

Abstract

C,D-Ring symmetric chlorins 8 were prepared in 47-85% yield, on scales up to several hundred milligrams, by condensation of appropriately substituted bis-formyldihydro-dipyrrins 6 and dihydropyrromethane bis-carboxylic acids 7 in 5% TFA/CH2Cl2 (25 examples). Target chlorins were chosen to systematically probe the effect of lipophilic and hydrophilic substituents on tissue partitioning and cellular membrane penetration in photodynamic therapy (PDT).
Introduction
The chlorins are a class of 18π-electron aromatic tetrapyrroles formally derived from porphyrins by saturation of a peripheral double bond (cf. ring A, Figure 1). Chlorophyll a (1, R = phytyl) is the most ubiquitous example, serving as a light harvesting chromophore in photosynthetic plants, algae, and cyanobacteria (certain bacteriochlorophylls perform a similar function in phototrophic bacteria).1 However, a number of lesser known chlorins also play important biological roles. Cyclopheophorbide (2) and related species are thought to inhibit damaging oxidative processes in certain marine species, including Darwinella oxeata (a sponge), the short-necked clam Ruditapes philippinarum and the scallop Patinopecten yessoensis.2 Also, the structurally unique chlorin bonellin (3) is a hormone responsible for sexual differentiation in the marine worm Bonella viridis.3 Finally, in addition to their natural functions synthetically derived chlorins have attracted significant attention in both medical and materials sciences. Due to their favorable photophysical properties chlorins show promise in tumor photodynamic therapy (PDT), a technique that employs photostimulated production of singlet oxygen to selectively eradicate malignant tissue.4 In a relatively new area of exploration materials engineers have studied this ring system as a chromophore in artificial photosynthesis.5
FIGURE 1.
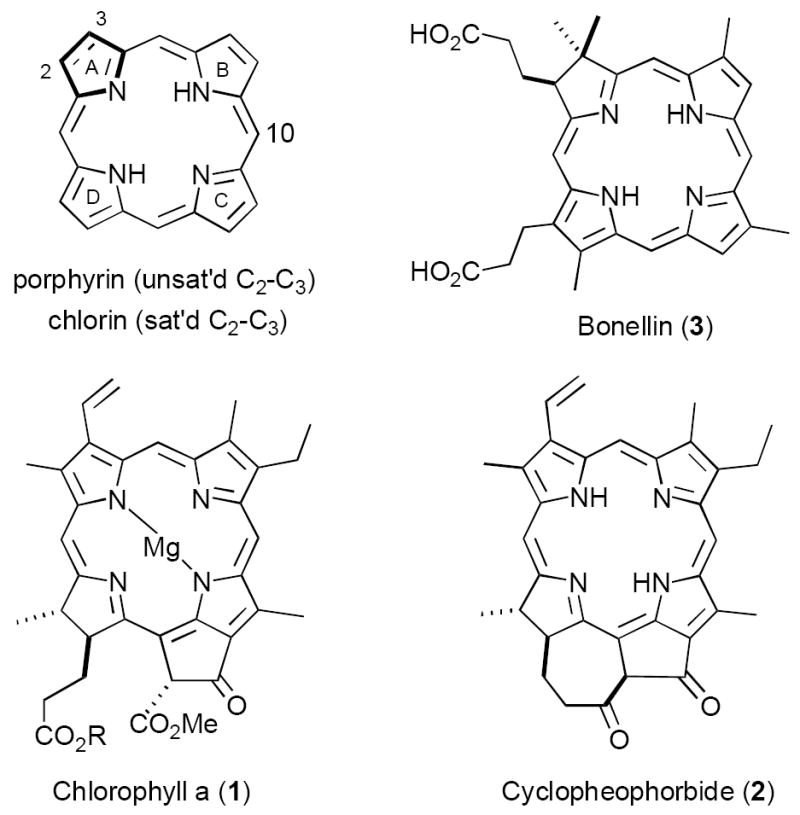
Some naturally occurring chlorins
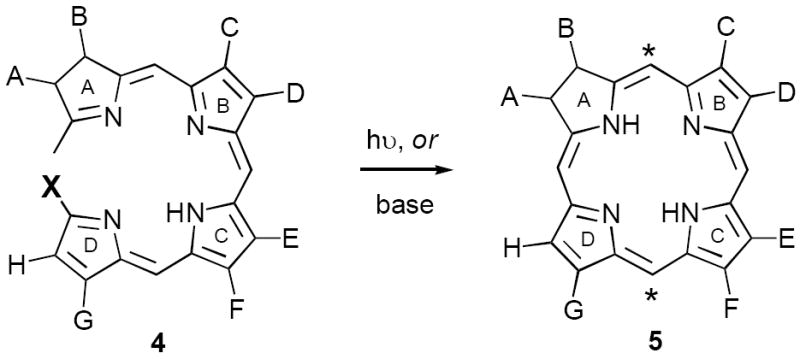
Most de novo syntheses of chlorins are modeled on the methodology of Battersby6a and Montforts,6b involving either photochemical or alkali induced ring closure of properly substituted linear tetrapyrroles 4 (Scheme 1; X = OMe, Br, etc.).7 Tetrapyrroles 4 are derived from simpler ring systems employing techniques such as sulfide contraction,6b,8 thio-Wittig reaction,9 and reductive cyclization of pyrrole-substituted nitroketones.10 While elegant in concept the cyclization of 4 to 5 can be problematic and is typically carried out on small scales, employing metal templates, and affording 5 in modest - good yields.6,8
SCHEME 1.
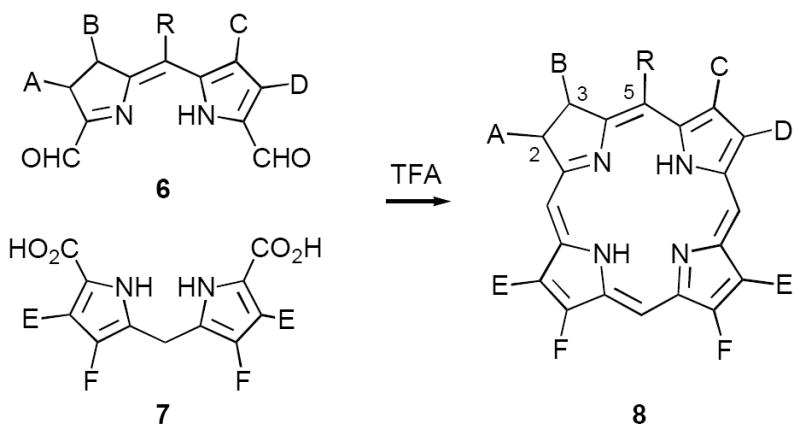
In 2001 we described in communication form a new synthesis of chlorins based upon a variant of the MacDonald porphyrin synthesis, involving condensation of bis-formyl-dihydrodipyrrins 6 with symmetrical dipyrromethanes 7 (Scheme 2).11 Simple dissolution of 6 and 7 in neat TFA was found to afford chlorins 8 in 35-45% yield with no special precautions against air or light and without metal complexation. A key feature of this approach is that the chlorin chromophore is obtained directly in the proper oxidation state and with no need for subsequent isomerization. We believe this characteristic accounts in large part for the simplicity of the experimental conditions. Since dipyrromethanes of type 7 are readily available,12 we have devoted much effort since our original paper developing improved synthetic routes to A,B-ring dialdehydes 6, incorporating diverse substituents A-D and in particular meso-groups R (C5 in 8). Typically meso-substituents have been the most challenging to introduce employing existing methodology, which is unfortunate since their properties can significantly influence tissue partitioning in PDT.13 In this paper we describe versatile new syntheses of these materials as well as greatly improved conditions for their conversion to C,D-ring symmetric chlorins 8.
SCHEME 2.
Results and Discussion
I. Synthesis of bis-Formyl Derivatives 6
Method A
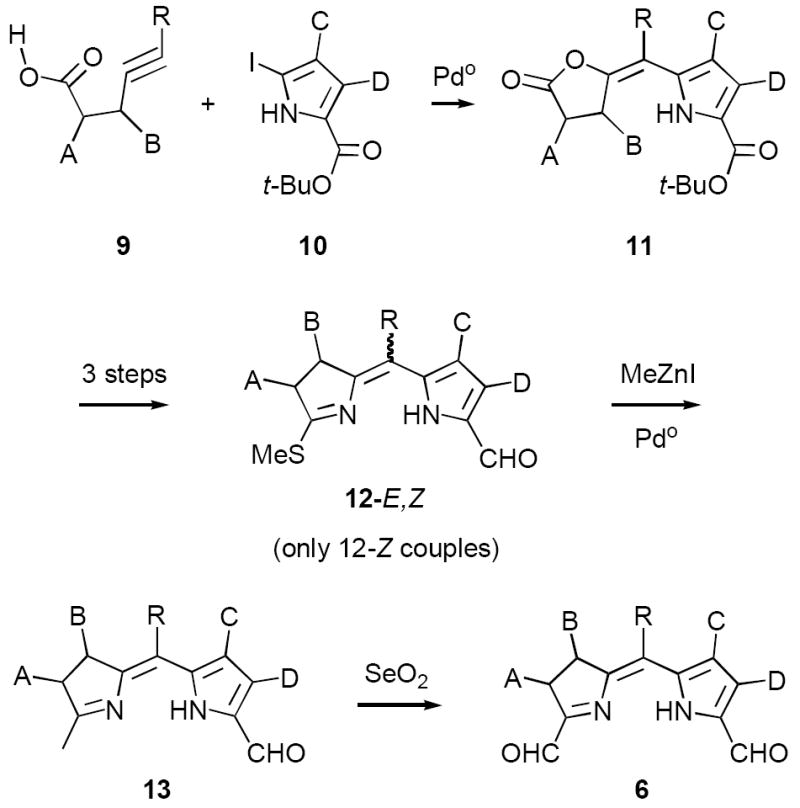
Our original route to bis-formyl-dihydrodipyrrins 6 built upon the ready availability of enelactones 11, prepared in 70-96% yield from alkyne acids 9 and iodopyrroles 10 (Scheme 3).11 Lactones 11 were then converted to E,Z-mixtures of thioimidates 12 by a three step sequence consisting of (1) aminolysis, (2) thiolactam formation, and (3) concomitant decarboxylative formylation/S-methylation employing trimethylorthoformate (TMOF) in neat TFA.9b We intended to convert both isomers of 12 to the corresponding dihydrodipyrrins 13 by transition metal-catalyzed methylation. In practice this transformation worked well with Z-thioimidates 12-Z employing the reagent system Pd(0)/MeZnI. The resulting Z-dihydrodipyrrins 13-Z were then oxidized in good yield to the desired diformyl derivatives 6. Surprisingly, however, the corresponding E-thioimidates 12-E were unreactive toward methylation using Pd(0)/MeZnI and most other commonly employed cross-coupling techniques (trace quantities of 13 were produced with Ni(II) catalysts). Eventually this difference was traced to a selective activating effect of Zn, which serves to polarize the thioimidate C-S bond in 12-Z by chelation.14 In the event this reactivity pattern had far reaching consequences, since the ratio of 12-Z:12-E decreased dramatically with increasing size of R (i.e. H > Me ≫ Ph), making it impractical to incorporate larger meso-substituents. Because of this we were able to prepare only a very limited number of chlorin precursors 6.
SCHEME 3.
Method B
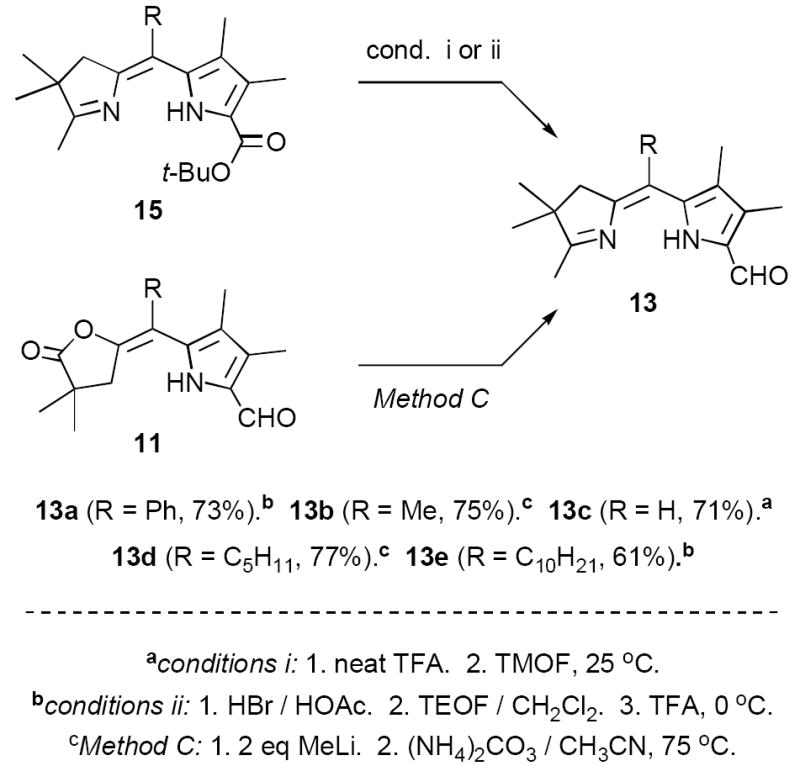
Subsequently we found that lactones 11 undergo clean condensation with the Petasis reagent (TiCp2Me2), affording high yields of enol ethers 14 incorporating a wide range of meso-substituents R (Scheme 4).15 This discovery provided the basis for a very streamlined synthesis of dihydrodipyrrins 15, which were formed in “one pot” by hydrolysis of 14 to the corresponding diketones, followed by condensation with an appropriate ammonia source. Importantly, the Petasis reagent was compatible with commonly occurring functional groups such as propionate esters (D = CH2CH2CO2Me), as well as regioisomeric substitution patterns in ring A (A,B = di-Me, di-H). Although prepared in a different context dihydrodipyrrins 15 were also attractive precursors to diformyl derivatives 6, via decarboxylative formylation to give the previously employed intermediates 13. As in Scheme 3 we expected this transformation to be straightforward, involving pre-treatment of 15 with TFA followed shortly thereafter with TMOF (conditions i). Unexpectedly, though, this seemingly well-precedented step proved to be highly capricious, affording 13 in yields ranging from 8% to 69% depending partly upon the nature of meso-substituents R. Only with R = H were we able to obtain reproducible yields in the range of ~70%. In part this behavior may be due to competitive protonation at the basic pyrroline ring nitrogen in 16, thereby inhibiting decarboxylation and/or formylation (i.e., 16-H+ → 17-H+ → 13). Battersby et al. experienced similar difficulties with a closely related ring system during the course of a synthesis of bonellin (3).10c
SCHEME 4.
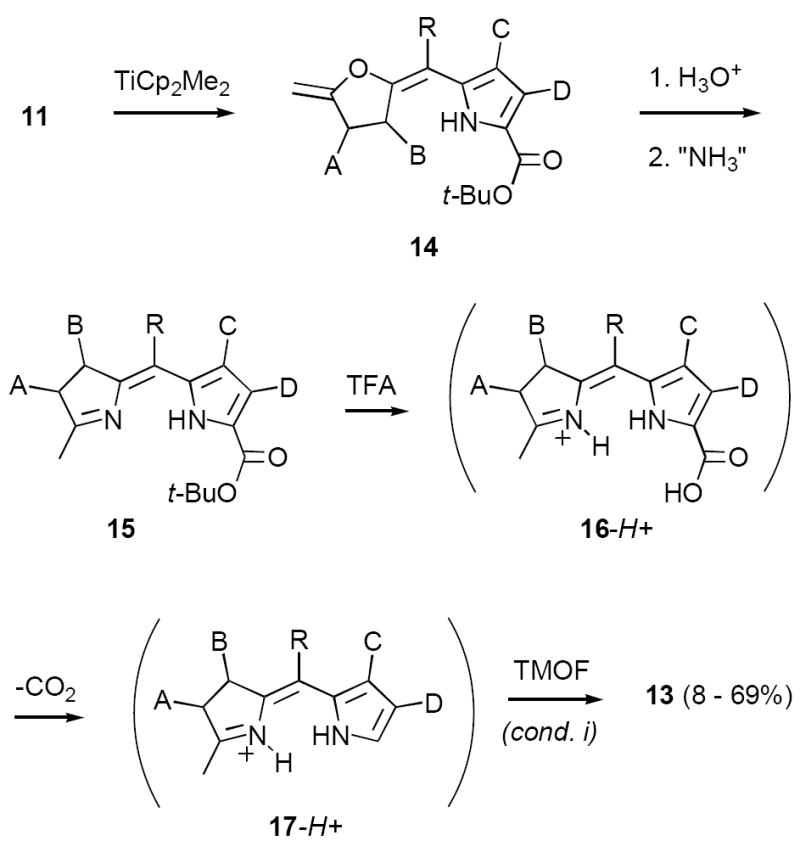
We explored many approaches for improving the conversion of 15 to 13, initially employing meso-phenyl substrate 15a in combination with variants of the standard protocol (TFA/TMOF, Scheme 5).16 As above, these experiments gave erratic yields of the desired formylated compound 13a accompanied by significant decomposition. In most cases the only identifiable by-product was the trifluoromethyl ketone 18a (13-18%). Interestingly, we could find no trace of the presumed intermediates 16a and 17a in the crude reaction mixtures, and it remains an open question whether formylation precedes or follows decarboxylation. Battersby et al. also observed trifluoromethyl ketone formation in their studies cited above,10c but the origin of this compound was not investigated. In the case of ketone 18a two observations provided insight into this question. First, 15a gave essentially identical yields of 18a upon treatment with TFA alone, seemingly eliminating any mechanistic role for TMOF in the formation of this compound. Second, 17a prepared independently (vide infra) reacted only very slowly with TFA under conditions that led to rapid conversion of 15a to 18a. Together these results suggest that a likely intermediate in the conversion of 15a to 18a is the mixed anhydride 19a, which might be transformed to 18a in either inter- or intramolecular fashion. In any event it was clear that decarboxylation of 15a with TFA would inevitably lead to substantial loss of starting material to this side reaction.
SCHEME 5.
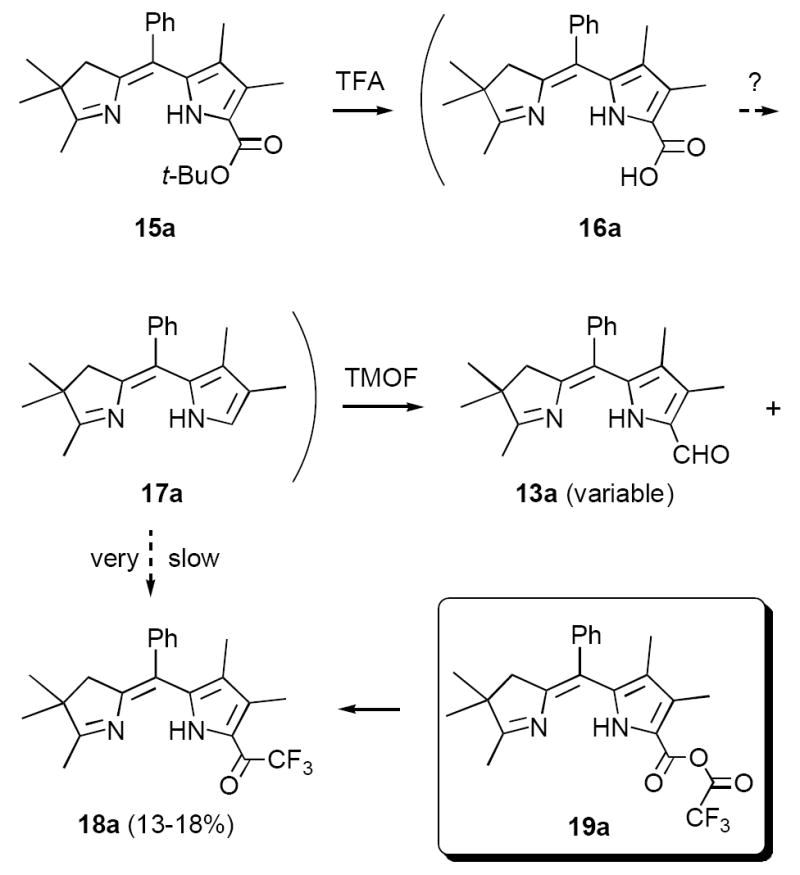
In an effort to circumvent this problem we briefly explored the possibility of reversing the order of decarboxylative formylation and oxidation (Scheme 6). Our thought was that the electron withdrawing formyl group in 20a would decrease the basicity of the pyrroline ring, thereby facilitating protonation of the pyrrole ring. However, although we obtained reasonable yields in the conversion of 15a to 20a (~65%), all attempts at decarboxylative formylation produced only the carboxylic acid 21a along with decomposition products. Decarboxylation also failed with very strong acid combinations such as HBr/HOAc.17 These results are reminiscent of those observed with simple pyrrole carboxylic acids bearing electron withdrawing groups.18
SCHEME 6.
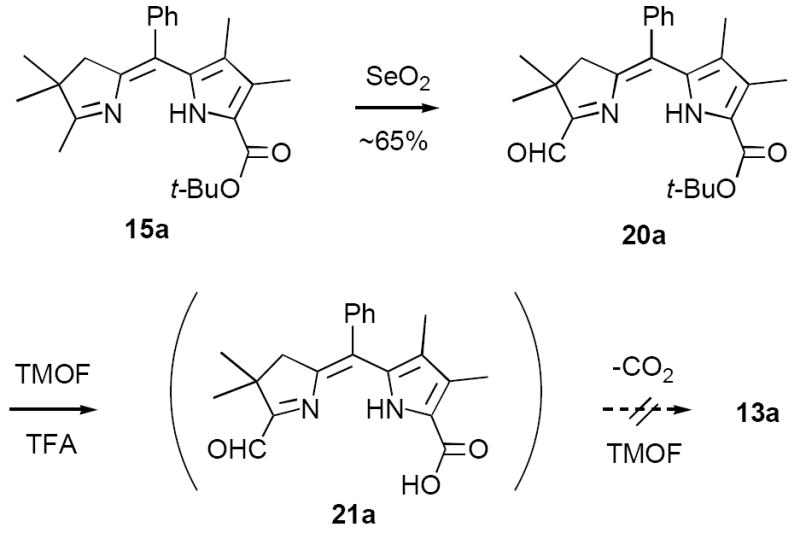
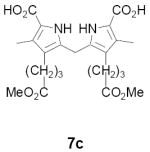
In contrast to formyl derivative 20a, the less electron deficient methylpyrroline 15a underwent clean decarboxylation with HBr/HOAc, affording a 96% yield of the α-free pyrrole 17a as a mixture of Z- and E-isomers (Table 1). With decarboxylation no longer an issue we examined a number of milder reagents for effecting the desired formylation of 17a to 13a. Of these, only the Vilsmeier reagent (POCl3/DMF) exhibited modest promise, affording a 43% yield of 13a (Z-isomer only).19 Employing TMOF/TFA the yields of 13a were actually lower than those obtained by direct decarboxylative formylation of 15a to 13a (vide supra). Mainly this was because 17a was unstable to prolonged exposure to TFA. In experiments to minimize this problem we found that 13a-Z,E was obtained in 73% yield when a solution of crude 17a in CH2Cl2/triethylorthoformate (TEOF) was added to pre-cooled TFA (conditions ii). These conditions provided reproducible results and were also satisfactory for preparing formyl derivatives 15d (R = C5H11, 57%) and 15e (R = C10H21, 61%). However, for substrates bearing small meso-substituents even these conditions proved to be too harsh (15b,c).
TABLE 1.
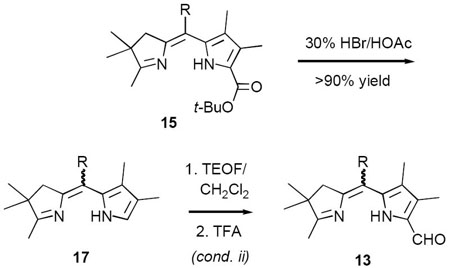 | ||
|---|---|---|
| 15 | R | Overall yield 13-Z,E |
| a | Ph | Z (64%) E (9%) |
| b | Me | 25% combined |
| c | H | <15% combined |
| d | C5H11 | Z only (57%) |
| e | C10H21 | Z only (61%) |
Method C
In parallel studies we were exploring a third strategy for synthesizing 13 that provided additional flexibility. Based on mechanistic considerations we expected that enelactones 11 would be good substrates for decarboxylative formylation, since in these compounds there is no pyrroline ring to compete for protonation (Method C, Scheme 7). In agreement, 11 routinely afforded 75-95% yields of formyl derivatives 22 with TEOF/TFA (E-isomers only). We further hoped that 22 might undergo a selective Petasis reaction at the lactone carbonyl group, as previously observed with ester-lactones 11 (cf. Scheme 4). However, all attempts at effecting this conversion produced complex mixtures. Previously we had examined the ring opening of ester-lactones 11 with alkyl lithium species Li-Nu, but in most cases were unable to prevent multiple addition (an exception was LiCH2TMS, which is very sterically hindered).15 Nevertheless we attempted the same transformation with formyl-lactones 22 and were pleased to find that these species underwent selective ring opening with a variety of nucleophiles Li-Nu (22 → 23 → 24). Of particular interest, reaction of 22c with lithiodithiane (Li-B) cleanly produced the protected glyoxaldehyde derivative 24cB (74%), while lithioanisole (Li-C) gave a 78% yield of ring-opened species 24bC. Both 24cB and 24bC held out the possibility of attaining the desired oxidation state of 6 directly.20 Even Li-Me (Li-A), possessing little if any steric hindrance, showed excellent chemoselectivity, affording 74-80% yields of 24aA-eA upon slow addition at -78° C. At present we have no rigorous explanation for this selectivity, although it is likely that the acidic pyrrole N-H group plays a role both in protecting the formyl group and in preventing multiple addition (two equivalents of Li-Nu are required for complete reaction). As a working hypothesis we suggest that the C-4 ketone deriving from 22 is stabilized in a chelated structure of type 23, maintaining the enolate tautomer until acid workup.
SCHEME 7.
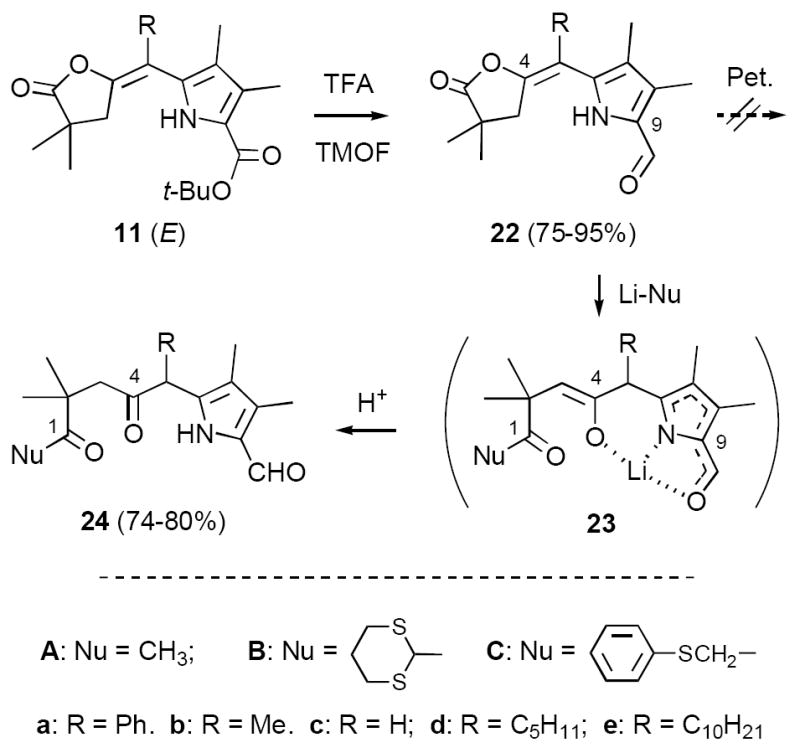
Unfortunately we were unable to effect the desired ring closure of diketones 24cB or 24bC despite intensive efforts over a period of many months (Scheme 8). Dithiane 24cB was typically recovered intact following aminolysis, most likely due to steric hindrance at the C-1 carbonyl group (X-ray analysis subsequently confirmed this suspicion; cf. Scheme 8 and Supporting Information). In contrast, thioanisole derivative 24bC underwent rapid aldol condensation to cyclopentenone 25 under mildly basic conditions (83%), reflecting the increased acidity of the C-20 methylene protons (chlorin numbering).
SCHEME 8.
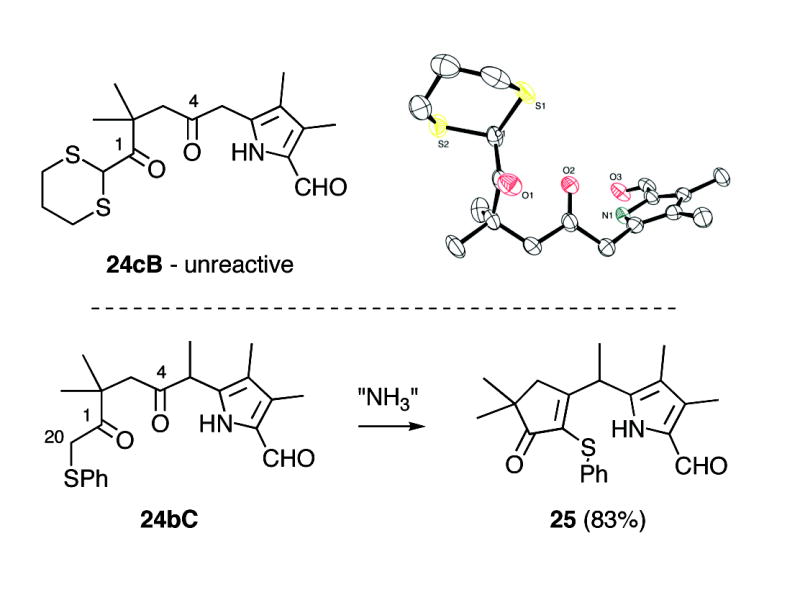
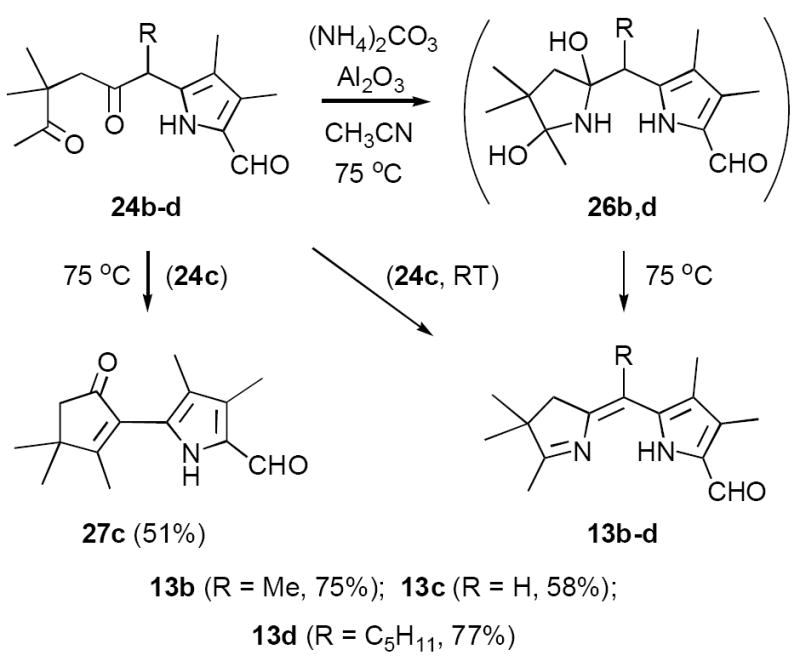
Even simple diketone derivatives 24a-e presented a number of hurdles to cyclization, partly because of the presence of the very sensitive formyl group (Scheme 9). Formylpyrroles as a class are generally unstable to acids,21 a property that was especially pronounced with 24a-e. These substrates underwent rapid decomposition with even weakly acidic ammonium salts. In contrast, basic or neutral sources of ammonia cleanly afforded mixtures of hemiaminal intermediates 26, but these species were resistant to dehydration and easily reverted to diketones 24. Working with substrate 24b (R = Me) we eventually found that the combination of (NH4)2CO3/basic Al2O3 in CH3CN at 75 °C gave ~75% yields of the corresponding dihydrodipyrrin 13b, a significant improvement over Method B (cf. Table 1). In analogous fashion diketone 24d (R = n-pentyl) afforded 13d in much improved yield (77%). However, the relatively unhindered substrate 24c (R = H) required special care. This material was susceptible to competing aldol condensation, and upon aminolysis at 75 °C gave predominantly the aldol product 27c (51%) along with only 18% of dihydrodipyrrin 13c. After further experimentation we found that the aldol pathway was greatly reduced by effecting aminolysis at RT for 24 hrs before briefly heating to 75°C, affording 13c in 58% yield along with 17% of 27c. However, 13c was still best prepared using conditions i (cf. Scheme 4). Finally, we were surprised to find that the meso-Ph diketone 24a underwent significant decomposition upon attempted aminolysis, since in related examples the Ph-substituent appeared to have a stabilizing influence.15 In any event dihydrodipyrrin 13a was best prepared using conditions ii, as was the n-decyl derivative 13e (cf. Table 1). All of these results are summarized in Scheme 10. Because of the unpredictable effect of meso-substituents R on substrate reactivity/stability a truly general synthesis of formyl derivatives 13 remains elusive. However, this investigation culminated in two complementary routes to these key intermediates that together are far more efficient than our original approach.11
SCHEME 9.
SCHEME 10.
Oxidation of Pyrrolines 13 to bis-Formyl-dihydrodipyrrins 6
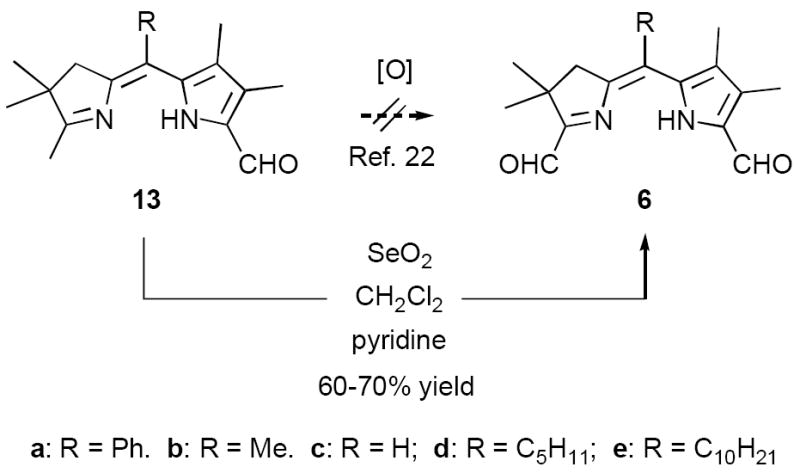
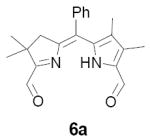
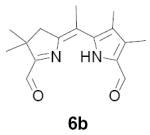
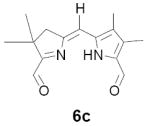
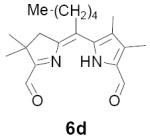
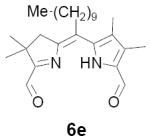
In our original studies we carried out the conversion of formyldihydrodipyrrins 13a-c to dialdehydes 6a-c by oxidation with selenium dioxide (cf. Scheme 3).11 While this procedure generally gave satisfactory yields of 6a-c, the final products were invariably contaminated with selenium metal that was very difficult to separate. Among other consequences this made obtaining accurate analytical data difficult and no doubt also impacted on subsequent steps. Because of this we spent considerable time exploring other means of accomplishing the desired oxidation. The literature contains many examples of similar oxidations employing a wide variety of reagents. Of these we screened 15-20 procedures that appeared to offer the most promise (see reference 22 for a complete listing).22-24 Generally speaking these fell into three categories: (1) direct oxidation using powerful reagents of the Cr(VI), Co(II), Cu(II), and Pb(IV) families;23 (2) oxidation initiated by bis-halogenation (NCS, NBS, SOCl2, etc.) followed by hydrolysis;24 and (3) methyl anion generation followed by in situ capture with oxidants including diselenides, disulfides, NBS, etc.24b In several cases we examined both stoichiometric and catalytic variants of the literature procedures. The results were uniformly discouraging, with some reagents exhibiting little reactivity while others caused rapid decomposition (Scheme 11). Ultimately we returned to the SeO2 procedure, exploring the effect of reagent purity, additives, and co-oxidants. To summarize briefly, freshly sublimed SeO2 provided no apparent advantage,25 nor did so-called “wet” solvents described in the literature as having beneficial effects (in our case even trace amounts of water accelerated decomposition).26 We did, however, observe very clean reactions using dry CH2Cl2 as solvent with 1-2 equivalents of pyridine as additive (the rate accelerating effect of pyridine is well documented).26 Employing 13b (R = Me) as a substrate we consistently obtained yields of 6b in the range of 65-70% following this protocol. While promising, though, ICP-MS analysis indicated that the product was still contaminated with up to 0.61% by weight (2 mol%) selenium. At this time we became aware of a report describing the removal of colloidal selenium by brief heating in DMF, which produces a black metallic allotrope that can be easily removed by filtration.27 When this step was applied to the crude oxidation product prepared as above, the resultant precipitate was easily removed, the overall yield remained essentially the same, and 6b was obtained in a high state of purity. In analogous fashion diformyl derivatives 6a-e were prepared in 60-70% yield without further optimization.
SCHEME 11.
II. Chlorin Formation
The experiments described above, while affording bis-formyl-dihydrodipyrrins 6 in much improved yields, would be of lesser value if the final condensation of 6 and dipyrromethanes 7 to give chlorins 8 were not of comparable efficiency. Our original conditions for this reaction called for dissolution of 6 and 7 in neat TFA, which was deemed necessary to initiate decarboxylation of 7 to the presumably more reactive α-unsubstituted derivatives (conditions a, cf. Scheme 2).11 This protocol afforded 35-45% yields of a limited number of chlorins 8. However, with greater quantities of 6 available, we began more detailed studies that revealed that our assumption pertaining to decarboxylation was not valid.
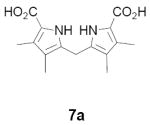
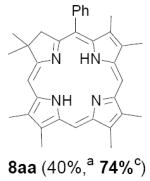
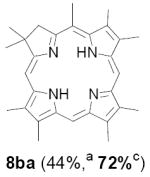
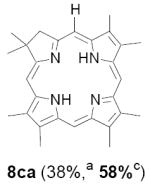
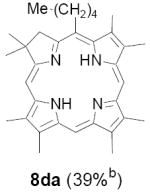
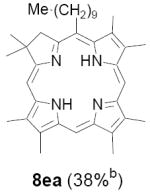
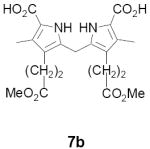
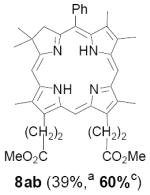
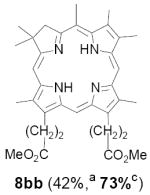
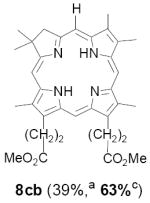
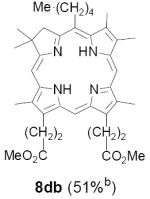
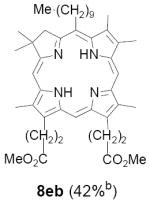
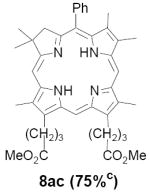
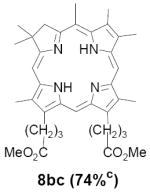
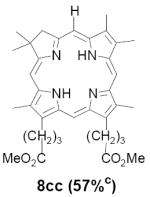
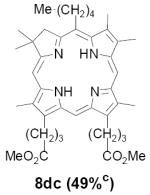
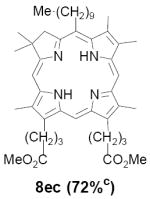
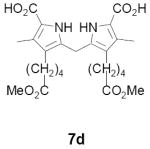
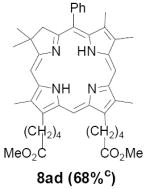
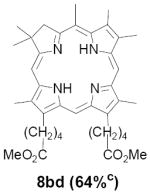
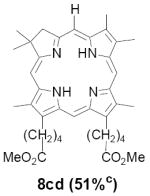
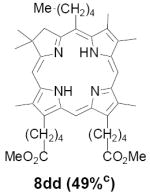
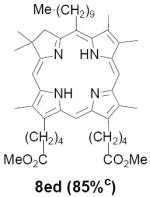
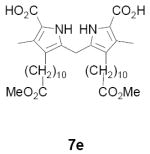
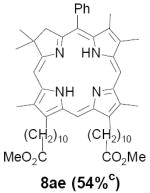
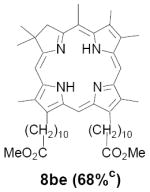
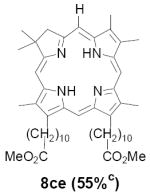
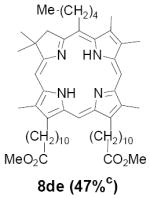
As representative substrates we first examined the stability of dihydrodipyrrin 6b and dipyrromethane 7b in neat TFA, now aware of the potential side reactions that this solvent might induce (Scheme 12). Under these conditions 6b decomposed within minutes at ambient temperature. Dipyrromethane 7b afforded moderate yields of the bis-decarboxylated product 29b along with significant quantities of trifluoroacylated derivative 28b,28 mirroring our experience with dihydrodipyrrins 15 outlined in Scheme 5. Interestingly, though, isolated and purified dipyrromethane 29b, for which we expected trifluoroacylation to be minimized (vide supra), gave lower yields of chlorin 8bb than dicarboxylic acid 7b under otherwise identical conditions (33% vs 42%). To explore this result further we screened numerous solvent and acid combinations with 29b, including such Lewis acids as TiCl4, BF3, and Sc(OTf)3 that have been successfully employed in similar condensations.29 In the present case these catalysts caused significant decomposition and little or no chlorin 8bb could be detected by TLC and/or UV analysis. More promising results were obtained with Bronsted acids, in particular TsOH in MeOH/CH2Cl2 (conditions b),30 although these conditions proved unreliable for larger scale syntheses of 8bb. Based on these experiments it appeared that decarboxylation was at the least not a prerequisite to chlorin formation, raising the possibility that dicarboxylic acid 7b might be a superior condensation partner under more refined conditions. Working from this premise we eventually identified the combination of 5% TFA in CH2Cl2 as a particularly effective medium for condensation,31 affording chlorin 8bb in a remarkably high state of purity following washing with either 3M NH4OH or saturated KHCO3 (conditions c). Final purification was by silica gel chromatography, which consistently afforded 65-75% yields of 8bb on scales up to several hundred milligrams (typically, however, reactions were run on 25-50 mg scale for convenience). In analogous fashion we prepared chlorin derivatives 8aa - 8ee bearing lipophilic substituents ranging up to n-decyl at C5, along with aliphatic esters of chain length 2-10 at C13 and C17 (Table 2; the potential usefulness of a carboxylate functionality is described below). Yields employing conditions c ranged from 47-85% and are shown in bold. Where applicable yields employing conditions a and b are also included.
SCHEME 12.
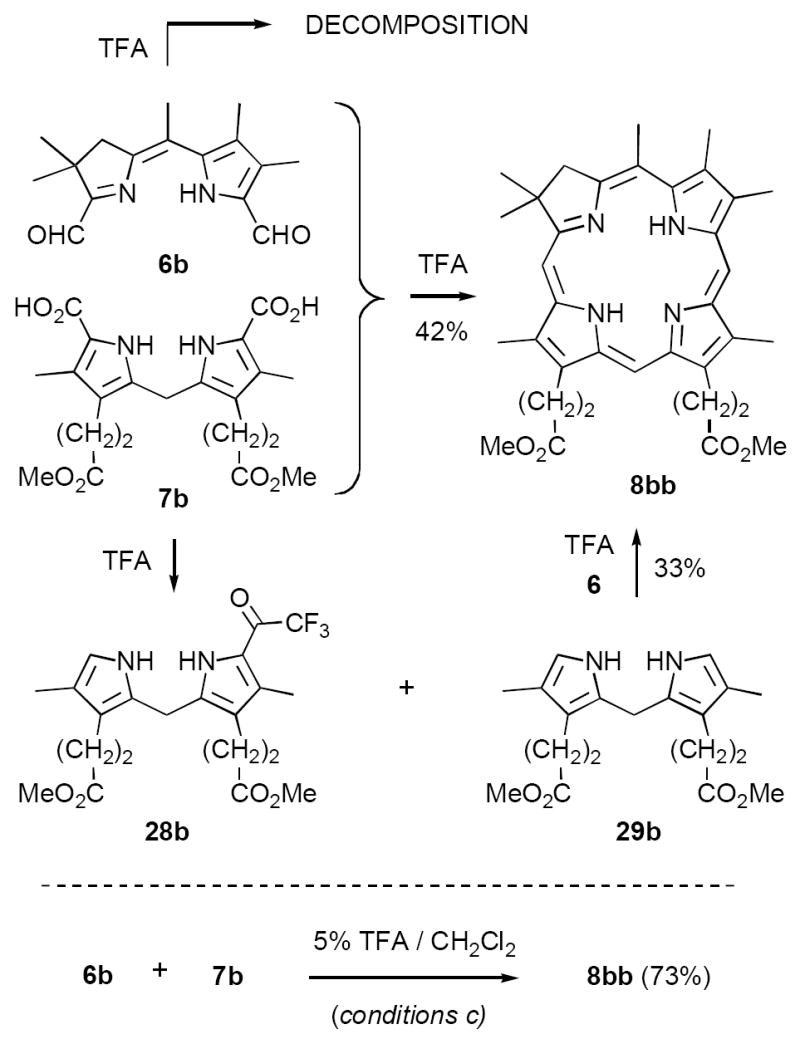
TABLE 2.
| A-B rings
C-D rings |
 |
 |
 |
 |
 |
|---|---|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
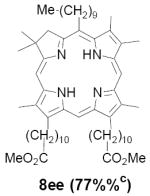
|
Conditions: a: neat TFA. b: TsOH/MeOH/CH2Cl2. c: 5% TFA in CH2Cl2.
An important component of these studies was to prepare C,D-ring symmetric chlorins 8 of a substitution pattern that could be useful in probing structure-activity relationships in photodynamic therapy (PDT, vide supra). It has been known for many years that lipophilic substituents can exert a strong influence on such critical parameters as drug uptake, intracellular location and photosensitizing efficacy.13 Moreover, as recently shown by Ehrenberg et al.,32 carboxylate “anchors” of the type incorporated in rings C and D in 8 might be tailored in length to either minimize or maximize cellular membrane penetration, and thus overall exposure to photogenerated 1O2 (Figure 2). Chlorins 8aa-ee in Table 2, incorporating both a lipophilic “head” and a hydrophilic “tail”, provide an attractive starting point for testing this hypothesis. Finally, the described methodology should also be suitable for preparing chlorins of potential interest in materials science. It is worth noting, though, that with C,D-ring symmetric chlorins 8 our synthetic objectives were simplified by the fact that it was not necessary to differentiate either the formyl groups in 6 or the α-pyrrole positions in dipyrromethanes 7. The application of this “2 + 2” strategy to the synthesis of unsymmetrical chlorins presents additional challenges that will be the subject of a forthcoming paper.
FIGURE 2.
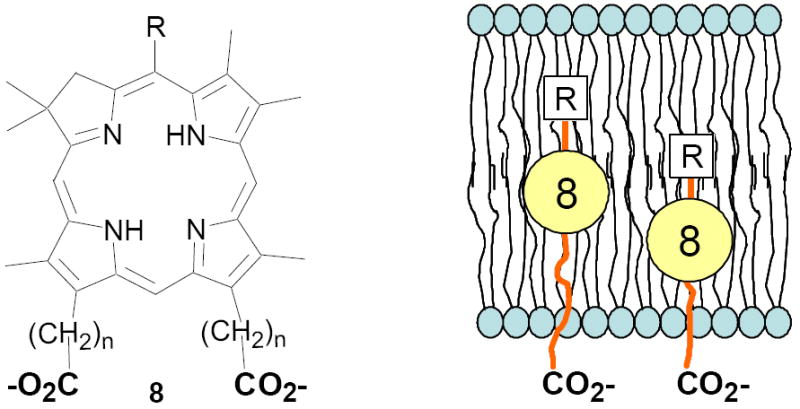
Schematic representation of the effect of tether length -(CH2)n- on cellular membrane penetration. The hydrophilic carboxylate groups “anchor” at the aqueous interface.
Experimental Section
Representative procedures for the synthesis of chlorins 8
5-[1-(4,4-Dimethyl-5-oxo-dihydro-furan-2-ylidene)-ethyl]-3,4-dimethyl-1H-pyrrole-2-carbaldehyde (22b)
TFA (8.80 mL, 118 mmol) was added to 11b (1.0 g, 3.0 mmol) under a nitrogen atmosphere, and the solution was stirred vigorously for 5 min. The reaction was cooled to 0°C for 10 min and then treated with triethylorthoformate (2.7 mL, 16.4 mmol). After 20 min, the reaction was poured into 10% aq KH2PO4 (40 ml) at 0°C and extracted with CH2Cl2 (3 × 15 ml). The combined organic extracts were washed sequentially with water and saturated aq NaHCO3, dried over Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography (silica gel, EtOAc:hexanes = 3:7) to give 22b (695 mg, 89%) as a colorless crystalline solid, mp 172.5-174 °C. Rf (4:6 EtOAc/hexanes) 0.41; IR(thin film) 3253, 1799, 1700, 1628, 1079 cm-1; 1H NMR (500 MHz, CDCl3) δ 1.27 (s, 6H), 1.91 (s, 3H), 2.01 (t, J=1.71, 3H), 2.27 (s, 3H), 2.58 (q, J=1.71, 2H), 9.54 (s, 1H), 9.74 (br s, 1H); 13C NMR (500 MHz, CDCl3) δ 9.2, 9.7, 16.1, 25.0, 40.5, 40.5, 104.9, 119.2, 129.2, 132.5, 136.3, 146.7, 177.0, 180.0. Anal. Calcd for C15H19NO3: C, 68.94; H, 7.33; N, 5.36; O, 18.37. Found: C, 68.54; H, 7.28; N, 5.33.
3,4-Dimethyl-5-(1,4,4-trimethyl-2,5-dioxo-hexyl)-1H-pyrrole-2-carbaldehyde (24b)
A solution of 22b (1.77 g, 6.77 mmol) in THF (130 mL) was cooled to -78°C and treated dropwise with 1.6M MeLi in Et2O (8.40 mL, 13.5 mmol) over 20 min. The reaction was then quenched by addition to 200 mL of saturated aq NH4Cl. After allowing the mixture to warm to rt, Et2O (50 mL) was added and the layers were separated. The organic layer was washed sequentially with water and brine, dried over Na2SO4, filtered and concentrated. The resulting yellow oil was purified by flash chromatography (silica gel, EtOAc:hexanes = 3:7) to give 24b (1.50 g, 80%) as a colorless crystalline solid, mp 103-104 °C. Rf (2:3 EtOAc/hexanes) 0.25; IR(thin film) 3253, 1706, 1628 cm-1; 1H NMR (500 MHz, CDCl3) 1.12 (s, 3H), 1.13 (s, 3H), 1.36 (d, J=7.3, 3H), 1.93 (s, 3H), 2.16 (s, 3H), 2.25 (s, 3H), 2.59 (d, J=17.9, 1H), 2.87 (d, J=17.9, 1H), 3.89 (q, J=7.1, 1H), 9.53 (s, 1H), 9.74 (br s, 1H); 13C NMR (500 MHz, CDCl3) δ 8.6, 9.0, 15.7, 25.1, 25.5, 25.7, 44.5, 45.9, 51.2, 118.5, 128.9, 132.4, 136.1, 176.8, 207.5, 213.3. HRMS (EI) Calcd for C16H23NO3: 277.1678; found: 277.1678; Anal. Calcd for C16H23NO3: C, 69.29; H, 8.36; N, 5.05; O, 17.31. Found: C, 69.44; H, 8.35; N, 4.96.
3,4-Dimethyl-5-[1-(4,4,5-trimethyl-3,4-dihydro-pyrrol-2-ylidene)-ethyl]-1H-pyrrole-2-carbaldehyde (13b)
A solution of 24b (1.35 g, 4.9 mmol) in CH3CN (53 mL) was treated with (NH4)2CO3 (4.6 g, 49 mmol) and basic alumina (6.7 g, Grade I), flushed with nitrogen, and heated to 75°C in a sealed flask for 8 hrs. At the end of this period, the mixture was cooled to rt, filtered, and concentrated. The residue was dissolved in CH2Cl2, washed with water, dried over Na2SO4, filtered and concentrated. The resulting oil was purified by flash chromatography (silica gel, EtOAc:hexanes = 1:2) to give 13b (940 mg, 75%) as a crystalline solid, mp 156-157 °C. Rf (1:2 EtOAc/hexanes) 0.27; IR(thin film) 3252, 1635, 1589 cm-1; 1H NMR (500 MHz, CDCl3) 1.20 (s, 6H), 2.11 (s, 3H), 2.16 (s, 3H), 2.17 (s, 3H), 2.26 (s, 3H), 2.58 (s, 2H), 9.58 (s, 1H), 11.77 (s, 1H); 13C NMR (500 MHz, CDCl3) δ 8.8, 11.2, 15.7, 17.9, 26.0, 43.8, 47.8, 113.3, 118.9, 128.3, 131.4, 136.9, 151.6, 176.0, 187.2. HRMS (EI) Calcd for C16H22N2O: 258.1732; found: 258.1726; Anal. Calcd for C16H22N2O: C, 74.38; H, 8.58; N, 10.84; O, 6.19. Found: C, 74.41; H, 8.72; N, 10.78.11
5-[1-(5-Formyl-4,4-dimethyl-3,4-dihydro-pyrrol-2-ylidene)-ethyl]-3,4-dimethyl-1H-pyrrole-2-carbaldehyde (6b)
A solution of 13b (0.80 g, 3.1 mmol) in CH2Cl2 (62 mL) and pyridine (0.37 mL, 3.7 mmol) was treated with SeO2 (0.41 g, 3.7 mmol) and stirred at rt for 2 h. The solvent was then removed by rotary evaporation, and the residue was redissolved in DMF (30 mL) and heated to 80°C for 15 min. The reaction mixture was cooled to rt, filtered, and poured into water (100 mL). The solution was extracted with CH2Cl2 (4 × 30 mL), and the combined organic extracts washed sequentially with saturated aq NaHCO3 and brine, dried over Na2SO4, filtered, and concentrated. The resulting oil was purified by flash chromatography (silica gel, EtOAc:hexanes = 2:3) to give 6b (599 mg, 71%) as a yellow crystalline solid, mp 135-137 °C. Rf(1:2 EtOAc/hexanes) 0.34; IR (thin film) 3302, 1687, 1635 cm-1; 1H NMR (500 MHz, CDCl3) 1.37 (s, 6H), 2.20 (s, 3H), 2.22 (s, 3H), 2.28 (s, 3H), 2.73 (s, 2H), 9.66 (s, 1H), 9.94 (s, 1H), 11.32 (br s, 1H); 13C NMR (500 MHz, CDCl3) δ 8.9, 11.8, 19.3, 26.1, 46.0, 46.3, 122.0, 125.3, 130.0, 131.2, 135.3, 152.0, 177.4, 177.5, 190.5; HRMS (EI) Calcd for C16H19N2O2: 271.1447; found: 271.1450; Anal. Calcd for C16H20N2O2: C, 70.56; H, 7.40; N, 10.29; O, 11.75. Found:C, 70.45; H, 7.42; N, 10.12.11
3-[18-(2-Methoxycarbonyl-ethyl)-3,7,8,10,13,13,17-heptamethyl-12,13,22,24-tetrahydro-porphin-2-yl]-propionic acid methyl ester (8bb)
Nitrogen was bubbled through a suspension of 6b (201 mg, 0.74 mmol) and 7b (321 mg, 0.74 mmol) in CH2Cl2 (67 mL) for 10 min. The mixture was treated with TFA (3.36 mL) and stirred at rt in the dark for 24 h. The reaction was then poured into 3M aqueous NH4OH (30 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (30 mL). The combined organic layers were washed sequentially with water and brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography (silica gel, EtOAc:hexanes = 1:5) to give 8bb (313 mg, 70%) as a green crystalline solid that was identical to the literature compound.11
Supporting Information Available
Experimental details, characterization data and NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
Financial support of this work by the National Institutes of Health, NIGMS Grant No. GM38913 is gratefully acknowledged. We are grateful to Victor G. Young, Jr. and the X-Ray Crystallographic Laboratory, Department of Chemistry, University of Minnesota for X-ray analysis of 24cB. William G. O’Neal is the recipient of an NSF predoctoral fellowship.
References
- 1.Montforts F-P, Glasenapp-Breiling M. In: Progress in the Chemisty of Organic Natural Products. Herz W, Falk H, Kirby GW, editors. Vol. 84. SpringerWienNew York; New York: 2002. pp. 1–51. [DOI] [PubMed] [Google Scholar]
- 2.(a) Karuso P, Berguquist PR, Buckleton JS, Cambie RC, Clark GR, Rickard CEF. Tetrahedron Lett. 1986;27:2177–2178. [Google Scholar]; (b) Watanabe N, Yamamoto K, Ihshikawa H, Yagi A. J Nat Prod. 1993;56:305–317. [Google Scholar]
- 3.Agius L, Ballantine JA, Ferrito V, Jaccarini V, Murray-Rust P, Pelter A, Psaila AF, Schembri PJ. Pure Appl Chem. 1979;51:1847. [Google Scholar]
- 4.Bonnett R. Chemical Aspects of Photodynamic Therapy. Gordon and Breach; Amsterdam: 2000. [Google Scholar]
- 5.(a) Wasielewski Michael R, Wiederrecht Gary P, Svec Walter A, Niemczyk Mark P. Sol Energy Mater Sol Cells. 1995;38:127–134. [Google Scholar]; (b) Tamiaki H, Yagai S. J Photosci. 2002;9:66–69. [Google Scholar]
- 6.(a) Battersby AR, Dutton CJ, Fookes CJR, Turner SPD. J Chem Soc Perkin Trans. 1988;1:1557–1567. [Google Scholar]; (b) Montforts F-P. Angew Chem Int Ed Engl. 1981;20:778–779. [Google Scholar]; (c) Montforts F-P, Gerlach B, Höper F. Chem Rev. 1994;94:327–347. [Google Scholar]; (d) Montforts F-P, Glasenapp-Breiling M. Prog Heterocycl Chem. 1998;10:1–24. [Google Scholar]
- 7.For other representative synthetic approaches to the chlorin macrocycle see: Burns DH, Li YH, Shi DC, Caldwell TM. J Org Chem. 2002;67:4536. doi: 10.1021/jo020105f.Gryko DT, Galęzowski M. Org Lett. 2005;7:1749–1752. doi: 10.1021/ol050327a.
- 8.Montforts F-P, Schwartz UM. Liebigs Ann Chem. 1991:609. [Google Scholar]
- 9.(a) Battersby AR, Turner SPD, Block MH, Sheng Z-C, Zimmerman SC. J Chem Soc Perkin Trans. 1988;1:1577. [Google Scholar]; (b) Arnott DM, Harrison PJ, Henderson GB, Sheng Z-C, Leeper FJ, Battersby AR. J Chem Soc Perkin Trans. 1989;1:265. [Google Scholar]
- 10.(a) Harrison PJ, Sheng Z-C, Fookes CJR, Battersby AR. J Chem Soc Perkin Trans. 1987;1:1667. [Google Scholar]; (b) Battersby AR, Cutton CJ, Fookes CJR, Turner SPD. J Chem Soc Perkin Trans. 1988;1:1557. [Google Scholar]; (c) Battersby AR, Dutton CJ, Fookes CJR. J Chem Soc Perkin Trans. 1988;1:1569. [Google Scholar]; (d) Strachan J-P, O’Shea DF, Balasubramanian T, Lindsey JS. J Org Chem. 2000;65:3160. doi: 10.1021/jo991942t. [DOI] [PubMed] [Google Scholar]; (e) Balasubramanian T, Strachan J-P, Boyle PD, Lindsey JS. J Org Chem. 2000;65:7919. doi: 10.1021/jo000913b. [DOI] [PubMed] [Google Scholar]; (f) Taniguchi M, Ra D, Mo G, Balasubramanian T, Lindsey JS. J Org Chem. 2001;66:7342. doi: 10.1021/jo0104835. [DOI] [PubMed] [Google Scholar]; (g) Ptaszek M, Bhaumik J, Kim H-J, Taniguchi M, Lindsey JS. Org Process Res Dev. 2005;9:651–659. doi: 10.1021/op050087e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacobi PA, Lanz S, Ghosh I, Leung SH, Lower F, Pippin D. Org Lett. 2001;3(6):831. doi: 10.1021/ol006983m. [DOI] [PubMed] [Google Scholar]
- 12.For an overview see: Smith KM. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 1. Academic Press; San Diego: 2000. p. 4.Jackson AH, Pandey RK, Smith KM. J Chem Soc Perkin Trans. 1987;1:299–305.Fischer H, Orth H. Die Chemie des Pyrrols. Vol. 1. Akademische Verlagsgesellschaft; Leipzig: 1934. p. 333.Mironov AF, Ovsepyan TR, Evstigneeva RP, Preobrazenskii NA. Zh Obshch Khim. 1965;35:324.Freeman BA, Smith KM. Synth Commun. 1999;29:1843–1855.
- 13.Pandey RK, Zheng G. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 6. Academic Press; San Diego: 2000. pp. 157–230. [Google Scholar]
- 14.Ghosh I, Jacobi PA. J Org Chem. 2002;67:9304. doi: 10.1021/jo026510o. [DOI] [PubMed] [Google Scholar]
- 15.O’Neal WG, Roberts WP, Ghosh I, Jacobi PA. J Org Chem. 2005;70:7243–7251. doi: 10.1021/jo050907l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clezy PS, Fookes CJR, Liepa AJ. Aust J Chem. 1972;25:1979–1990. [Google Scholar]
- 17.Neya S, Ohyama K, Funasaki N. Tetrahedron Lett. 1997;38:4113–4116. [Google Scholar]
- 18.Jackson AH. The Chemistry of Heterocyclic Compounds. In: Jones RA, editor. Pyrroles. Part 1. Vol. 48. John Wiley & Sons; New York: 1990. p. 315. [Google Scholar]
- 19.Mueller-Westerhoff UT, Swiegers GF. Synth Commun. 1994;24:1389–1393. [Google Scholar]
- 20.(a) Greene TW, Wuts PGM. Protective Groups in Organic Synthesis. John Wiley & Sons; New York: 1999. pp. 333–340. [Google Scholar]; (b) Bakuzis P, Bakuzis MLF, Fortes CC, Santos R. J Org Chem. 1976;41:2769–2770. [Google Scholar]
- 21.Jackson AH. The Chemistry of Heterocyclic Compounds. In: Jones RA, editor. Pyrroles. Part 1. Vol. 48. John Wiley & Sons; New York: 1990. p. 316. [Google Scholar]
- 22.(a) Reagents that were screened include: PCC, PDC, CAN, DDQ, MnO2, CrO3, Pb(OAc)4, Co(OAc)2, CuI/TBHP, CuCl2/TBHP, Pd/TBHP, Pd(OAc)2/ PhI(OAc)2, (SePh)2/PhIO2, t-BuI/FeCl2/DMSO, CuCl2/LiCl, NCS/NBS, SO2Cl2. Leading references are included for certain reagents.
- 23.Co(II): Salvador JAR, Clark JH. Chem Commun. 2001:33–34. Cu(I): Salvador JAR, Melo MLSá e, Neves ASC. Tetrahedron Lett. 1997;38:119–122. Cu(I): Arsenou ES, Koutsourea AI, Fousteris MA, Nikolaropoulos SS. Steroids. 2003;68:407–414. doi: 10.1016/s0039-128x(03)00042-4. Cu(II): Rothenburg G, Feldberg L, Wiener H, Sasson Y. J Chem Soc Perkin Trans. 1998;2:2429–2434. Pd/TBHP: Yu J-Q, Corey EJ. Org Lett. 2002;4:2727–2730. doi: 10.1021/ol0262340. Pd(OAc)2/ PhI(OAc)2: Dick AR, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:2300–2301. doi: 10.1021/ja031543m. (SePh)2/PhIO2 Barton DHR, Crich D. Tetrahedron. 1985;41:4359. t-BuI/FeCl2/DMSO: Vismara E, Fontana F, Minisci F. Gazz Chim Ital. 1987;117:135–136.
- 24.CuCl2/LiCl: Nobrega JA, Gonçalves SMC, Peppe C. Synth Commun. 2002;32:3711–3717. NCS/NBS/SO2Cl2: Tehrani KA, Borremans D, De Kimpe N. Tetrahedron. 1999;55:4133–4152.
- 25.Kaplan H. J Am Chem Soc. 1941;63:2654–2655. [Google Scholar]
- 26.Trachtenberg EN. In: Oxidation. Augustine RL, editor. Marcel Dekker; New York: 1969. pp. 19–187. [Google Scholar]
- 27.Milstein SR, Coats EA. Aldrichimica Acta. 1978;11:10. [Google Scholar]
- 28.Xie H, Lee DA, Wallace DM, Senge MO, Smith KM. J Org Chem. 1996;61:8508–8517. [Google Scholar]
- 29.Geier GR, III, Callinan JB, Rao PD, Lindsey JS. J Porphyr Phthalocya. 2001;5:810–823. [Google Scholar]
- 30.(a) Burns DH, Li YH, Shi DC, Caldwell TM. J Org Chem. 2002;67:4536–4546. doi: 10.1021/jo020105f. [DOI] [PubMed] [Google Scholar]; (b) Nguyen LT, Senge MO, Smith KM. J Org Chem. 1996;61:998–1003. [Google Scholar]
- 31.For use of TFA/CH2Cl2 in a related context see Lash TD. J Porphyr Phthalocya. 1997;1:29–44.Lash TD. Chem Eur J. 1996;2:1197–1200.
- 32.(a) Lavi A, Weitman H, Holmes RT, Smith KM, Ehrenberg B. Biophys J. 2002;82:2101–2110. doi: 10.1016/S0006-3495(02)75557-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bronshtein I, Afri M, Weitman H, Frimer AA, Smith KM, Ehrenberg B. Biophys J. 2004;87:1155–1164. doi: 10.1529/biophysj.104.041434. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details, characterization data and NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.