

Abstract
We investigated the role of Aurora kinase A (AURKA) in regulating p73-dependent apoptosis utilizing p53-deficient cancer cell lines; H1299, TE7, and HCT116p53−/−. Overexpression of AURKA led to down-regulation of the TAp73-induced activation of the p53/p73-dependent luciferase reporter plasmid (pG13-luc). The reduction in the TAp73 transcription activity was confirmed by measuring the activity of luciferase reporters for p21/WAF1, and Puma. The siRNA knockdown of endogenous AURKA reversed these effects and Western blot analysis demonstrated a significant increase in the protein level of TAp73 and its downstream transcription targets; Puma, Noxa, and p21/WAF1. The co-expression of AURKA together with TAp73 inhibited the activation of the pG13-luc, Puma-luc, and p21/WAF1-luc reporter plasmids with reduction in the protein levels of TAp73 transcription targets. Treatment with AURKA-selective small molecule inhibitor, MLN8054 (Millennium Pharmaceuticals, Cambridge, MA) led to a significant increase in the activities of pG13-luc, Puma-luc, and p21/WAF1-luc reporter plasmids. This effect was accompanied by a significant increase in the mRNA and protein levels of several TAp73 transcription targets; p21/WAF1, PUMA, and NOXA. Flow cytometry cell cycle analysis, following MLN8054 treatment showed more than a two-fold increase in cell death. The apoptotic outcome was corroborated by showing an increase in cleaved caspase-3 protein levels by Western blot. Using TUNEL assay, we demonstrated that the expression of dominant-negative mutant TAp73 expression plasmid (p73DD) counteracted the MLN8054-induced cell death. Taken together, our results indicate that AURKA regulates TAp73-dependent apoptosis and highlight the potential of the AURKA inhibitor MLN8054 in treating cancers that are defective in p53 signaling.
Keywords: AURKA, MLN8054, p73, apoptosis, cancer
INTRODUCTION
Aurora Kinase A (AURKA) belongs to a conserved family of serine/threonine protein kinases that also comprises Aurora kinase B (AURKB) and C (AURKC). AURKA gene encodes a centrosome associated cell cycle regulated serine/threonine kinase (1) that functions to establish mitotic spindles by regulating centrosome duplication and separation, as well as microtubule-kinetochore attachment, spindle checkpoint and cytokinesis. Cytological analysis revealed that over-expression of AURKA results in centrosome amplification, cytokinesis failure and aneuploidy (2). AURKA is located at chromosome 20q13, a region that is frequently amplified in a number of human adenocarcinomas derived from breast, ovarian, colon, gastrointestinal, esophageal, and prostate tissues (2–6). Gene amplification of AURKA is implicated in oncogenesis and tumor progression (2, 7) and its overexpression correlates with genomic instability and clinically aggressive disease showing resistance to chemotherapy (8–12).
The p53 tumor suppressor gene regulates the expression of several genes that are involved in apoptosis, DNA repair, and growth arrest, in response to cellular stress such as DNA damage induced by several chemotherapeutic agents (13–15). Several reports indicate that AURKA interacts with the p53 protein at multiple levels. AURKA phosphorylates p53 at Ser-315 to facilitate HDM2-mediated degradation of p53 (16) and at Ser-215 to suppress its transcriptional activity (17). In addition, AURKA regulates p53 through AKT/HDM2 Mechanisms (5). The loss of functional p53, due to deletions or mutations, occurs in over 50% of human cancers (18) and is a known risk factor related to failure of chemotherapy and radiotherapy treatments in a subset of cancer patients (19, 20). Recently, TAp73 was characterized as a p53 family member that plays an important role in tumorigenesis (21–23). In fact, TAp73 is a pro-apoptotic protein, with structural similarity to p53 that mimics many of the p53’s biological activities (21, 24). The p73 protein is expressed as multiple variants arising from an alternative splicing of the primary TAp73 transcript. The TAp73α is the longest form, which contains a sterile a motif domain (SAM domain) and an extreme COOH-terminal region, whereas TAp73β lacks the extreme COOH-terminal tail and most of the SAM domain. At the cellular level, the TAp73 protein can bind to the p53 consensus-binding sites. The resemblance of TAp73 to p53 in terms of transcriptional activity is translated into a similar biological outcome. This includes transactivation of an overlapping set of target genes such as p21/WAF1, BAX, PUMA, NOXA, 14-3-3-α, p53AIP1; induction of apoptosis, cell cycle arrest and cellular senescence (25–28). Similarly, the TAp73 is activated by DNA-damaging agents such as γ-irradiation or treatment with chemotherapeutic drugs, including; cisplatin, camptothecin, etoposide, doxorubicin and taxol (29, 30). Several studies have shown that the COOH-terminal splicing variants display different transcriptional and biological properties (24, 31). A number of reports have indicated that the ability of the TAp73β protein to transactivate the p53/TAp73 target genes and to induce apoptosis in cancer cells is stronger than the TAp73α (24, 31, 32). Taken together, these data suggest that activation of TAp73 could be a plausible approach for chemotherapy and radiotherapy, especially in cancers with non-functional p53.
MLN8054 is a recently developed selective inhibitor of AURKA (33). The MLN8054 inhibits AURKA phosphorylation on Thr-288 without affecting its degradation (33, 34). To date, there are no data available regarding the regulation of TAp73 by AURKA. In this study we investigated the role of AURKA in regulating TAp73-dependent apoptosis and the effects of AURKA perturbation in p53-deficient cancer cells.
MATERIALS AND METHODS
Tissue culture, vectors, transfection, and MLN8054 treatment
In this study, three p53-deficient cancer cell lines were used; H1299, TE7 and HCT116p53−/−. The H1299 cells were obtained from American Tissue Culture Collection (ATCC, Manassas, VA); HCT116p53−/− and TE7 were gifts from Dr. Bert Vogelstein (Johns Hopkins University, Baltimore, MD) and Hiroshi Nakagawa (University of Pennsylvania, Philadelphia, PA), respectively. Cells were cultured in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% fetal bovine serum (Invitrogen Life Technologies, Carlsbad, CA) at 37°C in an atmosphere containing 5% CO2. The full-length coding sequence of AURKA was cloned in-frame into pcDNA3.1 (5). A synthetic Flag tag sequence was added at the N-terminus of AURKA. The pcDNA3-flag-TAp73β, p73DD-IRES-GFP, and MSCV-IRES-GFP were previously described (35). The luciferase reporter plasmids pG13-luc, MG-15-luc, Puma-luc, and p21/WAF1-luc, were described elsewhere (5, 35, 36). For knockdown of AURKA, small-interfering RNA (siRNA) oligonucleotides for AURKA were used (#427, Ambion, Austin, TX). Transient transfections were performed using Lipofectamine 2000 (Invitrogen Life Technologies, Carlsbad, CA) following the manufacturers’ protocols. MLN8054 (Millennium Pharmaceuticals, Cambridge, MA) was dissolved in DMSO.
Luciferase assays
Luciferase activity assays were performed using pG13-luc, MG-15-luc, Puma-luc and p21/WAF1-luc reporter plasmids in p53-deficient cells. The pG13-luc is a p53/TAp73 response reporter plasmid containing 13 tandem repeats of the p53/TAp73 consensus DNA binding sites and is used to measure the transcriptional activity of p53/TAp73 (36). The MG-15-luc is a modified pG13-luc with mutations on the p53/TAp73 binding sites (36). Puma-luc and p21/WAF1-luc vectors were used to as a measure of the promoter activity of Puma and p21/WAF1 (5, 36). Luciferase activity was determined using a Dual-luciferase Reporter Assay kit (Promega, Madison, WI). The results were averaged from three independent experiments and expressed as mean values ± standard deviation (SD).
Cell viability assay and fluorescence assorted cell sorting (FACS)
Cell survival was measured using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega, Madison, WI). This is a homogeneous method of determining the number of viable cells in culture based on quantification of the ATP present, an indicator of metabolically active cells. 5 × 103 cells per well were seeded onto a 96-well plate and the analysis was performed following the manufacturers’ protocol. The cell cycle analysis was performed using flow cytometry. Cells were synchronized by growing them in the presence of 1% fetal bovine serum for 24 hrs. Cells were trypsinized, washed twice in 1x ice-cold phosphate buffered saline (PBS), and then re-suspended in 0.2 mL ice-cold PBS. Cells were fixed in 1mL ice-cold 70% ethanol for 1 hour. Following centrifugation, cells were re-suspended in 1mL PBS and treated with propidium iodide (50 μg/mL) and RNase A (100 μg/mL) for 30 minutes at 37°C. Flow cytometry was performed using a BD LSR II cytometer (BD Biosciences, San Jose, CA) and the data were analyzed with the BD FACS Diva software. Apoptotic cells were calculated as the percentage of SubG1 cells (SubG1/0 cell DNA content) after drug treatment.
Western blot analysis
Cell lysates were prepared in PBS containing 1x protease cocktail inhibitor (Pierce, Rockford, IL) and centrifuged at 1500 rpm for 10 minutes at 4°C. Protein concentration was measured using a Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA). Proteins (10–15 μg) from each sample were subjected to SDS-PAGE and transferred onto nitro-cellulose membranes. Target proteins were detected by using specific antibodies against, AURKA (TransGenic Inc., Japan); p53 and p21/WAF1 (Oncogene, San Diego, CA); Puma, actin, cleaved Caspase-3, and Flag (Cell Signaling Technology, Boston, MA); Noxa (Imgenex, San Diego, CA) and TAp73 (Bethyl laboratories, Montgomery, TX).
TUNEL (terminal deoxynucleotidyl transferase mediated nick-end labeling) assay
HCT116p53−/− cells were seeded into 8-well chamber slides and transfected with 500 ng of p73DD-IRES-GFP (dominant-negative mutant of TAp73) per well. Cells were treated either with MLN8054 (2 μM) or DMSO (vehicle, control) for 24 hrs. The cells were then stained by terminal deoxynucleotidyl transferase mediated nick-end labeling (TUNEL), according to the manufacturers’ instructions (Roche Diagnostics, Indianapolis, IN), and visualized by TRITC (red fluorescence) (37). The expression of p73DD was visualized by GFP. The nuclei were stained with DAPI (blue fluorescence) and TUNEL positive cells visualized by red fluorescence. More than 200 GFP positive and GFP negative cells were examined in 20 random fields and TUNEL positive cells were counted.
RESULTS
AURKA regulates TAp73 protein and its transcription targets in p53-deficient cells
In this study we utilized p53-deficient cancer cell lines (TE7, H1299, and HCT116p53−/−) to rule out any effect of p53 expression on apoptosis mediated through TAp73. The pG13-luc reporter was used to measure the transcriptional activity of TAp73 in cells. Transient transfection with AURKA for 48 hours downregulated the pG13-luc activity in both the H1299 (P<.005) and TE7 (P<.005) cells (Figure 1A–B). Consistent with the reduction in the pG13-luc activity, the promoter activity of two TAp73 downstream targets; Puma and p21/WAF1 were also significantly diminished by overexpression of AURKA (Figure 1A–B). Depletion of endogenous AURKA in these cells using siRNA transfection for 48 hours had the opposite effect on TAp73 activity and led to a 3.5- and 2.5-fold induction in the pG13-luc activity in H1299 cells (P<.005) and TE7 cells (P<0.005), respectively (Figure 1A–B). Western blot analysis indicated up-regulation of the endogenous TAp73β isoform protein expression (the TAp73α isoform was undetectable), following AURKA knockdown, in both cell lines (Figure 1C–D). Consistent with this finding, we detected up-regulation of the protein levels of TAp73 targets; p21/WAF1, Puma and Noxa (Figure 1C–D). Similar results were obtained in HCT116p53−/− cells (data not shown). These findings demonstrate that AURKA affects endogenous TAp73 protein expression and its transcriptional activity in p53-deficient cancer cells.
Figure 1. AURKA expression interferes with the transcriptional activity TAp73.
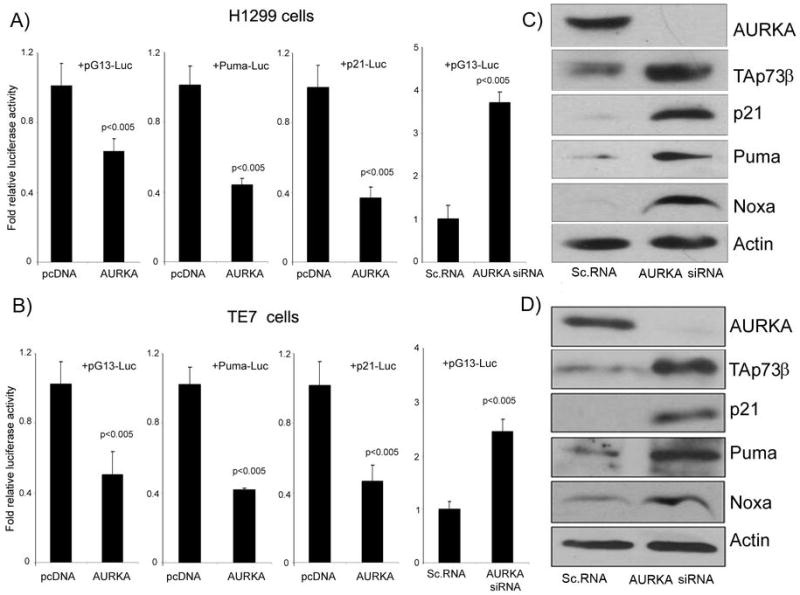
A–B) Luciferase assays using pG13-luc, Puma-luc and p21/WAF1-luc reporter vectors were used to study the effect of AURKA expression on TAp73 in H1299 (A) and TE7 (B) p53-deficient cancer cell lines. The cells were transfected with pcDNA3-AURKA (AURKA), empty vector control (pcDNA3), AURKA-siRNA, or scrambled siRNA (control). Overexpression of AURKA led to reduction in the luciferase activity of the three reporters whereas AURKA-siRNA led to an induction of the pG13-luc reporter. C–D) H1299 (C) and TE7 (D) cells were transiently transfected with scrambled siRNA or AURKA siRNA oligonucleotides. Cell lysates were analyzed by Western blotting for AURKA, TAp73 p21/WAF1, Puma, Noxa, and β–actin. The knockdown of AURKA with siRNA up-regulated the protein expression of endogenous TAp73β and its downstream targets; p21/WAF1, Puma, and Noxa.
We next examined the role of AURKA in regulating ectopically over-expressed TAp73 in p53-deficient cells. We performed these experiments in HCT116p53−/− since it has relatively lower expression of AURKA as compared to TE7 and H1299. The co-transfection of AURKA and TAp73β plasmids in HCT116p53−/− cells demonstrated AURKA-dependent reduction in the luciferase activity of pG13-luc, Puma-luc, and p21/WAF1-luc reporters (Figure 2A–D). On the other hand, the co-transfection of TAp73β with AURKA led to a significant reduction in the TAp73β protein level, and suppressed the induction of its transcription targets, p21/WAF1, Puma and Noxa, in HCT116p53−/− cells (Figure 2D).
Figure 2. AURKA regulates exogenous TAp73 and its targets.

A–C) The luciferase assays were performed on HCT116p53−/− cells that were transfected with pcDNA3-AURKA at different ratios (3:1 or 5:1) with TAp73β–pcDNA3, or empty vector control (pcDNA3) in combination with pG13-luc (A), Puma-luc (B), and p21/WAF1-luc (C) reporter plasmids. While TAp73β expression activated all three reporters, the co-expression of AURKA together with TAp73 reduced the promoter activities of the pG13-luc and the TAp73 downstream targets (Puma and p21/WAF1). D) HCT116p53−/− cells were co-transfected with GFP and TAp73β in combination with either empty vector (pcDNA3) or AURKA-pcDNA3 expression plasmid. Cell lysates were subjected to Western blot analysis using antibodies against Flag, for detection of the exogenous AURKA, TAp73, Puma, Noxa, p21/WAF1, β–Actin, and GFP. The transfection and gel loading were normalized for equal GFP and actin. In agreement with the reporter assays (A–C), the co-transfection with AURKA and TAp73β plasmids led to a significant decrease of the TAp73β, Puma, Noxa, and p21/WAF1 protein levels.
MLN8054 suppresses cell viability and induces TAp73 and its downstream pro-apoptotic targets promoting apoptosis in p53-deficient cancer cells
The above results indicated that knockdown of AURKA using siRNA can induce the TAp73 transcription activity and up-regulates its targets. To pursue the potential therapeutic significance of this finding, we utilized the AURKA-selective pharmacological inhibitor MLN8054. MLN8054 (2μM) treatment of HCT116p53−/− cells transfected with pG13-luc vector led to a significant time-dependent induction of the pG13-luc reporter activity (Figure 3A). The MLN8054 treatment had no effect on MG15-luc, a modified pG13-luc reporter with mutant p53/TAp73 binding sites, reporter activity (Figure 3A). We further observed activation of Puma-luc and p21/WAF1-luc reporters’ activities, downstream targets of p53 and TAp73 (Figure 3B–C). The qRT-PCR analysis demonstrated mRNA up-regulation of several TAp73 downstream targets (p21/WAF1, Noxa, Puma, and p53AIP1), following the treatment with MLN8054 (2 μM) for 24 hours (Figure 3D). Interestingly, the TAp73 mRNA expression remained unchanged, following the MLN8054 treatment (Figure 3D). Indeed, the treatment of H1299, TE7 and HCT116p53−/− cells with 2 μM MLN8054 for 48 hours led to approximately 50% decrease in their viability as compared to vehicle-treated cells (P<.01) (Figure 4A). To corroborate this effect, AURKA was knocked down by RNA interference in H1299, TE7 and HCT116p53−/− which led to a significant decrease in the cell viability in all three p53-deficient cancer cells (P<0.01) (Figure 4B). The Western blot analysis indicated up-regulation of TAp73β (the TAp73α isoform was neither expressed nor induced) and its downstream targets p21/WAF1, Puma and Noxa at the protein level in all three p53-deficient cancer cell lines (Figure 4C). The TAp63, a p53 family member, that regulates similar downstream targets, was neither expressed nor induced by MLN8054 treatment (data not shown). Taken together, these results confirm that AURKA inhibition by siRNA or MLN8054 treatment specifically induces TAp73β, at the protein and not at the mRNA level, and its downstream pro-apoptotic targets.
Figure 3. MLN8054 up-regulates promoter activity and mRNA levels of TAp73 target genes.

A) Luciferase assay of HCT116p53−/− cells transfected with pG13-luc reporter vector and treated with AURKA inhibitor MLN8054 (2μM) for different time periods as indicated. The pG13 promoter activity was significantly induced by MLN8054 (2μM in a time-dependent manner. The MG15-luc has mutated binding sites for p53 family members and was used as a negative control. The treatment with MLN8054 (2μM did not induce promoter activity of the MG15-luc (mutant of pG13-luc). B–C) The HCT116p53−/− cells were transfected with Puma-luc and p21/WAF1-luc reporter vectors, respectively, and treated with 2μM MLN8054 (M) or DMSO (D) for 24 hours. These cells exhibited induction of the Puma and p21/WAF1 promoter activities. D) Quantitative real time RT-PCR analysis demonstrated more than five fold increase in the expression of various downstream pro-apoptotic targets (p21/WAF1, NOXA, PUMA, and p53AIP1), after treatment with MLN8054 (2μM for 24 hrs). However, no effect on the mRNA level of TAp73 was observed.
Figure 4. The MLN8054 treatment suppresses cell survival and induces endogenous TAp73 and its target genes at the protein level in p53-deficient cells.

A) Cell viability assay of three p53-deficient cells (H1299, TE7, and HCT116p53−/−) was carried out by Cell Titer Glo assay after treating them with MLN8054 (2μM for 72 hours. These cells showed approximately 50% reduction in cell viability. B) The knockdown of AURKA by specific validated siRNA in H1299, TE7 and HCT116p53−/− showed a similar effect on cell viability. C) The H1299, TE7, and HCT116p53−/− cells were treated with vehicle (DMSO) or MLN8054 (2μM) for 24 hrs. Cell lysates were subjected to immunoblotting for p53, TAp73, p21/WAF1, Puma, Noxa, and β–Actin. Treatment with MLN8054 up-regulated TAp73β and its downstream apoptotic targets p21/WAF1, Puma and Noxa at protein levels. The TAp63 was neither expressed nor induced by MLN8054 treatment (data not shown). The results indicate that MLN8054 can specifically induce the expression of TAp73, but not TAp63, in these p53-deficient cancer cell lines.
Using flow cytometry cell cycle analysis we observed a substantial increase in the SubG1 cell population, indicative of apoptosis following 2μM and 5μM treatment with MLN8054 for 24 hours, as compared to DMSO control (Figure 5A–C). In addition to the increase in the number of cells in the SubG1 peak, we also observed a significant increase in cells in the G2/M and cells with more than 4N DNA content, indicating a delay in the mitosis process and polyploidy, which is known to occur upon AURKA inhibition (9, 33, 34, 38). The induction of apoptosis by MLN8054 treatment, as indicated by increased cells in the SubG1 peak, was further confirmed by the elevated expression of cleaved caspase-3 (Figure 5A–C). These results are consistent with the inhibition in cell viability following MLN8054 treatment (Figure 4) and demonstrate that MLN8054 induces cell death in p53-deficient cancer cell lines.
Figure 5. Cell cycle analysis following MLN8054 treatment in p53-deficient cancer cell lines.

The H1299 (A), TE7 (B), and HCT116p53−/− (C) were treated with two different concentrations of MLN8054 (2μM and (5μM) for 24 hours. A significant increase in the percentage of cells in the SubG1 peak was seen in H1299 and HCT116p53−/− cells, summarized in the bar graphs. In addition, the inhibition of AURKA by MLN8054 led to accumulation of cells at G2/M phase with a subsequent increase in the polyploidy of cells. The cell lysates from panels A–C were subjected to immunoblotting for cleaved caspase-3. Gel loading was normalized for equal β–actin. Cleaved caspase-3 protein was up-regulated in MLN8054- treated cells, confirming the induction of apoptosis as suggested by the increase of cells in the SubG1 peak.
The TAp73 protein is required for MLN8054-induced apoptosis in p53-deficient cancer cells
The aforementioned results indicate that the MLN8054 treatment induces expression of TAp73β and its pro-apoptotic targets, which in turn, leads to decreased cell survival and increased apoptosis in p53-deficient cancer cells. In order to ascertain the role of TAp73 in the MLN8054-induced cell death, we expressed the p73DD protein, a dominant-negative mutant of TAp73, in p53-deficient cells. The treatment with MLN8054 led to an approximately 50% reduction in cell viability in all cell lines. This effect was partially abrogated by overexpressing the p73DD (dominant-negative mutant of TAp73) where cells showed approximately a 20% decrease, instead of 50%, in cell viability (Figure 6A–C). These results were further supported by the TUNEL assay, which specifically indicated that the p73DD expressing cells (GFP positive) are resistant to MLN8054-induced apoptosis (Figure 6D). We observed a 50% reduction in the number of cells with positive TUNEL signals in the p73DD-expressing cells as compared to the non-p73DD-expressing cells (P<.001). These results confirm that TAp73 activity is required for the MLN8054-induced apoptosis in p53-deficient cancer cells.
Figure 6. The dominant-negative TAp73 mutant abrogates the MLN8054-induced cell death in p53-deficient cells.

A–C)The H1299 (A), TE7 (B), and HCT116p53−/− (C) cells were transfected with control vector (pcDNA3) or TAp73 dominant-negative (p73DD) expression plasmid and treated with vehicle (DMSO) or MLN8054 (2μM) for 24 hrs. Cell survival was determined by CellTiter Glo assay. While the control cells showed an approximately 50% reduction in cell viability, following ML8054 treatment, the p73DD- transfected cells had only 20% reduction in their cell viability. D) The HCT116p53−/− cells were transfected with control vector or p73DD-IRES-GFP plasmid, and then treated with vehicle (DMSO) or MLN8054 (2 μM) for 24 hrs. Apoptotic cells were detected by TUNEL assay (red fluorescence). The DAPI (blue fluorescence) was used as nuclear counterstain to count all cells. The majority of p73DD- expressing cells (green fluorescence, white arrows) resisted MLN8054 induced apoptosis as indicated by negative TUNEL staining. The bar graph summarizes the results and demonstrates a 50% reduction in the MLN8054-induced apoptosis level in p73DD- expressing cells as compared to control cells (P<.001).
DISCUSSION
In this study, we utilized the pG13-luc reporter as a measure of the transcriptional activity of p53/p73 (36) in p53-deficient cancer cell lines. Our findings that AURKA over-expression led to down-regulation of the activity of the pG13-luc in p53-deficient cells suggested that AURKA regulated other members of the p53 family protein. TAp73 is a pro-apoptotic protein that mimics many of p53’s biological activities and belongs to p53 family proteins (21). At the cellular level, the resemblance of TAp73 to p53 in terms of transcriptional activity is translated into a similar biological outcome (21, 27). This includes transactivation of an overlapping set of target genes, that are involved in cell cycle regulation and apoptosis, such as p21/WAF1, Puma, Noxa, and p53AIP1 (for review see (21, 28)). Indeed, we demonstrated activation of the p21/WAF1-luc and Puma-luc reporters. Conversely, the expression of AURKA together with TAp73 led to reduction in the activity of pG13-luc, p21/WAF1-luc, and Puma-luc reporters. Along with the reduction in the luciferase reporters’ activities, the Western blot analysis confirmed the down regulation of TAp73, Puma, Noxa, and p21/WAF1 proteins. The AURKA knockdown confirmed our hypothesis and we observed more than four fold induction of the activity of pG13-luc reporter that was accompanied by up-regulation in the protein level of TAp73, p21/WAF1, Puma, and Noxa. We detected expression and induction of the TAp73β isoform in our in vitro cell models. Indeed, the ability of p73β to transactivate a variety of p53/TAp73 target genes and to induce apoptotic cell death in cancerous cells is stronger than those of the full-length p73α (24, 31, 32). Our results are in agreement with earlier studies reporting that TAp73 can induce Puma and p21/WAF1 and induce apoptosis irrespective of the p53 status (39–42). Furthermore, our findings provide an explanation for a recent study that showed enhancement of radiation response by inhibition of AURKA using siRNA in p53-deficient cancer cells (43). In fact, our experimental data indicate that AURKA expression can interfere with the TAp73 protein and the levels of its pro-apoptotic targets.
Several clinical studies have shown that loss of functional p53 is associated with resistance to chemotherapy (44, 45) like resistance to cisplatin in small–cell lung cancer (46) and doxorubicin in breast cancer patients (47). The p53 protein is non-functional (deficient or mutant) in more than 50% of the human tumors (18). Our aforementioned results encouraged us to test the effect of a recently developed AURKA specific inhibitor MLN8054 in p53-deficient cancer cell lines. Indeed, the MLN8054 induced the activity of the pG13-luc reporter, but not its mutant MG15-luc. Along with this finding, this treatment led to a significant increase in the protein level of TAp73, p21/WAF1, Puma, and Noxa. The qRT-PCR results demonstrated a 4 to 10 fold increase in the mRNA levels of several TAp73 transcription targets; p21/WAF1, Noxa, Puma, and p53AIP1. However, the TAp73 mRNA levels remained unchanged suggesting that the increase in the protein level was unlikely a transcriptional effect. Furthermore, we did not observe induction of TAp63, another p53 family member, by MLN8054 treatment suggesting that the induction of pro-apoptotic targets is specifically due to TAp73. We next compared the knockdown of AURKA using siRNA versus the biochemical inhibition with MLN8054 treatment on cancer cell survival. Both approaches revealed a significant reduction in cell survival in p53-deficient cells. We also observed a considerable decrease in the cell survival and accumulation of G2/M and 4N cells both by MLN8054 treatment and AURKA knockdown. The AURKA knockdown is reported to inhibit cell proliferation (8) and cause spindle pole and chromosome congression defects leading to deleterious aneuploidy and mitotic catastrophe (34, 48). The Polo-like kinase-1 (PLK1) is an essential mitotic kinase regulating multiple aspects of the cell division process. A recent report has shown that PLK1 activation occurs several hours before entry into mitosis, and requires AURKA-dependent phosphorylation of Thr-210 suggesting that the initial activation of PLK1 is a primary function of AURKA (49). Taken together, our results are in agreement with these recent reports. In addition, the treatment of cells with MLN8054 led to a substantial increase in the percentage of cells in SubG1 peak, indicative of cell death, and an increase in the expression level of cleaved caspase-3; thus, supporting our findings of the induction of the several pro-apoptotic proteins and consistent with the decrease in cell survival. We have further confirmed that the MLN8054-mediated apoptosis through TAp73 in p53-deficient cells using TUNEL assay and demonstrated a significant reduction in apoptosis rate in cells transfected with the dominant-negative mutant TAp73 (p73DD). Taken together, these results indicate that AURKA can regulate the p73-dependent apoptosis and this inhibitor can have a dual effect by inducing massive polyploidy and mitotic catastrophe (33, 34) and also by induction of TAp73-dependent apoptosis. The ability to induce TAp73-dependent apoptosis by knockdown or inhibition of AURKA is an attractive approach that can have a significant impact especially in tumors that have lost the functional p53 activities. The mechanisms by which AURKA interferes with the TAp73 protein level remain to be elucidated in future studies.
Acknowledgments
This study was supported by the National Cancer Institute Grants R01CA106176 (WER) and the GI SPORE CA 95103. The contents of this work are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute or Vanderbilt University.
References
- 1.Giet R, McLean D, Descamps S, et al. Drosophila Aurora A kinase is required to localize D-TACC to centrosomes and to regulate astral microtubules. J Cell Biol. 2002;156:437–51. doi: 10.1083/jcb.200108135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou H, Kuang J, Zhong L, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet. 1998;20:189–93. doi: 10.1038/2496. [DOI] [PubMed] [Google Scholar]
- 3.Glover DM, Leibowitz MH, McLean DA, Parry H. Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell. 1995;81:95–105. doi: 10.1016/0092-8674(95)90374-7. [DOI] [PubMed] [Google Scholar]
- 4.Kallioniemi A, Kallioniemi OP, Piper J, et al. Detection and mapping of amplified DNA sequences in breast cancer by comparative genomic hybridization. Proc Natl Acad Sci (USA) 1994;91:2156–60. doi: 10.1073/pnas.91.6.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dar AA, Zaika A, Piazuelo MB, et al. Frequent overexpression of Aurora Kinase A in upper gastrointestinal adenocarcinomas correlates with potent antiapoptotic functions. Cancer. 2008;112:1688–98. doi: 10.1002/cncr.23371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamada K, Yamada Y, Hirao T, et al. Amplification/overexpression of Aurora-A in human gastric carcinoma: potential role in differentiated type gastric carcinogenesis. Oncol Rep. 2004;12:593–9. [PubMed] [Google Scholar]
- 7.Goepfert TM, Adigun YE, Zhong L, et al. Centrosome amplification and overexpression of aurora A are early events in rat mammary carcinogenesis. Cancer Res. 2002;62:4115–22. [PubMed] [Google Scholar]
- 8.Tanaka E, Hashimoto Y, Ito T, et al. The clinical significance of Aurora-A/STK15/BTAK expression in human esophageal squamous cell carcinoma. Clin Cancer Res. 2005;11:1827–34. doi: 10.1158/1078-0432.CCR-04-1627. [DOI] [PubMed] [Google Scholar]
- 9.Ogawa E, Takenaka K, Katakura H, et al. Perimembrane Aurora-A expression is a significant prognostic factor in correlation with proliferative activity in non-small-cell lung cancer (NSCLC) Ann Surg Oncol. 2008;15:547–54. doi: 10.1245/s10434-007-9653-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guan Z, Wang XR, Zhu XF, et al. Aurora-A, a negative prognostic marker, increases migration and decreases radiosensitivity in cancer cells. Cancer Res. 2007;67:10436–44. doi: 10.1158/0008-5472.CAN-07-1379. [DOI] [PubMed] [Google Scholar]
- 11.Landen CN, Jr, Lin YG, Immaneni A, et al. Overexpression of the centrosomal protein Aurora-A kinase is associated with poor prognosis in epithelial ovarian cancer patients. Clin Cancer Res. 2007;13:4098–104. doi: 10.1158/1078-0432.CCR-07-0431. [DOI] [PubMed] [Google Scholar]
- 12.Reiter R, Gais P, Jutting U, et al. Aurora kinase A messenger RNA overexpression is correlated with tumor progression and shortened survival in head and neck squamous cell carcinoma. Clin Cancer Res. 2006;12:5136–41. doi: 10.1158/1078-0432.CCR-05-1650. [DOI] [PubMed] [Google Scholar]
- 13.Ko LJ, Prives C. p53: puzzle and paradigm. Genes Dev. 1996;10:1054–72. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- 14.Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51:6304–11. [PubMed] [Google Scholar]
- 15.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–64. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 16.Katayama H, Sasai K, Kawai H, et al. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet. 2004;36:55–62. doi: 10.1038/ng1279. [DOI] [PubMed] [Google Scholar]
- 17.Liu Q, Kaneko S, Yang L, et al. Aurora-A abrogation of p53 DNA binding and transactivation activity by phosphorylation of serine 215. J Biol Chem. 2004;279:52175–82. doi: 10.1074/jbc.M406802200. [DOI] [PubMed] [Google Scholar]
- 18.Marks JR, Davidoff AM, Kerns BJ, et al. Overexpression and mutation of p53 in epithelial ovarian cancer. Cancer Res. 1991;51:2979–84. [PubMed] [Google Scholar]
- 19.Lee JM, Bernstein A. p53 mutations increase resistance to ionizing radiation. Proc Natl Acad Sci U S A. 1993;90:5742–6. doi: 10.1073/pnas.90.12.5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mueller H, Eppenberger U. The dual role of mutant p53 protein in chemosensitivity of human cancers. Anticancer Res. 1996;16:3845–8. [PubMed] [Google Scholar]
- 21.Zaika AI, El-Rifai W. The role of p53 protein family in gastrointestinal malignancies. Cell Death Differ. 2006;13:935–40. doi: 10.1038/sj.cdd.4401897. [DOI] [PubMed] [Google Scholar]
- 22.Flores ER, Tsai KY, Crowley D, et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002;416:560–4. doi: 10.1038/416560a. [DOI] [PubMed] [Google Scholar]
- 23.Mills AA. p63: oncogene or tumor suppressor? Curr Opin Genet Dev. 2006;16:38–44. doi: 10.1016/j.gde.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 24.Ozaki T, Nakagawara A. p73, a sophisticated p53 family member in the cancer world. Cancer Sci. 2005;96:729–37. doi: 10.1111/j.1349-7006.2005.00116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaghad M, Bonnet H, Yang A, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90:809–19. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- 26.Osada M, Ohba M, Kawahara C, et al. Cloning and functional analysis of human p51, which structurally and functionally resembles p53. Nat Med. 1998;4:839–43. doi: 10.1038/nm0798-839. [DOI] [PubMed] [Google Scholar]
- 27.Lokshin M, Tanaka T, Prives C. Transcriptional regulation by p53 and p73. Cold Spring Harb Symp Quant Biol. 2005;70:121–8. doi: 10.1101/sqb.2005.70.046. [DOI] [PubMed] [Google Scholar]
- 28.Harms K, Nozell S, Chen X. The common and distinct target genes of the p53 family transcription factors. Cell Mol Life Sci. 2004;61:822–42. doi: 10.1007/s00018-003-3304-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Irwin MS, Miller FD. p73: regulator in cancer and neural development. Cell Death Differ. 2004;11:S17–22. doi: 10.1038/sj.cdd.4401452. [DOI] [PubMed] [Google Scholar]
- 30.Shimodaira H, Yoshioka-Yamashita A, Kolodner RD, Wang JY. Interaction of mismatch repair protein PMS2 and the p53-related transcription factor p73 in apoptosis response to cisplatin. Proc Natl Acad Sci U S A. 2003;100:2420–5. doi: 10.1073/pnas.0438031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ueda Y, Hijikata M, Takagi S, Chiba T, Shimotohno K. New p73 variants with altered C-terminal structures have varied transcriptional activities. Oncogene. 1999;18:4993–8. doi: 10.1038/sj.onc.1202817. [DOI] [PubMed] [Google Scholar]
- 32.Lee CW, La Thangue NB. Promoter specificity and stability control of the p53-related protein p73. Oncogene. 1999;18:4171–81. doi: 10.1038/sj.onc.1202793. [DOI] [PubMed] [Google Scholar]
- 33.Manfredi MG, Ecsedy JA, Meetze KA, et al. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc Natl Acad Sci U S A. 2007;104:4106–11. doi: 10.1073/pnas.0608798104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoar K, Chakravarty A, Rabino C, et al. MLN8054, a small-molecule inhibitor of Aurora A, causes spindle pole and chromosome congression defects leading to aneuploidy. Mol Cell Biol. 2007;27:4513–25. doi: 10.1128/MCB.02364-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei J, O’Brien D, Vilgelm A, et al. Interaction of Helicobacter pylori with gastric epithelial cells is mediated by the p53 protein family. Gastroenterology. 2008;134:1412–23. doi: 10.1053/j.gastro.2008.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tomkova K, Belkhiri A, El-Rifai W, Zaika AI. p73 isoforms can induce T-cell factor-dependent transcription in gastrointestinal cells. Cancer Res. 2004;64:6390–3. doi: 10.1158/0008-5472.CAN-04-2176. [DOI] [PubMed] [Google Scholar]
- 37.Belkhiri A, Zaika A, Pidkovka N, et al. Darpp-32: a novel antiapoptotic gene in upper gastrointestinal carcinomas. Cancer Res. 2005;65:6583–92. doi: 10.1158/0008-5472.CAN-05-1433. [DOI] [PubMed] [Google Scholar]
- 38.Marumoto T, Honda S, Hara T, et al. Aurora-A kinase maintains the fidelity of early and late mitotic events in HeLa cells. J Biol Chem. 2003;278:51786–95. doi: 10.1074/jbc.M306275200. [DOI] [PubMed] [Google Scholar]
- 39.Melino G, Lu X, Gasco M, Crook T, Knight RA. Functional regulation of p73 and p63: development and cancer. Trends Biochem Sci. 2003;28:663–70. doi: 10.1016/j.tibs.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 40.Melino G, De Laurenzi V, Vousden KH. p73: Friend or foe in tumorigenesis. Nat Rev Cancer. 2002;2:605–15. doi: 10.1038/nrc861. [DOI] [PubMed] [Google Scholar]
- 41.Melino G, Bernassola F, Ranalli M, et al. p73 Induces apoptosis via PUMA transactivation and Bax mitochondrial translocation. J Biol Chem. 2004;279:8076–83. doi: 10.1074/jbc.M307469200. [DOI] [PubMed] [Google Scholar]
- 42.Zhu J, Jiang J, Zhou W, Chen X. The potential tumor suppressor p73 differentially regulates cellular p53 target genes. Cancer Res. 1998;58:5061–5. [PubMed] [Google Scholar]
- 43.Tao Y, Zhang P, Frascogna V, et al. Enhancement of radiation response by inhibition of Aurora-A kinase using siRNA or a selective Aurora kinase inhibitor PHA680632 in p53-deficient cancer cells. Br J Cancer. 2007;97:1664–72. doi: 10.1038/sj.bjc.6604083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buttitta F, Marchetti A, Gadducci A, et al. p53 alterations are predictive of chemoresistance and aggressiveness in ovarian carcinomas: a molecular and immunohistochemical study. Br J Cancer. 1997;75:230–5. doi: 10.1038/bjc.1997.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Righetti SC, Della Torre G, Pilotti S, et al. A comparative study of p53 gene mutations, protein accumulation, and response to cisplatin-based chemotherapy in advanced ovarian carcinoma. Cancer Res. 1996;56:689–93. [PubMed] [Google Scholar]
- 46.Rusch V, Klimstra D, Venkatraman E, et al. Aberrant p53 expression predicts clinical resistance to cisplatin-based chemotherapy in locally advanced non-small cell lung cancer. Cancer Res. 1995;55:5038–42. [PubMed] [Google Scholar]
- 47.Aas T, Borresen AL, Geisler S, et al. Specific P53 mutations are associated with de novo resistance to doxorubicin in breast cancer patients. Nat Med. 1996;2:811–4. doi: 10.1038/nm0796-811. [DOI] [PubMed] [Google Scholar]
- 48.Hirota T, Kunitoku N, Sasayama T, et al. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell. 2003;114:585–98. doi: 10.1016/s0092-8674(03)00642-1. [DOI] [PubMed] [Google Scholar]
- 49.Macurek L, Lindqvist A, Lim D, et al. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature. 2008 doi: 10.1038/nature07185. [DOI] [PubMed] [Google Scholar]