

Abstract
Gambogic acid (GA), the main active compound of Gamboge Hanburyi, has been previously reported to activate apoptosis in many types of cancer cell lines by targeting transferrin receptor and modulating NF-kappa B signaling pathway. Whether GA inhibits angiogenesis, which is crucial for cancer and other human diseases, remains unknown. Here we found that GA significantly inhibited human umbilical vein endothelial cell (HUVEC) proliferation, migration, invasion, tube formation, and micro-vessel growth at nM concentration. In a xenograft prostate tumor model, we found that GA effectively inhibited tumor angiogenesis and suppressed tumor growth with low side effects using metronomic chemotherapy with GA. GA was more effective in activating apoptosis and inhibiting proliferation and migration in HUVECs than in human prostate cancer cells (PC3), suggesting GA might be a potential drug candidate in cancer therapy through angioprevention with low chemotoxicity. Furthermore, we demonstrated that GA inhibited the activations of vascular endothelial growth factor receptor 2 (VEGFR2) and its downstream protein kinases, c-Src, FAK and AKT. Together, these data suggest that GA inhibits angiogenesis and may be a viable drug candidate in anti-angiogenesis and anti-cancer therapies.
Keywords: Gambogic acid, anti-angiogenesis, tumor angiogenesis, VEGF receptor 2 inhibitor, prostate cancer
Introduction
Gambogic acid (GA, C38H44O8; MW 628.76), a polyprenylated xanthone, is the main active compound of Gamboge Hanburyi (a traditional Chinese medicine) used as detoxification, homeostasis, anti-inflammatory and parasiticide medicines for thousands of years (1). Previous studies reported that GA activated apoptosis in many cancer cell lines and inhibited human hepatoma SMMC-7721 tumor growth in vivo in a nude mouse model (2-7). Recent reports demonstrated the molecular mechanism of apoptosis activation effect of GA by binding to the transferrin receptor (8) and suppressing nuclear factor-kappa B (NF-kappa B) signaling pathway (9). Whether GA inhibits angiogenesis, a crucial step in tumor growth and metastasis, is still unknown. Here we examined the effects of GA on migration, invasion, and tube formation of endothelial cells and angiogenesis in vitro and in vivo.
Recently, increased interests focused on metronomic chemotherapy with comparatively low doses of drug on a frequent or continuous schedule based on anti-angiogenesis. In the chemotherapy with angiogenesis inhibitors, endothelial cells were reported to be the first to undergo apoptosis compared to cancer cells in the tumor (10). In preclinical models of tumor treatment, metronomic chemotherapy showed its advantages of no tumor cell resistance to the same chemotherapeutics and less acute toxicity (11, 12). There are several angiogenesis inhibitors in phase I or phase II clinical trials, including VEGF receptor 2 (VEGFR2) inhibitors (10). In order to test whether GA inhibits tumor-angiogenesis and tumor growth, we used metronomic chemotherapy method with GA in a xenograft prostate tumor model and found GA inhibited tumor-angiogenesis and prevented tumor growth.
Vascular endothelial growth factor receptors (VEGFRs) and their signaling pathways represent rate-limiting steps in physiological angiogenesis (13, 14). VEGFRs undergo ligand-induced homodimerization or heterodimerization that activates their intrinsic tyrosine kinase activities (15). VEGFR1 is poorly autophosphorylated in response to VEGF in endothelial cells and is weakly involved in transducing the VEGF angiogenic signals (16). VEGFR3 mainly functions in the establishment and maintenance of lymphatics(15). In contrast, homodimerization of VEGFR2 leads to a strong autophosphorylation of several tyrosine residues of VEGFR2. VEGFR2 is the primary receptor mediating the angiogenic activity of VEGF through distinct signal transduction pathways that regulate endothelial cell proliferation, migration, differentiation, and tube formation. Major autophosphorylation sites on VEGFR2 have been described as Y1175 and Y951(15). In particular, phosphorylation of Tyr-1175 is required for activation of AKT, which is critical for subsequent activation of endothelial cell survival, migration and proliferation (17). Phosphorylation of Y951 is necessary for activation of c-Src which regulates cell migration. VEGFR2 regulates cell migration and focal adhesion turnover by mediating the activation of FAK (15, 16, 18). Here we examined the effect of GA on the activation of VEGFR2 and its downstream kinases: c-Src, FAK and AKT.
In this study, we investigated the effects of GA on angiogenesis, tumor-angiogenesis in the prostate tumor model, and the underlying molecular mechanisms. We found that GA significantly inhibited endothelial cell proliferation, migration, invasion, tube formation, angiogenesis in vitro and in vivo as well as tumor-angiogenesis. GA acts as a VGFR2 inhibitor and suppressed the VEGFR2 signaling pathway and its downstream kinases.
Materials and methods
Cell lines, cell culture and reagents
GA was ordered from Gaia Chemical Corp. (Gaylordsville, CT) 98% by TLC/HPLC. A 25 mM solution of GA was prepared in dimethyl sulfoxide, stored at -20°C, and then diluted as needed. Human umbilical vein endothelial cells (HUVECs) were kindly gifted from Dr. Xinli Wang (Cardiothoracic Surgery Division of the Michael E. DeBakey Department of Surgery at Baylor College of Medicine Hospital). The human prostate cancer cell line (PC3) was purchased from the American Type Culture Collection (Manassas, A). VEGF was obtained from NIH experimental branch. HTScan® VEGF receptor 2 kinase assay kit was ordered from Cell Signaling Technology. HRP labeled secondary antibody, TMB substrate and stop solution were kindly gifted by Cell Signaling Technology. Streptavidin coated yellow 96-well plates were kindly gifted by PerkinElmer Life Sciences. Matrigel was ordered from BD Biosciences, Bedford, MA. Mitomycin C was ordered from Roche, Germany.
Proliferation assay
HUVEC and PC3 cell proliferation assays with different concentrations of GA were followed the manual of CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega) with VERSAmax microplate reader (Molecular Devices).
Flow cytometry FACS analysis
About 2×106 HUVEC and PC3 cells were treated with GA at 37°C, 5% CO2 incubator for 24 hours. The cells were collected and analyzed in a FACS Vantage SE DiVa flow cytometer (Becton Dickinson) with Propidium Iodide staining. The cell population percentages at Sub G1 were defined as apoptotic cell percentages.
Migration assay
HUVECs were allowed to grow into full confluence in 6-well plates precoated with 0.1% gelatin and then incubated with 10μg/ml mitomycin C at 37°C, 5% CO2 for 2 hours to inactivate HUVECs. Monolayer inactivated HUVECs were wounded by scratching with 1 ml pipette tip. Fresh endothelial cell growth medium (ECGM) was added with or without 4ng/ml VEGF and different concentrations of GA. Images were taken by Nikon digital camera after 7-10 hours incubation at 37°C, 5% CO2. For PC3 cell migration, inactivated PC3 cells (with the same inactivation treatment as HUVECs) were performed migration assays as in HUVECs. The migrated cells were quantified by manual counting and percent inhibition was expressed using untreated wells as 100% (t-test, P<0.005). At least three independent experiments were performed.
Transwell migration assay
The transwell (Corning Incorporated, NY, USA) were coated with 0.1% gelatin (Sigma) for 30 minutes at 37°C. After washed the transwells three times with 1×PBS, the bottom chambers (600μl) were filled with ECGM with 20% FBS supplemented with 4 ng/ml VEGF and the top chambers were seeded with 100μl ECGM and inactivated HUVECs (4×104 cell/well). The top and bottom chambers contained the same series of concentrations of Gambogic acid. HUVECs were allowed to migrate for 4 hours at 37°C, 5% CO2. After the incubation, cells on the top surface of the membrane (non-migrated) were scraped with a cotton swab. Cells on the bottom side of the membrane (migrated cells) were fixed with 4% paraformaldehyde for 20 minutes and washed three times with 1×PBS. The cells were stained by Hematoxylin and eosin (H&E) staining and then destained with 1×PBS. Images were obtained using an OLYMPUS inverted microscope and with invading cells being quantified by manual counting. Percent inhibition of invading cells was quantified and expressed on the basis of untreated cells (control) representing 100% (t-test, P<0.01).
Tube formation assay
Matrigel (BD Biosciences) were thawed at 4°C for overnight and each well of prechilled 24-well plates was coated with 100μl matrigel and incubated at 37°C for 45 minutes. HUVECs (4×104 cells) were added in 1 ml ECGM with various concentrations of GA. After 12-16 hours of incubation at 37°C, 5% CO2, endothelial cell tube formation was assessed with OLYMPUS inverted microscope. Tubular structures were quantified by manual counting of low power fields (25×) and inhibition percentage was expressed using untreated wells as 100% (t-test, P<0.001).
Aortic ring assay
Aortic ring assay was performed as previously described with some modifications (19, 20). Forty-eight-well plates were covered with 100 μl of matrigel at 4°C and incubated at 37°C, 5% CO2 for 30 minutes. Aortas isolated from mice were cleaned of periadventitial fat and connective tissues, and cut into about 1-1.5 mm-long rings. After being rinsed five times with endothelial cell-based medium, the aortas were placed on the matrigel-covered wells and covered with another 100 μl of matrigel. Artery rings were cultured in 1.5 ml of ECGM without serum for 24 hours, and then the medium was replaced with 1.5 ml of ECGM with or without GA. The medium was changed every two days with the exact composition as described above. After 4 days incubation, the micro-vessel growth was quantified by taking photographs with Olympus IX 70 invert microscope with a 4×objective lens. After images were acquired, the outgrowth area was delineated and measured with the Pro plus software (Media Cybernetics).
Matrigel plug assay
Matrigel (0.5 ml/plug) with no VEGF or GA, VEGF (4 ng/ml) but no GA, VEGF (4 ng/ml) and 0.1/0.2 μM GA in liquid form at 4°C, respectively, were injected subcutaneously in the midventral abdominal region of 5-6 week C57BL/6 mice (five mice for each group). After 7 days, the mice were sacrificed and the plugs were removed. Each group had 4-5 matrigel plugs. The matrigel plugs were fixed with formalin and embedded with paraffin. The 5μm sections were stained with H&E staining. The number of erythrocyte-filled blood vessels in high power field (HPF, 200×) was counted (plug number = 4-5, t- test, P<0.005).
Xenograft Mouse Model
The 5 - 6 week old severe combined immune deficiency (SCID) male mice (ordered from NIH) weighing about 20 g were divided into groups with five mice per group. PC3 cells were subcutaneously injected (2 ×106 cell per mouse) into the mice (20). After the tumors had become established (about 50 mm3), the mice were subcutaneously injected with or without 3 mg/kg GA every day. The mice body weights and tumor sizes were recorded every day and the tumor sizes were determined by vernier caliper measurements and calculated as length × width × height. After 15 days, mice with s.c. tumors no greater than 1.5 cm in diameter were sacrificed (21).
Histology and Immuno-histochemistry
Tumors were removed and fixed with Histochoice® MB (Molecular Biology) tissue fixative (Amresco®) and embedded with paraffin. The 5μm sections were performed specific blood vessel staining with CHEMICON’s Blood Vessel Staining Kit (von Willebrand Factor, Chemicon International). Images were taken with ZEISS Axioskop 40 photo microscope. The number of blood vessels was counted (plug number = 4-5, t-test, P<0.005).
VEGF receptor 2 inhibition assay
A 12.5 μl of the 4× reaction cocktail containing 100 ng VEGF Receptor 2 (supplied from the HTScan® VEGF receptor 2 kinase assay kit, Cell Signaling Technology, USA) was incubated with 12.5 μl/tube of GA for 5 minutes at room temperature. A 25 μl of 2× ATP/substrate peptide cocktail was added to the pre-incubated reaction cocktail/GA compound. After incubation at room temperature for 30 minutes, a 50 μl stop buffer (50 mM EDTA, pH 8) was added per tube to stop the reaction. Then 25 μl of each reaction was transferred with 75 μl H2O/well to a 96-well streptavidin-coated plate (PerkinElmer Life Sciences, USA) and incubated at room temperature for 60 minutes. After washed the wells three times with 200 μl/well PBS/T (0.05% Tween-20 in 1 × PBS), a 100 μl primary antibody (Phosphor-Tyrosine Monoclonal Antibody (P-Tyr-100), 1:1000 in PBS/T with 1% BSA) was added per well. After being incubated at room temperature for 60 minutes, the wells were washed three times with 200 μl PBS/T. A 100 μl diluted HRP labeled anti-mouse IgG (1:500 in PBS/T with 1% BSA) was added per well. After incubation at room temperature for 30 minutes, the wells were washed five times with 200 μl PBS/T per well. Then a 100 μl/well TMB substrate was added per well and the plate was incubated at room temperature for 15 minutes. The stop solution (100μl/well) was added and mixed followed incubation for at room temperature 15 minutes. The plate was then detected at 405 nm with VERSAmax microplate reader (Molecular Devices) and the data (mean ± SEM, n = 3) were repeated 3 times.
Western immuno-blotting
HUVECs pretreated with or without 4 nM VEGF for 5 min were treated with or without GA for another 5 minutes. A 200μg total cellular protein of each sample was performed immunoprecipitation with anti-c-Src, anti-FAK and anti-AKT antibodies (Santa Cruz Biotech) and then subjected to the Western blotting. The pTyr-antibody (Santa Cruz Biotech) was used for detecting c-Src phosphorylation and pFAK397 antibody (Cell Signaling) was blotted for c-Src-associated FAK phosphorylation at multiple tyrosine residues. In addition, AKT phosphorylation was examined using pSer473-AKT antibody (Cell Signaling). Anti-cleaved Caspase3 antibody (Santa Cruz Biotech) was used for detecting cleaved Caspase 3 and poly ADP ribose polymerase (PARP) cleavage was detected by anti-PARP p85 fragment (Pormega) in apoptosis assay.
Statistical Analysis
The data (mean ± SEM, n = 3) were repeated 3 times for cell proliferation, apoptosis, migration, invasion, and aortic ring assays. Statistical significance of differences between control and sample groups was determined by t- test. The minimal level of significance was P < 0.05.
Results
Gambogic acid (GA) inhibits HUVEC migration, invasion and tube formation
As cell migration is necessary for endothelial cells in angiogenesis and for cancer cells in tumor growth and metastasis (22, 23), we performed wound-healing migration assays to determine the effects of GA (Fig.1A) on HUVEC migration and found 10 nM GA strongly inhibited the migration of inactivated HUVECs (Fig.1B). Cell invasion is a critical aspect of endothelial cells in angiogenesis (24), we performed transwell assays to evaluate the ability of inactivated HUVECs to pass through the membrane barrier of the transwell in the presence of GA. As shown in Fig. 1C, 40 nM GA inhibited almost all invasion activities of inactivated HUVECs, suggesting that GA significantly inhibited the invasion properties of endothelial cells at very low concentrations (nM).
Figure 1.
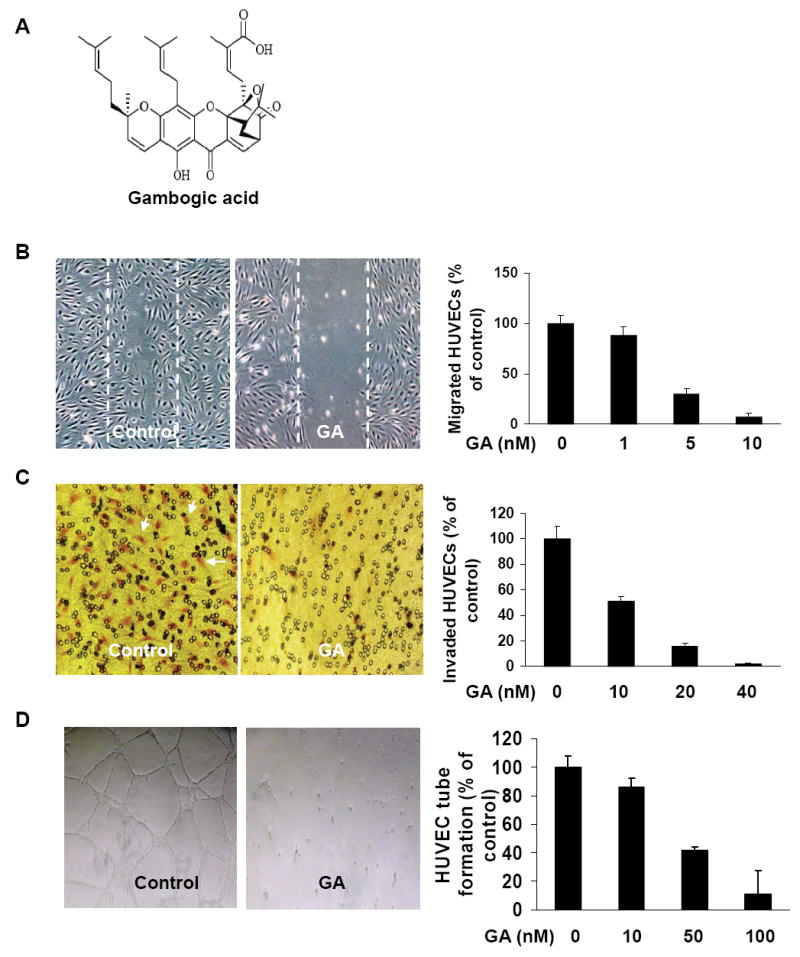
GA inhibits HUVEC migration, invasion and tube formation. A, the chemical structure of Gambogic acid (GA). B, effects of GA on HUVEC migration in wound-migration assays. HUVECs were inactivated by incubating with 10 μg/ml mitomycin C and migration assays were performed as described in Materials and Methods described (mean ± SEM, n = 3, t-test, P<0.005). C, effects of GA on HUVEC invasion in transwell assay. The bottom chambers (600μl) of the transwells were filled with ECGM and 4ng/ml VEGF while the top chambers were seeded with 100μl ECGM containing HUVECs (4×104 cell/ well) in the presence or absence of GA at the indicated concentrations. HUVECs were allowed to migrate for 4 hours. The red cells with irregular shape in the images were invaded cells attached on outside surface of the top chamber (mean ± SEM, n = 3, t- test, P < 0.01). D, effects of GA on HUVEC tube formation. HUVECs (4×104 cells) and GA were added on Matrigel layers. After 12-16 hours of incubation, HUVEC tube-like formation was assessed with an inverted photomicroscope. Tubular structures were quantified by manual counting of low power fields (25×) and percent inhibition was expressed using untreated wells as 100% (mean ± SEM, n = 3, t- test, P < 0.001).
Although angiogenesis is a complex procedure of several kinds of cells, tube formation of endothelial cells is the key step (25). To further investigate the effect of GA on endothelial cell tube formation, we added HUVECs (4×104 cells) in 1 ml ECGM with different concentration of GA onto matrigel layers. After 12-16 hours incubation, the ability of endothelial cells forming tube-like structures was assessed with an inverted photomicroscope. Approximately 50 nM GA inhibited 50% tube formation of HEVEC cells on matrigel assays and 100 nM GA completely inhibited the tube-formation ability of HUVECs on matrigel (Fig. 1D).
GA inhibits angiogenesis in vitro and in vivo
To examine the inhibitory effect of GA on angiogenesis, we performed aortic ring assays using isolated aortas from mice. The 1-1.5 mm-long aortic rings were put on matrigel and covered by another matrigel layer and ECGM with or without GA. After 4 days incubation, the numbers of micro-vessel growth of the aortic rings were quantified and compared in the presence or absence of GA. We found that 10 nM GA (higher concentration not shown) inhibited almost all new micro-vessel growth (Fig. 2A), suggesting GA dramatically inhibited angiogenesis in vitro.
Figure 2.
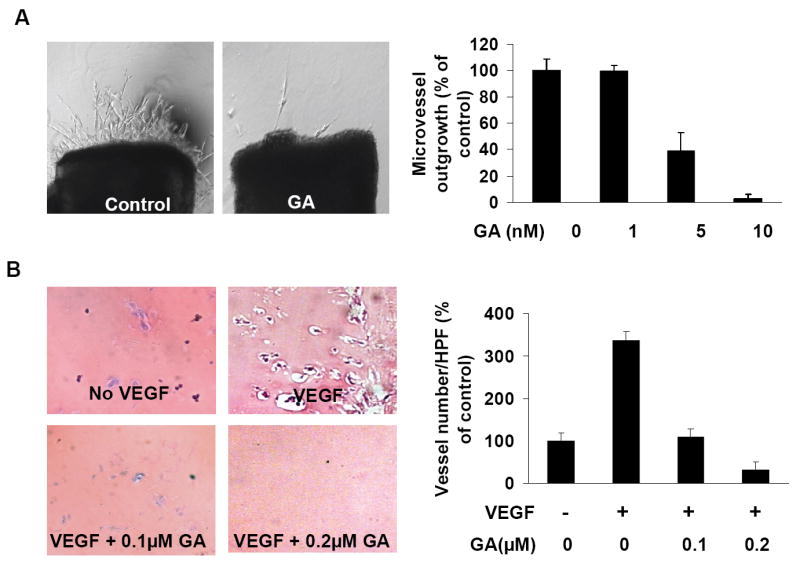
GA inhibits angiogenesis in vitro and in vivo. A, effects of GA on angiogenesis in the aortic ring assays. About 1-1.5 mm-long cleaned mice aortic rings were placed in the matrigel-covered wells with 100 μl of matrigel. After 4 days incubation with 1.5 ml of ECGM with or without GA, images were taken with Olympus IX 70 invert microscope. The number of microvessels was counted as described in the Methods (mean ± SEM, n = 4, t- test, P < 0.05). B, effects of GA on angiogenesis in vivo in Matrigel plug assay. Matrigel plugs (0.5 ml/plug) with neither GA nor VEGF, VEGF (4ng/ ml) but no GA, VEGF (4ng/ ml) and 0.1μM GA, VEGF (4ng/ ml) and 0.2μM GA, were injected subcutaneously in the midventral abdominal region of 5-6 week C57BL/6 mice (five mice for each group). After 7 days, the Matrigel plugs were removed, fixed with formalin, and the 5μm sections were stained with H&E staining. The number of erythrocyte-filled blood vessels in high power field (HPF, 200×) was counted (mean ± SEM, n = 4, t- test, P < 0.005).
To further verify the inhibitory effect of GA on angiogenesis, we used matrigel plug assay for anti-angiogenesis effect of GA in vivo. We subcutaneously injected matrigel (0.5 ml/plug) with no VEGF, VEGF (4ng/ml), VEGF (4ng/ml) and 0.1μM GA or 0.2 μM GA in the midventral abdominal region of 5-6 week C57BL/6 mice (five mice for each group). After 7 days, the mice were sacrificed and the matrigel plugs were removed, sectioned, and H&E stained. As shown in Fig. 2B, 100 nM GA inhibited VEGF dependent angiogenesis while 200 nM GA totally abolished angiogenesis in the Matrigel plug assays, indicating GA strongly inhibited angiogenesis in vivo. Based on the above analyses, we concluded that GA inhibited angiogenesis in vitro and in vivo using different angiogenesis assays.
GA inhibits tumor-angiogenesis and tumor growth in vivo
Tumor angiogenesis provides oxygen, nutrients and main routes for tumor growth and metastasis and acts as a rate limiting step in tumor procedures(26). Metronomic chemotherapy is an effective method to suppress tumor growth for angiogenesis inhibitors with low dose of drug (10). To determine the effect of GA on tumor-angiogenesis and tumor growth at low dosage, we used a xenograft mouse prostate tumor model. It has been demonstrated that 4 mg/kg of GA at a frequency of 1 treatment every two days is a non-toxic dosage (27). We injected s.c. (2×106 PC3 cell per mouse) into the mice. After the tumors had become established (about 50 mm3), the mice were subcutaneously injected with or without 3 mg/kg GA every day. The mouse body weights and tumor sizes were recorded every day and the tumor sizes were measured by vernier caliper and calculated as length × width × height as previous described (21). After 15 days, mice were sacrificed and the tumors were removed. As shown in Fig. 3A, at day 15 after injection of tumor cells, the average tumor size of control group was 1144 ± 169 mm3 while that of GA treated group was 169.1 ± 25.6 mm3. The average tumor weight of control group was 0.28 ± 0.08 g while that of GA treated group was 0.012 ± 0.0008 g (Fig. 3B), indicating GA significantly inhibited tumor growth with metronomic chemotherapy.
Figure 3.
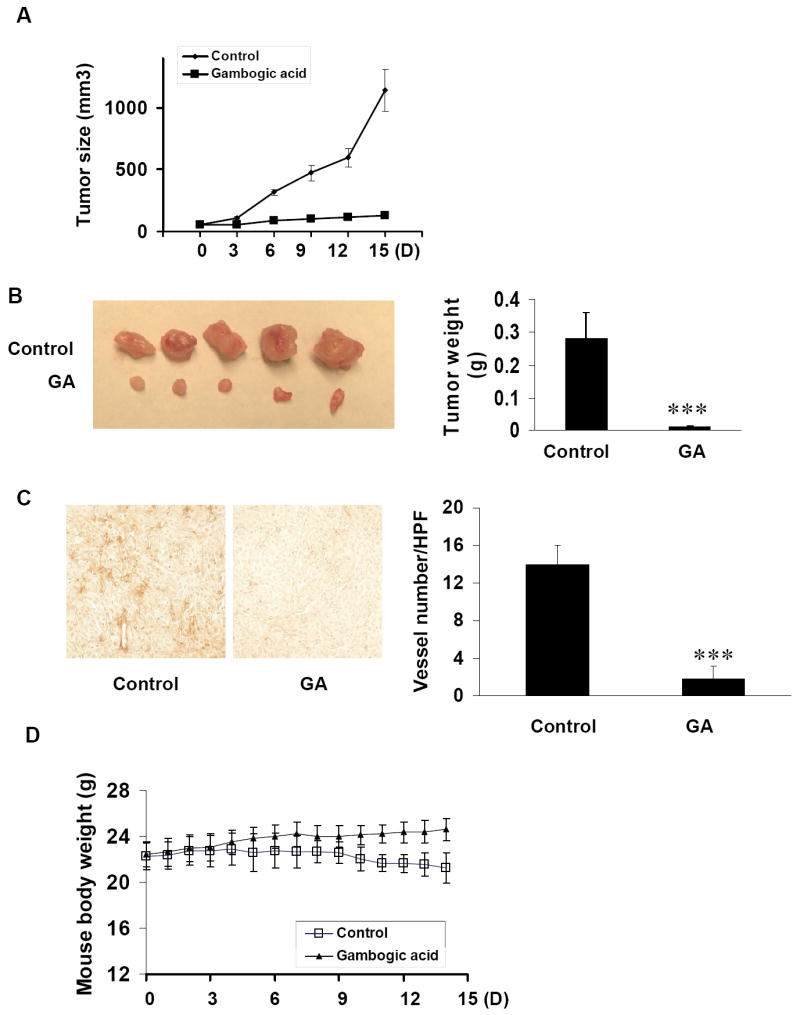
GA inhibits tumor-angiogenesis and tumor growth in vivo. PC3 cells were injected s.c. (2×106 cell per mouse) into the 5-6 week old SCID male mice. After the tumors have established (about 50 mm3), the mice were injected with or without 3 mg/kg/day GA. After 15 days, mice were sacrificed and tumors were removed and taken images by Nikon camera. A, tumors of control group increased from 51.18 ± 5.3 mm3 to 1144 ± 169 mm3 while tumors from GA treated group increased only from 51.74 ± 3.8 mm3 to 127.4 ± 25.6 mm3. B, tumors from the mice with GA treatment were significantly smaller than that from control group. The average weight of tumors from control group was 0.28 ± 0.08g while that from GA treated groups was 0.012 ± 0.0008g (mean ± SEM, n = 5, t- test, P < 0.001). C, effects of GA on tumor angiogenesis in xenograft mouse tumor model. Tumors were fixed with Histochoice® MB (Molecular Biology) tissue fixative (Amresco®) and embedded with paraffin. The 5μm sections were performed with specific blood vessel staining. The average vessel number in tumors of control group was 14 ± 2 /high performance field (HPF, 200×) while that in GA treated group was 1.8 ± 1.3 /HPF(mean ± SEM, n = 5, t- test, P < 0.001). D, The average mouse body weight of control group decreased from 22.3 ± 1.2 g to 21.2 ± 1.3g while that of GA treated group increased from 22.4 ± 1g to 24.6 ± 0.9g.
To examine the inhibitory effect of GA on tumor-angiogenesis, we stained the 5μm tumor sections with specific blood vessel staining kit. The average vessel number in tumors of control group was 14 ± 2 (HPF) while that in GA treated group was 1.8 ± 1.3 (HPF) (Fig. 3C), indicating that GA significantly inhibited tumor-angiogenesis and prevented prostate tumor growth. To evaluate the side effect or chemotoxicity of GA on mice normal growth, we recorded the mice body weights everyday. During the 15 days, the average body weight of control group decreased 1 ± 1.3g while that of GA treated group increased 3.2 ± 0.9g (Fig. 3D), indicating that 3 mg/kg/day GA for mice is not a toxic dosage or at least low-toxic dosage. The observed mouse body weight decrease in control group is probably due to the tumor burden compared to the GA-treated group.
GA is more effective in inhibiting cell migration and proliferation and activating apoptosis in HUVECs than in PC3 cancer cells
To better understand the inhibitory effects of GA on angiogenesis/tumorangiogenesis and tumor growth, it is important to explore whether GA has different effects on cell migration, proliferation and apoptosis in endothelial and tumor cells. Considering that VEGFR2 is the primary receptor through which VEGFs regulate the angiogenic activities in both normal angiogenesis and tumor-angiogenesis and that HUVECs express VEGFR2 (KDR/Flk-1) but PC3 cancer cells do not express VEGFR2(28), we firstly examined the effects of GA on VEGF dependent cell migration in HUVEC and PC3 cells. We found 10 nM GA strongly inhibited VEGF dependent migration of HUVECs (Fig. 4A), while more than 100 nM GA was required to inhibit VEGF dependent migration for PC3 cancer cells (Fig. 4B), indicating that GA suppressed VEGF-induced cell migration for both endothelial cells and cancer cells and that endothelial cells were more sensitive to GA inhibition compared to PC3 cancer cells. Then we performed proliferation assays of HUVEC and PC3 cancer cells with GA. We found that 80 nM GA inhibited 50% HUVEC cell proliferation (Fig. 4C) while more than 400 nM GA was required to obtain the same inhibitory effect on PC3 cancer cells (Fig. 4D), indicating that GA was more effective in inhibiting proliferation of endothelial cells than that of PC3 cancer cells. Before we examined the effects of GA in activating apoptosis in HUVEC and PC3 cancer cells, we identified GA promoted apoptosis in HUVECs with anti-cleaved caspase 3 and anti-cleaved PARP antibodies (Fig. 4E). Finally, we measured the apoptotic population of HUVEC and PC3 cancer cells treated with GA by flow cytometry analysis (FACS assays). As shown in Table 1, 80 nM GA induced 40% HUVECs into apoptosis while only 4% of PC3 cancer cells were induced into apoptosis by the same concentration of GA, indicating that GA was much more effective on endothelial cells than on PC3 cancer cells in promoting cell apoptosis. Taken together, the above different effects of GA on HUVECs and PC3 cancer cells suggest VEGFR2 is primarily if not exclusively expressed on endothelial cells. Thus, the inhibitory effects seen with PC-3 cell xenografts in mice (Fig. 3) are due to inhibition of angiogenesis and GA regulates the activity of VEGFR2.
Figure 4.
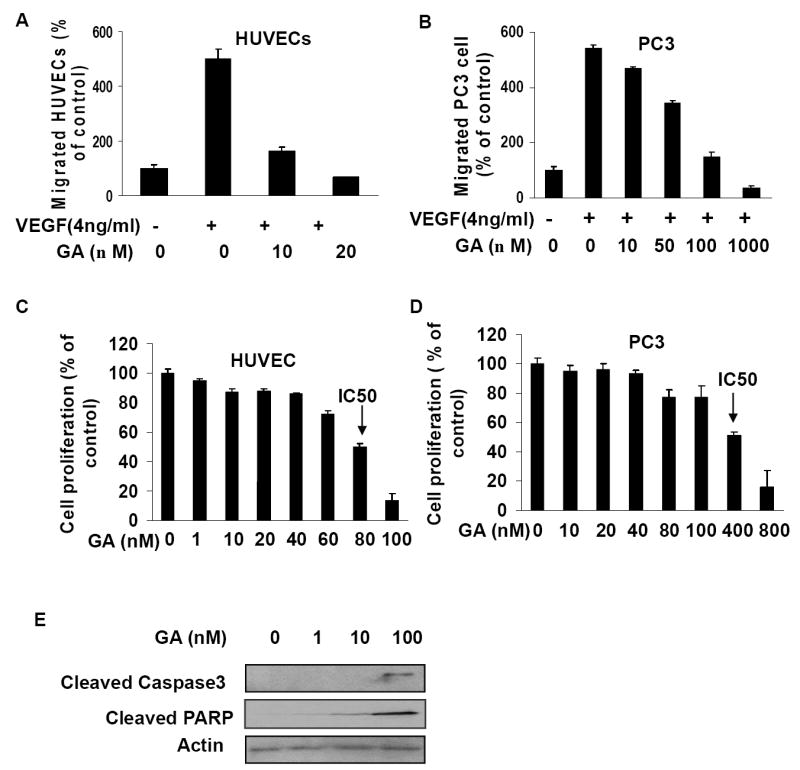
GA is more effective in activating apoptosis and inhibiting cell proliferation and migration in HUVECs than in PC3 cells. A-B, effects of GA on VEGF induced migration of HUVECs and PC3 cells (B) (mean ± SEM, n = 3, t- test, P < 0.01). C-D, effects of GA on cell proliferation in HUVECs and PC3 cells (D) (mean ± SEM, n = 3, t-test, P < 0.01). IC50 was identified as the GA concentration to inhibit 50% cell proliferation. E, GA activates apoptosis in HUVECs. HUVECs were treated with GA for 24 hours and whole cell proteins were analyzed by Western blotting with anti-cleaved caspase 3 antibodies and anti-cleaved PARP p85 antibody.
Table 1.
GA activates apoptosis in HUVECs and PC3 cells.
| Apoptotic population (% of total) | |||||
|---|---|---|---|---|---|
| GA (nM) | 0 | 20 | 40 | 80 | 100 |
| PC3 | 1 ± 0.3 | 1.6 ± 0.3 | 2.9 ± 0.4 | 4.3 ± 0.5 | 13.8 ± 3.6 |
| HUVECs | 2.3 ± 0.2 | 11.6 ± 1.1 | 14.4 ± 2.3 | 40 ± 3.6 | 57.6 ± 4.7 |
GA is an inhibitor of VEGF receptor 2
VEGFR2 is the primary receptor in VEGF signaling pathway that regulates endothelial cell proliferation, migration, differentiation, tube formation, and angiogenesis (14, 15). To understand the molecular mechanism of GA-mediated anti-angiogenesis effect, we examined whether GA inhibits the activation of VEGFR2. We found GA dramatically inhibited the phosphorylation of VEGFR2 in the assays (Fig. 5A), suggesting that GA is a potential inhibitor of VEGFR2. To verify the inhibitory effect of GA on VEGFR2, we further examined the effects of different concentrations of GA on the specific activation of VEGFR2 using HTScan® VEGF receptor 2 kinase assay kit according to suggested methods (Cell Signaling Technology and PerkinElmer Life Sciences, USA). We found that GA inhibited VEGFR2 kinase activity with the IC50 of 12 pM (Fig. 5B), indicating GA is a potent VEGFR2 inhibitor.
Figure 5.
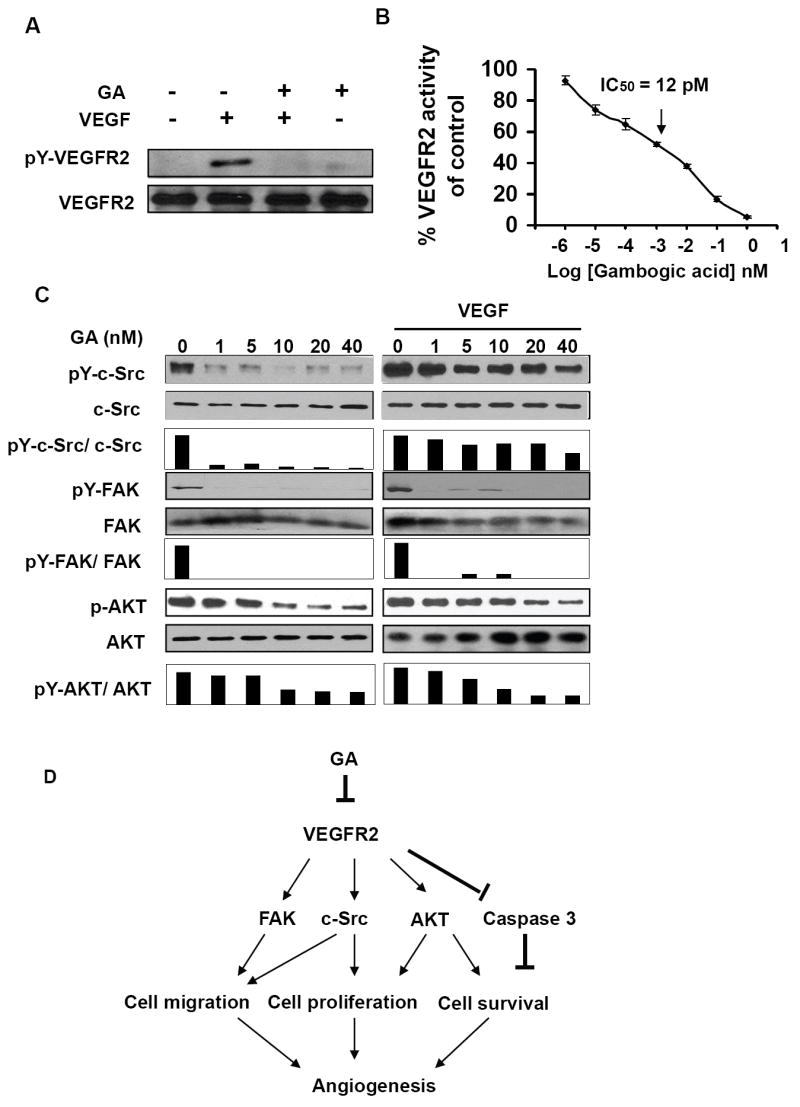
GA inhibits the activation of VEGFR2 and its downstream protein kinases. A, 1nM GA strongly inhibited VEGF-induced phosphorylation and activation of VEGFR2. After starvation in ECGM without serum for overnight, HUVECs were washed with 1×PBS twice followed with incubation in M199 medium. Then HUVECs treated with 1nM of GA in the presence or absence of 4nM of VEGF for 5 minutes. VEGFR2 was immunoprecipitated using anti-VEGFR2 antibody. Anti-phospho-tyrosine antibody was used for the detection of phosphorylation of tyrosine residue of VEGF receptor 2. B, inhibition of GA on VEGFR2 activation in a specific VEGFR2 inhibition assay. The calculated IC50 = 12 pM and the IC50 was identified as the concentration of GA to inhibit 50% of the activity of 100 ng VEGFR2. The data (mean ± SEM, n = 3) repeat 3 times. C, GA suppressed VEGFR2-mediated protein kinase activation of c-Src, FAK and AKT. HUVECs, pretreated with or without 4 nM VEGF for 5 minutes, were treated with GA for another 5 minutes. (D) Diagram of signaling pathways for GA mediated anti-angiogenesis.
GA inhibits VEGFR2 signaling pathway
VEGFR2 regulates cell migration and proliferation by regulating the activation of focal adhesion kinase (FAK) and c-Src (29, 30, 31). To understand the inhibitory effects of GA on cell proliferation and migration, we examined the effects of GA on the phosphorylation of Src and FAK. As shown in Fig. 5C, GA significantly inhibited the phosphorylation of both c-Src and FAK but not affected the total protein expression of c-Src or FAK, suggesting that GA inhibited cell migration and proliferation by inhibiting VEGFR2 and its down-stream protein kinase activities. The activation of c-Src with VEGF pretreatment (Fig. 5C, left) was stronger than that without VEGF treatment (Fig. 5C, right), which was consistent with VEGF promoted c-Src activation by stimulating VEGFR2.
The phosphorylation of Y1175 of VEGFR2 mediates the activation of AKT to regulate cell proliferation(32). To further examine the down-stream signaling pathways mediated by VEGFR2, we examined the activation of the serine/threonine kinase AKT (PKB). As shown in figure 5C, GA inhibited AKT phosphorylation (Fig. 5C), suggesting GA inhibited cell proliferation by regulating the activation of AKT signaling pathways.
Discussion
We identified GA as a novel VEGFR2 inhibitor and comprehensively demonstrated that GA inhibited angiogenesis and tumor progression. Our work focuses on GA’s inhibitory effects on HUVEC proliferation, migration, invasion, and tube formation, four key characteristics of endothelial cells in angiogenesis. By directly blocking VEGFR2 phosphorylation and activation, GA suppressed the AKT signaling pathway and inhibited cellular proliferation. As a consequence of VEGFR2 inhibition by GA, the phosphorylation and activation of c-Src and FAK were blocked. Using non-toxic dosage of GA, we demonstrated that GA can inhibit tumor angiogenesis and prostate tumor growth in SCID mouse models.
We demonstrated that GA is a potent VEGFR2 inhibitor, which could provide a new mechanism of actions for xanthone family members (33). We observed GA was more effective in activating apoptosis and inhibiting migration and proliferation in HUVECs than that in PC3 cancer cells, which was in agreement with previous reports that HUVECs express VEGFR2 but PC3 cancer cells do not express it (28). GA also arrested the migration and proliferation of PC3 cancer cells at relatively high concentration, indicating that GA may not specifically inhibit VEGFR2 and GA may attenuate the actions of other RTKs at high doses. As compounds that act as RTK inhibitors always show inhibitory characteristics for multiple kinases (34), we will further investigate whether GA can inhibit other receptor tyrosine kinases. Interestingly, we also found that GA was more effective in inhibiting HUVEC migration (10 nM) and invasion (40 nM) than in inhibiting HUVEC proliferation (100 nM) and activating apoptosis, suggesting that GA could inhibit endothelial cell migration and invasion before its reported effects on cell proliferation and apoptosis during angioprevention. These findings may provide a molecular mechanism for GA as a new anti-angiogenesis candidate with low chemotoxicity during chemotherapy.
Phosphorylation of VEGFR2 at Tyr-1175 is required for the activation of AKT and endothelial cell proliferation (17). We found that GA inhibited the activation of VEGFR2 and AKT as well as cell proliferation. Phosphorylation of Tyr-951 of VEGFR2 is required for the binding site of TSAD which mediates its substrate of c-Src and then regulates cell migration. VEGFR2 also directly regulates the phosphorylation of FAK and mediates cell migration (22, 35, 36). In this study, we demonstrate that GA inhibits the activation of FAK and c-Src and cell migration.
Although the relationship between anti-inflammation and angioprevention has not been fully understood, recent studies have reported several “classic” anti-inflammatory drugs lead to angioprevention (37), such as epigallocatechin gallate in green tea (38), through mechanisms that include interference with VEGF signaling pathway (39, 40). In this study, we showed significant inhibitory effects of GA, a traditional Chinese medicine used for anti-inflammatory for thousands of years, on angiogenesis in vitro and in vivo. We identified GA as a VEGFR2 inhibitor for the first time, which might add a new “classic” drug member with both anti-inflammatory and anti-angiogenesis functions.
Pandey MK et al. demonstrated GA potentated tumor necrosis factor (TNF)-induced apoptosis and inhibits TNF-induced cellular invasion through NF-kappa B signaling pathway (9). TNF, via its receptor 2 (tumor necrosis factor receptor 2, TNFR2), regulates endothelial cell migration, invasion and tube formation, but TNFR2 lacks an intrinsic kinase activity (41, 42). Interestingly, Zhang R et al. reported that the activation of TNFR2 was VEGFR2 dependent and VEGFR2 inhibitor suppressed the activation of VEGFR2 and then inhibited TNFR2’s activation (17). We found that GA could function as a VEGFR2 inhibitor, significantly blocking endothelial cell migration, invasion, and tube formation. Therefore, it is possible that GA regulates NF-kappa B signaling pathway by inhibiting the activation of VEGFR2 and TNFR2.
In summary, our studies demonstrate an axis of action by GA, e.g. GA functions as an inhibitor of VEGFR2 and its signaling pathway, leading to the inhibition of angiogenesis and tumorigenesis (Fig. 5D). We demonstrated the previously unreported inhibition of GA on HUVEC cell proliferation, migration, and tube formation, as well as the anti-angiogenesis activity of GA in vitro and in vivo. Our data suggest a new mechanism of action for the well known xanthone family and their potential as anti-angiogenesis and anti-cancer drugs.
Acknowledgments
This work is supported partially by a grant (1R01CA106479 to M Liu) from National Cancer Institute, National Institutes of Health (NIH).
References
- 1.Guo QL, Lin SS, You QD, et al. Inhibition of human telomerase reverse transcriptase gene expression by gambogic acid in human hepatoma SMMC-7721 cells. Life Sci. 2006;78(11):1238–45. doi: 10.1016/j.lfs.2005.06.046. [DOI] [PubMed] [Google Scholar]
- 2.Liu W, Guo QL, You QD, Zhao L, Gu HY, Yuan ST. Anticancer effect and apoptosis induction of gambogic acid in human gastric cancer line BGC-823. World J Gastroenterol. 2005;11(24):3655–9. doi: 10.3748/wjg.v11.i24.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao L, Guo QL, You QD, Wu ZQ, Gu HY. Gambogic acid induces apoptosis and regulates expressions of Bax and Bcl-2 protein in human gastric carcinoma MGC-803 cells. Biol Pharm Bull. 2004;27(7):998–1003. doi: 10.1248/bpb.27.998. [DOI] [PubMed] [Google Scholar]
- 4.Zhang HZ, Kasibhatla S, Wang Y, et al. Discovery, characterization and SAR of gambogic acid as a potent apoptosis inducer by a HTS assay. Bioorg Med Chem. 2004;12(2):309–17. doi: 10.1016/j.bmc.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 5.Guo QL, You QD, Wu ZQ, Yuan ST, Zhao L. General gambogic acids inhibited growth of human hepatoma SMMC-7721 cells in vitro and in nude mice. Acta Pharmacol Sin. 2004;25(6):769–74. [PubMed] [Google Scholar]
- 6.Wu ZQ, Guo QL, You QD, Zhao L, Gu HY. Gambogic acid inhibits proliferation of human lung carcinoma SPC-A1 cells in vivo and in vitro and represses telomerase activity and telomerase reverse transcriptase mRNA expression in the cells. Biol Pharm Bull. 2004;27(11):1769–74. doi: 10.1248/bpb.27.1769. [DOI] [PubMed] [Google Scholar]
- 7.Yu J, Guo QL, You QD, et al. Gambogic acid-induced G2/M phase cell-cycle arrest via disturbing CDK7-mediated phosphorylation of CDC2/p34 in human gastric carcinoma BGC-823 cells. Carcinogenesis. 2007;28(3):632–8. doi: 10.1093/carcin/bgl168. [DOI] [PubMed] [Google Scholar]
- 8.Kasibhatla S, Jessen KA, Maliartchouk S, et al. A role for transferrin receptor in triggering apoptosis when targeted with gambogic acid. Proc Natl Acad Sci U S A. 2005;102(34):12095–100. doi: 10.1073/pnas.0406731102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pandey MK, Sung B, Ahn KS, Kunnumakkara AB, Chaturvedi MM, Aggarwal BB. Gambogic acid, a novel ligand for transferrin receptor, potentiates TNF-induced apoptosis through modulation of the nuclear factor-{kappa}B signaling pathway. Blood. 2007 doi: 10.1182/blood-2007-03-079616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kerbel RS, Kamen BA. The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer. 2004;4(6):423–36. doi: 10.1038/nrc1369. [DOI] [PubMed] [Google Scholar]
- 11.Bello L, Carrabba G, Giussani C, et al. Low-dose chemotherapy combined with an antiangiogenic drug reduces human glioma growth in vivo. Cancer Res. 2001;61(20):7501–6. [PubMed] [Google Scholar]
- 12.Man S, Bocci G, Francia G, et al. Antitumor effects in mice of low-dose (metronomic) cyclophosphamide administered continuously through the drinking water. Cancer Res. 2002;62(10):2731–5. [PubMed] [Google Scholar]
- 13.Brunelleschi S, Penengo L, Santoro MM, Gaudino G. Receptor tyrosine kinases as target for anti-cancer therapy. Curr Pharm Des. 2002;8(22):1959–72. doi: 10.2174/1381612023393530. [DOI] [PubMed] [Google Scholar]
- 14.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9(6):669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 15.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol. 2006;7(5):359–71. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 16.Masson-Gadais B, Houle F, Laferriere J, Huot J. Integrin alphavbeta3, requirement for VEGFR2-mediated activation of SAPK2/p38 and for Hsp90-dependent phosphorylation of focal adhesion kinase in endothelial cells activated by VEGF. Cell Stress Chaperones. 2003;8(1):37–52. doi: 10.1379/1466-1268(2003)8<37:ivrfva>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang R, Xu Y, Ekman N, et al. Etk/Bmx transactivates vascular endothelial growth factor 2 and recruits phosphatidylinositol 3-kinase to mediate the tumor necrosis factor-induced angiogenic pathway. J Biol Chem. 2003;278(51):51267–76. doi: 10.1074/jbc.M310678200. [DOI] [PubMed] [Google Scholar]
- 18.Holmes K, Roberts OL, Thomas AM, Cross MJ. Vascular endothelial growth factor receptor-2: Structure, function, intracellular signalling and therapeutic inhibition. Cell Signal. 2007;19(10):2003–12. doi: 10.1016/j.cellsig.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 19.Masson VV, Devy L, Grignet-Debrus C, et al. Mouse Aortic Ring Assay: A New Approach of the Molecular Genetics of Angiogenesis. Biol Proced Online. 2002;4:24–31. doi: 10.1251/bpo30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaseb AO, Chinnakannu K, Chen D, et al. Androgen receptor and E2F-1 targeted thymoquinone therapy for hormone-refractory prostate cancer. Cancer Res. 2007;67(16):7782–8. doi: 10.1158/0008-5472.CAN-07-1483. [DOI] [PubMed] [Google Scholar]
- 21.Saffran DC, Raitano AB, Hubert RS, Witte ON, Reiter RE, Jakobovits A. Anti-PSCA mAbs inhibit tumor growth and metastasis formation and prolong the survival of mice bearing human prostate cancer xenografts. Proc Natl Acad Sci U S A. 2001;98(5):2658–63. doi: 10.1073/pnas.051624698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shibuya M. Vascular endothelial growth factor (VEGF)-Receptor2: its biological functions, major signaling pathway, and specific ligand VEGF-E. Endothelium. 2006;13(2):63–9. doi: 10.1080/10623320600697955. [DOI] [PubMed] [Google Scholar]
- 23.Nikitenko L, Boshoff C. Endothelial cells and cancer. Handb Exp Pharmacol. 2006;176(Pt 2):307–34. doi: 10.1007/3-540-36028-x_10. [DOI] [PubMed] [Google Scholar]
- 24.Petrovic N, Schacke W, Gahagan JR, et al. CD13/APN regulates endothelial invasion and filopodia formation. Blood. 2007;110(1):142–50. doi: 10.1182/blood-2006-02-002931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patan S. Vasculogenesis and angiogenesis. Cancer Treat Res. 2004;117:3–32. doi: 10.1007/978-1-4419-8871-3_1. [DOI] [PubMed] [Google Scholar]
- 26.Tozer GM, Kanthou C, Baguley BC. Disrupting tumour blood vessels. Nat Rev Cancer. 2005;5(6):423–35. doi: 10.1038/nrc1628. [DOI] [PubMed] [Google Scholar]
- 27.Guo Q, Qi Q, You Q, Gu H, Zhao L, Wu Z. Toxicological studies of gambogic acid and its potential targets in experimental animals. Basic Clin Pharmacol Toxicol. 2006;99(2):178–84. doi: 10.1111/j.1742-7843.2006.pto_485.x. [DOI] [PubMed] [Google Scholar]
- 28.Kitagawa Y, Dai J, Zhang J, et al. Vascular endothelial growth factor contributes to prostate cancer-mediated osteoblastic activity. Cancer Res. 2005;65(23):10921–9. doi: 10.1158/0008-5472.CAN-05-1809. [DOI] [PubMed] [Google Scholar]
- 29.Le Boeuf F, Houle F, Huot J. Regulation of vascular endothelial growth factor receptor 2-mediated phosphorylation of focal adhesion kinase by heat shock protein 90 and Src kinase activities. J Biol Chem. 2004;279(37):39175–85. doi: 10.1074/jbc.M405493200. [DOI] [PubMed] [Google Scholar]
- 30.Rousseau S, Houle F, Kotanides H, et al. Vascular endothelial growth factor (VEGF)-driven actin-based motility is mediated by VEGFR2 and requires concerted activation of stress-activated protein kinase 2 (SAPK2/p38) and geldanamycin-sensitive phosphorylation of focal adhesion kinase. J Biol Chem. 2000;275(14):10661–72. doi: 10.1074/jbc.275.14.10661. [DOI] [PubMed] [Google Scholar]
- 31.Lamalice L, Le Boeuf F, Huot J. Endothelial cell migration during angiogenesis. Circ Res. 2007;100(6):782–94. doi: 10.1161/01.RES.0000259593.07661.1e. [DOI] [PubMed] [Google Scholar]
- 32.Brazil DP, Park J, Hemmings BA. PKB binding proteins. Getting in on the Akt. Cell. 2002;111(3):293–303. doi: 10.1016/s0092-8674(02)01083-8. [DOI] [PubMed] [Google Scholar]
- 33.Pinto MM, Sousa ME, Nascimento MS. Xanthone derivatives: new insights in biological activities. Curr Med Chem. 2005;12(21):2517–38. doi: 10.2174/092986705774370691. [DOI] [PubMed] [Google Scholar]
- 34.Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4(5):361–70. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- 35.Laramee M, Chabot C, Cloutier M, et al. The scaffolding adapter Gab1 mediates vascular endothelial growth factor signaling and is required for endothelial cell migration and capillary formation. J Biol Chem. 2007;282(11):7758–69. doi: 10.1074/jbc.M611327200. [DOI] [PubMed] [Google Scholar]
- 36.Wilhelm S, Carter C, Lynch M, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5(10):835–44. doi: 10.1038/nrd2130. [DOI] [PubMed] [Google Scholar]
- 37.Brown JR, DuBois RN. COX-2: a molecular target for colorectal cancer prevention. J Clin Oncol. 2005;23(12):2840–55. doi: 10.1200/JCO.2005.09.051. [DOI] [PubMed] [Google Scholar]
- 38.Garbisa S, Biggin S, Cavallarin N, Sartor L, Benelli R, Albini A. Tumor invasion: molecular shears blunted by green tea. Nat Med. 1999;5(11):1216. doi: 10.1038/15145. [DOI] [PubMed] [Google Scholar]
- 39.Fassina G, Vene R, Morini M, et al. Mechanisms of inhibition of tumor angiogenesis and vascular tumor growth by epigallocatechin-3-gallate. Clin Cancer Res. 2004;10(14):4865–73. doi: 10.1158/1078-0432.CCR-03-0672. [DOI] [PubMed] [Google Scholar]
- 40.Albini A, Tosetti F, Benelli R, Noonan DM. Tumor inflammatory angiogenesis and its chemoprevention. Cancer Res. 2005;65(23):10637–41. doi: 10.1158/0008-5472.CAN-05-3473. [DOI] [PubMed] [Google Scholar]
- 41.Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3(9):745–56. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 42.He Y, Luo Y, Tang S, et al. Critical function of Bmx/Etk in ischemia-mediated arteriogenesis and angiogenesis. J Clin Invest. 2006;116(9):2344–55. doi: 10.1172/JCI28123. [DOI] [PMC free article] [PubMed] [Google Scholar]