

Abstract
Misfolded or unfolded proteins are often refolded with the help of chaperones or degraded by the 26S proteasome. An alternative fate of these proteins is the aggresome pathway. The microtubule-organizing center (MTOC) transports unfolded proteins to lysosomes and are degraded through autophagy. Histone deacetylase 6 (HDAC6) deacetylates α-tubulin, which is thought to be a component of the MTOC. Recently, two small molecule inhibitors of the aggresome pathway and HDAC6 have been described. One inhibitor, tubacin, prevents deacetylation of α-tubulin and produces accumulation of polyubiquitinated proteins and apoptosis. Tubacin acts synergistically with the proteasome inhibitor, bortezomib, to induce cytotoxicity in one type of hematologic malignancy, multiple myeloma. The other, LBH589, is a pan HDAC inhibitor and hydroxamic acid derivative that induces apoptosis of multiple myeloma cells resistant to conventional therapies. In this review, we summarize recent reports on targeting the aggresome pathway and HDAC6 in hematologic malignancies.
Keywords: aggresomes, protein degradation, multiple myeloma, apoptosis, HDAC6, α-tubulin
Protein folding
Proteins must overcome several obstacles on their way to becoming functional molecules. Small proteins fold through a sequence of intermediates. Partially folded proteins expose hydrophobic domains that lead to inappropriate associations and protein aggregation. Aggregation of proteins is toxic to cells due to high concentrations of macromolecules causing a significant increase in the association constants of unfolded polypeptides over those in dilute solution. The effects of protein aggregation are accentuated by the fact that stable folding of a domain cannot occur until the entire protein is synthesized. This is particularly important during synthesis of identical nascent polypeptides on polysomes, where numerous polypeptides expose the same aggregation-prone domains leading to increased aggregation [1].
The fate of a protein is either correct folding or degradation of misfolded proteins by the proteasome or autophagy [1]. Whether a polypeptide folds correctly or whether it aggregates is dependent on particular mutations, modification, errors during translation, or unequal synthesis of subunits [1–4]. Misfolding can result from alterations in pH, temperature, ionic strength, and redox environment. A certain level of protein misfolding cannot be avoided. Therefore, cells have acquired quality control mechanisms to eliminate misfolded proteins and aggregation [5, 6].
Chaperones and ubiquitin-proteasome system
Molecular chaperones assist with folding of newly synthesized proteins and refolding of proteins damaged by stress and cellular injury. Chaperones bind to and stabilize exposed hydrophobic residues through ATP-dependent interactions, thereby allowing the protein to achieve proper folding [7]. Chaperones do not appear to catalyze folding, but rather, they prevent inter- and intra-molecular interactions between partially folded or misfolded polypeptides. This is an evolutionarily conserved mechanism from bacteria to eukaryotes to maintain proper folding after protein synthesis [1]. Second, misfolded proteins are targeted for degradation by the ubiquitin-proteasome system (UPS). The proteasome is a multisubunit complex found in the cytosol and nucleus that degrades cytosolic, nuclear, secretory, and transmembrane proteins into smaller peptides [8–10]. Misfolded secretory and transmembrane proteins are retained in the lumen or membrane of the ER, then retrotranslocated back to the cytosol and delivered to the proteasome. When correct folding is difficult or impossible and degradation is not performed rapidly, proteins interact with other unfolded or partially folded proteins, leading to the formation of aggregates [6]. Cells then destroy protein aggregates through the aggresome pathway resulting in autophagic degradation of the inclusion bodies.
The Unfolded Protein Response
Another important pathway in cells to regulate misfolded proteins is the Unfolded Protein Response (UPR). Proteins synthesized in the endoplasmic reticulum (ER) are properly folded with the help of ER chaperones. Misfolded proteins are disposed of by ER associated protein degradation (ERAD). When the level of misfolded proteins exceeds the folding capacity of the ER, cells activate a feedback mechanism known as the ER stress response [11]. Expression of ER chaperones and ER associated proteins are induced to decrease protein synthesis and hence, the burden on the ER. There are four classes of agents that induce ER stress: reducing agents, hypoxia and inhibitors of glycosylation and calcium metabolism. Finally, the ER stress response can result in activation of apoptosis [12].
Three transmembrane proteins regulate the mammalian ER stress response. PERK is a transmembrane kinase; ATF6 is a transmembrane transcription factor; and IRE1 is a transmembrane RNase. These three proteins integrate the stress response and are critical for cell survival. ER stress-induced apoptotic pathways act through pro-apoptotic and anti-apoptotic proteins, such as bcl-2, p53, and c-abl. Stress activated protein kinase and Jun kinase are also activated [12].
The UPR appears to also be involved with tumorigenesis [13]. As tumors increase in size, cells are exposed to environmental stressors, such as hypoxia, limited nutrients, and acidosis. Chemotherapy also activates the UPR resulting following exposure to DNA-crosslinking agents, e.g. cisplatin, however, activation of the stress response could confer resistance to drugs, e.g. topoisomerase II inhibitors [13]. Thus, the role of the ERAD and ER stress response is complex due to the heterogeneity of tumor response.
The aggresome pathway
Studies have demonstrated a proteasome-independent pathway that eliminates misfolded proteins, known as the aggresome pathway, resulting in autophagic degradation of inclusion bodies (Figure 1) [1, 14–19]. The initial process appears to occur as nascent polypeptide chains are coming off the polysome. If nascent peptides do not fold correctly, they will co-aggregate to form aggresomal particles. In cells, these particles are equal in size, supporting the idea that a fixed number of proteins aggregate to form a single particle [1]. After their formation, the particles are transported towards the microtubule (MT) organizing center or MTOC, where they are sequestered into a single large cellular garbage bin-like structure known as the aggresome [1]. Movement of the aggresome is an active process and requires intact microtubules and association with motor dynein. In cells treated with the microtubule depolarizing agent, nocodazole, the pre-aggresomal inclusion bodies remains distributed throughout the cytosol. Electron microscopy examination revealed that these particles consist of multiple loosely associated particles.
Figure 1.
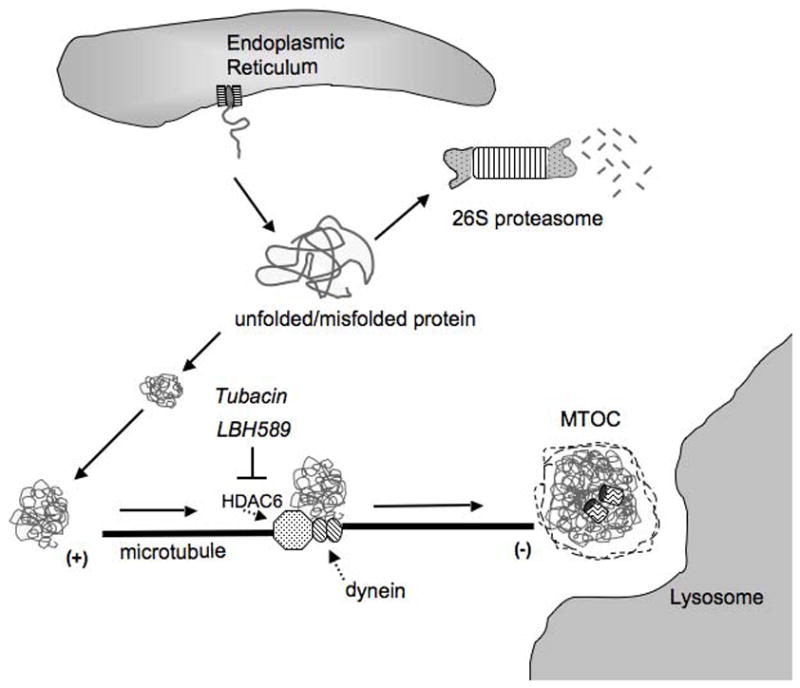
The aggresome pathway prevents accumulation of misfolded proteins. Unfolded or misfolded protein can originate from translating polysomes, from proteins retrotranslocated from the ER to the cytosol, or from proteins damaged by stress. If such proteins fail to fold correctly and are not degraded by the proteasome, they can form aggregates in cells. These aggregates are transported by the microtubule and require the dynein/dynactin motor complex. HDAC6 (dotted arrow) deacetylates α-tubulin and associates with dynein (dotted arrow) to facilitate transport of aggresomes through the cytosol for degradation. Tubacin and LBH589 are small molecule inhibitors that target HDAC6 or pan HDACs, respectively, resulting in increased acetylation of α-tubulin, accumulation of polyubiquitinated proteins, and apoptosis [25, 31].
Aggresomes are recruited to the degradation machinery
Aggresomes are not only static compartments that store misfolded proteins, but they also recruit cytosolic components such as chaperones, E3 ubiquitin ligases, and proteasomes to facilitate clearance of aggregated proteins [1]. Several chaperones have been identified, e.g. HSC70, HSP40, HSP70, and some are targeted for cancer therapy. Misfolded proteins associate with ER chaperones first, followed by association with cytosolic chaperones [1]. Similar to chaperones, proteasomes associate with aggresomes. Proteasomes associate with aggresomes relatively late, after small aggresomal particles are delivered to the MTOC. Autophagy is a pathway by which large structures such as mitochondria and peroxisomes are degraded in cells [1].
Autophagy is one of the cellular stress responses. How do cells recognize aggregated proteins resulting in the formation of aggresomes? Previous reports suggest that the sequestration of aggregated proteins into single aggresome may be induced by cellular signaling mechanisms. Activation of the Mitogen Activated Protein Kinase Kinase (MEK1) increases formation of aggresomes [20]. MEKK1 appears to act at a relatively late stage of aggresome formation and affects sequestration of particles into peri-centriolar aggresomes. The activity of MEKK1 is required; suggesting that phosphorylation of downstream substrates is critical for aggresome formation. Previous studies demonstrated that MEKK1 regulated pathways involve Jun Kinase (JNK), extracellular signal regulated kinase (ERK) and p38. Interestingly, the effect of MEKK1 on aggresome formation does not seem to involve any of these kinases, suggesting that there are yet unidentified signaling pathways activated by cellular stress resulting from aggresomes.
Histone Deacetylase 6
There are 18 histone deacetylases (HDACs) reported in humans. They are not redundant in function and are classified into three categories based on their homology to yeast proteins. Although histones are important targets of HDACs, more than 50 nonhistone targets have been identified. These nonhistone proteins play roles in proliferation, cell migration, and death [21] Histone Deacetylase 6 (HDAC6) is a class II HDAC protein deacetylates α-tubulin and increases cell motility. HDAC6 is localized in the cytoplasm and has two catalytic domains with deacetylase activity. The C-terminal domain is able to deacetylate α-tubulin both in vitro and in vivo, and this activity is reversibly inhibited by trichosatin A (TSA) [22, 23]. HDAC6 plays an essential role in aggresomal protein degradation because it can bind to both polyubiquitinated proteins and dynein proteins, thereby recruiting protein cargo to dynein motors to transport misfolded proteins to aggresomes [24]. HDAC6 appears to constitutively binds both polyubiquitinated proteins and dynein [25]. The proteasome inhibitor bortezomib has been used to treat various malignancies, including multiple myeloma. Bortezomib is a dipeptidyl boronic acid that binds to the serine residue of the beta subunit that confers the proteasomal chymotryptic proteolytic activity [26]. Published work suggests that targeting both proteasome-dependent pathways with bortezomib and the aggresome pathway in tumor cells induces greater accumulation of polyubiquitinated proteins resulting in increased cell stress and apoptosis [25].
Inhibitors of HDAC6
Inhibitors of HDAC6 have been identified recently. Stuart Schreiber’s laboratory at MIT/Broad Institute isolated a small molecule inhibitor of HDAC6 in a chemical genetic screen of 7,392 small molecules and cell-based assay [27, 28]. This inhibitor, known as tubacin, inhibits α-tubulin deacetylation in mammalian cells. Unlike trichostatin A (TSA), which is a broad spectrum HDAC inhibitor, tubacin is specific for the tubulin deacetylase activity of HDAC6. These compounds inhibit the HDAC6 deacetylase activity by chelating a Zn++ cation and appears to alter the formation of complexes of HDAC6 with other intracellular proteins, such as HSP90, Dia2, and protein phosphatase-1 [22]. Tubacin does not affect global histone deacetylation, gene-expression profiling, or cell cycle progression [27, 28]. Recent evidence suggests that tubacin does not affect microtubule stability but rather, cell motility in lymphocytes [28]. More selective inhibitors of HDAC6 has been identified [29].
The effects of tubacin has been studied in hematologic malignancies, such as multiple myeloma and leukemia cell lines [25]. Overexpression of HDAC6 in primary lymphocytes and T cell lines increase cell migration in response to cytokines. Knockdown of HDAC6 in T cells decreased chemotactic mobility independent of its enzymatic activity [22]. Furthermore, treatment of multiple myeloma cells with tubacin resulted in decreased cell growth at an IC50 of 2–5μM [25]. In terms of toxicity to normal bone marrow and blood cells, treatment of peripheral blood mononuclear cells (PBMC) and bone marrow progenitor cells (BMPC) with tubacin (5μM) for 12 hours showed that constitutive expression of HDAC6 is higher in BMPCs than PBMCs. Furthermore, acetylation of α-tubulin was markedly enhanced by tubacin in BMPCs but not in PBMCs [25].
Tubacin in combination with the proteasome inhibitor bortezomib resulted in increased α-tubulin acetylation and accumulation of polyubiquitinated proteins in multiple myeloma cells [25]. Peak acetylation occurred after 12 hours. Multiple myeloma cells undergo apoptosis following tubacin treatment through a caspase-8 dependent pathway at an IC50 5–20μM at 72 hours. Poly-(ADP-ribose) polymerase (PARP) cleavage was also noted after treating multiple myeloma cells with tubacin and bortezomib. Tubacin was also found to inhibit interaction of HDAC6 with dynein and augmented activation of c-Jun NH2-terminal kinase and caspase−3, −8 and −9. Both bortezomib and tubacin together induced synergistic antitumor activity in multiple myeloma cells and primary bone marrow plasma cells [25]. Published data therefore provide support for combined therapy in clinical trials for patients with multiple myeloma.
Another interesting observation was that tubacin combined with bortezomib inhibited paracrine stimulation of multiple myeloma cells. Tubacin enhanced bortezomib-induced inhibition of proliferation of adherent multiple myeloma cells. Combined treatment of tubacin with bortezomib induced synergistic selective antitumor activity against multiple myeloma cells in the bone marrow milieu, thereby overcoming cell adhesion-induced resistance to conventional therapies [25]. These results provide further rationale to treat patients with multiple myeloma that is resistant to bortezomib.
Another inhibitor of HDAC6 has been recently described. LBH589 is a novel hydroxamic acid derivative that induces apoptosis at low nanomolar concentrations in multiple myeloma cells resistant to other therapies. Similar to tubacin, LBH589, induced apoptosis through a caspase and PARP-dependent manner [30]. Synergistic cytotoxicity was observed with LBH589 in combination with bortezomib against multiple myeloma cells that were sensitive and resistant to dexamethasone. This inhibitor also induced α-tubulin hyperacetylation. Interestingly, aggresome size was smaller in multiple myeloma cells treated with LBH589 and bortezomib. These results further support the use of LBH589 with bortezomib in resistant multiple myeloma [31].
Conclusions
Studies on the aggresome pathway and specific inhibitors in multiple myeloma suggest the possibility that HDAC6 is a rational target for other hematologic malignancies, such as acute lymphoblastic leukemia (ALL). Since tubacin has been demonstrated to act synergistically with other agents in multiple myeloma cells to induced apoptosis, it is possible that tubacin may have greater effects in combination with bortezomib or chemotherapy in ALL cells. Further experiments are necessary to elucidate the pathways that regulate HDAC6-induced apoptosis in malignant hematopoietic cells.
Acknowledgments
T.S.W. is funded by a grant from the American Academy of Pediatrics. A.R.G. is supported by the Department of Defense (W81XWH-06-1-0192) and the MEC/Fulbright fellowship (EX2005-0517). T.L. is funded by the NIH postdoctoral fellowship (T32HL086345). A.K.I. is supported by the NIH postdoctoral fellowship (T32CA9056). K.M.S. is supported by the National Institutes of Health grants HL75826 and HL83077, American Cancer Society grant RSG-99-081-01-LIB, Department of Defense (CM050077), and the Leukemia and Lymphoma Society Translational Research Grant (6019-07). K.M.S. is a Scholar of the Leukemia and Lymphoma Society.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Garcia-Mata R, Gao YS, Sztul E. Hassles with taking out the garbage: aggravating aggresomes. Traffic. 2002;3:388–96. doi: 10.1034/j.1600-0854.2002.30602.x. [DOI] [PubMed] [Google Scholar]
- 2.Bonifacino JS, Suzuki CK, Lippincott-Schwartz J, Weissman AM, Klausner RD. Pre-Golgi degradation of newly synthesized T-cell antigen receptor chains: intrinsic sensitivity and the role of subunit assembly. J Cell Biol. 1989;109:73–83. doi: 10.1083/jcb.109.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hurle MR, Helms LR, Li L, Chan W, Wetzel R. A role for destabilizing amino acid replacements in light-chain amyloidosis. Proc Natl Acad Sci U S A. 1994;91:5446–50. doi: 10.1073/pnas.91.12.5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wetzel R. Mutations and off-pathway aggregation of proteins. Trends Biotechnol. 1994;12:193–8. doi: 10.1016/0167-7799(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 5.Gottesman S, Wickner S, Maurizi MR. Protein quality control: triage by chaperones and proteases. Genes Dev. 1997;11:815–23. doi: 10.1101/gad.11.7.815. [DOI] [PubMed] [Google Scholar]
- 6.Wickner S, Maurizi MR, Gottesman S. Posttranslational quality control: folding, refolding, and degrading proteins. Science. 1999;286:1888–93. doi: 10.1126/science.286.5446.1888. [DOI] [PubMed] [Google Scholar]
- 7.Netzer WJ, Hartl FU. Protein folding in the cytosol: chaperonin-dependent and -independent mechanisms. Trends Biochem Sci. 1998;23:68–73. doi: 10.1016/s0968-0004(97)01171-7. [DOI] [PubMed] [Google Scholar]
- 8.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–79. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 9.Hirsch C, Ploegh HL. Intracellular targeting of the proteasome. Trends Cell Biol. 2000;10:268–72. doi: 10.1016/s0962-8924(00)01768-2. [DOI] [PubMed] [Google Scholar]
- 10.Verma R, Deshaies RJ. Assaying degradation and deubiquitination of a ubiquitinated substrate by purified 26S proteasomes. Methods Enzymol. 2005;398:391–9. doi: 10.1016/S0076-6879(05)98032-4. [DOI] [PubMed] [Google Scholar]
- 11.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 12.Yoshida H. ER stress and diseases. Febs J. 2007;274:630–58. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
- 13.Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: friend or foe? Nat Rev Cancer. 2004;4:966–77. doi: 10.1038/nrc1505. [DOI] [PubMed] [Google Scholar]
- 14.Bennett EJ, Bence NF, Jayakumar R, Kopito RR. Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol Cell. 2005;17:351–65. doi: 10.1016/j.molcel.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 15.Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–30. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 16.Heath CM, Windsor M, Wileman T. Aggresomes resemble sites specialized for virus assembly. J Cell Biol. 2001;153:449–55. doi: 10.1083/jcb.153.3.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnston JA, Illing ME, Kopito RR. Cytoplasmic dynein/dynactin mediates the assembly of aggresomes. Cell Motil Cytoskeleton. 2002;53:26–38. doi: 10.1002/cm.10057. [DOI] [PubMed] [Google Scholar]
- 18.Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–98. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kopito RR, Sitia R. Aggresomes and Russell bodies. Symptoms of cellular indigestion? EMBO Rep. 2000;1:225–31. doi: 10.1093/embo-reports/kvd052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meriin AB, Mabuchi K, Gabai VL, Yaglom JA, Kazantsev A, Sherman MY. Intracellular aggregation of polypeptides with expanded polyglutamine domain is stimulated by stress-activated kinase MEKK1. J Cell Biol. 2001;153:851–64. doi: 10.1083/jcb.153.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 2007;5:981–9. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- 22.Cabrero JR, Serrador JM, Barreiro O, Mittelbrunn M, Naranjo-Suarez S, Martin-Cofreces N, Vicente-Manzanares M, Mazitschek R, Bradner JE, Avila J, Valenzuela-Fernandez A, Sanchez-Madrid F. Lymphocyte chemotaxis is regulated by histone deacetylase 6, independently of its deacetylase activity. Mol Biol Cell. 2006;17:3435–45. doi: 10.1091/mbc.E06-01-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–8. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 24.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–38. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 25.Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, Schreiber SL, Anderson KC. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci U S A. 2005;102:8567–72. doi: 10.1073/pnas.0503221102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell. 2004;5:417–21. doi: 10.1016/s1535-6108(04)00120-5. [DOI] [PubMed] [Google Scholar]
- 27.Haggarty SJ, Koeller KM, Wong JC, Butcher RA, Schreiber SL. Multidimensional chemical genetic analysis of diversity-oriented synthesis-derived deacetylase inhibitors using cell-based assays. Chem Biol. 2003;10:383–96. doi: 10.1016/s1074-5521(03)00095-4. [DOI] [PubMed] [Google Scholar]
- 28.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A. 2003;100:4389–94. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Itoh Y, Suzuki T, Kouketsu A, Suzuki N, Maeda S, Yoshida M, Nakagawa H, Miyata N. Design, Synthesis, Structure-Selectivity Relationship, and Effect on Human Cancer Cells of a Novel Series of Histone Deacetylase 6-Selective Inhibitors. J Med Chem. 2007 doi: 10.1021/jm7009217. [DOI] [PubMed] [Google Scholar]
- 30.Catley L, Weisberg E, Tai YT, Atadja P, Remiszewski S, Hideshima T, Mitsiades N, Shringarpure R, LeBlanc R, Chauhan D, Munshi NC, Schlossman R, Richardson P, Griffin J, Anderson KC. NVP-LAQ824 is a potent novel histone deacetylase inhibitor with significant activity against multiple myeloma. Blood. 2003;102:2615–22. doi: 10.1182/blood-2003-01-0233. [DOI] [PubMed] [Google Scholar]
- 31.Catley L, Weisberg E, Kiziltepe T, Tai YT, Hideshima T, Neri P, Tassone P, Atadja P, Chauhan D, Munshi NC, Anderson KC. Aggresome induction by proteasome inhibitor bortezomib and alpha-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood. 2006;108:3441–9. doi: 10.1182/blood-2006-04-016055. [DOI] [PMC free article] [PubMed] [Google Scholar]