recently by Hudlicky and Rinner in 2005.4 Lycoricidine (1),5 the structurally related Narciclasine (2),6 as well as Pancratistatin (3)7 and 7-Deoxypancratistatin (4)8 are popular synthetic targets primarily because their heterocyclic framework provides a means to demonstrate the utility of new synthetic strategies.4 In addition, the narcissus alkaloids are available only in small quantities from natural sources9 and their use as therapeutic agents10 depends on their ready availability. The principle hurdles to their synthesis include the introduction of the aryl group and the stereocontrolled construction of the fused BC ring system.

A major strategy that is routinely employed for the synthesis of the lycorine group consists of constructing two fragments representing the A and C cycles which are then coupled together to form the B ring. This approach often involves formation of the 10a-10b carbon-carbon bond by either a palladium-based11 or photocyclization6d reaction, although other coupling methods have also been used.3,4 An alternate synthetic route would be the formation of the C-ring from a suitable precursor in which the 10a-10b bond of the target is already present.12,13 It is against this background that we report a short and efficient synthesis of (±)-Lycoricidine. Our approach derives from a general program underway in our laboratory that is designed to exploit the facile Diels-Alder reaction of amidofurans for the purposes of natural product synthesis.14 Our retrosynthetic analysis of (±)-Lycoricidine is shown in Scheme 1 and makes use of a tandem cascade sequence consisting of a Stille coupling15 followed by a spontaneous intramolecular [4+2]-cycloaddition of an amidofuran (IMDAF). The resulting cycloadduct 6 is then used for the stereocontrolled installation of the other functionality present in the C-ring of Lycoricidine. The carbomethoxy substituent is utilized as a critical control element not only to facilitate the [4+2]-cycloaddition but also to provide a handle for the introduction of the required π-bond and to set the stereochemistry at the C4a-ring juncture.
Scheme 1.
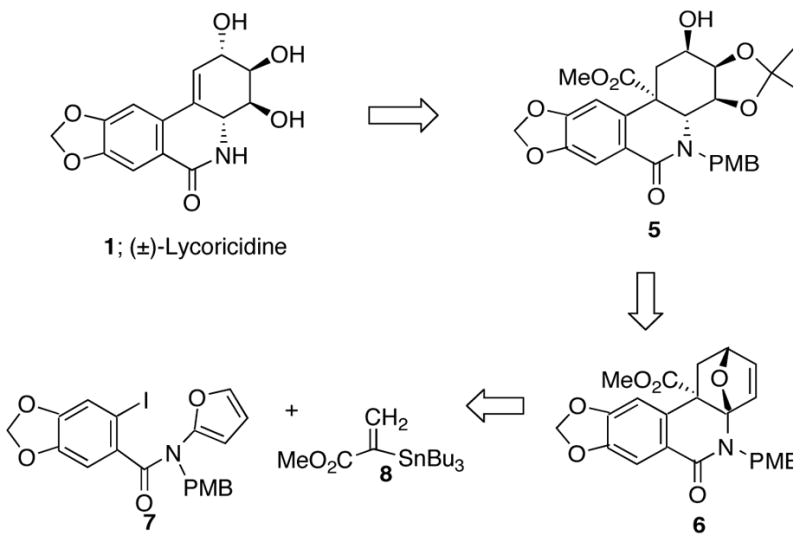
Our synthesis of amidofuran 7 began by coupling the known acid chloride 916 with the lithiated carbamate 10b derived by treating furanyl-2-ylcarbamic acid tert-butyl ester (10a) with n-BuLi. Removal of the t-Boc-protecting group from the resulting carbamate 11 with Mg(ClO4)2 afforded NH amide 12 in 75% yield and this was followed by reaction with NaH and p-methoxybenzyl chloride to give 7 in 83% yield.17 The methyl acrylate moiety was introduced by means of a Stille coupling15 using methyl 2-tri-n-butylstannylacrylate18 (Scheme 2). The optimum conditions for this reaction were eventually determined to be those described by Corey which utilize a combination of CuCl/Pd(0)/LiCl for the key coupling.19 The use of DMSO with rigorous exclusion of oxygen and moisture at 60 °C gave the best results. The expected cross-coupled amidofuran 13, however, was not isolated as it spontaneously underwent an intramolecular [4+2]-cycloaddition to furnish cycloadduct 6 in 82% overall yield for the two-step cascade. The increase in reactivity of 13 when compared to related furanyl carbamates20 (>150 °C) is due to both the placement of the carbonyl center within the dienophilic tether as well as the presence of the carbomethoxy group which lowers the LUMO energy of the π-bond thereby facilitating the cycloaddition. Dramatic effects on the rate of the Diels-Alder reaction were previously noted to occur when an amido group was used to anchor the diene and dienophile.21 Our ability to isolate oxabicyclic adduct 6 is presumably a result of the lower reaction temperature employed (i.e., 60 °C) as well as the presence of the amido carbonyl group, which diminishes the basicity of the nitrogen atom thereby retarding the ring cleavage/rearrangement reaction generally encountered with these systems.20 Moreover, the IMDAF cycloaddition proceeds by a transition state where the sidearm of the tethered vinyl group is oriented exo with respect to the oxygen bridge.22 As a consequence of this preferred orientation, the carbomethoxy group and oxy-bridge are disposed in an anti relationship in the resulting cycloadduct 6.
Scheme 2.
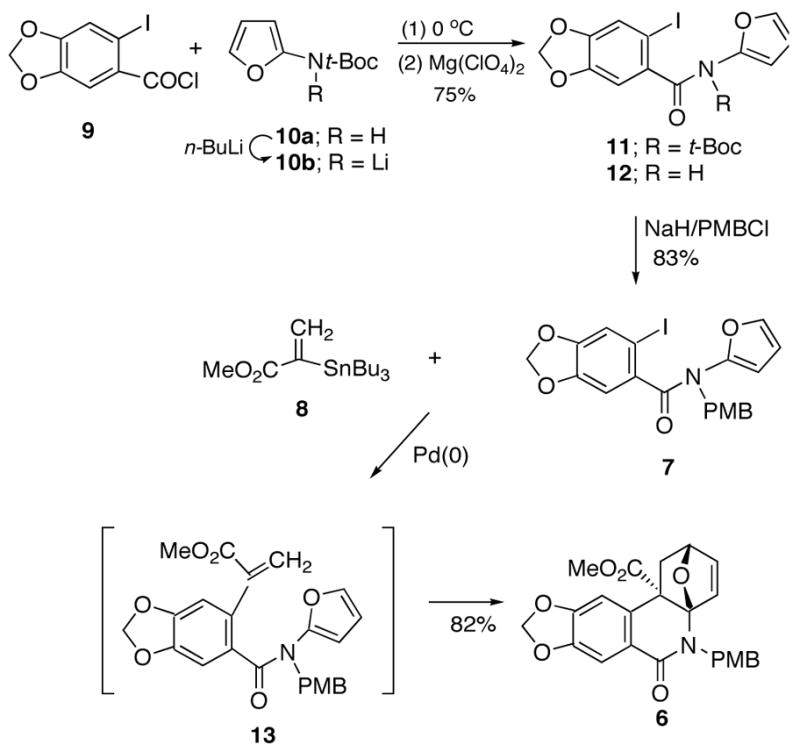
With the rapid construction of the Lycoricidine framework in hand, installation of the other functional groups present on the C-ring with the correct relative stereochemistry was investigated. To continue the synthesis, cycloadduct 6 was transformed to diol 14 by reaction with catalytic OsO4 in the presence of 4-methyl-morpholine-N -oxide. The dihydroxylation reaction occurred exclusively from the less hindered exo face producing 14 in 98% yield (Scheme 3). Having introduced the correct cis-stereochemistry of the hydroxyl groups at the C3,C4 positions, we then proceeded to set the stereochemistry at the C4a position, insert the remaining α-hydroxyl group at C2 and ultimately introduce the required π-bond. All of these operations were facilitated by making use of the available carbomethoxy group (vide infra). First, diol 14 was converted to the corresponding acetonide 15 in 80% yield by treatment with 2,2-dimethoxypropane and catalytic pyridinium p-toluenesulfonate. The uniquely functionalized oxabicyclic adduct 15 contains a “masked” N-acyliminium ion which can be released by treatment with a Lewis acid such as TMSOTf. When the resulting ring opened iminium ion was treated with Zn(BH4)2, 23 alcohol 5 was obtained in 74% yield.
Scheme 3.
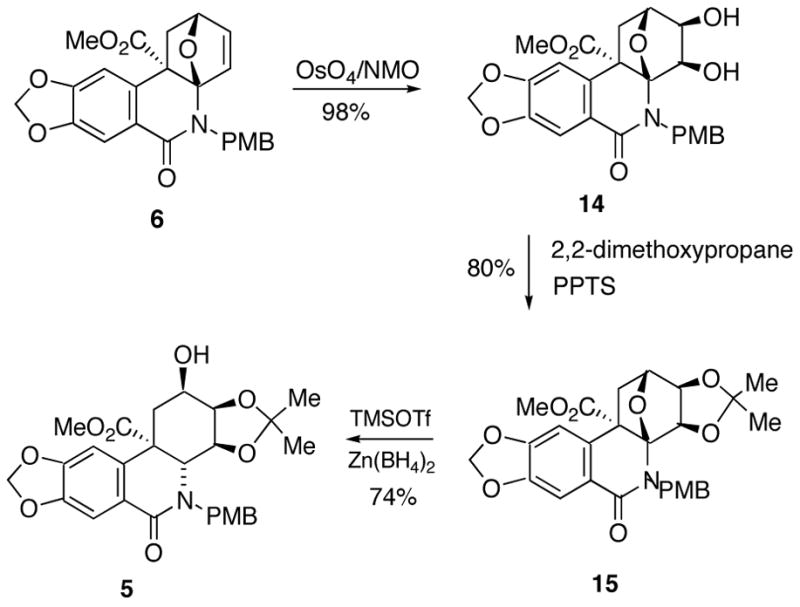
What was required for the end game leading to Lycoricidine (1) was to invert the stereochemistry of the C2-hydroxyl group, remove the carbomethoxy moiety and generate a double bond between the C1-C10b position of the C-ring. To this end, compound 5 was treated with NaH followed by the addition of CS2 and MeI to give the corresponding xanthate ester which, upon heating at reflux in 1,2-dichlorobenzene for 12 h, afforded the expected olefin derived from a Chugaev elimination24 in 94% yield (Scheme 4). Since the β-face of the π-bond of 16 was blocked by the bulky acetonide, a dihydroxylation reaction was expected to take place from the less hindered α-face, syn to the carbomethoxy group, thereby setting the correct stereochemistry of the C2-hydroxyl group. Indeed, when 16 was treated with OsO4/NMO, the desired diol 17 was formed as a transient species but underwent spontaneous cyclization with the adjacent carbomethoxy group to deliver γ-lactone 18. A subsequent mesylation reaction afforded mesylate 19 in 76% yield for the two-step sequence starting from 16. The γ-lactonization of 17 to 18 permits the selective activation of the C1-hydroxyl group. The PMB group was removed by the reaction of 19 with PdCl2 in the presence of acetic acid25 to furnish the deprotected amide. Gratifyingly, the reaction of this amide with LiOH in aqueous THF induced a novel tandem hydrolysis/decarboxylation/elimination sequence26 to furnish allylic alcohol 20. Deprotection of the acetonide with TFA afforded (±)-Lycoricidine in 90% yield.
Scheme 4.
In summary, we have described here a novel and highly efficient synthesis of (±)-Lycoricidine which was achieved in 14 steps with a 12.6% overall yield. The synthesis illustrates (a) the use of a one-pot Stille-IMDAF cycloaddition cascade to construct the ring skeleton, (b) a stereocontrolled dihydroxylation and N-acyliminium ion reduction to set the correct stereochemistry at carbon atoms C3, C4 and C4a, and (c) the novel introduction of the C1-C10b π-bond by a one-pot hydrolysis-decarboxylation-elimination sequence. The application of this approach to other members of the lycorine family of alkaloids is currently under investigation, the results of which will be disclosed in due course.
Supporting Information Available
Spectroscopic data and experimental details for the preparation of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We appreciate the financial support provided the The National Institutes of Health (GM059384) and The National Science Foundation (CHE-0450779).
References
- 1.(a) Hartwell JL. Lloydia. 1967;30:379. [Google Scholar]; (b) Fitzgerald DB, Hartwell JL, Leiter J. J Nat Cancer Inst. 1958;20:763. [PubMed] [Google Scholar]; (c) Pettit GR, Gaddamidi V, Herald DL, Singh SB, Cragg GM, Schmidt JM, Boettner FE, Williams M, Sagawa Y. J Nat Prod. 1986;49:995. doi: 10.1021/np50048a005. [DOI] [PubMed] [Google Scholar]; (d) Ceriotti G. Nature. 1967;213:595. doi: 10.1038/213595a0. [DOI] [PubMed] [Google Scholar]; (e) Okamoto T, Torii Y, Isogai Y. Chem Pharm Bull. 1968;16:1860. doi: 10.1248/cpb.16.1860. [DOI] [PubMed] [Google Scholar]
- 2.For some leading references, see: Hudlicky T. J Heterocycl Chem. 2000;37:535.Martin SF. In: The Amaryllidaceae Alkaloids In The Alkaloids. Chapter 3. Brossi A, editor. Vol. 30. Academic Press; New York: 1987. pp. 251–376.
- 3.(a) Polt R. In: Organic Synthesis: Theory and Applications. Hudlicky T, editor. Vol. 3. JAI Press; Greenwich, CT: 1997. p. 109. [Google Scholar]; (b) Hoshino O. In: The Alkaloids. Cordell GA, editor. Vol. 51. Academic Press; New York: 1998. p. 323. [Google Scholar]; (c) Jin Z. Nat Prod Rep. 2003;20:606. doi: 10.1039/b304144c. [DOI] [PubMed] [Google Scholar]
- 4.Rinner U, Hudlicky T. Synlett. 2005:365. [Google Scholar]
- 5.For syntheses of Lycoricidine, see: Ohta S, Kimoto S. Chem Pharm Bull. 1976;24:2977.Paulsen H, Stubbe M. Tetrahedron Lett. 1982;23:3171.Paulsen H, Stubbe M, Liebigs Ann Chem. 1983:535.Chida N, Ohtsuka M, Ogawa S. Tetrahedron Lett. 1991;32:4525.Hudlicky T, Olivo HF. J Am Chem Soc. 1992;114:9694.Chida N, Ohtsuka M, Ogawa S. J Org Chem. 1993;58:4441.Martin SF, Tso HH. Heterocycles. 1993;35:85.Hudlicky T, Olivo HF, McKibben B. J Am Chem Soc. 1994;116:5108.Keck GE, Wager TT. J Org Chem. 1996;61:8366.Elango S, Yan TH. Tetrahedron. 2002;58:7375.
- 6.For syntheses of Narciclasine, see: Mondon A, Krohn K. Tetrahedron Lett. 1972;13:2085.Mondon A, Krohn K. Chem Ber. 1975;108:445.Krohn K, Mondon A. Chem Ber. 1976;109:855.Rigby JH, Mateo ME. J Am Chem Soc. 1997;119:12655.Rigby JH, Maharoof USM, Mateo ME. J Am Chem Soc. 2000;122:6624.Gonzalez D, Martinot T, Hudlicky T. Tetrahedron Lett. 1999;40:3077.
- 7.For syntheses of Pancratistatin, see: Danishefsky S, Lee JY. J Am Chem Soc. 1989;111:4829.Tian X, Hudlicky T, Königsberger K. J Am Chem Soc. 1995;117:3643.Trost BM, Pulley SR. J Am Chem Soc. 1995;117:10143.Keck GE, McHardy SF, Murry JA. J Am Chem Soc. 1995;117:7289.Hudlicky T, Tian XR, Königsberger K, Mauray R, Rouden J, Fan B. J Am Chem Soc. 1996;118:10752.Chida N, Jitsuoka M, Yamamoto Y, Ohtsuka M, Ogawa S. Heterocycles. 1996;43:1385.Doyle TJ, Hendrix M, VanDerveer D, Javanmard S, Haseltine J. Tetrahedron. 1997;53:11153.Magnus P, Sebhat IK. Tetrahedron. 1998;54:15509.Magnus P, Sebhat IK. J Am Chem Soc. 1998;120:5341.Pettit GR, Melody N, Herald DL. J Org Chem. 2001;66:2583. doi: 10.1021/jo000710n.Kim S, Ko H, Kim E, Kim D. Org Lett. 2002;4:1343. doi: 10.1021/ol0256419.Ko HJ, Park JE, Kim S. J Org Chem. 2004;69:112. doi: 10.1021/jo035371n.
- 8.(a) Akgun H, Hudlicky T. Tetrahedron Lett. 1999;40:3081. [Google Scholar]; (b) Rinner U, Siengalewicz P, Hudlicky T. Org Lett. 2002;4:115. doi: 10.1021/ol0169877. [DOI] [PubMed] [Google Scholar]; (c) Tian X, Maurya R, Königsberger K. Synlett. 1995:1125. [Google Scholar]; (d) Keck GE, Wager TT, McHardy SF. J Org Chem. 1998;63:9164. [Google Scholar]; (e) Keck GE, McHardy SF, Murry JA. J Am Chem Soc. 1995;117:7289. [Google Scholar]; (f) Acena JL, Arjona O, Leon ML, Plumet J. Org Lett. 2000;2:3683. doi: 10.1021/ol000268v. [DOI] [PubMed] [Google Scholar]
- 9.Pettit GR, Backhaus RA, Boettner FE. J Nat Prod. 1995;58:37. doi: 10.1021/np50115a004. [DOI] [PubMed] [Google Scholar]
- 10.(a) Pettit GR, Freeman S, Simpson MJ, Thompson MA, Boyd MR, Williams GR, Pettit GR, III, Doubek DL. Anti-Cancer Drug Des. 1995;10:243. [PubMed] [Google Scholar]; (b) Pettit GR, Orr S, Ducki S. Anti-Cancer Drug Des. 2000;15:389. [PubMed] [Google Scholar]
- 11.Grubb LM, Dowdy AL, Blanchette HS, Friestad GK, Branchaud BP. Tetrahedron Lett. 1999;40:2691. [Google Scholar]
- 12.Keck GE, Wager TT, Rodriquez JFD. J Am Chem Soc. 1999;121:5176. [Google Scholar]
- 13.Khaldi M, Chre'tien F, Chapleur Y. Tetrahedron Lett. 1995;36:3003. [Google Scholar]
- 14.(a) Wang Q, Padwa A. Org Lett. 2004;6:2189. doi: 10.1021/ol049348f. [DOI] [PubMed] [Google Scholar]; (b) Padwa A, Ginn JD. J Org Chem. 2005;70:5197. doi: 10.1021/jo050515e. [DOI] [PubMed] [Google Scholar]; (c) Padwa A, Bur SK, Zhang H. J Org Chem. 2005;70:6833. doi: 10.1021/jo0508797. [DOI] [PubMed] [Google Scholar]
- 15.Farina V, Krishnamurphy V, Scott WJ. Org React. 1997;50:1. [Google Scholar]
- 16.McIntosh MC, Weinreb SM. J Org Chem. 1993;58:4823. [Google Scholar]
- 17.Replacement of the t-Boc group with the PMB functionality was necessary for the subsequent Stille reaction.
- 18.(a) Zhang HX, Guiibe F, Balavoine G. J Org Chem. 1990;55:1857. [Google Scholar]; (b) Miyake H, Yamamura K. Chem Lett. 1989:981. [Google Scholar]; (c) Cochran JC, Bronk BS, Terrence KM, Philips HK. Tetrahedron Lett. 1990;31:6621. [Google Scholar]
- 19.Han X, Stoltz BM, Corey EJ. J Am Chem Soc. 1999;121:7600. [Google Scholar]
- 20.(a) Padwa A, Brodney MA, Dimitroff M. J Org Chem. 1998;63:5304. doi: 10.1021/jo010020z. [DOI] [PubMed] [Google Scholar]; (b) Bur SK, Lynch SM, Padwa A. Org Lett. 2002;4:473. doi: 10.1021/ol016804g. [DOI] [PubMed] [Google Scholar]; (c) Ginn JD, Padwa A. Org Lett. 2002;4:1515. doi: 10.1021/ol025746b. [DOI] [PubMed] [Google Scholar]
- 21.(a) Oppolzer W, Fröstl W. Helv Chem Acta. 1975;58:590. doi: 10.1002/hlca.19750580232. [DOI] [PubMed] [Google Scholar]; (b) White JD, Demnitz FWJ, Oda H, Hassler C, Snyder JP. Org Lett. 2000;2:3313. doi: 10.1021/ol000200f. [DOI] [PubMed] [Google Scholar]; (c) Padwa A, Ginn JD, Bur SK, Eidell CK, Lynch SM. J Org Chem. 2002;67:3412. doi: 10.1021/jo0111816. [DOI] [PubMed] [Google Scholar]; (d) Tantillo DJ, Houk KN, Jung ME. J Org Chem. 2001;66:1938. doi: 10.1021/jo001172h. [DOI] [PubMed] [Google Scholar]
- 22.Padwa A, Brodney MA, Satake K, Straub CS. J Org Chem. 1999;64:4617. doi: 10.1021/jo982061+. [DOI] [PubMed] [Google Scholar]
- 23.Gensler WJ, Johnson F, Sloan ADB. J Am Chem Soc. 1960;82:6074. [Google Scholar]
- 24.Nace HR. Org React. 1962;12:57. [Google Scholar]
- 25.Keck GE, Boden E, Sonnewald U. Tetrahedron Lett. 1981;22:2615. [Google Scholar]
- 26.For a related fragmentation reaction, see: Khim S, Dai M, Zhang X, Chen L, Pettus L, Thakkar K, Schultz AG. J Org Chem. 2004;69:7728. doi: 10.1021/jo0490853.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Spectroscopic data and experimental details for the preparation of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.