

Abstract
Insufficient efficacy and/or specificity of antisense oligonucleotides limit their in vivo usefulness. We demonstrate here that a high-affinity DNA analog, locked nucleic acid (LNA), confers several desired properties to antisense agents. Unlike DNA, LNA/DNA copolymers were not degraded readily in blood serum and cell extracts. However, like DNA, the LNA/DNA copolymers were capable of activating RNase H, an important antisense mechanism of action. In contrast to phosphorothioate-containing oligonucleotides, isosequential LNA analogs did not cause detectable toxic reactions in rat brain. LNA/DNA copolymers exhibited potent antisense activity on assay systems as disparate as a G-protein-coupled receptor in living rat brain and an Escherichia coli reporter gene. LNA-containing oligonucleotides will likely be useful for many antisense applications.
Antisense oligonucleotides designed according to straightforward base-pairing rules have been useful in functional genomics efforts, and there also has been recent clinical progress in developing antisense drugs (1–5). The key objective in the field, however, remains the identification of oligonucleotide analogs that provide the possibility to achieve high in vivo efficacy in the absence of significant toxicity (1–3).
To date, all human antisense studies, as well as the vast majority of studies on other species, have relied on the use of phosphorothioate DNA analogs (where one nonbridging phosphate oxygen has been replaced). Although phosphorothioates are markedly more resistant to degrading enzymes than DNA, their DNA-binding capacity (relating to potency when used as antisense agents) is low, and they are well known to cause nonspecific protein binding, largely because of their polyanionic nature. The latter phenomenon contributes to a toxicity profile that limits many applications (6, 7). For example, when injected into the brain, phosphorothioates can cause severe tissue damage, especially with repeated or prolonged administration schedules (7, 8). Such phosphorothioate-induced toxic reactions are thought to be reduced but not absent in second-generation antisense agents, like mixed backbone oligonucleotides (containing phosphorothioates in combination with oligodeoxyribonucleotides or oligoribonucleotides) (9).
Interestingly, conformational restriction has been successfully applied in recent years to the design of high-affinity oligonucleotides. Several analogs containing bi- and tricyclic carbohydrate moieties have displayed enhanced duplex stability (10–20) and most notably so locked nucleic acids (LNA) (Fig. 1). LNA induces unprecedented increases in the thermal stability (melting temperature, Tm) of duplexes toward complementary DNA and RNA (ΔTm/LNA monomer = + 3 to + 11°C compared with the corresponding DNA reference). By virtue of their bicyclic structure, the furanose ring of the LNA monomers is locked in a 3′-endo conformation, thus structurally mimicking the standard RNA monomers. Moreover, LNA/LNA duplex formation has been shown to constitute the most stable Watson–Crick base-pairing system yet developed (19).
Figure 1.

Two representations of the chemical structure of LNA (one nucleotide monomer shown). We have defined LNA as an oligonucleotide containing one or more 2′-C,4′-C-oxy-methylene-linked bicyclic ribonucleotide monomers (LNA monomers). The representation shown to the left highlights the chemical connectivities of an LNA monomer. The representation shown to the right highlights the locked 3′-endo (3E) conformation of the furanose ring of an LNA monomer.
The very high Tm values of all-LNA oligonucleotides prompted us to pursue biological antisense experiments not with molecules consisting solely of LNA but with combinations, or copolymers, of LNA and DNA: (i) LNA/DNA mix-mers, and (ii) LNA/DNA/LNA gap-mers. Throughout this study we used 15-mer oligonucleotides (nine LNA and six DNA monomers, i.e., 60% LNA content for mix-mers and gap-mers) for antisense purposes. Stringently mismatched control oligonucleotides were used in every in vivo antisense experiment.
Results from the present study suggest that LNA oligonucleotides have high in vivo efficacy in the absence of significant toxicity and are therefore likely to be useful for functional genomics as well as therapeutic applications.
Methods
Locked Nucleic Acids.
LNA-containing sequences were synthesized on an automated DNA synthesizer as described (16, 17). The oligonucleotides were analyzed as >90% pure by reversed phase–HPLC and in some cases also analyzed by capillary electrophoresis. The hybridization properties of these LNA-containing antisense oligonucleotides were evaluated in thermal denaturation experiments toward complementary single-stranded DNA and RNA. In addition, the possible formation of LNA homocomplexes by self association was evaluated in similar experiments without a complementary strand.
To target the rat delta opioid receptor (DOR) mRNA, two antisense sequences, DOR-AS-1 and DOR-AS-2, were used for in vitro (stability and RNase H) and in vivo (efficacy and toxicity) experiments as all-phosphodiester, phosphorothioate (fully or partially substituted), all-LNA, LNA/DNA mix-mer, or LNA/DNA/LNA gap-mer. The latter two sequence designs were: gTgTCCgAgACgTTg and GTGTccgagaCGTTG for DOR-AS-1 and CAcGggCaGaAGgCA and CACGggcagaAGGCA for DOR-AS-2, respectively (LNA monomers represented by uppercase letters and DNA monomers by lowercase letters). The database submission reference used for the rat DOR gene is U00475, and the sequence coordinates for the antisense targets sites are 99–113 for DOR-AS-1 and 59–73 for DOR-AS-2. As control sequences for antisense activity, four base mismatches were introduced in the same corresponding position in every (including all mix-mers and gap-mers) DOR-AS-1 and DOR-AS-2 analog (here exemplified by phosphodiesters): gttgccgagactgtg and cgcaggcagacggaa, respectively. The results of studies on melting temperatures (Tm values) with DOR-AS-1 are displayed in Table 1 (analogous results were obtained with DOR-AS-2 and are not shown).
Table 1.
Results of thermal denaturation experiments with DOR-AS-1
| LNAs synthesized | DNA complement Tm, °C | RNA complement Tm, °C | No complement*Tm, °C |
|---|---|---|---|
| All-DNA 5′-gtgtccgagacgttg | 59 | 56 | n.t.† |
| All-LNA 5′-GTGTCCGAGACGTTG | >90‡ | >90‡ | >90‡ |
| LNA-DNA mix-mer 5′-gTgTCCgAgACgTTg | 83 | >90‡ | 73 |
| LNA-DNA gap-mer 5′-GTGTccgagaCGTTG | 72 | 83 | n.t.† |
LNA monomers are shown in capital letters, whereas standard DNA monomers are shown in lower-case letters. Melting temperatures (Tm values) were obtained from the maxima of the first derivatives of the melting curves (A260 vs. temperature; 10–90°C with a 1°C increase per min), recorded in medium salt buffer (10 mM sodium phosphate/100 mM NaCl/0.1 mM EDTA, pH 7.0) by using 1.5-μM concentrations of the two complementary strands, assuming identical extinction coefficients for modified and unmodified nucleotides with the same nucleobase.
3.0 μM concentrations of LNAs used.
† n.t., no transition detected; as no transition was detected, and Tm values were detected for the homo-complexes, self pairing above 10°C is precluded.
‡Transitions above 90°C assumed.
Uptake of LNA into MCF-7 Cells.
Experiment in brief: MCF-7 cells were cultured in standard medium supplemented with 1% FCS. At the start of the experiment, cells were approximately 40% confluent, and the serum-containing medium was removed and replaced with serum-free medium. Optimal transfection was obtained when Lipofectin (GIBCO/BRL) was diluted ×40 in medium without serum and combined with the oligonucleotide to a concentration of 750 nM oligonucleotide. The oligonucleotide–Lipofectin complex was allowed to form for 15 min at room temperature and was further diluted with serum-free medium to a final concentration of 250 nM oligonucleotide/0.8 μg/ml Lipofectin. Then the medium was removed from the cells and replaced with the medium containing oligonucleotide–Lipofectin complex. The cells were incubated at 37°C for 6 h, rinsed once with medium without serum, and incubated for a further 18 h in DME/F12 with 1% FCS at 37°C. The result of the experiment was evaluated by using a Leica Das Microskop Research Biology fluorescence microscope equipped with a high-resolution charge-coupled device camera either directly on living cells in culture flasks or on cells cultured in slide flasks and fixed in 4% ice-cold paraformaldehyde. Before fixation, the growth medium was removed, the cells were rinsed once with ice-cold PBS and fixed on ice with ice-cold 4% paraformaldehyde (wt/vol) in PBS for 15–30 min. After fixation, the paraformaldehyde–PBS was removed and the cells were washed three times with ice-cold PBS, mounted with coverglass, and analyzed.
Serum Stability Assay.
Samples of whole blood were taken from adult Sprague–Dawley rats. The four isosequential oligomers were then investigated in parallel in a final concentration of ≈0.05 μg/μl. The samples were immediately incubated at 37°C. At time points 0, 10, 30, 60, 180, 300, and 1,200 min, 20-μl aliquots were taken and added to tubes containing formamide loading buffer (95% formamide/×1 Tris/borate/EDTA electrophoresis buffer (90 mM Tris/64.6 mM boric acid/2.5 mM EDTA, pH 8.3)/0.04% xylene cyanol/0.04% bromophenol blue) to quench nucleolytic activity and then stored at −70°C. The samples were heated to 60°C for 3 min and full-length (intact) and digested oligomers were then separated on a denaturing 15% polyacrylamide gel (7 M urea) run at 43°C. The gels from the stability tests were stained with SYBR Gold Nucleic Acid Gel Stain (S-11494, Molecular Probes), which was found to be sufficiently sensitive for direct visualization of the oligonucleotides. The gels were exposed to UV light and photographed. Densitometric calculations were done with easy win imaging software (Herolab, Wiesloch, Germany). The volume density of the major band, corresponding to intact oligomer (stability tests) and intact target RNA probe (RNase H assay) respectively, was calculated in each lane with correction for background. The volume density for the time 0 sample was set as reference value for each incubation. Relative values for the other time-point samples in the corresponding incubation were calculated on the basis of these reference values.
RNase H Activation Assay.
To obtain target RNA corresponding to the antisense (DOR-AS-1) targeted portion of the rat DOR, two complementary 41-mer oligonucleotides, making up a linearized double-stranded template for subsequent T7 polymerase run-off transcription, were used as illustrated below. In the 5′ end of the sense template strand, the promoter sequence for T7 polymerase recognition and initiation of transcription was contained, followed by the DNA sequence coding for the target-RNA sequence. The two complementary oligonucleotides were heated to 80°C for 10 min to produce the linearized double-stranded template. A 20-μl in vitro transcription reaction containing 500 μM each of ATP, GTP, and CTP, 12 μM of UTP, ≈50 μCi of [α-32P] UTP, ×1 transcription buffer (Tris⋅HCl, pH 7.5), 10 mM DTT, 1% BSA, 20 units of RNasin ribonuclease inhibitor (Promega), 0.2 μl template, and 240 units T7 RNA polymerase. The inclusion of RNasin was to prevent degradation of the target RNA from ribonucleases. The reactions were carried out at 37°C for 2 h to produce the desired 24-mer 32U-labeled RNA run-off transcript. For target RNA purification, 1.5 μl (1.5 units) of DNase I was added, the mixture was incubated at room temperature for 30 min and then resolved in a 15% polyacrylamide gel containing 7 M urea, and the correctly sized fragment was excised from the gel, dispensed in elution buffer (0.1% SDS/0.5 M ammonium acetate/10 mM Mg-acetate) and incubated at room temperature overnight. The target RNA sequence was then purified by precipitation with 1/10 volume of 3 M sodium acetate (pH 5.2) and 2.5 volumes of 95% ethanol followed by centrifugation at 12,000 × g. The pellets were diluted in TE buffer and subsequently subjected to RNase H digestion assay. Herein, the decrease of intact substrate, i.e., the 24-mer [α-32P] UTP-labeled target RNA sequence, was assayed over time as follows. The reactions were carried out in a total volume of 110 μl and contained (added in the mentioned order): ×1 nuclease-free buffer (20 mM Tris⋅HCl, pH 7.5/40 mM KCl/8 mM MgCl2/0.03 mg/ml BSA), 10 mM DTT/4% glycerol/100 nM of oligomer/3 units RNasin inhibitor/labeled target RNA strand/0.1 units of E. coli RNaseH. An excess of antisense oligomer was added to each reaction mixture to ensure full hybridization of the RNA target sequences. Two negative controls were also included (not shown) and were prepared as above but (i) with no antisense DNA-oligonucleotide, or (ii) with no RNase H added to the reaction mixture. All of the reactions were incubated at 37°C. At time points 0, 10, 20, 40, and 60 min, 10-μl aliquots were taken and immediately added to ice-cold formamide loading buffer to quench the reaction and stored at −20°C. The samples were heated to 85°C for 5 min before loading and running on a 15% polyacrylamide gel containing 7 M urea. The following is the composition of the sense and antisense template strands, corresponding to DOR-AS-1, used as doubled-stranded DNA template for the in vitro transcription reaction and the 24-mer labeled RNA run-off transcript (results displayed in Fig. 3B):
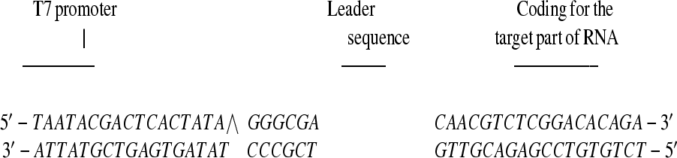 |
Produced RNA run off transcript: 5′-GGGCGA CAACG32UC32 UCGGACAC AGA-3′. The same approach was taken to study RNase H activation induced by DOR-AS-2 (see Fig. 4 for further information).
Figure 3.

Stability in rat blood serum (A) and RNase H activation (B) of DOR-AS-1 oligonucleotides. LNA monomers are represented by uppercase letters, DNA monomers by lowercase letters, and phosphorothioate (PS) monomers by italic lowercase letters. The location of the full-length RNA is indicated (arrow), and the relative volume densities for single measurements are shown as line graphs.
Figure 4.

RNase H cleavage of a DOR-2 RNA target sequence with DNA and DNA/LNA copolymers. (Upper) RNase H cleavage of a 15-mer synthetic RNA (5′UGCCUUCUGCCCGUG) lowercase letters corresponding to the DOR-2 target. (Right) Control incubations lacking added antisense DNA or RNase H, thus demonstrating that the degradation visible (Upper) is mediated by added RNase H and antisense DNA. LNA monomers are represented by uppercase letters, DNA monomers by lowercase letter, and phosphorothioate (PS) monomers by italic lowercase letters. The location of the full-length RNA is indicated (arrow), and the relative volume densities for single measurements are shown as line graphs.
Animals.
Adult male Sprague–Dawley rats (280–320 g) (B & K Universal, Sollentuna, Sweden) were used. The animals arrived in the laboratory at least 1 wk before experiments and were housed four per cage (Makrolon IV) under controlled conditions of light–dark cycle (12 h:12 h, lights off 7:30 p.m.), relative humidity (≈55%) and temperature (21°C ± 1°C). Food (R36, Ewos, Södertälje, Sweden) and tap water were available ad libitum. All experiments were approved by the Local Committee on Ethics of Animal Experimentation, Stockholm, Sweden.
Animal Surgery and Brain Infusions.
Three different routes of administration were used in the present study: intrathecal, intracerebroventricular, and intrastriatal. For intrathecal injections, rats were anaesthetized and implanted with an intrathecal polyethylene (PE-10) catheter as described by Yaksh and Rudy (21). For intracerebral injections, a guide cannula (25 gauge) was stereotaxically mounted in the skull bone in anaesthetized rats and placed against the dura for unilateral injections, either into the lateral ventricle (efficacy study) or caudate-putamen (toxicity study). The coordinates, relative bregma, were: AP −1.0 mm and L −1.3 mm for the lateral ventricle and AP + 0.7 mm and L −2.7 mm for the caudate-putamen (22). The injection needle (31 gauge) penetrated 4.0 mm (lateral ventricle) and 5.0 mm (caudate-putamen) below the dura.
Intrathecal injections were spaced over a period of 30 s, and the compounds were injected by using a 50-μl Hamilton syringe in a 5-μl volume followed by a 10-μl saline flush of the catheter. Intracerebroventricular and intrastriatal injections were done by means of a 10-μl Hamilton syringe, and the compounds were infused in a volume of 5 μl (lateral ventricle) or 1 μl (caudate-putamen) over 90 s. The injection needle was left in situ for another 180 s.
Behavioral Measurements.
For the tail-flick test, the oligonucleotides were administered in a 5-μl volume twice daily (0800 and 1700 h) for 3 days. The day after the last injection, the rats were injected with [D-Ala2]deltorphin II (60 μg, intrathecal) and tested in the warm water (52°C) tail-flick test (e.g., ref. 23) for DOR-mediated antinociception. After [D-Ala2]deltorphin II administration, the maximal possible latency allowed was 10 s. Rats not responding within 10 s with tail withdrawal were removed from the water, and a score of 100% was recorded. Scores for antinociception were calculated according to the following formula: antinociception = ×100 (test latency − control latency)/(10-control latency).
Core temperature was measured in a temperature-controlled room (ambient temperature 21.0°C ± 1.0°C). Rectal core temperature was recorded by means of a flexible probe connected to an automated telethermometer that was activated when the temperature had stabilized (± 0.1°C) (24). In the toxicity studies, rats were injected into the caudate-putamen with phosphorothioate oligodeoxynucleotides, gap-mer DNA/phosphorothioate oligodeoxynucleotides or LNA oligodeoxynucleotides, on day 1 and day 3 (10 nmol/day), and temperature was recorded 2 h after the last injection. In the antisense efficacy studies, DOR-AS-2 (50 μg injected twice daily for 4 days) and [D-Ala2]deltorphin II (100 μg injected on day 5) were administered intracerebroventriculary, and core temperature was recorded 20 and 60 min after administration of [D-Ala2]deltorphin II.
Radioreceptor Binding Analysis.
Lumbar spinal cords were dissected and quickly frozen on dry ice. Tissues were pooled from each group of six to eight animals and homogenized with a Polytron (Brinkmann) in a homogenization buffer (50 mM Tris/0.1 mM PMSF, pH 7.4). The membranes were pelleted at 40,000 × g for 30 min at 4°C. Membrane pellets were resuspended in Tris buffer in a total volume of 42 ml and stored in 4-ml and 8-ml aliquots at −80°C. Protein contents were determined by the method of Lowry. Total membrane yields from each group ranged from 34 to 44 mg. In saturation assays, 163–325 μg of membrane/assay tube was used. Binding buffer was 5 mM Tris/0.5% BSA/0.1 mM PMSF, pH 7.4. As radioligand, [3H]naltrindole, specific activity 32 Ci/mmol (New England Nuclear), was used in concentrations ranging from 15 pM to 6.1 nM. Nonspecific binding was defined in the presence of 0.01 mM naloxone. Reactions (in 1-ml final volume) were terminated by rapid filtration through Whatman GF/B filters (Whatman).
Immunohistochemistry.
Some of the rats used in the temperature experiments were subsequently perfused transcardially with formalin-picric acid as described previously (25). Cryostat-cut brain sections of 14 μm thickness were incubated with polyclonal rabbit antiserum to tyrosine hydroxylase (26), rinsed, and incubated with FITC-conjugated secondary antibodies (Jackson ImmunoResearch), as described (25). The sections were then rinsed, mounted, and examined in a Nikon Microphot FX microscope equipped for epifluoroscence (Nikon). Kodak T.Max 400 film (Kodak) was used for photomicrographs.
Stability and Effects of LNA/DNA Copolymers in E. coli.
An E. coli (strain K12) cell extract was prepared by using freshly grown cells that were washed twice in 20 mM Tris⋅HCl, pH 7.4/40 mM KCL/20 mM NaCl/10 mM MgCl2/1 mM DTT and lysed with the addition of chicken egg-white lysozyme to 0.5 mg/ml. The lysed extract was cleared by centrifugation at 12,000 × g for 15 min, and the supernatant was stored frozen at −70°C. The DNA and DNA/LNA copolymers were incubated in the cell lysate and analyzed as described for blood serum (above). In vitro antisense effects in E. coli for LNA/DNA mix-mer antisense oligonucleotide CtCataCtCttCC (10 μM) targeted to the E. coli beta-lactamase gene and control LNA oligonucleotide gTTgCCgAgACTgTg were determined by using a S-30 E. coli transcription and translation system (27, 28).
Results and Discussion
This study was undertaken to investigate whether LNA could provide a useful component of antisense oligonucleotides. The chemical structure of LNA is shown in Fig. 1, and we asked whether this DNA analogue enters cells efficiently. Fig. 2 shows the cellular uptake of an all-LNA 15-mer oligonucleotide in the human MCF-7 cells, and identical localization was seen in both living and fixed cells.
Figure 2.

Uptake of FITC-labeled all-LNA 15-mer oligonucleotide into human MCF-7 breast cancer cells. (A–C) Low-magnification pictures of living cells. A, light microscopy; C, fluorescence microscopy; B, superimposed view of A and C. (D–F) High magnification of fixed cells. D is phase contrast; F is fluorescence; E is a superimposed view of F and E.
Importantly, a systemically useful antisense oligonucleotide must exhibit stability in blood serum that is far greater than DNA itself. This was found with the DOR-AS-1 LNA/DNA mix-mer and, to a lesser degree, also with the DOR-AS-1 LNA/DNA/LNA gap-mer (Fig. 3A). In fact, the predicted half-life of the mix-mer in serum was greater than isosequential all-phosphorothioate (Fig. 3A). The lesser stability of the gap-mer is likely because of involvement of serum endonucleases. Similar results were obtained with DOR-AS-2.
It is thought that an antisense agent should have at least a 2-fold mechanism of action: (i) causing translational arrest at a ribosomal level, and (ii) activating RNase H, a ubiquitous enzyme that degrades the RNA strand in a RNA/DNA duplex (29). With respect to steric hindrance of ribosomal translation, LNA-containing oligonucleotides have excellent binding affinities and potential to block RNA-processing enzymes. As shown in Table 1, target-bound LNA oligonucleotides show superior thermal stabilities. Indeed, in none of the experiments involving the fully modified LNA could a melting transition be detected, which we attribute to the formation of a very stable LNA/LNA homoduplex with melting temperatures above the applied temperature range (10–90°C). With respect to RNase H activation, which often accounts for the major part of the antisense efficacy (1–8, 29), significant activation has to date been achieved only with oligonucleotides consisting of DNA or phosphorothioates (at least six contiguous nucleotides of either kind seem to be required) (1, 2, 6, 29, 30). No other oligonucleotide analogs (or mimics), e.g., peptide nucleic acids (31), confer the ability to activate RNase H in the absence of contiguous stretches of DNA or phosphorothioate.
To ask whether the LNA-containing oligonucleotides would activate RNase H, an in vitro RNase H cleavage assay was performed. As shown in Fig. 3B, both the DOR-AS-1 LNA/DNA mix-mer and the DOR-AS-1 LNA/DNA/LNA gap-mer were able to activate RNase H, as were the corresponding DNA and phosphorothioate analogs. LNA/DNA mix-mer and LNA/DNA/LNA gap-mer of the independent antisense sequence DOR-AS-2 were also able to recruit RNase H (Fig. 4). All-LNA analogs of DOR-AS-1 as well as DOR-AS-2 showed slow but significant RNase H activities. These results, particularly with the mix-mers, were unexpected and indicate that the activity of RNase H is not necessarily contingent on a contiguous stretch of six DNA or phosphorothioate nucleotides when LNA is used as a component of the oligonucleotide. Structural studies by NMR support this hypothesis (32). However, the exact design criteria of LNA/DNA mix-mers for optimal recruitment of RNase H and cleavage remain to be elucidated.
As a next step in this study, we investigated whether the LNA-containing oligonucleotides would elicit toxic actions on the rat brain when injected into the parenchyma (the striatum). The assays were core body temperature and histological examination of brain tissue. In no type of assay did the DOR-AS-1 LNA-containing oligonucleotides (all-LNA, LNA/DNA mix-mer, or LNA/DNA/LNA gap-mer) differ from their all-DNA counterparts, and we therefore suggest that the liability of LNA toxicity is minimal in the living rat brain. In contrast, and what is well known (7, 8, 25), the all-phosphorothioate oligonucleotides induced severe tissue damage, especially when given into parenchyma (Fig. 5). Signs of toxicity, however less severe, were produced also with DOR-AS-1 oligonucleotides containing only five phosphorothioated bases in the center of the 15-mer (Fig. 5) as well as with DNA oligonucleotides “end-capped” with two phosphorothioate nucleotides at each end of the 15-mer (not shown).
Figure 5.

Rat core temperature and immunofluorescence micrographs from rat caudate-putamen. Rats were injected into the caudate-putamen with phosphorothioate (PS) oligodeoxynucleotides (A), gap-mer DNA/phosphorothioate oligodeoxynucleotides (B), or LNA oligodeoxynucleotides (C), on day 1 and day 3 (10 nmol/day), and core temperature was measured 2 h after last injection. Temperature data are presented as means ± SEM based on three animals per group. A pooled group of DNA-injected animals served as controls (n = 24). Statistical analysis was done by means of an independent Student's t test (nsP > 0.05, *P < 0.05, **P < 0.01). Fluorescence micrographs from sections stained for tyrosine hydroxylase from caudate-putamen (same rats as used in temperature experiments). Arrow indicates lesion with absence of tyrosine hydroxylase staining. (Scale bar, as shown in A, = 100 μm) (CPu, caudate-putamen; cc, corpus callosum)
As the next part of this study, we investigated the antisense efficacy of LNA-containing oligonucleotides. Here we focused on the central nervous system of the living rat and injected oligonucleotides that had been characterized in vitro directly into the cerebrospinal fluid (CSF), (i) at the spinal cord level, and (ii) at the level of the lateral ventricle. CSF administration is particularly useful for testing in vivo antisense activities, because CSF contains very low levels of nucleases and all-phosphodiester (unmodified DNA) oligonucleotides are therefore often sufficiently stable and efficacious as antisense compounds (33–35). Many central nervous system antisense studies have concerned knockdown of G-protein-coupled receptors and have monitored alterations in behavioral and/or biochemical parameters (8, 33–36).
In this study, we targeted the DOR (37–40), a G-protein-coupled receptor that, when activated by the selective agonist, deltorphin II, normally induces, (i) a spinal antinociceptive response in the tail-flick test, and (ii) hypothermia. Efficacious antisense compounds will thus inhibit these in vivo responses to DOR stimulation by deltorphin II. Indeed, we found DOR-AS-1 and DOR-AS-2 (both as LNA/DNA mix-mers and as LNA/DNA/LNA gap-mers) targeting DOR to be highly efficacious and potent.
As shown in Fig. 6, phosphodiester DOR-AS-1 inhibited the spinal antinociceptive response to deltorphin II in a dose-dependent manner. Treatment was performed twice daily for 3 days, which is a sufficiently long time to achieve DOR knockdown (37–40). Both the DOR-AS-1 LNA/DNA mix-mer and the DOR-AS-1 LNA/DNA/LNA gap-mer were highly efficacious, with potencies exceeding that of the isosequential phosphodiester. In control rats, we used instead stringently mismatched DNA or LNA/DNA mix-mer sequences and found both of these to be without effect even at higher doses (cf. Fig. 6). In receptor-binding experiments, we subsequently analyzed DOR densities in the spinal cords of these same groups of rats, as described previously (38). Animals treated with behaviorally maximally efficacious doses of DOR-AS-1 LNA/DNA mix-mer, LNA/DNA/LNA gap-mer, or phosphodiester displayed reductions of receptor densities by 35–55% (each group was significantly reduced at P < 0.05 when compared with control animals, but there was no significant difference between the three groups).
Figure 6.

Blockade by DOR-AS-1 antisense oligonucleotides of [D-Ala2]Deltorphin-(Deltorphin II)-induced antinociception in the warm water tail-flick test in rats. The oligonucleotides were administered in a 5-μl volume twice daily (intrathecal) (0800 and 1700 h) for 3 days. The day after the last injection, the rats were injected with [D-Ala2]deltorphin (60 μg, intrathecal) and tested in the warm water (52°C) tail-flick test for DOR-mediated antinociception. Data are presented as means ± SEM based on six to eight animals per group. The shaded area shows means ± SEM for deltorphin-treated controls. Student's t test was used for comparisons with deltorphin-treated controls, and all differences were considered as statistically significant at P < 0.05 (as indicated by the asterisk).
An important approach in antisense experiments is to use two (or more) different oligonucleotides targeting different regions of the same mRNA. The second set of rat in vivo experiments thus involved a different antisense oligonucleotide (DOR-AS-2) to the same DOR target, and core body temperature was used as a supraspinal likely hypothalamic site-of-action assay. Interestingly, by the use of DOR-AS-2 LNA/DNA mix-mer, we could inhibit the hypothermic response to DOR stimulation. Treatment with 50 μg mix-mer twice daily for 4 days blocked the hypothermia produced by deltorphin II (100 μg into the lateral ventricle) when compared with mismatched control at 20 min (means ± SEM): 37.7°C ± 0.2°C vs. 36.9 ± 0.3°C (P < 0.05, Student's t test; n = 9).
Finally, to determine the nuclease resistance and the selectivity of antisense properties of LNA-containing oligonucleotides in a different biological system distant from the rat brain, we evaluated the stability and antisense effects of a LNA/DNA mix-mer (60% LNA) in E. coli cell lysates. The stability against nucleases was better than that of DNA and all-phosphorothioate oligonucleotides (not shown). Also, antisense LNA/DNA mix-mer targeted to a reporter gene showed potent and sequence-specific gene inhibition in an S-30 transcription and translation system (not shown).
In conclusion, this study introduces a component, LNA, in antisense oligonucleotides. The molecules tested for antisense activity, which contained only LNA and DNA nucleotides, seem to offer an attractive set of properties when compared with many other antisense oligonucleotides: (i) biological stability, (ii) RNase H activation, (iii) lack of detectable toxicity, and (iv) potent biological activities. The present LNA-based strategies (or variations thereof) will likely expand the use of antisense oligonucleotides in research and potentially therapeutics.
Acknowledgments
This study was supported in part by grants to C.W. from the Pharmacia Corporation and the Swedish Medical Research Council.
Abbreviations
- LNA
locked nucleic acid
- DOR
delta opioid receptor
References
- 1.Wahlestedt C, Good L. Curr Opin Drug Disc Dev. 1999;2:142–146. [PubMed] [Google Scholar]
- 2.Stein C A. Nat Biotechnol. 1999;17:209. doi: 10.1038/6909. [DOI] [PubMed] [Google Scholar]
- 3.Branch A D. Trends Biochem Sci. 1998;23:45–50. doi: 10.1016/s0968-0004(97)01155-9. [DOI] [PubMed] [Google Scholar]
- 4.Crooke S T. Antisense Nucleic Acid Drug Dev. 1998;8:vii–viii. doi: 10.1089/oli.1.1998.8.vii. [DOI] [PubMed] [Google Scholar]
- 5.Perry C M, Balfour J A. Drugs. 1999;57:375–380. doi: 10.2165/00003495-199957030-00010. [DOI] [PubMed] [Google Scholar]
- 6.Agrawal S, Zhao O. Curr Opin Chem Biol. 1998;2:519–528. doi: 10.1016/s1367-5931(98)80129-4. [DOI] [PubMed] [Google Scholar]
- 7.Engelhard H H, Egli M, Rozental J M. Pediatr Neurosurg. 1998;28:279–285. doi: 10.1159/000028665. [DOI] [PubMed] [Google Scholar]
- 8.Ho S P, Livanov V, Zhang W, Lie J-H, Lesher T. Mol Brain Res. 1998;62:1–11. doi: 10.1016/s0169-328x(98)00185-5. [DOI] [PubMed] [Google Scholar]
- 9.Zhou W, Agrawal S. Bioorg Med Chem Lett. 1998;8:3269–3274. doi: 10.1016/s0960-894x(98)00591-5. [DOI] [PubMed] [Google Scholar]
- 10.Tarköy M, Leumann C. Angew Chem Int Ed Engl. 1993;32:1432–1435. [Google Scholar]
- 11.Altmann K-H, Kesselring R, Francotte E, Rihs G. Tetrahedron Lett. 1994;35:2331–2332. [Google Scholar]
- 12.Marquez V E, Siddiqui M A, Ezzitouni A, Russ P, Wang J, Wagner R W, Matteucci M D. J Med Chem. 1996;39:3739–3747. doi: 10.1021/jm960306+. [DOI] [PubMed] [Google Scholar]
- 13.Steffens R, Leumann C J. J Am Chem Soc. 1997;119:11548–11550. [Google Scholar]
- 14.Nielsen P, Pfundheller H M, Olsen C E, Wengel J. J Chem Soc, Perkin Trans. 1997;1:3423–3433. [Google Scholar]
- 15.Christensen N K, Petersen M, Nielsen P, Jacobsen J P, Olsen C E, Wengel J. J Am Chem Soc. 1998;120:5458–5461. [Google Scholar]
- 16.Singh S K, Nielsen P, Koshkin A A, Wengel J. Chem Commun. 1998;1998:455–456. [Google Scholar]
- 17.Koshkin A A, Singh S K, Nielsen P, Rajwanshi V K, Kumar R, Meldgaard M, Olsen C E, Wengel J. Tetrahedron. 1998;54:3607–3630. [Google Scholar]
- 18.Obika S, Nanbu D, Hari Y, Andoh J, Morio K, Doi T, Imanishi T. Tetrahedron Lett. 1998;39:5401–5407. [Google Scholar]
- 19.Koshkin A A, Nielsen P, Meldgaard M, Rajwanshi V K, Singh S K, Wengel J. J Am Chem Soc. 1998;120:13252–13260. [Google Scholar]
- 20.Wengel J. Acc Chem Res. 1999;32:301–302. [Google Scholar]
- 21.Yaksh T L, Rudy T A. J Pharmacol Exp Ther. 1977;202:411–428. [PubMed] [Google Scholar]
- 22.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. London: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- 23.Ossipov M H, Kovelowski C J, Nichols M L, Hruby V J, Porreca F. Pain. 1995;62:287–293. doi: 10.1016/0304-3959(94)00231-3. [DOI] [PubMed] [Google Scholar]
- 24.Salmi P, Karlsson T, Ahlenius S. Eur J Pharmacol. 1994;253:67–73. doi: 10.1016/0014-2999(94)90758-7. [DOI] [PubMed] [Google Scholar]
- 25.Broberger C, Nylander I, Geijer T, Terenius L, Hökfelt T, Georgieva J. Mol Brain Res. 2000;75:25–45. doi: 10.1016/s0169-328x(99)00276-4. [DOI] [PubMed] [Google Scholar]
- 26.Markey K A, Kondo S, Shenkman L, Goldstein M. Mol Pharmacol. 1980;17:79–85. [PubMed] [Google Scholar]
- 27.Thorson J S, Cornish V W, Barrett J E, Cload S T, Yano T, Schultz P G. Methods Mol Biol. 1998;77:43–73. doi: 10.1385/0-89603-397-X:43. [DOI] [PubMed] [Google Scholar]
- 28.O'Callaghan C H, Morris A, Kirby S M, Shingler A H. Antimicrob Agents Chemother. 1972;1:283–287. doi: 10.1128/aac.1.4.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uhlmann E, Peyman A, Will D W. In: Encyclopedia of Cancer. Bertino J R, editor. Vol. 1. San Diego: Academic; 1997. pp. 64–81. [Google Scholar]
- 30.Uhlmann E. Biol Chem. 1998;379:1045–1052. [PubMed] [Google Scholar]
- 31.Nielsen P E. Curr Opin Biotechnol. 1999;10:71–75. doi: 10.1016/s0958-1669(99)80013-5. [DOI] [PubMed] [Google Scholar]
- 32.Bondensgaard, K., Petersen, M., Singh, S. K., Rajwanshi, V. K., Wengel, J. & Jacobsen, J. P. (2000) Chemistry - A European Journal, in press. [DOI] [PubMed]
- 33.Wahlestedt C, Pich E M, Koob G F, Yee F, Heilig M. Science. 1993;259:528–531. doi: 10.1126/science.8380941. [DOI] [PubMed] [Google Scholar]
- 34.Wahlestedt C, Golanov E, Yamamoto S, Yee F, Ericson H, Yoo H, Inturrisi C E, Reis D J. Nature (London) 1993;363:260–263. doi: 10.1038/363260a0. [DOI] [PubMed] [Google Scholar]
- 35.Wahlestedt C. Trends Pharmacol Sci. 1994;15:42–46. doi: 10.1016/0165-6147(94)90107-4. [DOI] [PubMed] [Google Scholar]
- 36.Szklarczyk A W, Kaczmarek L. Antisense Nucleic Acid Drug Dev. 1999;9:105–116. doi: 10.1089/oli.1.1999.9.105. [DOI] [PubMed] [Google Scholar]
- 37.Standifer K M, Chien C C, Wahlestedt C, Brown G P, Pasternak G W. Neuron. 1994;12:805–810. doi: 10.1016/0896-6273(94)90333-6. [DOI] [PubMed] [Google Scholar]
- 38.Bilsky E J, Bernstein R N, Hruby V J, Rothman R B, Lai J, Porreca F. J Pharmacol Exp Ther. 1996;277:491–501. [PubMed] [Google Scholar]
- 39.Kest B, Lee C E, McLemore G L, Inturrisi C E. Brain Res Bull. 1996;39:185–188. doi: 10.1016/0361-9230(95)02092-6. [DOI] [PubMed] [Google Scholar]
- 40.Tseng L F, Collins K A, Kampine J P. Eur J Pharmacol. 1994;258:R1–R3. doi: 10.1016/0014-2999(94)90072-8. [DOI] [PubMed] [Google Scholar]