

Summary
Mdm2 regulates the p53 tumor suppressor by promoting its proteasome-mediated degradation. Mdm2 and p53 engage in an autoregulatory feedback loop that maintains low p53 activity in non-stressed cells. We now report that Mdm2 regulates p53 levels also by targeting ribosomal protein L26. L26 binds p53 mRNA and augments its translation. Mdm2 binds L26 and drives its polyubiquitylation and proteasomal degradation. In addition, the binding of Mdm2 to L26 attenuates the association of L26 with p53 mRNA and represses L26-mediated augmentation of p53 protein synthesis. Under non-stressed conditions, both mechanisms help maintain low cellular p53 levels by constitutively tuning down p53 translation. In response to genotoxic stress the inhibitory effect of Mdm2 on L26 is attenuated, enabling a rapid increase in p53 synthesis. The Mdm2-L26 interaction thus represents an additional important component of the autoregulatory feedback loop that dictates cellular p53 levels and activity.
Introduction
The p53 tumor suppressor protein is a pivotal regulator of cell fate, particularly under conditions of stress (Aylon and Oren, 2007; Harris and Levine, 2005; Marine et al., 2006; Poyurovsky and Prives, 2006; Riley et al., 2008). p53 is subject to exquisite regulation. One key regulator of p53 is the Mdm2 (mouse double minute 2) protein, which binds specifically to p53 and inhibits many of p53’s biochemical activities (Marine et al., 2006; Michael and Oren, 2003). Furthermore, as a p53-selective E3-ubiquitin ligase, Mdm2 promotes p53 polyubiquitylation and targets p53 to degradation by the 26S proteasome (Fang et al., 2000; Haupt et al., 1997; Honda et al., 1997; Kubbutat et al., 1997). As the Mdm2 gene is a transcriptional target of p53, Mdm2 and p53 form a negative feedback loop, which ensures that p53 is maintained at low levels under normal conditions (Barak et al., 1993; Lahav et al., 2004; Wu et al., 1993) and is of vital importance to cellular homeostasis. Numerous mechanisms regulate the p53-Mdm2 axis, enabling optimal coupling of the particular triggering stress with the ensuing cellular response. Under stress conditions, various mechanisms render p53 less affected by Mdm2. Such mechanisms include enhanced Mdm2 degradation, post-translational modifications on p53 and Mdm2, altered binding to other proteins that modulate the p53-Mdm2 interaction and its consequences, and altered sub-cellular localization of p53 and Mdm2 (reviewed in (Marine et al., 2006; Oren, 2003; Toledo and Wahl, 2006)).
The Mdm2 protein comprises several distinct, highly conserved regions. The N-terminal domain harbors the main p53 binding interface. Two other notable regions of Mdm2 are the central domain (amino acids ~200–300), often referred to as the acidic domain (AD), and the C-terminal RING domain (amino acids 438–478). The latter is the enzymatic heart of Mdm2, enabling its E3-ubiquitin ligase activity, while the acidic domain is a hub for many protein-protein interactions that regulate Mdm2 function (Oren 2003). The acidic domain contributes to p53 degradation in at least two distinct ways. On the one hand, it harbors an additional p53 binding site (Kulikov et al., 2006; Ma et al., 2006; Wallace et al., 2006; Yu et al., 2006), required for efficient p53 polyubiquitylation, while on the other hand it mediates a post-ubiquitylation step required for proteasomal degradation of p53 (Argentini et al., 2001), which may involve direct binding of Mdm2 to the proteasome (Sdek et al., 2005).
Mdm2 interacts with a variety of ribosomal proteins, including L5, L11, L23 and S7 (Chen et al., 2007; Dai and Lu, 2004; Dai et al., 2004; Jin et al., 2004; Lindstrom et al., 2007; Lohrum et al., 2003; Marechal et al., 1994; Zhang et al., 2003). These interactions, which typically involve the acidic domain and sometimes the adjacent zinc finger of Mdm2, interfere with the inhibitory functions of this region of Mdm2 and contribute to p53 activation. As first exemplified for L11 (Lohrum et al., 2003), these interactions increase when ribosome biogenesis is disrupted, a situation termed “ribosomal biogenesis stress” or “nucleolar stress” (Pestov et al., 2001; Rubbi and Milner, 2003). Such stress can be induced by drugs that inhibit RNA polymerase I, e.g., low levels of actinomycin D (Bhat et al., 2004; Lohrum et al., 2003), 5′-FU (Gilkes et al., 2006), or other growth inhibitory conditions such as serum starvation and contact inhibition (Bhat et al., 2004). Mechanistically, ribosomal stress causes translocation of free ribosomal proteins from the nucleolus to the nucleoplasm (Bhat et al., 2004; Lam et al., 2007), where they bind Mdm2 (Bhat et al., 2004). The increased binding of ribosomal proteins to Mdm2 augments cellular p53 activity, leading to growth arrest and coupling deficient protein synthesis with cessation of cell proliferation.
We have previously described the use of a yeast two-hybrid screen (Y2H) to discover proteins that interact with the Mdm2 acidic domain, leading to identification of the Lats2 tumor suppressor as a novel regulator of the p53-Mdm2 axis (Aylon et al., 2006). We now report, using the same Y2H approach, that the ribosomal protein L26 (RPL26) also interacts specifically with Mdm2. Interestingly, L26 is an important positive regulator of p53 expression, which binds the 5′-UTR of p53 mRNA and augments its translation exposure to DNA damage, where it contributes significantly to the stress-induced increase in p53 protein and p53 activity (Takagi et al., 2005).
We show that L26 is a substrate for Mdm2-mediated ubiquitylation and proteasomal degradation. In addition to promoting L26 degradation, the binding of Mdm2 to L26 diminishes the interaction of L26 with the 5′-UTR of p53 mRNA and compromises L26-mediated enhancement of p53 translation. Together, these findings reveal an additional mechanism whereby Mdm2 restricts cellular p53 activity and identify L26 as a novel component of the p53-Mdm2 autoregulatory loop.
Results
L26 and Mdm2 interact in mammalian cells
In search of novel Mdm2-interacting proteins, we employed the Ras recruitment yeast two hybrid (Y2H) screen (Aronheim, 2001), utilizing the central acidic domain of human Mdm2 (amino acids 229–310) as bait. One of the interacting clones contained the complete open reading frame (ORF) of the L26 protein, a component of the large ribosomal subunit. This was of particular interest in light of the finding that L26 binds to the 5′ untranslated region (5′-UTR) of p53 mRNA and regulates p53 translation in response to DNA damage (Takagi et al., 2005).
To validate the proposed interaction in a mammalian cellular environment, plasmids expressing Flag-tagged human L26 and wild type (wt) human Mdm2 were co-transfected into HEK293 cells, and cell extracts were subjected to reciprocal co-immunoprecipitation (Co-IP) analysis. As seen in Fig. 1A (top panels; input whole cell extracts shown in bottom panels), a specific interaction between Flag-L26 and transfected Mdm2 was readily apparent (lanes 4,8). Furthermore, an interaction between endogenous L26 and endogenous Mdm2 could be demonstrated in SJSA-1 cells, which express elevated amounts of Mdm2 protein owing to Mdm2 gene amplification (Oliner et al., 1992). Under standard culture conditions this interaction was rather weak, and was observable only when the immunoprecipitation was performed with anti-L26 antibodies (Fig. 1B, upper panels, lane 2). However, when the proteasome inhibitor MG132 was applied to the cells, the interaction became very robust (lanes 3,6). The increase in coprecipitation upon MG132 treatment greatly exceeded the relative overall increase in L26 and Mdm2 protein levels as assessed by analysis of whole cell extracts (WCE, lower panels, compare lane 2 to 3 and lane 5 to 6). This suggests that inhibition of the proteasome greatly enhances either the strength of the interaction or the stability of the interacting molecules.
Figure 1. Mdm2 physically interacts with L26 in mammalian cells.
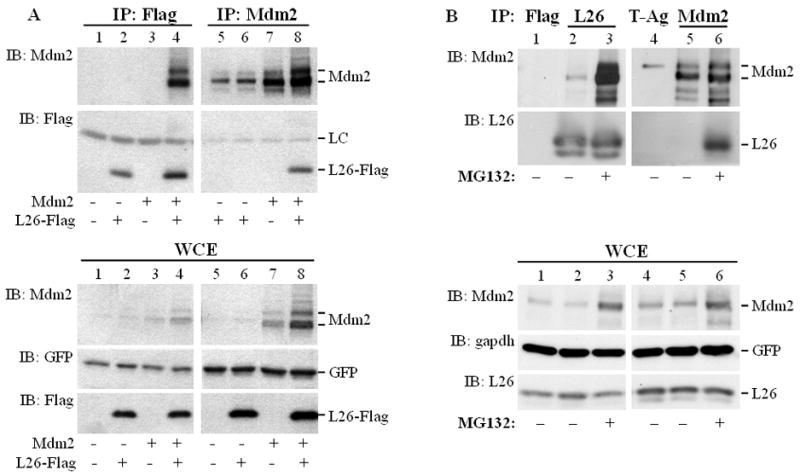
A. HEK293 cells (1.75×106 cells/10cm dish) were transiently co-transfected with the indicated combinations of expression plasmids encoding L26-Flag (6μg/dish), wild type human Mdm2 (6μg/dish), and GFP (0.3μg/dish) as a transfection control. Cells were harvested and extracted 48 hours later. Top panel: Extracts were immunoprecipitated (IP) with anti-Flag or anti-Mdm2 antibodies, and the immunoprecipitated material was resolved by SDS-PAGE and subjected to Western immunoblot analysis (IB) with the indicated antibodies. LC: antibody light chain. Bottom panel: 2.5% of each whole cell extract (WCE) was resolved by SDS-PAGE and subjected to Western blot analysis with the indicated antibodies.
B. SJSA-1 cell cultures were treated with 8μM MG132 overnight where indicated, or left untreated. The next day, cells were harvested and subjected to protein extractions. Extracts were immunoprecipitated (IP) with control (anti-Flag rabbit polyclonal, upper panel, lane 1) or anti-L26 (lanes 2, 3) antibody (two 15cm dishes/lane). For the reciprocal reaction, extracts were immunoprecipitated with control (anti-SV40 large T antigen, lane 4) or anti-Mdm2 (lanes 5, 6) antibodies (four 15cm dishes/lane). The immunoprecipitated material was resolved by SDS-PAGE and subjected to Western blot analysis with the indicated antibodies (IB). Lower panel: 1.7% of each whole cell extract was analyzed directly by SDS-PAGE, followed by Western blot analysis (IB) with the indicated antibodies.
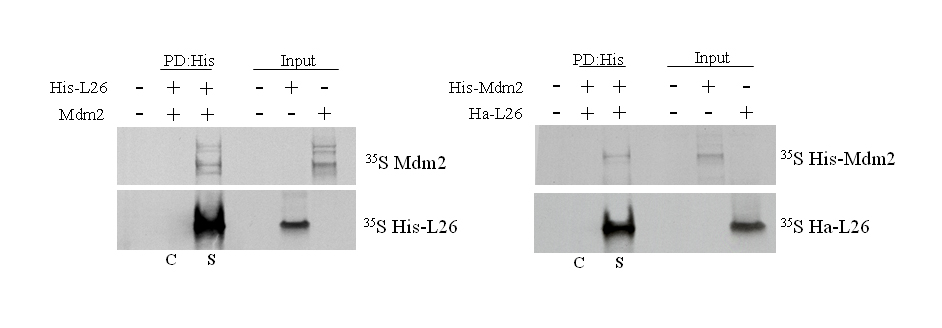
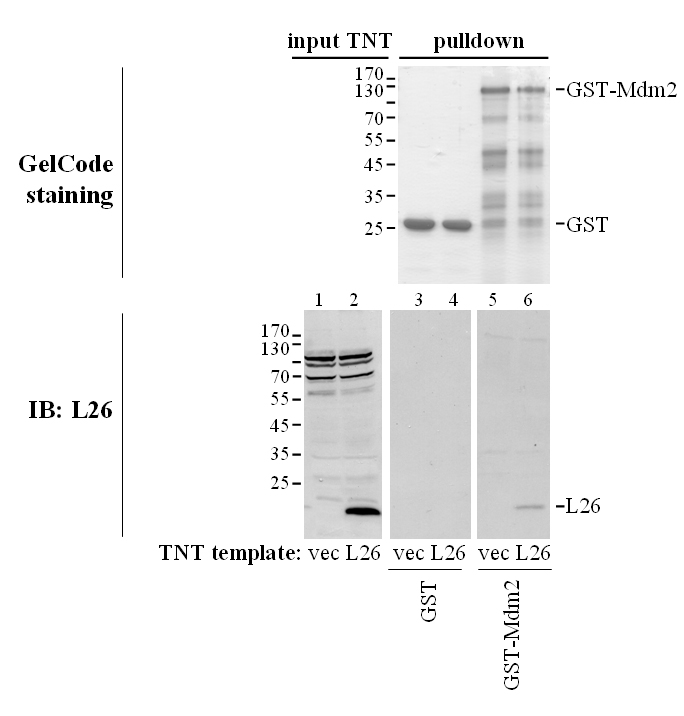
When L26 and Mdm2 were produced by in vitro translation in a reticulocyte lysate and mixed together, either protein could be coprecipitated with antibodies directed against the other one (Supplemental Fig. S1). Furthermore, GST pull-down analysis revealed that in vitro translated L26 was bound by GST-Mdm2 but not by GST (Supplemental Fig. S2); it is of note that in vitro translation was performed in that case in a wheat germ extract, to avoid the presence of any mammalian proteins in the binding reaction. Together, these results strongly support the existence of a direct protein-protein interaction between Mdm2 and L26.
To characterize the Mdm2 interaction domain, we performed Co-IP assays with two Mdm2 deletion mutants, one lacking the C-terminal RING domain (deletion of amino acids 441–491; Mdm2ΔR) and the other lacking the acidic domain (deletion of amino acids 239–301; Mdm2ΔA). Mdm2ΔR co-precipitated with Flag-L26 at least as efficiently as wtMdm2 (Supplemental Fig. S3, upper panel; compare lanes 7,14 to lanes 3,10, respectively). In contrast, deletion of the acidic domain significantly attenuated the interaction of Mdm2 with Flag-L26. When anti-Flag antibody was used in the IP, an interaction of Mdm2ΔA with Flag-L26 was detectable (Supplemental Fig. S3 upper panel, lane 5), but its intensity was somewhat reduced relative to wtMdm2 and Mdm2ΔR (compare to relative inputs, bottom panel). Strikingly, in the reciprocal experiment using anti-Mdm2 in the IP, no Flag-L26 signal was detected at all (lane 12). This suggests that Mdm2 may harbor two L26 interaction sites: a major site within the acidic domain and a weaker site elsewhere within the protein. Next, a variety of L26 deletion mutants were used in Co-IP assays, revealing that an internal domain of L26, immediately adjacent to the KOW motif (76-120aa), is critical for Mdm2 interaction (mutant D6; Supplemental Fig. S4), while neither the amino terminal portion nor the carboxyl terminal portion of L26 is required. In sum, Mdm2 associates with L26 in human cells, and this is mediated primarily via the acidic domain of Mdm2.
L26 is polyubiquitylated by Mdm2
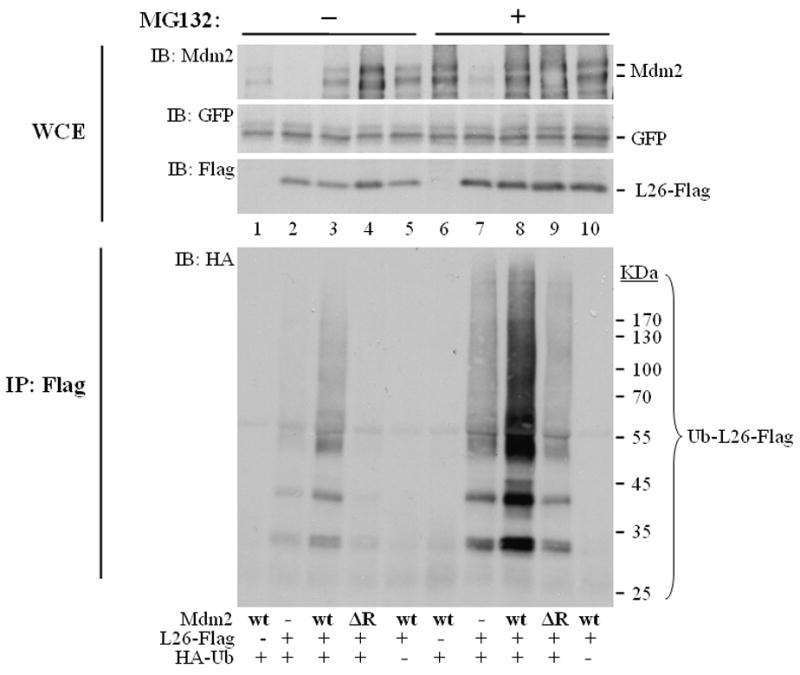
Mdm2 is an E3 ubiquitin ligase, known primarily for its ability to target p53. We noted that cotransfection of Mdm2 induced the appearance of a slower migrating form of L26, whose mobility was suggestive of monoubiquitylated L26 (asterisk in Supplemental Fig. S3, lane 3), raising the possibility that L26, too, is an Mdm2 substrate. To further explore this possibility, U2OS cells were co-transfected with L26-Flag and HA-ubiquitin together with either wtMdm2 or Mdm2ΔR, treated with MG132, and subjected to analysis of in vivo ubiquitylation of L26-Flag. WtMdm2 greatly increased L26 mono- and polyubiquitylation (Ub-L26-Flag, Fig. 2A, lane 3). In contrast, the E3-deficient Mdm2ΔR mutant failed to do so. Hence, L26 is a substrate for the E3 ligase activity of Mdm2.
Figure 2. Mdm2 promotes L26 polyubiquitylation.

A. U2OS cells were seeded at 800,000 cells/10cm dish. Twenty four hours later, cells were transfected with expression plasmids encoding L26-Flag (6μg/dish), human wtMdm2 (wt, 3μg/dish), Mdm2ΔR (ΔR, 3μg/dish), HA-ubiquitin (1.5μg/dish), and GFP (0.4μg/dish) as transfection control. After two days, cells were treated with 25μM MG132 for 4 hours, harvested and extracted in SDS lysis buffer under denaturing conditions. WCE: 5% of the extract was resolved by SDS-PAGE and subjected to Western blot analysis with the indicated antibodies. IP: the rest of the extract was immunoprecipitated with anti-Flag antibodies, and subjected to SDS-PAGE followed by Western blot analysis with anti-HA antibodies (IB: HA). The membrane was re-blotted with anti-Flag antibodies to detect immunoprecipitated non-ubiquitylated L26 (bottom panel). Positions of molecular size markers are indicated on the right.
B. U2OS cells (2×106 cells/15cm dish) were co-transfected with expression plasmids encoding L26-Flag (6.5μg/dish), Mdm2 (3.2μg/dish), HA-ubiquitin (2.7μg/dish), and GFP (0.5μg/dish) as transfection control. One day later, cells were trypsinized and separated into nuclear and cytoplasmic fractions. Extract: 4% of each fraction was resolved by SDS-PAGE and subjected to Western blot analysis with the indicated antibodies. NUC: nucleus; CYTO: cytoplasm. IP: the rest of each extract was subjected to denaturing conditions (SDS lysis buffer), immunoprecipitated with anti-Flag antibodies, and subjected to SDS-PAGE followed by Western blot analysis with anti-HA antibodies and reblotting with anti-Flag antibodies. Ub-L26-Flag: ubiquitylated L26-Flag. Positions of molecular size markers are indicated on the right.
A substantial fraction of cellular L26 is in the nucleolus, where ribosomal assembly occurs, whereas the rest is found within the cytoplasm and nucleoplasm (Takagi et al., 2005). In U2OS cells, endogenous L26 is distributed roughly equally between cytoplasm and nucleus (data not shown). Similarly, Mdm2 is distributed between cytoplasm, nucleoplasm and nucleolus. In transfected U2OS cells, most of the exogenous Mdm2 was cytoplasmic, whereas much of the exogenous L26 was in the nuclear fraction, presumably mainly in the nucleolus (Fig. 2B upper panels). Mdm2 did not affect noticeably the cytoplasmic/nuclear distribution of total Flag-L26 protein (upper panels). Consistent with the continuous ubiquitylation and degradation of ribosomal proteins within the nucleus (Lam et al., 2007), basal levels of ubiquitylated L26 were detectable in the nuclear fraction (lane 1, Fig. 2B middle panel), increasing moderately in the presence of exogenous Mdm2 (lane 2). In the absence of exogenous Mdm2, the cytoplasm was practically devoid of ubiquitylated L26 (lane 3). However, co-transfection of Mdm2 led to a dramatic increase in cytoplasmic ubiquitylated L26 (lane 4), which becomes even more impressive when one considers the overall lower amounts of L26 in that compartment (Fig. 2B, lower panel). Although it remains possible that ubiquitylation of L26 in the nucleus by Mdm2 promotes its export into the cytoplasm, the data favors the conclusion that Mdm2 preferentially targets cytoplasmic L26.
To explore whether Mdm2-mediated L26 polyubiquitylation leads to its proteasomal degradation, we compared the accumulation of ubiquitylated L26 in the absence and in the presence of MG132. Consistent with the data in Fig. 2A, cotransfection with wild type Mdm2 but not RING-deficient Mdm2 resulted in extensive accumulation of ubiquitylated L26 (Ub-L26) in presence of the proteasome inhibitor (Fig. 3, lane 8). However, without MG132 (lane 3) Ub-L26 was markedly reduced, indicating that it is subject to rapid proteasomal degradation. Total L26 was hardly affected under those conditions (IB: Flag, compare lane 2 to 3), even though it increased modestly in cells overexpressing the delta RING Mdm2 mutant, which presumably acts in a negative dominant fashion (lane 4). The reduction in Ub-L26 was more pronounced than the decrease in total L26 protein (IB: Flag, compare lane 3 to 8). Even more dramatic effects of MG132 on accumulation of ubiquitylated L26 were obtained in MCF7 breast cancer cells (Supplemental Fig. S5, compare lanes 4,5). These results are most consistent with a model wherein Mdm2 targets L26 for ubiquitylation and subsequent proteasomal degradation, but this activity is exerted only towards a minor, distinct L26 subpopulation, while the remainder of cellular L26 is not affected.
Figure 3. Mdm2 promotes the proteasomal degradation of polyubiquitylated L26.
U2OS cells were seeded at 8×105 cells/10cm dish. Twenty four hours later, cultures were transfected with expression plasmids encoding L26-Flag (3μg/dish), human wtMdm2 (wt, 2.5μg/dish), Mdm2ΔR (ΔR, 2.5μg/dish), HA-ubiquitin (1μg/dish), and GFP (0.25μg/dish) as internal transfection control. After one day, cells were treated with 8μM MG132 overnight, harvested and extracted in SDS lysis buffer under denaturing conditions. WCE: 2.5% of the extract was resolved by SDS-PAGE and subjected to Western blot analysis with the indicated antibodies. IP: the rest of the extract was immunoprecipitated with anti-Flag antibodies, and subjected to SDS-PAGE followed by Western blot analysis with anti-HA antibodies (IB: HA).
The experiment in Fig. 3 employed transfected Mdm2 and L26. We next sought to extend the findings to the endogenous setting. As shown in Fig. 4A, endogenous L26 was readily ubiquitylated by exogenous Mdm2; polyubiquitin conjugates were most prominent, but bands corresponding to the expected positions of mono- and diubiquitylated L26 (arrows) were also apparent, along with mono- and diubiquitylated p53 (Ub-p53). Furthermore, siRNA-mediated knockdown of endogenous Mdm2 greatly reduced the ubiquitylation of transfected L26 (Fig. 4B). Most notably, knockdown of endogenous Mdm2 quenched substantially the ubiquitylation of endogenous L26 (Fig. 4C); in fact, L26 polyubiquitylation (polyUb-L26) was reduced more than 2 fold in Mdm2-deprived cells. These findings argue that Mdm2 is constitutively engaged in L26 ubiquitylation, presumably driving the rapid decay of a particular subpopulation of L26. The fact that such activity was observed in both U2OS (Fig. 4A,C) and MCF7 cells (Fig. 4B) attests to the generality of this conclusion.
Figure 4. Endogenous L26 is polyubiquitylated by endogenous Mdm2.

A. Transfected Mdm2 augments endogenous L26 polyubiquitylation. U2OS cells were transfected with the indicated combinations of Mdm2 and HA-ubiquitin (HA-Ub) expression plasmids. Cells were extracted in SDS lysis buffer under denaturing conditions and subjected to immunoprecipitation (IP) with either L26-specific antibodies or anti-FLAG rabbit polyclonal antibodies as control, followed by Western blot analysis with antibodies directed against HA to detect ubiquitin conjugates. Arrows indicate bands migrating at the positions expected for monoubiquitylated and diubiquitylated L26. Upper panels: analysis of whole cell extracts (WCE) with the indicated antibodies. Ub-p53 indicates the positions of monoubiquitylated and diubiquitylated p53. GAPDH was used as loading control.
B. Knockdown of endogenous Mdm2 reduces the polyubiquitylation of transfected L26. MCF-7 cells were transfected with the indicated expression plasmid combinations, along with synthetic Mdm2-specific siRNA oligonucleotides or control siRNA. Where indicated, MG132 treatment was performed prior to cell harvesting. Following extraction in urea lysis buffer (see Experimental Procedures), lysates were precipitated with Ni-NTA magnetic agarose beads (Qiagen) and the immunoprecipitated material was resolved by SDS-PAGE and analyzed by Western blot analysis with anti-HA antibodies to visualize the precipitated ubiquitylated L26 (bottom panel, Pull-down). Upper panels: analysis of whole cell extracts (WCE).
C. Knockdown of endogenous Mdm2 reduces the polyubiquitylation of endogenous L26. U2OS cells were seeded at 1.25×106 cells/15cm dish. Twenty four hours later, cells were transfected with siRNA (SMART pool, Dharmacon) directed against Mdm2, or control siRNA (SMART control). After one day, cultures were transfected with plasmids encodig HA-ubiquitin (2.5μg/dish) and GFP. The next evening, cultures were treated with 10μM MG132 overnight. Cells were then harvested and extracted in SDS lysis buffer under denaturing conditions. WCE (upper panels): 1.8% of the extract was resolved by SDS-PAGE and subjected to Western blot analysis with the indicated antibodies. GFP was employed as a transfection efficiency control. IP (lower panels): Extracts were subjected to immunoprecipitation with anti-L26 antibodies followed by Western blot analysis with antibodies directed against HA to detect ubiquitin conjugates. The positions of total ubiquitylated L26 (Ub-L26) and of polyubiquitylated L26 are indicated. A short (SE) and long (LE) exposure of the same membrane are shown. The membrane was re-blotted with anti-L26 antibodies to quantify the total immunoprecipitated L26 protein (bottom panel, very short exposure shown). To calculate the relative extent of endogenous L26 polyubiquitylation, (Poly Ub-L26/relative), the polyubiquitination signal of L26 (polyUb-L26, short exposure) was quantified using ImageJ software, divided by the corresponding GFP signal and normalized to 1.0.
Functional consequences of Mdm2 binding to L26
To assess the impact of Mdm2 binding on L26 function, we examined whether Mdm2 could directly affect the interaction of L26 with the 5′-UTR of p53 mRNA. GFP-L26 was transfected into MCF7 cells in the absence or presence of cotransfected Mdm2 and the ability of L26 to bind to p53 mRNA was assessed by IP-RT-PCR. As reported (Takagi et al., 2005), binding of GFP-L26 to p53 mRNA in cells was readily detectable (Fig. 5, lane 2). Interestingly, in the absence of DNA damage, cotransfection of Mdm2 blunted the binding of GFP-L26 to p53 mRNA (lanes 3,4). Mdm2 itself did not associate detectably with p53 mRNA, and L26 did not bring down the irrelevant GAPDH mRNA (Supplemental Fig. S6), strongly suggesting that the binding of Mdm2 to L26 directly blocks the latter’s ability to bind p53 mRNA. Interestingly, in cells exposed to ionizing radiation (IR), which leads to rapid p53 upregulation, L26 remained bound to p53 mRNA even in the face of Mdm2 levels that were sufficient to hinder binding under basal conditions (Fig. 5, lane 7). It is conceivable that IR-induced modifications of Mdm2 (Blattner et al., 2002; Maya et al., 2001) may not only alter the impact of Mdm2 on p53 but also reduce the ability of Mdm2 to interfere with the binding of L26 to p53 mRNA.
Figure 5. Mdm2 inhibits binding of L26 to p53 mRNA.

MCF7 cells (3×106 cells/10cm dish) were co-transfected with expression plasmids encoding GFP (4μg/dish); lanes 1,5) or GFP-L26 (4μg/dish); lanes 2–4, 6–8), without or with increasing amounts of human Mdm2 (4μg or 8μg/dish) expression plasmid. The total amount of DNA was kept at 12μg by the inclusion of plain pCMV-Neo-Bam vector. Forty-eight hours post-transfection, cultures were exposed to ionizing radiation (IR; 10Gy) as indicated and harvested 30 minutes post-IR. Lysates were immunoprecipitated with anti-GFP antibodies and the amount of coprecipitated p53 mRNA was assessed by IP-RT-PCR, employing primers that map within the 5′-UTR of p53 mRNA and span an intron within the genomic human p53 DNA. PCR products were visualized by ethidium bromide staining (top panel). Protein input for each IP reaction was resolved by SDS-PAGE and assessed by Western blot analysis with the indicated antibodies (bottom panels).
Direct inhibition by Mdm2 of L26 binding to p53 mRNA may be predicted to abrogate L26-mediated enhancement of p53 translation. This possibility was tested in a rabbit reticulocyte system that recapitulates the positive effects of L26 protein on p53 translation. As previously noted (Takagi et al., 2005), addition of L26 markedly increased the expression of p53 in this in vitro transcription/translation system (Fig. 6A, lanes 5,6), but this did not occur with p53 mRNA lacking its 5′-UTR (lanes 1,2). Addition of Mdm2 reduced L26-enhanced translation of p53 in a dose-dependent manner (lanes 7,8). This effect of Mdm2 was not due to degradation of p53 protein since the reticulocyte lysates were pretreated with MG132; furthermore, Mdm2 did not affect the level of p53 protein synthesized from p53 mRNA lacking the 5′-UTR (lanes 3,4), which is translated independently of L26 binding (Takagi et al., 2005). Moreover, p53 mRNA levels were similar under all conditions (Supplemental Fig. S7). Hence, Mdm2 interferes specifically with the ability of L26 to augment the translation of p53 mRNA.
Figure 6. Mdm2 inhibits L26-mediated enhancement of p53 translation in vitro.

PCR products consisting of a T7 promoter-tagged p53 cDNA harboring a 1nt or 110nt 5′UTR, L26 cDNA and human Mdm2 cDNA (wild type (wt), RING domain deletion (ΔR), acidic domain deletion (ΔA)) were in vitro transcribed/translated in rabbit reticulocyte lysates in the presence of [35S] methionine. Reticulocyte lysates were pre-treated with 50μM MG132 for 15 minutes at room temperature prior to the addition of PCR-generated cDNAs. Lysates were resolved by SDS-PAGE and subjected to Western blot analysis with the indicated antibodies.
A. Effect of L26 on translation of p53 mRNA harboring a 1 or 110nt 5′UTR in the absence or presence of increasing wtMdm2 (1:1 and 1:2 ratio of L26 cDNA to Mdm2 cDNA). Amounts of total L26, including the endogenous L26 of the reticulocyte lysate, is shown in the middle panel. Equal loading was assessed by Ponceau staining of the membrane (bottom panel).
B. Effect of L26 on translation of p53 mRNA harboring a 110nt 5′UTR in the absence or presence of increasing Mdm2 (wild-type, ΔR, ΔA). Analysis was as in A.
The binding domain mapping experiments (Supplemental Fig. 3) indicated that the acidic domain of Mdm2, but not the RING domain, was critical for tight interaction of Mdm2 with L26. Remarkably, while the delta RING mutant (ΔR) was at least as effective as wild-type Mdm2 in curtailing L26 enhancement of p53 translation in a reconstituted rabbit reticulocyte lysate (Fig. 6B, compare lanes 5,6 to 3,4), the acidic domain mutant (ΔA) was severely compromised (Fig. 6B, lanes 7,8). Hence, tight binding of Mdm2 to L26 is required in order for the former to block L26-mediated enhancement of p53 translation.
The in vitro observations described above are suggestive of the following in vivo scenario: under normal conditions, Mdm2 constitutively hinders L26 binding to p53 mRNA; consequently, L26 is prevented from exerting its full positive effect on p53 translation. In response to genotoxic stress, such as IR, Mdm2 is rendered less potent (see Fig. 5), allowing L26 to effectively augment p53 protein synthesis. If this model is correct, then excessive elevation of Mdm2 should be able to compensate for its reduced potency and restore the suppression of L26-mediated p53 translation. This was indeed found to be the case: when assayed in a pulse-labeling/immunoprecipitation assay, the in vivo rate of p53 translation was markedly augmented by IR exposure of the cells (Fig. 7A, lanes 1,2), consistent with earlier findings (Takagi et al., 2005). Importantly, overexpression of Mdm2 completely blunted the stimulatory effect of IR on p53 protein synthesis (lanes 7,8). This was not due to enhanced p53 degradation, since the overall amount of immunoprecipitated p53 remained unaffected under those conditions (see WB of IP: p53, second row from top). Consistent with their in vitro effects, the delta RING mutant of Mdm2 blocked effectively the IR-induced increase in cellular p53 translation whereas the delta acidic domain mutant was severely compromised in that regard (lanes 3,4 and 5,6, respectively).
Figure 7. Excess Mdm2 inhibits DNA damage-induced enhancement of p53 translation in vivo.

A. MCF7 cells were transiently transfected with the corresponding Mdm2 expression plasmids and treated with 50μM MG132 for 30 minutes prior to exposure to 10Gy ionizing radiation (IR). Twenty five minutes after IR the cells were pulse-labeled for 5 minutes with [35S]methionine. p53 protein was immunoprecipitated (IP) and the level of immunoprecipitated radiolabeled protein was assessed by autoradiography (35S); Western blot analysis was employed to assess the total amount of immunoprecipitated p53 (IB:p53). Autoradiography of hole cell extracts (WCE, 35S, middle panel) confirm equal amounts of overall [35S]methionine incorporation into cellular proteins and Western blot analysis of WCE shows total levels of Mdm2 and p53. Numbers below upper panel indicate the relative increase in p53 synthesis upon IR treatment for each pair of samples.
B. Model depicting the involvement of L26 in the p53-Mdm2 autoregulatory loop. Transcription of the mdm2 gene is positively regulated by p53. In turn, the Mdm2 protein binds p53, inhibits its biochemical activities and targets it for ubiquitin-dependent proteasomal degradation. Translation of p53 mRNA is positively regulated by L26, but Mdm2 reduces this effect by promoting L26 degradation as well as interfering with the binding of L26 to p53 mRNA. Thus, Mdm2 downregulates p53 by both inhibiting its synthesis and augmenting its demise.
Together, our results strongly suggest that Mdm2 directly targets L26, inhibits its ability to bind to the 5′-UTR of p53 mRNA and blunts L26-mediated stimulation of p53 translation. These inhibitory effects depend on the ability of the acidic domain to mediate Mdm2 binding to L26, but do not require the RING domain of Mdm2 and are therefore independent of Mdm2’s ability to ubiquitylate L26. In addition, Mdm2 ubiquitylates L26, leading to its proteasome-mediated degradation. It is conceivable that when Mdm2 levels are limiting, as is the case in normal cells, both mechanisms contribute together to restraining the rate of p53 synthesis.
Discussion
The findings reported in this study reveal that Mdm2 interacts with ribosomal protein L26 (RPL26), primarily through the acidic domain of the former. Furthermore, L26 is a novel substrate for the E3 ligase activity of Mdm2: Mdm2 drives L26 polyubiquitylation, which is followed by proteasomal degradation. Mdm2-mediated L26 ubiquitylation occurs constitutively in non-stressed cells, but appears to impact only a small, discrete subpopulation of L26, which may be functionally distinct from the bulk of this ribosomal protein. In addition to promoting L26 ubiquitylation and degradation, Mdm2 also counteracts L26-mediated enhancement of p53 translation by blocking the interaction of L26 with the 5′-UTR of p53 mRNA; this does not require the E3 activity of Mdm2 but requires its acidic domain, implicating direct binding of Mdm2 to L26 in this inhibitory effect. The steady state levels of p53 are largely controlled through an autoregulatory feedback loop, which encompasses p53 and Mdm2 as its backbone constituents and regulates the rate of p53 degradation. Our findings delineate an additional component of this feedback loop, operating through regulation of p53 translation (Fig. 7B). The rapid accumulation of p53 in response to stress signals such as DNA damage is proposed to be driven by a combination of concomitant enhanced translation and reduced degradation, both relying on the stress-induced attenuation of Mdm2 activity.
L26 is not the first ribosomal protein to be described as a ubiquitylation target. Ribosomal protein L27a has been shown to undergo non-canonical K63-linked ubiquitylation in a cell-cycle dependent manner (Spence et al., 2000). This polyubiquitylation does not lead to L27a proteasomal degradation, but rather serves a regulatory role (Spence et al., 2000). Furthermore, it has recently been reported that while assembled ribosomal 40S and 60S subunits are very stable, free ribosomal proteins are continuously degraded by nuclear proteasomes (Lam et al., 2007). Such degradation may serve as a general mechanism to prevent undesirable accumulation of non-assembled ribosomal under normal conditions; the identity of the responsible E3 ubiquitin ligase(s) remains to be established. However, we would like to propose that L26 is not behaving like a generic ribosomal protein in that regard. Rather, because it carries a unique function as a specific facilitator of p53 translation (Takagi et al, 2005), it is biochemically integrated into the Mdm2-p53 autoregulatory feedback loop as a positive regulator of p53 and a target for the inhibitory action of Mdm2 (Fig. 7B). Our data further suggest that Mdm2 targets selectively the cytoplasmic fraction of L26 (Fig. 2B), which presumably includes those L26 molecules that bind p53 mRNA and augment its translation. We propose that under non-stressed conditions, cytoplasmic Mdm2 effectively restrains p53 protein synthesis both by reducing the levels of free cytoplasmic L26 and by interfering directly with the binding of L26 to the 5′-UTR of p53 mRNA. However, in response to various types of stress, the balance is shifted in favor of L26. Drugs such as Actinomycin D, which interfere directly with ribosomal biogenesis, as well as a variety of DNA damaging agents that compromise nucleolar integrity (Rubbi and Milner, 2003), may lead to increased accumulation of unassembled L26 in the cytoplasm. At the same time, Mdm2 is proposed to undergo DNA damage-induced modifications that attenuate its functionality, leading also to a reduced ability to disrupt L26-p53 mRNA binding. Together, these events are expected to enable L26 to gain the upper hand, bind efficiently to p53 mRNA and elevate its rate of translation in stressed cells, as indeed observed (Takagi et al., 2005).
Experimental Procedures
Plasmids
L26-Flag was generated by subcloning the L26 ORF from a human testis cDNA library with a C-terminal Flag tag (MDYKDDDDK). pCMV-Bam-Neo-Mdm2 and pCMV—Neo-Bam-p53 were kindly provided by B. Vogelstein. Mdm2 ΔRING (Δ441–491) and HA-ubiquitin expression plasmid (Treier et al., 1994) were kindly provided by A. Levine and D. Bohmann, respectively. Mdm2 ΔAcidic (Δ239–301) was created by site-directed mutagenesis. pEGFP-C1 was from Stratagene. GST-Mdm2 expression plasmid was from Y. Shiloh.
Cell culture and transfections
All cells were maintained at 37°C in a 5% CO2 humidified incubator. HEK293, U2OS and mouse embryonic fibroblasts were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) plus 10% heat-inactivated fetal bovine serum (HyClone) and antibiotics. MCF7 cells were maintained in DMEM supplemented with 10% FBS (GibcoBRL), 2mM L-glutamine (GibcoBRL) and antibiotics. HEK293 were transfected by the calcium phosphate method; U2OS cells were transfected using JetPEI (PolyPlus Transfection). MCF7 cells were transfected using Fugene 6 (Roche).
Yeast two-hybrid screen (Y2H)
Y2H was done as described (Aylon et al., 2006).
Antibodies and coimmunoprecipitation (Co-IP) analysis
Mdm2 was detected with a mix of monoclonal antibodies 4B2, 2A9 and 4B11. L26 was detected with rabbit polyclonal antibody against the N terminal region of L26 (Sigma, R0655). Unless otherwise indicated, the Flag epitope was detected with the M2 monoclonal antibody (Sigma); in Fig. 1B and 4A, rabbit polyclonal anti-Flag antibody (Sigma, F7425) was employed. p53 was detected with a mix of monoclonal antibodies PAb1801 and DO1. Monoclonal antibody PAb419, directed against SV40 large T antigen, served as control. For Co-IP analysis, cells were washed twice with ice-cold PBS and incubated on ice for 30 min in Hunt lysis buffer [20mM Tris-HCl at pH 8.0, 120mM NaCl, 1mM EDTA 0.5% NP-40, and protease inhibitor mix (Sigma)]. The cleared lysate was added to protein A Sepharose beads (Repligen) pre-incubated with either anti-Mdm2 antibodies, anti-Flag antibodies or anti-T-antigen antibodies. After incubation with the lysate for 2–4h at 4°C, beads were washed with Hunt buffer followed by buffer B [5% sucrose, 50mM Tris pH=7.4, 5mM EDTA, 0.5M NaCl and 0.5% NP-40]. Proteins released from beads by boiling were resolved by SDS-PAGE.
IP-RT-PCR
IP-RT-PCR was performed as previously described (Tenenbaum et al., 2002). Briefly, MCF7 cells were transiently co-transfected with pCMV-Neo-Bam-Mdm2 either alone or together with L26 pEGFP-C3. Lysates were immunoprecipitated with polyclonal anti-GFP (ab290; Abcam) or monoclonal anti-Mdm2 (sc-8334; Santa Cruz) antibodies. Immunoprecipitated complexes were split into two aliquots, one for Western blot analysis with monoclonal anti-Mdm2 (sc-965; Santa Cruz) and polyclonal anti-GFP (sc-8334; Santa Cruz) antibodies and the remaining lysate for RT-PCR. Total RNA was isolated using TRIzol reagent (Invitrogen). cDNA was synthesized with the SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen), and PCR-amplified with primers mapping within the 5′-UTR of p53 mRNA and spanning an intron within the genomic human p53 DNA (Forward: CCTTCTCAAAAGTCTAGAGCCAC; Reverse: ATGGCAGTGACCCGGAAGGCAGTC). PCR products, visualized by ethidium bromide staining, were quantified with ImageJ software.
In vitro transcription and translation
T7 RNA polymerase promoter-tagged p53 cDNA was PCR amplified from p53 pLPCX, L26 cDNA from L26 pEGFP-C3 and Mdm2 from pCMV-Neo-Bam-Mdm2. PCR products were in vitro transcribed/translated in the presence of 20μCi [35S]methionine (1000 Ci/mmol; Perkin-Elmer), using TnT T7 Quick for PCR DNA (Promega). Proteins were resolved by electrophoresis on a 4–12% Bis-Tris polyacrylamide gel (Invitrogen) and subjected to Western blot analysis with monoclonal anti-Mdm2 (Santa Cruz, sc-965), monoclonal anti-p53 (sc-126; Santa Cruz) and polyclonal anti-L26 antibodies (Takagi et al., 2005). Results were quantified with ImageJ software.
In vivo radiolabeling
In vivo 35S pulse radiolabeling and analysis of rates of p53 protein synthesis were performed as described (Takagi et al., 2005).
Cell fractionation
Cell pellets were resuspended in cold RSB buffer [10mM NaCl, 10mM Tris-HCl pH7.4, 15mM MgCl2, 100μg/ml heparin and protease inhibitor mix (Sigma)] supplemented with 1.5% Triton X-100 and 1.5% deoxycholate. Lysates were vortexed for 3 seconds, stored 3 minutes on ice and vortexed again. After centrifugation, the supernatant (cytoplasmic fraction) was separated from the pellet (nuclear fraction). The pellet was resuspended in RSB buffer supplemented with Triton X-100 and deoxycholate. Solutions were brought to room temperature and SDS was added to 1.5%.
In vivo ubiquitylation analysis
HA-ubiquitin protocol
Cells were transfected with HA-ubiquitin plus other expression plasmids as indicated. For denatured immunoprecipitation, cell pellets were lysed in SDS lysis buffer [1% SDS in TBS (10mM Tris-HCl, pH 7.5, 150mM NaCl, and protease inhibitor mix (Sigma)] by consecutive vigorous vortexing and boiling. After addition of two volumes of 1.5% triton X-100 in TBS, lysates were cleared by centrifugation and supernatants were added to protein A mix prepared in advance (see “co-immunoprecipitation”). Ubiquitin conjugates were detected with anti-HA antibodies (Santa Cruz).
6xHis-ubiquitin protocol
Cells were transfected with 6xHis-ubiquitin plus additional expression plasmids as indicated. Cells were incubated with 25μM MG132 prior to harvest. Cell pellets were lysed in urea lysis buffer [8M Urea, 0.1M NaH2PO4, 0.1M Tris HCl, pH 8.0, 0.05% Tween and 10mM imidazole, pH 8.0] at pH 8.0 at room temperature for 1 hour. The insoluble fraction was removed by centrifugation. Equal amounts of protein (determined by Bradford assay) were incubated with Ni-NTA magnetic agarose beads (Qiagen) at room temperature overnight. Beads were extensively washed with Denaturing Wash Buffer [8M Urea, 0.1M NaH2PO4, 0.1M Tris HCl, pH 8.0, 0.05% Tween, 20mM imidazole, pH 8.0] and a single wash with Native Wash Buffer [0.1M NaH2PO4, 0.1M Tris HCl pH 8.0, 0.05% Tween and 20mM imidazole, pH 8.0]. Protein complexes were eluted in 2X sample buffer (BioRad) and 200mM imidazole, resolved by SDS-PAGE and subjected to Western blot analysis with monoclonal anti-HA antibody (MMS-101R; Covance).
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We thank J. Wakim for help with the GST pull-down experiments, Y. Aylon for fruitful discussions, B. Vogelstein, A. Levine, Y. Shiloh and D. Bohmann for the gift of plasmids. Supported in part by grant R37 CA40099 from the National Cancer Institute, the Dr. Miriam and Sheldon Adelson Medical Research Foundation, a Prostate Cancer Foundation (Israel) Center of Excellence, and the Yad Abraham Center for Cancer Diagnosis and Therapy (to M.O.), grants R37ES05777 and 2P30CA021765 (to M.B.K.) and by the American Lebanese Syrian Associated Charities (ALSAC) of the St. Jude Children’s Research Hospital.
References
- Argentini M, Barboule N, Wasylyk B. The contribution of the acidic domain of MDM2 to p53 and MDM2 stability. Oncogene. 2001;20:1267–1275. doi: 10.1038/sj.onc.1204241. [DOI] [PubMed] [Google Scholar]
- Aronheim A. Membrane recruitment systems for analysis of protein-protein interactions. Methods Mol Biol. 2001;177:319–328. doi: 10.1385/1-59259-210-4:319. [DOI] [PubMed] [Google Scholar]
- Aylon Y, Michael D, Shmueli A, Yabuta N, Nojima H, Oren M. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev. 2006;20:2687–2700. doi: 10.1101/gad.1447006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylon Y, Oren M. Living with p53, dying of p53. Cell. 2007;130:597–600. doi: 10.1016/j.cell.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Barak Y, Juven T, Haffner R, Oren M. mdm2 expression is induced by wild type p53 activity. Embo J. 1993;12:461–468. doi: 10.1002/j.1460-2075.1993.tb05678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KP, Itahana K, Jin A, Zhang Y. Essential role of ribosomal protein L11 in mediating growth inhibition-induced p53 activation. Embo J. 2004;23:2402–2412. doi: 10.1038/sj.emboj.7600247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner C, Hay T, Meek DW, Lane DP. Hypophosphorylation of Mdm2 augments p53 stability. Mol Cell Biol. 2002;22:6170–6182. doi: 10.1128/MCB.22.17.6170-6182.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Zhang Z, Li M, Wang W, Li Y, Rayburn ER, Hill DL, Wang H, Zhang R. Ribosomal protein S7 as a novel modulator of p53-MDM2 interaction: binding to MDM2, stabilization of p53 protein, and activation of p53 function. Oncogene. 2007;26:5029–5037. doi: 10.1038/sj.onc.1210327. [DOI] [PubMed] [Google Scholar]
- Dai MS, Lu H. Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J Biol Chem. 2004;279:44475–44482. doi: 10.1074/jbc.M403722200. [DOI] [PubMed] [Google Scholar]
- Dai MS, Zeng SX, Jin Y, Sun XX, David L, Lu H. Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol Cell Biol. 2004;24:7654–7668. doi: 10.1128/MCB.24.17.7654-7668.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem. 2000;275:8945–8951. doi: 10.1074/jbc.275.12.8945. [DOI] [PubMed] [Google Scholar]
- Gilkes DM, Chen L, Chen J. MDMX regulation of p53 response to ribosomal stress. Embo J. 2006;25:5614–5625. doi: 10.1038/sj.emboj.7601424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–2908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- Jin A, Itahana K, O’Keefe K, Zhang Y. Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol Cell Biol. 2004;24:7669–7680. doi: 10.1128/MCB.24.17.7669-7680.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Kulikov R, Winter M, Blattner C. Binding of p53 to the central domain of Mdm2 is regulated by phosphorylation. J Biol Chem. 2006;281:28575–28583. doi: 10.1074/jbc.M513311200. [DOI] [PubMed] [Google Scholar]
- Lahav G, Rosenfeld N, Sigal A, Geva-Zatorsky N, Levine AJ, Elowitz MB, Alon U. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat Genet. 2004;36:147–150. doi: 10.1038/ng1293. [DOI] [PubMed] [Google Scholar]
- Lam YW, Lamond AI, Mann M, Andersen JS. Analysis of nucleolar protein dynamics reveals the nuclear degradation of ribosomal proteins. Curr Biol. 2007;17:749–760. doi: 10.1016/j.cub.2007.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrom MS, Deisenroth C, Zhang Y. Putting a finger on growth surveillance: insight into MDM2 zinc finger-ribosomal protein interactions. Cell Cycle. 2007;6:434–437. doi: 10.4161/cc.6.4.3861. [DOI] [PubMed] [Google Scholar]
- Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell. 2003;3:577–587. doi: 10.1016/s1535-6108(03)00134-x. [DOI] [PubMed] [Google Scholar]
- Ma J, Martin JD, Zhang H, Auger KR, Ho TF, Kirkpatrick RB, Grooms MH, Johanson KO, Tummino PJ, Copeland RA, Lai Z. A second p53 binding site in the central domain of Mdm2 is essential for p53 ubiquitination. Biochemistry. 2006;45:9238–9245. doi: 10.1021/bi060661u. [DOI] [PubMed] [Google Scholar]
- Marechal V, Elenbaas B, Piette J, Nicolas JC, Levine AJ. The ribosomal L5 protein is associated with mdm-2 and mdm-2-p53 complexes. Mol Cell Biol. 1994;14:7414–7420. doi: 10.1128/mcb.14.11.7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marine JC, Francoz S, Maetens M, Wahl G, Toledo F, Lozano G. Keeping p53 in check: essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ. 2006;13:927–934. doi: 10.1038/sj.cdd.4401912. [DOI] [PubMed] [Google Scholar]
- Maya R, Balass M, Kim ST, Shkedy D, Leal JF, Shifman O, Moas M, Buschmann T, Ronai Z, Shiloh Y, et al. ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev. 2001;15:1067–1077. doi: 10.1101/gad.886901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol. 2003;13:49–58. doi: 10.1016/s1044-579x(02)00099-8. [DOI] [PubMed] [Google Scholar]
- Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358:80–83. doi: 10.1038/358080a0. [DOI] [PubMed] [Google Scholar]
- Oren M. Decision making by p53: life, death and cancer. Cell Death Differ. 2003;10:431–442. doi: 10.1038/sj.cdd.4401183. [DOI] [PubMed] [Google Scholar]
- Pestov DG, Strezoska Z, Lau LF. Evidence of p53-dependent cross-talk between ribosome biogenesis and the cell cycle: effects of nucleolar protein Bop1 on G(1)/S transition. Mol Cell Biol. 2001;21:4246–4255. doi: 10.1128/MCB.21.13.4246-4255.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyurovsky MV, Prives C. Unleashing the power of p53: lessons from mice and men. Genes Dev. 2006;20:125–131. doi: 10.1101/gad.1397506. [DOI] [PubMed] [Google Scholar]
- Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–412. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- Rubbi CP, Milner J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. Embo J. 2003;22:6068–6077. doi: 10.1093/emboj/cdg579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sdek P, Ying H, Chang DL, Qiu W, Zheng H, Touitou R, Allday MJ, Xiao ZX. MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol Cell. 2005;20:699–708. doi: 10.1016/j.molcel.2005.10.017. [DOI] [PubMed] [Google Scholar]
- Spence J, Gali RR, Dittmar G, Sherman F, Karin M, Finley D. Cell cycle-regulated modification of the ribosome by a variant multiubiquitin chain. Cell. 2000;102:67–76. doi: 10.1016/s0092-8674(00)00011-8. [DOI] [PubMed] [Google Scholar]
- Takagi M, Absalon MJ, McLure KG, Kastan MB. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell. 2005;123:49–63. doi: 10.1016/j.cell.2005.07.034. [DOI] [PubMed] [Google Scholar]
- Tenenbaum SA, Lager PJ, Carson CC, Keene JD. Ribonomics: identifying mRNA subsets in mRNP complexes using antibodies to RNA-binding proteins and genomic arrays. Methods. 2002;26:191–198. doi: 10.1016/S1046-2023(02)00022-1. [DOI] [PubMed] [Google Scholar]
- Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6:909–923. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- Treier M, Staszewski LM, Bohmann D. Ubiquitin-dependent c-Jun degradation in vivo is mediated by the delta domain. Cell. 1994;78:787–798. doi: 10.1016/s0092-8674(94)90502-9. [DOI] [PubMed] [Google Scholar]
- Wallace M, Worrall E, Pettersson S, Hupp TR, Ball KL. Dual-site regulation of MDM2 E3-ubiquitin ligase activity. Mol Cell. 2006;23:251–263. doi: 10.1016/j.molcel.2006.05.029. [DOI] [PubMed] [Google Scholar]
- Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7:1126–1132. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- Yu GW, Rudiger S, Veprintsev D, Freund S, Fernandez-Fernandez MR, Fersht AR. The central region of HDM2 provides a second binding site for p53. Proc Natl Acad Sci U S A. 2006;103:1227–1232. doi: 10.1073/pnas.0510343103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wolf GW, Bhat K, Jin A, Allio T, Burkhart WA, Xiong Y. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol Cell Biol. 2003;23:8902–8912. doi: 10.1128/MCB.23.23.8902-8912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.