

Abstract
Phosphorylation of the regulatory (R) domain initiates cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel activity. To discover how the function of this domain is determined by its structure, we produced an R domain protein (R8) that spanned residues 708–831 of CFTR. Phosphorylated, but not unphosphorylated, R8 stimulated activity of CFTR channels lacking this domain, indicating that R8 is functional. Unexpectedly, this functional R8 was predominantly random coil, as revealed by CD and limited proteolysis. The CD spectra of both phosphorylated and nonphosphorylated R8 were similar in aqueous buffer. The folding agent trimethylamine N-oxide induced only a small increase in the helical content of nonphosphorylated R8 and even less change in the helical content of phosphorylated R8. These data, indicating that the R domain is predominantly random coil, may explain the seemingly complex way in which phosphorylation regulates CFTR channel activity.
Cystic fibrosis (CF) transmembrane conductance regulator (CFTR) is a phosphorylation-regulated Cl− channel that provides a pathway for Cl− movement across epithelial membranes in the airways, sweat gland, intestine, and pancreas (1, 2). Loss of CFTR function causes the genetic disease CF. CFTR Cl− channel activity is regulated by cAMP-dependent protein kinase (PKA) and protein kinase C phosphorylation of the regulatory (R) domain (3–8). After phosphorylation of the R domain, ATP binding and hydrolysis by the two nucleotide-binding domains (NBDs) gate the channel (7, 8).
The R domain has a unique primary sequence with no homology to any known protein (1). The R domain was originally defined as those residues encoded by exon 13 (residues 590–830) (1). However, the N-terminal half of exon 13 shares sequence similarity with the NBDs of other ATP-binding cassette (ABC) transporters (9, 10). Compelling evidence that the N-terminal portion of exon 13 is actually part of NBD1 came from the crystal structure of HisP, the NBD of the histidine permease ABC transporter (11). In CFTR, an NBD structure analogous to that of HisP would extend to approximately residue 660. The C-terminal half of exon 13 appears to be the functional R “regulatory” domain (9, 10). Consistent with this, deletion of residues 708–835 produces a CFTR channel with properties like those of wild-type CFTR, suggesting that residues 708–835 form a functional R domain that can be deleted (9, 12–14). More extensive deletions (residues 590–835, 656–835, 676–835, or 681–835) disrupted function (9), probably because they disrupt a portion of NBD1.
Exon 13 contains eight serines in consensus motifs for PKA phosphorylation; five of these serines, S660, S700, S737, S795, and S813, are phosphorylated in cells (15–17). The contribution of the various phosphoserines to channel activity is complex. No one serine is required for activity; mutation of single or multiple serines decreases, but does not abolish, phosphorylation-stimulated activity (6, 13, 15, 18–21). Phosphorylation of several different serines can stimulate channel activity, although not all of the serines have the same quantitative effect on channel activity. Mutating the consensus serines to aspartate, to mimic the negative charge imposed by phosphorylation, stimulated channel activity in the absence of phosphorylation (19), suggesting that negative charge plays a role in channel stimulation. Earlier work suggested that phosphorylation of the R domain stimulates channel activity by increasing the binding and/or hydrolysis of ATP by the NBDs (13, 22).
Additional clues to the mechanism by which the R domain regulates channel activity came from studies in which parts of the R domain were deleted and/or synthesized as separate proteins and then added back to the channel. An example is CFTR-ΔR/S660A, in which residues 708–835 are deleted and Ser-660 is mutated to alanine (12, 19, 23). CFTR-ΔR/S660A lacks the serines (660, 737, 795, and 813) shown to be responsible for the majority of PKA-stimulated channel activity (6, 15, 16, 19). This channel differs from wild-type CFTR in two important ways: it has a low level of constitutive Cl− channel activity in the absence of PKA-dependent phosphorylation, and its activity is not altered by addition of PKA (9, 12–14).
Constitutive activity of CFTR-ΔR/S660A suggests that the unphosphorylated R domain might have an inhibitory function (12). In support of this idea, application of an unphosphorylated R domain protein (residues 588–855) to wild-type CFTR in lipid bilayers was reported to inhibit activity (23). However, another study observed no effect of the unphosphorylated R domain protein R1 (residues 645–834) on wild-type CFTR channels in excised cell-free patches of membranes (13). Moreover, neither unphosphorylated R1 nor the unphosphorylated 588–855 protein had an effect on CFTR-ΔR/S660A (13, 14). Furthermore, if the function of the R domain were primarily inhibitory, the activity of CFTR-ΔR/S660A would be expected to be equal to or greater than that of the phosphorylated wild-type channels. In contrast to this prediction, the activity of CFTR-ΔR/S660A is approximately one-third that of phosphorylated wild-type CFTR (13, 19). Thus whether the R domain has an inhibitory role remains uncertain.
Other evidence indicates that the phosphorylated R domain has a stimulatory role. Recombinant protein containing residues 645–835 stimulated CFTR-ΔR/S660A activity in excised membrane patches and residues 588–855 stimulated CFTR-ΔR/S660A in lipid bilayers (13, 14). Phosphorylation was required for channel activation; the unphosphorylated R domain had no stimulatory effect.
All these data suggest that the R domain forms a unique structural and functional domain with boundaries from about residue 708 to approximately residue 830. The goal of this work was to determine the structure of an R domain construct comprising residues 708–831 (R8), which closely complements the deletion in CFTR-ΔR/S660A. We were encouraged by the finding that R8 retains stimulatory activity, suggesting that if the R domain has a specific structure, it is retained in this protein.
Experimental Procedures
Site-Directed Mutagenesis and Plasmids.
We amplified the R domain of wild-type CFTR and ligated the resultant fragment into the pET21d vector (Novagen). Vector splice sites and the insert coding sequence were verified by sequencing (University of Iowa DNA Core Facility). The amino acid sequence of R8 is MG-CFTR residues 708–831-LEHHHHHH.
Bacterial Expression and Purification.
pET plasmids and pDC952 (encoding arginine tRNA) (a gift of James Walker, University of Texas, Austin, TX) were cotransformed into BL21 (DE3) (Novagen) on LB plus carbenicillin/chloramphenicol plates. We used the parental BL21 (DE3) cell line and not the derivative BL21 (DE3) pLysS, because the pLysS cells, but not the parental cells, produce sufficient levels of chloramphenicol acetyltransferase to interfere with subsequent purification of R8. Cultures were grown at 37°C to 0.6 OD at 600 nm, induced with 0.1 mM isopropyl-d-thiogalactoside, and grown for 3 h at 37°C. Cells were centrifuged at 3,500 × g, resuspended in binding buffer (BB) (20 mM Tris⋅HCl, pH 7.9/0.5 M NaCl/1 mM imidazole) and sonicated. After sonication, inclusion bodies were isolated by centrifugation at 39,000 × g for 20 min. The inclusion bodies were resuspended in BB with 6 M urea, solubilized overnight at 4°C, and centrifuged at 39,000 × g for 20 min to separate the urea-soluble and urea-insoluble pellet.
Proteins in the urea-soluble fraction were bound to Talon cobalt columns (CLONTECH) in BB and purified by using a step gradient of imidazole (10 mM to 500 mM) in BB. Protein concentrations were determined by the Bradford assay (Bio-Rad).
Identification of R8.
R8 was analyzed by SDS/PAGE under nonreducing conditions. The composition of R8 was confirmed by amino acid analysis, sequencing of proteolytic fragments, and reactivity with the anti-CFTR antibody 13–1 (Genzyme).
Phosphorylation of R8.
Purified protein was phosphorylated with 40 nM PKA/1 mM MgATP/3 mM MgCl2 in 10 mM potassium phosphate (KPi), pH 6.8, at 30°C.
Proteolysis of R8.
R8 dialyzed into Hepes/NMDG-Cl buffer [in mM: 140 N-methyl-d-glucamine (NMDG), 3 MgCl2/1 CsEGTA/10 Hepes (pH 7.3 with HCl; Cl− concentration, 140 mM)] was incubated at room temperature for the indicated time with either trypsin or papain (sequencing grade; Boehringer-Mannheim) (1:100, w:w, protease:R8). At each time point, 1 volume of ×2 sample buffer (62.5 mM Tris, pH 6.8/4% SDS/100 mM DTT/20% glycerol/0.005% bromophenol blue) was added to an aliquot of the reaction and boiled immediately for at least 3 min to inactivate proteases (24). For some experiments, 1 mM PMSF was added before sample buffer. Results did not differ with or without PMSF. After completion of the last time point, samples were electrophoresed on SDS/PAGE and stained with Coomassie blue. For sequencing, proteolytic fragments of R8 were quantitatively transferred to ProBlot nitrocellulose (IBI) (25) and sequenced by Edman degradation.
Patch-Clamp Analysis.
We expressed CFTR-ΔR/S660A in HeLa cells by using the vaccinia virus/T7 hybrid expression system (19). Methods for excised inside-out patches were as previously described at 37°C and −40 mV (13). Pipette solution contained (in mM): 140 NMDG/2 MgCl2/5 CaCl2/100 l-aspartic acid/10 Hepes (pH 7.3 with HCl; Cl− concentration, 50 mM). Bath solutions were: Hepes/NMDG-Cl buffer or 10 KPi, pH 7.3, with 100 NaCl. Similar results were obtained with both solutions. PKA (75 nM) and 1 mM ATP were present throughout patch recording. Data points are mean current during successive 10-s intervals.
Analytical Ultracentrifugation.
Sedimentation equilibrium was conducted at 25°C on a Beckman XL-I ultracentrifuge (Beckman Coulter) equipped with an An-60 Ti rotor. Concentrations of R8 were 40, 20, and 10 μM in 10 mM glycine and 0.1 M NaCl, pH 9.0, similar to conditions used for some CD studies.
Samples were centrifuged at 17,000, 24,000, and 35,000 rpm. Absorbance was monitored at 236 nm. Data were analyzed by using origin software (Ver. 3.78, Microcal Software, Northampton, MA). The partial specific volume, ν̄, of R8 was calculated to be 0.711 ml/g on the basis of amino acid composition (26). The calculated solution density, ρ, for the samples was 1.004 g/ml (27). The apparent molecular weight of R8, M, was determined by the equation for an ideal solution containing a single species (28):
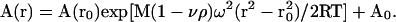 |
1 |
CD Analysis.
CD spectra were recorded on an Aviv CD Model 62DS (Aviv Associates, Lakewood, NJ) in an 0.2-cm pathlength quartz cuvette (Hellma, Forest Hills, NY) thermostated at 22°C. Spectra were collected from 260 to 210 nm at a bandwidth of 1 nm every 0.25 nm with a 2-s averaging time. Each spectrum is the result of three summed spectra corrected for solute buffer.
R8 was phosphorylated as described above in 1 mM glycine, pH 9.0, with 1 mM ATP, dialyzed to remove ATP and MgCl2, and centrifuged at 200,000 × g. Parallel incubations and dialyses were carried out in the absence of PKA for nonphosphorylated R8. Spectra were obtained from nonphosphorylated and phosphorylated R8 diluted in the following buffers: (i) 1 mM glycine, pH 9; (ii) 10 mM KPi, pH 7.3; or (iii) 10 mM KPi, 100 mM NaCl, pH 7.3. Similar CD spectra were obtained with each buffer. The inclusion of 5 mM MgCl2 or MnCl2 had no effect on the spectrum. To analyze the effect of lipid, R8 was incorporated into a sonicated phospholipid mixture (PC/PS, 3:1).
For CD spectra in trimethylamine oxide (TMAO), concentrated R8 dialyzed into 1 mM glycine was diluted to 0.15 mg/ml in 10 mM KPi, pH 7.3, or into 2.8 M recrystallized TMAO. Titration curves with TMAO were carried out at 220 nm, the wavelength of greatest difference between the R8 spectra in KPi and TMAO.
Results
Expression and Purification of R8.
We expressed and purified R8 by using the pET vector system and metal-affinity chromatography. To alleviate the problem of poor expression and protein truncation caused by the infrequent usage of the arginine codons AGA and AGG by Escherichia coli, we coexpressed R8 with the tRNA synthase for the AGA codon (29). R8 contains no cysteines, thus preventing dimerization and aggregation of the purified protein in the absence of reducing agents. We partially purified R8, which was sequestered in inclusion bodies, by isolating the inclusion body pellet (Fig. 1). After solubilization of the inclusion bodies in urea (lane 4), R8 was purified by metal-affinity chromatography (lanes 6–8). Typical 500-ml cultures yielded ≥10 mg R8 that was 94–97% pure.
Figure 1.

Purification of R8. Coomassie-stained gel. Lane 1 (UI): before induction by IPTG. Lane 2 (Total): 3 h after IPTG. Lane 3 (Sup): supernatant from induced culture. Lane 4 (Urea Sol): urea-soluble fraction from inclusion bodies. Lane 5 (Urea Insol): urea-insoluble pellet. Lane 6 (FT): flow through from metal-affinity column. Lane 7 (Wash): wash with 1 mM imidazole. Lane 8 (Imid): elution with 200 mM imidazole. Arrow indicates R8.
After affinity purification, we dialyzed R8 to remove urea and imidazole and to promote protein folding. R8 was soluble (3–4 mg/ml) in 1 mM glycine or 10 mM Tris at pH 9 and (5–6 mg/ml) in Hepes/NMDG-Cl buffer (pH 7.3).
Functional Studies of R8.
R8 was phosphorylated by PKA in vitro as indicated by the shift in electrophoretic migration of R8 on SDS/PAGE (Fig. 2). This shift is similar to that seen in other studies of R domain proteins (16, 30).
Figure 2.

Effect of phosphorylation on electrophoretic mobility of R8. R8 was phosphorylated with PKA and ATP at 30°C for time indicated and electrophoresed on SDS/PAGE. Arrows indicate nonphosphorylated and phosphorylated R8.
Earlier work showed that R1 (CFTR residues 645–834) stimulated CFTR-ΔR/S660A channel activity in excised membrane patches (13). Fig. 3 shows that adding phosphorylated R8 to the cytosolic surface of CFTR-ΔR/S660A reversibly stimulated current 33.4 ± 3.7% (n = 9). This stimulation is similar to that which we previously reported with the R1 construct (43.2 ± 3.4%) (13). The small difference may be caused by the presence of Ser-660 in R1. Like wild-type CFTR, current depended on ATP (Fig. 3). Nonphosphorylated R8 had no effect, consistent with an earlier study showing no effect of nonphosphorylated R1 (13). Similar results were obtained with both NMDG-Cl and NaCl buffers. These data indicate that purified recombinant R8 retains stimulatory activity and that stimulation requires phosphorylation.
Figure 3.

R8 stimulation of CFTR-ΔR/S660A Cl− channel activity. (A) Current from excised inside-out patch containing many channels. Bars indicate exposure to ATP (1 mM) and phosphorylated R8 (1.5 μM). PKA (75 nM) was present throughout. (B) Change in current produced by unphosphorylated (n = 4) and phosphorylated R8 (n = 9).
Structural Studies of R8.
Sedimentation equilibrium showed that soluble R8 was a monomer, both phosphorylated and nonphosphorylated (Fig. 4).
Figure 4.

Sedimentation equilibrium ultracentrifugation of R8. Smooth lines (Middle) are fit of data to Eq. 1. Fitted molecular weight for nonphosphorylated R domain is 16,640 ± 680 and A0 = 0.022; that for phosphorylated R domain is 15,250 ± 470 and A0 = −0.003. Predicted molecular weight of nonphosphorylated R8 based on amino acid composition is 15,270. Residuals of fit are shown (Top and Bottom) for phosphorylated and nonphosphorylated R8, respectively.
We used limited proteolysis to determine whether portions of R8 would be inaccessible to proteases, suggesting conformational protection. We chose proteases with multiple potential consensus sites within R8, trypsin (10 sites), and papain (3 sites), which are evenly distributed throughout the protein. The tryptic patterns of phosphorylated and nonphosphorylated R8 (Fig. 5A) show that most of the protein is susceptible to trypsin. Similar results were found after papain digestion (Fig. 5B). In addition, the tryptic pattern after an 80-min digestion was similar in phosphorylated and nonphosphorylated R8 after correcting for the size difference because of phosphorylation. We sequenced proteolytic fragments from nonphosphorylated R8 and found fragments that corresponded to most of the predicted proteolytic sites. For example, trypsin cut at R810/811, R792, and R764/765/766. We also sequenced three proteolytic fragments from phosphorylated R8 that corresponded to nonphosphorylated R8 (Fig. 5). These data suggest that most of R8 was accessible to both proteases and that phosphorylation did not induce a global conformational change in R8, but they do not eliminate the possibility of small discrete changes undetectable by these methods.
Figure 5.

Proteolytic patterns of R8. Equal amounts of R8 were incubated with either trypsin or papain for indicated times. Lanes 1–5 are nonphosphorylated and lanes 6–10 are phosphorylated R8. Arrow indicates intact R8. Sequences of indicated proteolytic fragments are: (●) GIRKFS1; (*)RQSVLNLM; and (○)SVNQGQNI.
CD was used to examine the conformation of R8. Fig. 6A shows that R8 was a random coil with little well-ordered secondary structure. The results were the same irrespective of whether spectra were obtained in high pH, low salt, or the buffers used for patch clamp. The negative ellipticity at 222 nm is consistent with a helical content of about 5% (31) with the balance being random coil. Phosphorylated and nonphosphorylated R8 had similar CD spectra in the aqueous buffers (Fig. 6A), suggesting that phosphorylation did not induce a large structural change in R8.
Figure 6.

CD spectra of R8. (A) R8 (0.15 mg/ml) in 10 mM KPi, pH 7.3. (B) R8 (0.15 mg/ml) dialyzed into 2.8 M TMAO. Other conditions were similar to those in A. Spectra extend only to 210 nm because TMAO interferes with signal below 210 nm. (C) Equal concentrations of phosphorylated and nonphosphorylated R8 (0.15 mg/ml) in 1 mM glycine, pH 9.0, were diluted into increasing concentrations of TMAO and ellipticity measured at 220 nm.
We considered the possibility that interaction of R8 with lipid components of the plasma membrane might alter its conformation. To test this, we incorporated R8 in phospholipid micelles. This did not alter the CD spectrum.
To test whether R8 has the potential to fold, we examined the effect of TMAO. TMAO is a natural osmolyte that promotes folding of proteins; many unfolded proteins have been shown to adopt their native structure in TMAO (32). As shown in Fig. 6B, TMAO altered the CD spectra of nonphosphorylated R8 (compare Fig. 6 A and B), consistent with a small decrease in random coil and a modest increase in helical content to about 10%. This relatively small TMAO-induced increase in helical content was saturated at 2.8 M as shown by the TMAO titration curve (Fig. 6C). Interestingly, the helical content of phosphorylated R8 was only slightly increased in TMAO and was less than the change observed for nonphosphorylated R8 in TMAO. Thus, TMAO had only minimal effects on the secondary structure of R8; R8 retained a predominantly random coil conformation, whether phosphorylated or not.
Discussion
The data show that R8 (residues 708–831 of CFTR) is functional; it stimulated CFTR-ΔR/S660A channel activity. Surprisingly, R8 is predominantly unstructured, exhibiting a CD spectrum characteristic of random coil, both when phosphorylated and nonphosphorylated. Because R8, like wild-type CFTR, must be phosphorylated before it can activate CFTR-ΔR/S660A, either the phosphorylated R8 binds and stimulates CFTR while it is a random coil, or R8 adopts a more ordered structure on contact with the rest of CFTR. Interestingly, TMAO that promotes folding of other unstructured proteins had only minimal effects on R8.
The conclusion that the functional R domain is predominantly random coil is consistent with three other observations. First, we evaluated R8 with three structural prediction algorithms: Chou and Fasman (33), Profile Network Prediction of Secondary Structure (34, 35), and jpred (36). The programs did not agree in the assignment of α-helix and β-sheet secondary structure. Second, the sequence of R8 is not well conserved in CFTR from different species (refs. 9 and 10 and unpublished work). Most highly structured proteins show substantial sequence conservation; examples are the NBDs of CFTR. Moreover, many of the conserved residues in such proteins have hydrophobic side chains buried within the protein core, where they help maintain structure (37). In contrast, the conserved sequences in R8 are the consensus PKA phosphorylation motifs, which are expected to be solvent exposed where they can serve a functional rather than a structural role. Third, although more than 850 mutations have been reported in the CFTR gene [http://www.genet.sickkids.on.ca (1999) Website], there are relatively few missense mutations in the region encoding R8. Within exon 13, missense mutations have been observed in 17% of the codons in residues 590–708, but in only 6% of the codons in residues 708–830. From this region, the mutations R792G, A800G, E822K, and E826K increase or decrease current, but have not been reported to alter channel properties (38, 39). These observations are consistent with the notion that an amino acid change in a region that is predominantly random coil may be less likely to disrupt function and thereby cause CF.
Earlier studies of larger R domain proteins had suggested that the R domain adopts an ordered structure (30, 40). The CD spectrum of a construct (595–831) encoding most of exon 13 suggested a structural content of 10% α-helix and 30% β-sheet (40). A study of an even larger construct (404–830) predicted 19% α-helix and 43% β-sheet (30). Both of those constructs include significant portions of NBD1 based on sequence comparisons and the crystal structure of HisP (see Introduction and ref. 11). Therefore, it seems likely that the α-helices and β-sheets were localized in NBD1 and the random coil was localized in the R domain. Thus, the results of those studies may not be so different from those reported here.
If phosphorylation does not increase R8 structure, how does it activate the channel? Phosphorylation might induce a conformational change only in discrete regions of R8, resulting in small spectral shifts masked by the larger signal from the random coil. Such discrete structural changes might be sufficient for a functional effect. Second, phosphorylation might simply introduce negative charge into R8. This interpretation would be consistent with the finding that mutation of phosphorylatable serines to aspartate activated the channel (19). Finally, phosphorylation may not directly change the conformation of R8, but may simply be required for an interaction of R8 with other sites within CFTR. Perhaps the regions immediately surrounding the conserved PKA phosphorylation sites are involved in interaction with and stimulation of CFTR.
This study also has some limitations. First, in the Introduction, we described the rationale for choosing the boundaries of R8. It could be that the actual boundaries of the R domain extend beyond residues 708–831. Importantly, however, more extensive deletions could not be made without destroying channel function. The ability to delete residues 708–831 and yet retain channel function, coupled with the ability of these same residues to partially restore phosphorylation-regulated activity, suggests that R8 represents a “functional” domain. Second, whereas our data indicate that R8 is primarily random coil even under conditions designed to allow folding, it is possible that R8 could adopt a more structured conformation in the presence of the rest of CFTR.
The advantages of unfolded proteins for signaling or regulation are becoming more apparent (41, 42). Examples include the cell-cycle kinase inhibitor p21waf1/Cip1/Sdi1 (43), the FlgM flagellar protein of Salmonella typhimurium (44), and the fibronectin receptor-binding protein (45). The fast inactivation domain of the Shaker B K+ channel acts in a structure-independent manner (46, 47). An R domain composed predominantly of random coil has an analogy in the kinase-inducible domain (KID) of the cAMP-responsive element-binding protein (CREB). Several structural parameters indicate that KID is unstructured (48–51). Like R8, serine phosphorylation of KID does not increase structure (48, 51). However, just as phosphorylation allows R8 to activate the Cl− channel, phosphorylation allows KID to bind and activate CREB-binding protein (CBP). This binding induces a conformational change in KID that is limited to the phosphoserine and a few neighboring amino acids (51, 52).
An R domain that is predominantly random coil with a few conserved phosphorylation sites may explain some seemingly puzzling aspects of CFTR function. Earlier studies have shown that no single phosphorylation site is required for stimulation, that phosphorylation of several different sites can stimulate, and that not all phosphorylated sites have the same effect (6–8, 13, 15, 18–21). Fig. 7 shows some speculative models of how the R domain might stimulate CFTR and emphasize the functional advantages of an R domain with few structural constraints.
Figure 7.
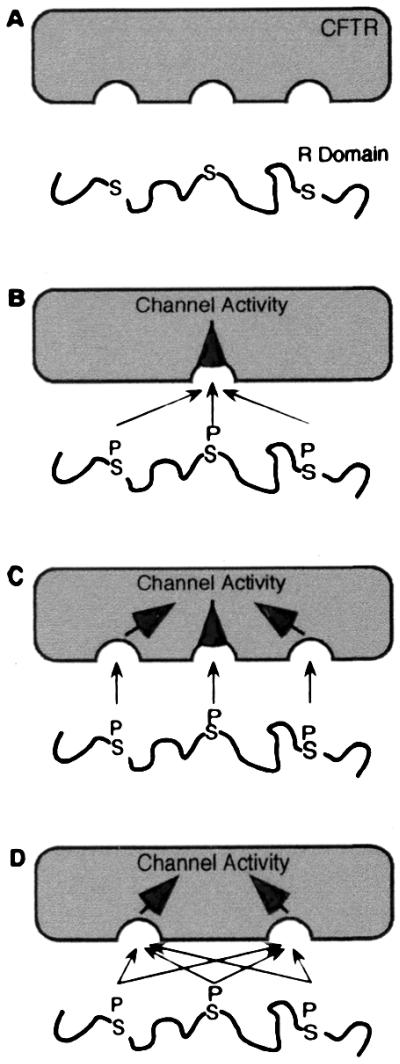
Models of interaction of R domain with the rest of CFTR (in gray). See text for details.
Model A shows the unphosphorylated R domain as random coil. Without phosphorylation, it does not stimulate activity.
Model B shows a phosphorylated R domain. Although we show three phosphoserines in the R domain, the actual number phosphorylated in an individual CFTR molecule is not certain. Because the R domain is unstructured, any of several different phosphoserines could interact with the single site shown on CFTR to stimulate channel activity. Model B could also explain the ability of multiple different phosphoserines to stimulate, possibly with quantitatively different effects on activity. However, this model does not explain how phosphorylation of more than one serine would give a graded increase in activity.
Model C shows multiple phosphoserines interacting with multiple sites in the rest of the protein. This model can account for increasing activity with increasing phosphorylation. Model C predicts that if there are three or four or five phosphoserines in the R domain, there must be three or four or five interaction sites for those phosphoserines. This model could also account for quantitatively different effects generated by phosphorylation of different serines.
Model D is the one we favor. This model shows multiple phosphoserines in the R domain interacting with multiple binding sites in CFTR. Although we do not know the number of sites with which the R domain interacts, here we show two, suggesting the possibility of at least one interaction site at each NBD. However, there may be more interaction sites, as evidenced by the observation that regions within the R domain and the N terminus interact (53). Irrespective of the number of interaction sites, model D could account for the apparent promiscuous relationship between phosphorylation sites and stimulation of activity. In such a model, a relatively unstructured R domain would allow multiple different phosphoserines to stimulate, no one phosphoserine would be required, different phosphoserines could have quantitatively different effects, and the greater the degree of phosphorylation, the greater the activity.
The finding that the R domain is predominantly random coil is consistent with the evolving recognition that many protein domains and full length proteins are intrinsically unstructured (41, 42). It is also consistent with the emerging appreciation that unstructured proteins and domains hold certain advantages for signaling functions.
Acknowledgments
We thank Suzy Dietz, Lisa Einwalter, Pary Weber, and Theresa Mayhew for excellent assistance, Dr. Michael Winter for discussions, and Dr. James Walker for the pDC952 plasmid. We thank the University of Iowa DNA Core for sequencing help, and the University of Iowa Molecular Analysis Facility for protein sequencing and amino acid analysis. This work was supported by the National Heart Lung and Blood Institute (HL42385) and the Howard Hughes Medical Institute (HHMI). M.J.W. is an Investigator of the HHMI.
Abbreviations
- CFTR
cystic fibrosis transmembrane conductance regulator
- CF
cystic fibrosis
- R domain
regulatory domain
- NBD
nucleotide-binding domain
- PKA
cAMP-dependent protein kinase
- NMDG
N-methyl-d-glucamine
- TMAO
trimethylamine oxide
- KID
kinase-inducible domain
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.100588797.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.100588797
References
- 1.Riordan J R, Rommens J M, Kerem B-S, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou J-L, et al. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 2.Welsh M J, Tsui L-C, Boat T F, Beaudet A L. In: The Metabolic and Molecular Basis of Inherited Disease. Scriver C R, Beaudet A L, Sly W S, Valle D, editors. New York: McGraw–Hill; 1995. pp. 3799–3876. [Google Scholar]
- 3.Tabcharani J A, Chang X-B, Riordan J R, Hanrahan J W. Nature (London) 1991;352:628–631. doi: 10.1038/352628a0. [DOI] [PubMed] [Google Scholar]
- 4.Berger H A, Travis S M, Welsh M J. J Biol Chem. 1993;268:2037–2047. [PubMed] [Google Scholar]
- 5.Hwang T-C, Nagel G, Nairn A C, Gadsby D C. Proc Natl Acad Sci USA. 1994;91:4698–4702. doi: 10.1073/pnas.91.11.4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilkinson D J, Strong T V, Mansoura M K, Wood D L, Smith S S, Collins F S, Dawson D C. Am J Physiol. 1997;273:L127–L133. doi: 10.1152/ajplung.1997.273.1.L127. [DOI] [PubMed] [Google Scholar]
- 7.Gadsby D C, Narin A C. Physiol Rev. 1999;79:S77–S107. doi: 10.1152/physrev.1999.79.1.S77. [DOI] [PubMed] [Google Scholar]
- 8.Sheppard D N, Welsh M J. Physiol Rev. 1999;79:S43–S45. doi: 10.1152/physrev.1999.79.1.S23. [DOI] [PubMed] [Google Scholar]
- 9.Rich D P, Gregory R J, Cheng S H, Smith A E, Welsh M J. Recept Channels. 1993;1:221–232. [PubMed] [Google Scholar]
- 10.Dulhanty A M, Riordan J R. FEBS Lett. 1994;343:109–114. doi: 10.1016/0014-5793(94)80300-5. [DOI] [PubMed] [Google Scholar]
- 11.Hung L-W, Wang I X, Nikaido K, Liu P-Q, Ames G F-L, Kim S-H. Nature (London) 1998;396:703–707. doi: 10.1038/25393. [DOI] [PubMed] [Google Scholar]
- 12.Rich D P, Gregory R J, Anderson M P, Manavalan P, Smith A E, Welsh M J. Science. 1991;253:205–207. doi: 10.1126/science.1712985. [DOI] [PubMed] [Google Scholar]
- 13.Winter M C, Welsh M J. Nature (London) 1997;389:294–296. doi: 10.1038/38514. [DOI] [PubMed] [Google Scholar]
- 14.Ma J, Zhao J, Drumm M L, Xie J, Davis P B. J Biol Chem. 1997;272:28133–28141. doi: 10.1074/jbc.272.44.28133. [DOI] [PubMed] [Google Scholar]
- 15.Cheng S H, Rich D P, Marshall J, Gregory R J, Welsh M J, Smith A E. Cell. 1991;66:1027–1036. doi: 10.1016/0092-8674(91)90446-6. [DOI] [PubMed] [Google Scholar]
- 16.Picciotto M R, Cohn J A, Bertuzzi G, Greengard P, Nairn A C. J Biol Chem. 1992;267:12742–12752. [PubMed] [Google Scholar]
- 17.Cohn J A, Nairn A C, Marino C R, Melhus O, Kole J. Proc Natl Acad Sci USA. 1992;89:2340–2344. doi: 10.1073/pnas.89.6.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang X-B, Tabcharani J A, Hou Y-X, Jensen T J, Kartner N, Alon N, Hanrahan J W, Riordan J R. J Biol Chem. 1993;268:11304–11311. [PubMed] [Google Scholar]
- 19.Rich D P, Berger H A, Cheng S H, Travis S M, Saxena M, Smith A E, Welsh M J. J Biol Chem. 1993;268:20259–20267. [PubMed] [Google Scholar]
- 20.Seibert F S, Tabcharani J A, Chang X-B, Dulhanty A M, Mathews C, Hanrahan J W, Riordan J R. J Biol Chem. 1995;270:2158–2162. doi: 10.1074/jbc.270.5.2158. [DOI] [PubMed] [Google Scholar]
- 21.Mathews C J, Tabcharani J A, Chang X-B, Jensen T J, Riordan J R, Hanrahan J W. J Physiol. 1998;508:365–377. doi: 10.1111/j.1469-7793.1998.365bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramjeesingh M, Li C, Garami E, Huan L-J, Galley K, Wang Y, Bear C E. Biochemistry. 1999;38:1463–1468. doi: 10.1021/bi982243y. [DOI] [PubMed] [Google Scholar]
- 23.Ma J, Tasch J E, Tao T, Zhao J, Xie J, Drumm M L, Davis P B. J Biol Chem. 1996;271:7351–7356. doi: 10.1074/jbc.271.13.7351. [DOI] [PubMed] [Google Scholar]
- 24.Price N C, Johnson C M. In: Proteolytic Enzymes; A Practical Approach. Beynon R J, Bond J S, editors. Washington, D.C.: IRL; 1989. pp. 163–180. [Google Scholar]
- 25.Matsudaira P. J Biol Chem. 1987;262:10035–10038. [PubMed] [Google Scholar]
- 26.Zamyatnin A A. Annu Rev Biophys Bioeng. 1984;13:145–165. doi: 10.1146/annurev.bb.13.060184.001045. [DOI] [PubMed] [Google Scholar]
- 27.Laue T M, Shah B D, Ridgeway T M, Pelletier S L. Analytical Ultracentrifugation in Biochemistry and Polymer Science. Cambridge: The Royal Society of Chemistry; 1992. [Google Scholar]
- 28.Hansen J C, Lebowitz J, Demeler B. Biochemistry. 1994;33:13155–13163. doi: 10.1021/bi00249a001. [DOI] [PubMed] [Google Scholar]
- 29.Spanjaard R A, Chen K, Walker J R, van Duin J. Nucleic Acids Res. 1990;18:5031–5036. doi: 10.1093/nar/18.17.5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neville D C A, Rozanas C R, Tulk B M, Townsend R R, Verkman A S. Biochemistry. 1998;37:2401–2409. doi: 10.1021/bi972021k. [DOI] [PubMed] [Google Scholar]
- 31.Scholtz J M, Qian H, York E J, Stewart J M, Baldwin R L. Biopolymers. 1991;31:1463–1470. doi: 10.1002/bip.360311304. [DOI] [PubMed] [Google Scholar]
- 32.Baskakov I, Bolen D W. J Biol Chem. 1998;273:4831–4834. doi: 10.1074/jbc.273.9.4831. [DOI] [PubMed] [Google Scholar]
- 33.Chou P Y, Fasman G D. Adv Enzymol Relat Areas Mol Biol. 1978;47:45–148. doi: 10.1002/9780470122921.ch2. [DOI] [PubMed] [Google Scholar]
- 34.Rost B, Sander C. J Mol Biol. 1993;232:584–599. doi: 10.1006/jmbi.1993.1413. [DOI] [PubMed] [Google Scholar]
- 35.Rost B, Sander C. Proteins. 1994;19:55–72. doi: 10.1002/prot.340190108. [DOI] [PubMed] [Google Scholar]
- 36.Cuff J A, Barton G J. Proteins. 1999;34:508–519. doi: 10.1002/(sici)1097-0134(19990301)34:4<508::aid-prot10>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 37.Bowie J U, Reidhaar-Olson J F, Lim W A, Sauer R T. Science. 1990;247:1306–1310. doi: 10.1126/science.2315699. [DOI] [PubMed] [Google Scholar]
- 38.Wei L, Vankeerberghen A, Cuppens H, Droogmans G, Cassiman J-J, Nilius B. FEBS Lett. 1998;439:121–126. doi: 10.1016/s0014-5793(98)01351-9. [DOI] [PubMed] [Google Scholar]
- 39.Vankeerberghen A, Wei L, Jaspers M, Cassiman J J, Nilius B, Cuppens H. Hum Mol Genet. 1998;7:1761–1769. doi: 10.1093/hmg/7.11.1761. [DOI] [PubMed] [Google Scholar]
- 40.Dulhanty A M, Riordan J R. Biochemistry. 1994;33:4072–4079. doi: 10.1021/bi00179a036. [DOI] [PubMed] [Google Scholar]
- 41.Wright P E, Dyson H J. J Mol Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 42.Plaxco K W, Groß M. Nature (London) 1997;386:657–659. doi: 10.1038/386657a0. [DOI] [PubMed] [Google Scholar]
- 43.Kriwacki R W, Hengst L, Tennant L, Reed S I, Wright P E. Proc Natl Acad Sci USA. 1996;93:11504–11509. doi: 10.1073/pnas.93.21.11504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Daughdrill G W, Chadsey M S, Karlinsey J E, Hughes K T, Dahlquist F W. Nat Struct Biol. 1997;4:285–291. doi: 10.1038/nsb0497-285. [DOI] [PubMed] [Google Scholar]
- 45.Penkett C J, Redfield C, Dodd I, Hubbard J, McBay D L, Mossakowska D E, Smith R A G, Dobson C M, Smith L J. J Mol Biol. 1997;274:152–159. doi: 10.1006/jmbi.1997.1369. [DOI] [PubMed] [Google Scholar]
- 46.Murrell-Lagnado R D, Aldrich R W. J Gen Physiol. 1993;102:949–975. doi: 10.1085/jgp.102.6.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murrell-Lagnado R D, Aldrich R W. J Gen Physiol. 1993;102:977–1003. doi: 10.1085/jgp.102.6.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Richards J P, Bächinger H P, Goodman R H, Brennan R G. J Biol Chem. 1996;271:13716–13723. doi: 10.1074/jbc.271.23.13716. [DOI] [PubMed] [Google Scholar]
- 49.Parker D, Rivera M, Zor T, Henrion-Caude A, Radhakrishnan I, Kumar A, Shapiro L H, Wright P E, Montminy M, Brindle P K. Mol Cell Biol. 1999;19:5601–5607. doi: 10.1128/mcb.19.8.5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Radhakrishnan I, Perez-Alvarado G C, Parker D, Dyson H J, Montminy M R, Wright P E. Cell. 1997;91:741–752. doi: 10.1016/s0092-8674(00)80463-8. [DOI] [PubMed] [Google Scholar]
- 51.Radhakrishnan I, Perez-Alvarado G C, Dyson H J, Wright P E. FEBS Lett. 1998;430:317–322. doi: 10.1016/s0014-5793(98)00680-2. [DOI] [PubMed] [Google Scholar]
- 52.Parker D, Ferreri K, Nakajima T, LaMorte V J, Evans R, Koerber S C, Hoeger C, Montminy M R. Mol Cell Biol. 1996;16:694–703. doi: 10.1128/mcb.16.2.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Naren A P, Cormet-Boyaka E, Fu J, Villain M, Blalock J E, Quick M W, Kirk K L. Science. 1999;286:544–548. doi: 10.1126/science.286.5439.544. [DOI] [PubMed] [Google Scholar]