

Abstract
In this study, we examine the effects of reperfusion on activation of matrix metalloproteinase (MMP) and assess the relationship between MMP activation during reperfusion and neurovascular injury. Ischemia was produced using suture-induced middle cerebral artery occlusion in rats. The MMP activation was examined with in situ and gel zymography. Injury to cerebral endothelial cells and basal lamina was assessed using endothelial barrier antigen (EBA) and collagen IV immunohistochemistry. Injury to neurons and glial cells was assessed using Cresyl violet staining. These were examined at 3 hours after reperfusion (8 h after initiation of ischemia) and compared with permanent ischemia at the same time points to assess the effects of reperfusion. A broad-spectrum MMP inhibitor, AHA (p-aminobenzoyl-gly-pro-d-leu-d-ala-hydoxamate, 50 mg/kg intravenously) was administered 30 minutes before reperfusion to assess the roles of MMPs in activating gelatinolytic enzymes and in reperfusion-induced injury. We found that reperfusion accelerated and potentiated MMP-9 and MMP-2 activation and injury to EBA and collagen IV immunopositive microvasculature and to neurons and glial cells in ischemic cortex and striatum relative to permanent ischemia. Administering AHA 30 minutes before reperfusion decreased MMP-9 activation and neurovascular injury in ischemic cerebral cortex.
Keywords: Cerebral ischemia, Brain damage, Neuroprotection, Rats
Introduction
Recently, the occluded intracranial large vessels were directly opened with endovascular embolectomy device (Merci Retriever) in acute stroke patients. These treatments promoted recanalization and produced favorable outcome in the patients within 8 hours of the onset of stroke symptoms. However, intracranial hemorrhage occurred in 35.8% to 46.7 % of patients. Symptomatic intracranial hemorrhage occurred in 6.7% -9.9% of patients (Smith et al., 2005; Smith, 2006). Although it has been known that prolonged ischemia is important in hemorrhage transformation (HT) occurrence, the role of reperfusion itself on HT and related mechanism have not been well studied (Molina et al., 2001). In this report, in order to investigate the HT mechanisms, we have studied the effect of reperfusion on activation of matrix metalloproteinases (MMPs) after prolonged ischemia and its link to neurovascular injury.
MMPs have an important role in ischemia reperfusion injury after transient ischemia. In non-human primates, increased proMMP-2 coincided with early extravasations of plasma constituents and neuronal injury and increased proMMP-9 was observed in subjects with HT (Heo et al., 1999; del Zoppo and Mabuchi, 2003). Nonspecific MMP inhibitors decreased Evans blue leakage and brain edema (Rosenberg et al. 1998; Gasche et al., 2001; Pfefferkorn and Rosenberg 2003). A targeted knockout of MMP-9 decreased infarct volume (Asahi et al., 2001a, b). However, it is controversial whether MMP-9 and -2 are activated after transient ischemia. In some studies, activated MMP-9 was observed in mice and rats after ischemia (Fujimura et al. 1999; Planas et al., 2000, 2001; Gasche et al., 2001), while in others, no activated form of MMP-9 was reliably detected in rat, mice and baboons (Rosenberg et al., 1998; Heo et al., 1999;Asahi et al. 2001a; Pfefferkorn and Rosenberg, 2003). A few studies observed increase of active MMP-2 after ischemia, especially in the SOD1 knockout mouse (Heo et al., 1999; Planas et al., 2000; Gasche et al. 2001), but other studies did not detect activated MMP-2 (Rosenberg et al., 1998; Fujimura et al., 1999; Asahi et al., 2001a; Pfefferkorn and Rosenberg, 2003). Moreover, MMP changes after transient ischemia are impacted by two factors: ischemia and reperfusion injury. It is unclear whether reperfusion potentiates MMP activation as compared to ischemia alone. Since administering protective drugs in conjunction with endovascular embolectomy (just before reperfusion) is the most practicable time point for preventing HT in clinical practice, it is important to distinguish between these two mechanisms.
In the present study, we therefore tested the hypothesis that reperfusion, after prolonged cerebral ischemia, potentiates MMP activation relative to permanent ischemia that causes neurovascular injury. A rat suture middle cerebral artery occlusion model (MCAO) was used to precisely control reperfusion time. Permanent ischemia was used as the control to assess the effects of reperfusion. A water soluble, broad-spectrum MMP inhibitor, AHA (MMP-1, -3, -8 and -9 blocker), was administered to a subset of rats 30 minutes before reperfusion to assess the effects of MMPs on neurovascular reperfusion injury and the feasibility of AHA protection.
Methods and Materials
Rat Focal Cerebral Ischemia Model
Animal protocols were approved by the University of Cincinnati animal Care Committee and conform to the National Institutes of Health Guide for Care and Use of Laboratory Animals. Male Sprague-Dawley rats weighing approximately 290-320 grams had unrestricted access to food and water and were housed with a 12-hour light-dark cycle.
Focal ischemia was produced as described in our previous studies (Lu et al., 2002). Briefly, rats were anaesthetized with 3% isoflurane and maintained with 1.5% isoflurane in a mixture of 70% N2 and 28.5% O2. Rectal temperature was monitored and maintained at 37 ± 0.5° C with a feedback-controlled heating blanket. Blood pressure, blood gases (pO2, pCO2, and pH) and blood glucose concentration were monitored by tail artery catheter and maintained in the normal range. The left common carotid artery, external carotid artery, and internal carotid artery were isolated via a ventral midline incision. A 3/0 monofilament nylon suture was used to occlude the middle cerebral artery. The suture was inserted into the external carotid artery and advanced into the internal carotid artery until the tip occluded the junction of the middle cerebral artery and anterior cerebral artery. Cerebral blood flow was measured using Laser Doppler (5 mm lateral and 2 mm posterior to bregma) (Nito et al., 2004). The wound was closed and the suture was kept in place for 5 hours. Rats were re-anesthetized, and the filament was pulled back to achieve reperfusion. After the operation, rats were transferred to a temperature-controlled incubator at 37 °C for 30 minutes until animals completely woke up, and then to cages with the Delta Phase Isothermal Pad (Braintree scientific, Inc) to prevent hypothermia. Four groups of animals were studied (n = 4). (1) The sham operation group was given 0.9 % saline intravenously at 4.5 h with an infusion pump over a period of 30 minutes after sham operation, and sacrificed at 8 h. The sham operation was the same as the MCAO procedure except that there was no suture insertion and occlusion of middle cerebral artery. (2) The permanent ischemia group was administered 0.9 % saline at 4.5 h after MCAO using an infusion pump over a period of 30 min, and sacrificed at 5 h or 8 h after the onset of ischemia. (3) The ischemia-reperfusion group was administered 0.9 % saline intravenously at 4.5 h after MCAO using an infusion pump over a period of 30 min, reperfused at 5 h after MCAO, and sacrificed at 3 h after reperfusion (8 h after the onset of ischemia). (4) The ischemia-reperfusion with MMP inhibitor group was administered AHA (Calbiochem, CA, USA. 50 mg/kg in 0.9%saline) intravenously at 4.5 h after MCAO using an infusion pump over a period of 30 min, with reperfusion at 5 h after MCAO, and sacrificed at 3 h after reperfusion (8 h after the onset of ischemia). The dose of the AHA is from previous publication (Gasche et al., 2001).
Immunohistochemistry and Cresyl violet staining
At designated time points after MCAO, rats were deeply anesthetized with isoflurane and sacrificed. The brains were removed and frozen in 2-methylbutane. Coronal sections (20 μm) were cut in a cryostat (-20 °C) and stored at -80 °C. Sections were thawed and fixed in cold acetone or 2% Paraformaldehyde for 10 min and quenched in 3% hydrogen peroxide in PBS for 5 min. The sections were blocked for 1.5 hours in blocking buffer (2% serum/0.2% Triton X-100/0.1% BSA in 0.1 mol PB) and then incubated with primary antibody. After PB washes, the sections were incubated with a biotinylated secondary antibody for 1.5 hours (Vector Labs). After three 5-min PB washes, ABC reagent (Vector Labs) was applied. Diaminobenzidine was used to visualize the HRP. The primary antibodies used for immunohistochemistry included Mouse anti-Rat Endothelial barrier antigen (EBA) antibody (SMI71, 1: 1000; Sternberger Monoclonals. Lutherville, MD, USA), and Mouse anti-rat collagen IV monoclonal antibody (M3F7; 1: 200; Developmental Studies Hybridoma Bank at the University of Iowa, USA). EBA serves as a marker of the injury to endothelial cells, and collagen IV as a marker of the injury to cerebral microvascular basal lamina. Some sections were stained in 1% filtered cresyl violet solution to assess neuronal and glial cell injury.
For tissue measurements, we focused on sections at 0.2 mm anterior to bregma. For counting, two images were taken through parietal cerebral cortex (arrow, Fig 1.) where AHA showed the best protection; and four images were taken from the striatum (area numbers 1-4 on Fig 1). The average was calculated for measurements. Based on previously published data (Lin and Ginsberg, 2000), the following measurements were made: (1) The total area of the microvasculature per square millimeter of tissue (μm2/mm2) was measured and calculated for the EBA and Collagen IV stained sections; and the total area of the cells per square millimeter of tissue (μm2/mm2) was measured on cresyl violet stained sections. (2) In addition, the total numbers of separate EBA and Collagen stained microvascular profiles were counted in each square millimeter of tissue (numbers/mm2); and the total numbers of cresyl violet stained cells were counted in each square millimeter of tissue (numbers/mm2). These measurements were performed for the regions of parietal cortex shown in Figure 1 and in the areas of striatum shown in Figure 1 using the MCID automated imaging system. Finally, we calculated the average size of each microvascular EBA and Collagen IV stained profile by dividing the total area of the profiles (μm2/mm2) by the total numbers of profiles (numbers/mm2) to give the average area of each microvascular profile (μm2/microvascular profile). Similarly, we calculated the average area of each Cresyl violet stained cell by dividing the total area of the cells (μm2/mm2) by the total numbers of cells (numbers/mm2) to give the average area of each cell (μm2/cell).
Fig 1.
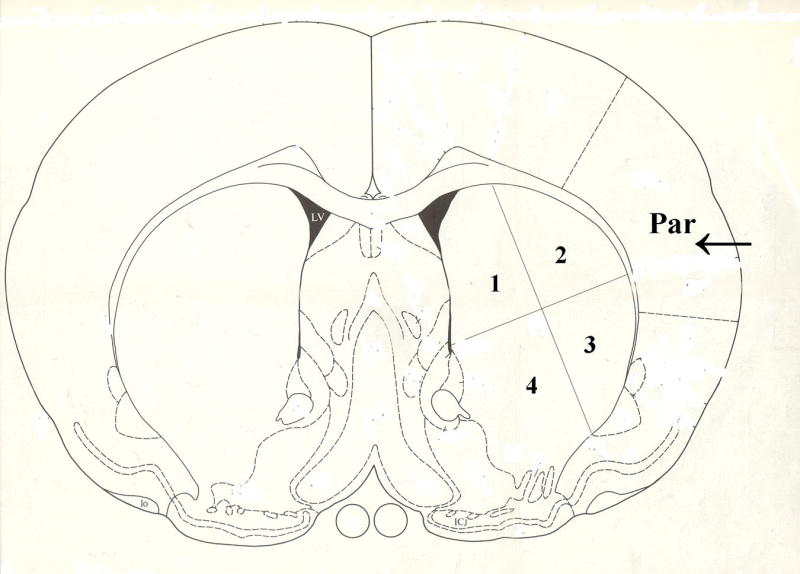
Quantification was performed in the regions of parietal cerebral cortex (Par, arrow) and the striatum (area number 1-4) shown in this coronal section of the rat brain. In addition, all of the images in the following Figures were taken from these regions of parietal cerebral cortex (arrow in Par area) and striatum (area number 1-4).
In situ zymography
The frozen sections were thawed and incubated with reaction buffer containing 100μg/ml FITC-labeled DQ gelatin for 10 hours at room temperature, and 2 hours at 37 °C. The sections were rinsed in PBS and then mounted in ProLong® Gold antifade reagent (Invitrogen, Carlsbad, CA, USA). The images were observed and pictures were taken using fluorescence microscopy. FITC-labeled DQ gelatin is intramolecularly quenched. Proteolysis by gelatinases yields cleaved gelatin-FITC peptides that are fluorescent. Incubation could vary from 2 hours to 24 hours and the fluorescence intensity increases with prolonged incubation times (Gasche et al., 2001; Jourquin et al., 2003). A constant exposure condition was used and the fluorescent intensity was visually estimated (Thiyagarajan et al., 2004).
MMP gel zymogram
Rats were deeply anesthetized with isoflurane and sacrificed. The brains were quickly removed and sliced into six 2-mm coronal slices at 4 °C. The cortex and striatum in the 2nd, 3rd, and 4th slices were dissected and frozen immediately in dry ice. Brain tissue extracts were prepared as previously described (Zhang and Gottschall, 1997). Briefly, brain samples were homogenized in “working buffer” (50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 5 mM CaCl2, 0.05% BRIJ-35, and 0.02% NaN3) and 1% Triton X-100, and then purified with gelatin-Sepharose 4B. The pellets were eluted with “elution buffer” (“working buffer” plus 10% DMSO). The proteins in the eluant were separated by electrophoresis through 10% polyacrylamide zymogram gels containing gelatin (Invitrogen, Carlsbad, California, USA). After washing in 2.5% Triton X-100 to remove SDS and then in 50 mM Tris-HCl, pH 7.5, the gels were incubated overnight in developing buffer (50 mM Tris-HCl, pH 7.5, 10 mM CaCl2, 1μM ZnCl2, and 0.05% BRIJ-35) at 37°C. Gels were stained with 0.1% Commassie blue and destained before being photographed and analyzed by the MCID automated imaging system.
Statistical analysis
Quantitative data were expressed as mean + SEM. Statistical comparisons were conducted using ANOVA for group comparisons. Differences with p < 0.05 were considered statistically significant.
Results
The change of cerebral blood flow after ischemia and reperfusion
Regional cerebral blood flow (rCBF) is measured with Laser Doppler flowmetry (LDF) in ischemia-reperfusion cortex. After ischemia, perfusion dropped to approximately 20% of baseline value (Table 1). The degree of ischemia was not statistically different between the groups. Reperfusion produced more than 90% recovery of average rCBF. AHA has no effect on rCBF (Table 1).
Table 1.
The change of regional cerebral blood flow after ischemia-reperfusion
| Group | Before MCAO | 10 min after MCAO | 5 h 10 min after MCAO (10 min after reperfusion) | 8 h after MCAO (3 h after reperfusion) |
|---|---|---|---|---|
| PMCAO 8 h | 100 | 20.6 ± 4.4** | 17.9 ± 8.1** | 17.2 ± 7.8** |
| Isch 5 h/R 3 h | 100 | 17.4 ± 6.8** | 93.6 ± 30.5 | 90.4 ± 23.8 |
| Isch 5 h/R 3 h + AHA | 100 | 21.9 ± 7.4** | 95.7 ± 34.1 | 91.9 ± 23.0 |
PMCAO: permanent middle cerebral artery occlusion. Isch: ischemia. R: reperfusion (n=4). AHA: MMP blocker.
p < 0.01 compared with before MCAO.
MMP activation and reperfusion injury of endothelial cells in cerebral blood vessels
EBA immunohistochemistry was performed to assess the injury to endothelial cells in cerebral blood vessels in rats following focal ischemia-reperfusion, and the relationship between reperfusion-induced MMP activation and the endothelial injury was defined by administering MMP inhibitor AHA just before reperfusion. The anti-EBA antibody is specific for a rat endothelial protein found in areas with blood-brain or blood-nerve barriers and is a marker of its integrity. EBA is decreased after brain trauma, inflammation, ischemia and hemorrhage (Lin and Ginsberg, 2000; Nishigaya et al., 2000; Fagan et al., 2003; Gursoy-Ozdemir et al., 2004). In the cortex, 8 hour PMCAO produced modest injury to cerebral blood vessel endothelium. Some EBA-immunoreactive microvessels appear short, with a tendency towards decreased microvascular area (μm2/mm2) and decreased average microvascular size relative to sham animals (p < 0.07, Fig 2B, E and F). In contrast, 5 hours of focal ischemia followed by 3 hour reperfusion (8 h total) accelerated and potentiated injury to blood vessel endothelium. Many of the EBA-immunoreactive microvessels disappeared or had degraded to short segments (Fig 2C). The total microvascular area and size of each microvascular profile were significantly decreased compared with permanent ischemia 8 hours (p < 0.05 and 0.01, respectively from Figs 2 E and F). When the broad-spectrum MMP inhibitor AHA (50 mg/kg) was injected intravenously 30 minutes before reperfusion (4.5 h after MCAO), reperfusion-induced injury of blood vessel endothelium was decreased in parietal cerebral cortex where the total microvascular area and size of each microvascular profile were significantly increased (p < 0.01) relative to PMCO 8 h (Figs 2 D, E and F). Injury in striatum evolved at a much faster rate. PMCAO 5h and 8h produced severe injury, and significantly decreased the total microvascular area (p < 0.05) and size of each microvascular profile (p < 0.05 or 0.01) compared with sham animals (Table 2). Reperfusion 3 h after 5 h MCAO worsened endothelial cell injury resulting in further decreases in the total microvascular area (Table 2: p < 0.05, relative to PMCAO 8 h group). The microvascular numbers were also decreased relative to PMCAO 8 h (data not shown, p < 0.05). This was not be prevented by administering AHA 30 minutes prior to reperfusion (Table 2, AHA 50 mg/kg, i.v. starting from 4.5 h after MCAO).
Fig 2.


EBA immunohistochemistry (Fig.2a; A-D) and quantification of the EBA staining (Fig.2b; E, F) in parietal cerebral cortex after focal MCA ischemia in adult rats. PMCAO: permanent middle cerebral artery occlusion. Isch: ischemia. R: reperfusion (n = 4). AHA: MMP blocker. (A) Sham. (B) PMCAO 8 h. (C) Isch 5 h/R 3 h. (D) Isch 5 h/R 3 h + AHA. *p < 0.05, **p < 0.01 compared with PMCO 8 h group. ##p < 0.01 compared with Ischemia 5 h/R3 h group. Reperfusion 3 h after 5 h MCAO accelerated and potentiated injury to blood vessel endothelium relative to PMCAO 8 h in parietal cortex (C, E and F). Administering the MMP inhibitor AHA just before reperfusion (30 min before) decreased the injury (D, E and F).
Table 2.
The effects of reperfusion and the drug AHA on EBA- and collagen IV-immunoreactive microvessels in striatum.
| EBA- immunoreactive | Collagen IV-immunoreactive | |||
|---|---|---|---|---|
| Groups | Total micro vascular area (μm2)/mm2 tissue | Average microvascular size (μm2/profile) | Total Micro vascular area (μm2)/mm2 tissue | Average microvascular size (μm2/profile) |
| Sham | 39.4 ± 14.7 | 0.17 ± 0.03 | 55.5 ± 8.6 | 0.23 ± 0.06 |
| PMCAO 5 h | 21.2 ± 3.9& | 0.11 ± 0.02& | 43.2 ± 2.7& | 0.19 ± 0.02 |
| PMCAO 8 h | 20.0 ± 3.6& | 0.09 ± 0.02&& | 42.0 ± 3.8& | 0.16 ± 0.01 |
| Isch 5 h/R 3 h | 12.9 ± 5.7* | 0.08 ± 0.01 | 31.3 ± 3.6** | 0.12 ± 0.02* |
| Isch 5 h/R 3 h + AHA | 19.1 ± 6.9 | 0.08 ± 0.02 | 35.0 ± 1.8 | 0.14 ± 0.02 |
PMCAO: permanent middle cerebral artery occlusion. Isch: ischemia. R: reperfusion (n=4). AHA: MMP blocker.
p < 0.05,
p < 0.01 compared with Sham group.
p < 0.05,
p < 0.01 compared with PMCAO 8h group.
MMP activation and reperfusion injury of cerebral microvascular basal lamina
Injury to the cerebral microvascular basal lamina was assessed using collagen IV immunohistochemistry in rats, and the relationship between reperfusion-induced MMP activation and the basal lamila injury was defined by administering MMP inhibitor AHA just before reperfusion. Type IV collagen is one of the main components of the basal lamina (Petty and Wettstein, 2001), and this protein disappears following focal ischemia and reperfusion in rats (Hamann et al., 2004). The effects of PMCAO and ischemia-reperfusion on Collagen IV immunoreactive microvessels in cortex and striatum were similar to those already described. In the cortex, PMCAO 8 h only slightly degraded Collagen IV of the basal lamina in parietal cortex. The average microvascular profile size was decreased compared with sham group (p < 0.05, Figs 3 B and F). However, 5 h ischemia followed by 3 hours of reperfusion accelerated and worsened the injury. The total microvascular area (μm2/mm2) and size of each microvascular profile were decreased further compared to PMCAO 8 h (p < 0.05, Fig 3 C, E and F). When the MMP inhibitor AHA (50 mg/kg) was infused intravenously 30 minutes before reperfusion (4.5 h after MCAO), these changes were partially reversed (p < 0.05 for the microvascular area and p < 0.01for the average microvascular size relative to the Isch5 h/R 3 h group) (Figs 3 D, E and F). In striatum, PMCAO of 5 h and 8 h led to definite damage to microvascular basal lamina assessed using Collagen IV immunoreactivity (Table 2). The total microvascular area was decreased compared with sham group (p < 0.05; Table 2). However, 5 hours of focal ischemia followed by 3 hour reperfusion (Ischemia 5 h/R 3 h) accelerated and worsened the injury, and further decreased the total microvascular area (p <0.01) and the size of each microvascular profile (p < 0.05) relative to PMCAO 8 hours (Table 2). AHA (50 mg/kg, i.v. starting at 4.5 h after MCAO) did not protect against the injury to the basal lamina in the striatum (Table 2).
Fig 3.


Collagen IV immunostaining (Fig.3a; A-D) and quantification of the Collagen IV staining (Fig. 3b; E, F) in parietal cerebral cortex of adult rats following MCA focal ischemia. PMCAO: permanent middle cerebral artery occlusion (MCAO). Isch: ischemia. R: reperfusion (n = 4). (A) Sham. (B) PMCAO 8 h. (C) Isch 5 h/R 3 h. (D) Isch 5 h/R 3 h + AHA. &p < 0.05 compared with Sham group. *p < 0.05 compared with PMCO 8h group. #p < 0.05, ##p < 0.01 compared with Isch 5 h/R 3h group. Reperfusion 3 h after 5 h MCAO accelerated and worsened the injury to the basal lamina relative to PMCAO 8 h in parietal cortex (C, E and F). Administering the MMP inhibitor AHA just before reperfusion (30 min before) decreased the injury (D, E and F).
MMP activation and reperfusion injury to neuronal cells
Injury to neuronal cells was assessed using Cresyl violet staining, and the relationship between reperfusion-induced MMP activation and neuronal cell injury was defined by administering MMP inhibitor AHA just before reperfusion. In cortex there was a modest decrease in the total area of Nissl stained cells following 8 h of permanent MCAO (p < 0.08 relative to sham animals, Figs 4 B and E). In contrast, 5 h MCAO followed by 3 h reperfusion markedly accelerated and worsened damage to cortex. The total number of cells (numbers/mm2) and the average size of each cell were significantly decreased relative to PMCAO 8 h group (p < 0.01 and 0.05, respectively, Figs 4 C, E and F). This was partially reversed by the MMP inhibitor AHA (50 mg/kg) when it was injected intravenously 30 minutes before reperfusion (4.5 h after MCAO; p < 0.05, Figs 4 D, E and F). These results in cortex again contrasted with those in striatum. There was more severe damage by 5h and 8 h PMCAO and decreased the total cell area and the average cell size relative to sham operation group (p < 0.01; Table 3). The 5 hours of focal ischemia followed by 3 h reperfusion worsened the damage and resulted in a further decrease relative to PMCAO 8 h (p < 0.01 for the cell area and p < 0.05 for the size). The total cell numbers were also decreased relative to PMCAO 8 h (p < 0.01; Table 3). AHA (50 mg/kg, i.v. starting at 4.5 h after MCAO) did not protect the Nissl substance loss in the neuronal cells in the striatum (Table 3).
Fig 4.


Cresyl violet staining (Fig. 4a; A-D) and quantification of the Cresyl violet staining (Fig. 4b; E, F) in parietal cerebral cortex after focal ischemia in adult rats. PMCAO: permanent middle cerebral artery occlusion. Isch: ischemia. R: reperfusion (n = 4). (A) Sham. (B) PMCAO 8 h. (C) Isch 5 h/R 3 h. (D) Isch 5 h/R 3 h + AHA. *p < 0.05, **p < 0.01 compared with PMCO 8 h group. #p < 0.05 compared with Isch 5 h/R 3h group. Reperfusion 3 h after 5 h MCAO accelerated and worsened the injury to neuronal and glial cells relative to PMCAO 8 h in parietal cortex (C, E and F). Administering the MMP inhibitor AHA just before reperfusion (30 min before) decreased the injury (D, E and F).
Table 3.
The effects of reperfusion and AHA on the injury to neuronal and glial cells assessed with Cresyl violet staining in striatum.
| Groups | Total cell area (μm2)/mm2 tissue | Cell numbers /mm2 tissue | Average cell size (μm2)/cell |
|---|---|---|---|
| Sham | 67.9 ± 7.8 | 1237.6 ± 38.3 | 0.055 ± 0.005 |
| PMCAO 5 h | 34.7 ± 3.6&& | 1205.4 ± 99.4 | 0.032 ± 0.004&& |
| PMCAO 8 h | 34.1 ± 2.1&& | 1239.6 ± 16.5 | 0.028 ± 0.002&& |
| Isch 5 h/R 3 h | 19.2 ± 4.8** | 991.1 ± 61.6** | 0.019 ± 0.004* |
| Isch 5 h/R 3 h + AHA | 22.5 ± 2.9 | 1114.6 ± 97.8 | 0.020 ± 0.003 |
PMCAO: permanent middle cerebral artery occlusion. Isch: ischemia. R: reperfusion (n=4). AHA: MMP blocker.
p < 0.01 compared with Sham group.
p < 0.05,
p < 0.01 compared with PMCAO 8h group.
Reperfusion and MMP activation after cerebral ischemia
The effects of ischemia, reperfusion and AHA on activation of gelatinolytic enzymes were estimated using in situ zymography in the parietal cortex and striatum (Fig 5 a). Permanent ischemia for 8 hours caused weak activation of gelatinolytic enzymes in cortex (Figs 5 B) and moderate activation in striatum (data not shown). In contrast, 5 hours of focal ischemia followed by 3 hours of reperfusion (Ischemia 5 h, R 3 h, 8 h total) markedly increased gelatinolytic enzyme activation in parietal cortex (Fig 5 C) and in striatum. The methods used for the in situ gelatin zymography are thought to mostly detect MMP-2 and -9 (Snoek-van Beurden and Von den Hoff, 2005). To confirm that activated gelatinolytic enzymes are MMPs that are blocked by AHA, AHA (50 mg/kg) was injected intravenously just before reperfusion (4.5 h after MCAO, reperfusion starting at 5 h after MCAO). AHA blocked the activation of gelatinolytic enzymes in parietal cortex (Fig 5 D), but AHA did not block their activation in striatum. The gelatinolytic activity was most likely located in neural cells (C, arrowhead) and the microvasculature (C, arrow).
Fig 5.


a. In situ gelatinolysis assays in parietal cerebral cortex (Fig. 5a; A to D) of adult rats after focal MCA ischemia. PMCAO: permanent middle cerebral artery occlusion. Isch = ischemia. R = reperfusion (n = 3). (A) Sham. (B) PMCAO 8 h. (C) Isch 5 h/R 3 h. (D) Isch 5 h/R 3 h + AHA. The gelatinolytic activity (green fluorescence) was located in neural cells (C, arrowhead) and the microvasculature (C, arrow).
b. Gel zymogram and quantification of band density in cerebral cortex (Fig. 5b; E to G) and in striatum (Fig. 5b; H to G) of adult rats after focal MCA ischemia. PMCAO: permanent middle cerebral artery occlusion. I = ischemia. R = reperfusion (n = 4). (1) Sham. (2) PMCAO 8 h. (3) Isch 5 h/R 3 h. (4) Isch 5 h/R 3 h + AHA. (5) pro-MMP-9 positive control. (6) pro-MMP-2 positive control. &p < 0.05, &&p < 0.01 compared with sham group. *p < 0.05, **p < 0.01 compared with PMCAO 8 h group. #p < 0.05, ##p < 0.01 compared with Isch 5 h/R 3 h group. 5 h of MCA ischemia followed by 3 h of reperfusion potentiated activation of MMP-9 in the cortex (E, G) and activation of MMP-2 in the striatum (H, J). Administering AHA just before reperfusion (30 min before) decreased MMP-9 activation in the cortex (E, G).
To further evaluate and quantify MMP activity of the extracted samples, gelatin gel zymagrams were performed (Fig 5 b). Active and latent forms of MMP-9 and their dimers were found in both MMP-9 positive control and ischemic samples. In the cortex, active MMP-9 (dimer) was statistically increased after 5 h ischemia followed by 3 h reperfusion relative to PMCAO group (p < 0.05, Figs 5 E and G). Administration of AHA just before reperfusion inhibited MMP-9 activation compared with ischemia 5 h/reperfusion 3 h group (p < 0.05,; Fig 5 E and G). Pro-MMP-9 was also increased after 8 h ischemia compared with sham animals (p < 0.01) and further enhanced after 5 hischemia/3 h reperfusion (p < 0.05), and decreased with AHA administration (p < 0.01, Fig 5 E and F). No significant change of MMP-2 was observed. In the striatum, Pro- and active MMP-9 were increased in both PMCAO group and ischemia 5h/reperfusion 3 h group compared with sham control (p < 0.01 and 0.05, Fig 5 H and I). The 3 h reperfusion after 5 h ischemia also increased active MMP-2 compared with sham control or PMCAO group (p < 0.01, Fig 5 H and J). AHA did not inhibit MMP-2 and MMP-9 activation in the striatum.
Discussion
Our results show that reperfusion worsens neurovascular injury as compared to ischemia alone. Previous studies have shown that specific degradation of collagen IV and laminin occurs after cerebral ischemia. The loss of extracellular matrix in microvessel is associated with the loss of endothelial cell reactivity during focal ischemia. Endothelial cell P-selectin and E-selectin, and β1-integrin expression within the ischemic territory occur only on those microvessels with an intact (laminin and collagen-containing) basal lamina. The degradation of parenchymal laminin may interrupt cell-matrix interactions and Akt survival-signaling pathways, which triggers endothelial cell and neuron anoikis or apoptosis (del Zoppo et al., 1998; Lo et al., 2002; del Zoppo and Mabuchi, 2003; Wagner et al., 2003; Hamann et al., 2004). These reports support our results. Especially, our results demonstrate that administration of AHA just before reperfusion decreased the injury to the microvessel endothelium, basal lamina and neural cells in the parietal cortex. This suggests that MMP activation is an important mediator of reperfusion-induced injury to the neurovascular unit, and blocking MMP activation caused by reperfusion is likely to be a feasible treatment to decrease injury to the neurovascular unit.
Various studies support the concept that reperfusion potentiates MMP activation. First, reactive oxygen radicals are a key mediator of tissue damage after reperfusion in many organs including heart, kidney and brain (Lo et al., 2003). Oxidative stress triggers activation of MMP-9 and MMP-2 within the ischemic mouse brain (Gasche et al., 2001). Second, reperfusion after focal cerebral ischemia caused neutrophil adhesion to blood vessel endothelium, infiltration into the ischemic area and provided a major source of pro-MMP9 (del Zoppo and Mabuchi, 2003; Justicia et al., 2003; Gidday et al., 2005). Neutrophil elastase breaks down TIMP and activates MMP9 and MMP2 (Itoh and Nagase, 1995; Rice and Banda, 1995; Delclaux et al., 1996; Okada et al., 1988; Kohyama et al. 2002). Third, reperfusion increased nNOS and iNOS expression and nNOS activation after focal ischemia (Sorrenti et al., 1999; Holtz et al., 2001). Nitric oxide can directly activate MMP9 through S-nitrosylation at the catalytic site (Gu et al., 2002). Fourth, focal ischemia increased plasminogen activation in the ischemic hemisphere, and plasmin has been shown to activate pro–MMP-2 and pro-MMP-9 (Pfefferkorn et al., 2000; del Zoppo and Mabuchi 2003). Our results provide the direct evidence of reperfusion accelerating and potentiating MMP-9 activation in the ischemic cortex and MMP-2 activation in the ischemic striatum relative to permanent ischemia. In our results (Fig 4), the MMP signal is most likely located in neural cells and the microvasculature. Previous studies showed that ischemia-reperfusion produced genatinolytic activity in capillary and neural cells. Many injured neural cells showed strong genatinolytic signals (Gasche et al., 2001; Rosell et al., 2006). These reports are consistent with our results reported here. It is conceivable that MMPs from neural cells may contribute to the injury of both neurons and microvessels. The current study also showed that stronger MMP activation in reperfusion group led to more severe loss of EBA, collagen IV and neural cells. The MMP blocker administered just before reperfusion decreased both MMP activation and neurovascular injury in the cortex. These results also support the concept that MMP activation during reperfusion exacerbates neurovascular injury.
Even with mechanical recanalization, symptomatic intracranial hemorrhage occurs in 6.7% -9.9% of patients (Smith et al., 2005; Smith, 2006). Our unpublished experiments showed that reperfusion after 5 hours of cerebral ischemia worsened intracerebral hemorrhage and neurological function relative to permanent ischemia. The peak time of the hemorrhage and animal death was between 8 to 12 hours after ischemia. Administration of AHA just before reperfusion decreased intracerebral hemorrhage and improved neurological function. However, neither infarction volumes and nor brain edema was significantly changed in ischemia (5 hours)/ reperfusion (19 hours) group as compared to permanent ischemia. This study showed that the neurovascular injury was accelerated by reperfusion, which may contributes to more severe hemorrhage with behavior exacerbation.
A few caveats are worth considering. First, AHA administered just before reperfusion did not protect the striatum from injury to the neurovascular unit. This may be because 5 hours of ischemia induced too severe an injury in striatum compared to the cortex in striatum, and/or the possibility that 3 hours of reperfusion after 5 hours of ischemia had induced a strong MMP-9 and MMP-2 activation that was not blocked by a dose of 50 mg/kg AHA in the striatum. Further studies are needed to answer these questions. Second, the assessments were done at 8 hour after ischemia. It is unclear whether the protective effect of MMP inhibition could be sustained behind this time window. A repeated administration of AHA might be necessary to sustain longer protective effects. However, the half-life time of AHA is not available at this time, so we do not know the required interval(s) for AHA administration. Future studies are also needed to clarify these issues. Third, we focused on brain sections at 0.2 mm to bregma for evaluate neurovascular injury. Although there are a good morphology of both striatum and cortex and reliable neurovascular injury in this section, assessments of more brain sections with a stereological method may increase precision of the results.
In summary, this is the first evidence that reperfusion, after prolonged ischemia, accelerated and potentiated the activation of MMP-9 and MMP-2 relative to permanent ischemia. Administering AHA just before reperfusion decreased MMP-9 activation and neurovascular injury in ischemic cortex. This work is clinically relevant, since no matter how reperfusion is achieved –using drugs or mechanical devices, this study suggests that the reperfusion per se injures the neurovascular unit after prolonged ischemia and blocking MMPs can decrease this injury.
Acknowledgments
This study was supported by NIH grants NS42774, NS43252, NS28167 (Sharp FR), NS44283 (Broderick J and Clark J), NS050569 (Clark J), NS049428 (Pyne-Geithman G), NS030652 (Wagner K) and NS57367 (Lu A).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Asahi M, Sumii T, Fini ME, Itohara S, Lo EH. Matrix metalloproteinase 2 gene knockout has no effect on acute brain injury after focal ischemia. Neuroreport. 2001a;12:3003–3007. doi: 10.1097/00001756-200109170-00050. [DOI] [PubMed] [Google Scholar]
- Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001b;21:7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Zoppo GJ, Mabuchi T. Cerebral microvessel responses to focal ischemia. J Cereb Blood Flow Metab. 2003;23:879–894. doi: 10.1097/01.WCB.0000078322.96027.78. [DOI] [PubMed] [Google Scholar]
- del Zoppo GJ, von Kummer R, Hamann GF. Ischaemic damage of brain microvessels: inherent risks for thrombolytic treatment in stroke. J Neurol Neurosurg Psychiatry. 1998;65:1–9. doi: 10.1136/jnnp.65.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delclaux C, Delacourt C, D’Ortho MP, Boyer V, Lafuma C, Harf A. Role of gelatinase B and elastase in human polymorphonuclear neutrophil migration across basement membrane. Am J Respir Cell Mol Biol. 1996;14:288–295. doi: 10.1165/ajrcmb.14.3.8845180. [DOI] [PubMed] [Google Scholar]
- Fagan SC, Nagaraja TN, Fenstermacher JD, Zheng J, Johnson M, Knight RA. Hemorrhagic transformation is related to the duration of occlusion and treatment with tissue plasminogen activator in a nonembolic stroke model. Neurol Res. 2003;25:377–382. doi: 10.1179/016164103101201526. [DOI] [PubMed] [Google Scholar]
- Fujimura M, Gasche Y, Morita-Fujimura Y, Massengale J, Kawase M, Chan PH. Early appearance of activated matrix metalloproteinase-9 and blood-brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain Res. 1999;842:92–100. doi: 10.1016/s0006-8993(99)01843-0. [DOI] [PubMed] [Google Scholar]
- Gasche Y, Copin JC, Sugawara T, Fujimura M, Chan PH. Matrix metalloproteinase inhibition prevents oxidative stress-associated blood-brain barrier disruption after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2001;21:1393–1400. doi: 10.1097/00004647-200112000-00003. [DOI] [PubMed] [Google Scholar]
- Gidday JM, Gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, Chan PH, Park TS. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol. 2005;289:H558–568. doi: 10.1152/ajpheart.01275.2004. [DOI] [PubMed] [Google Scholar]
- Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186–1190. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- Gursoy-Ozdemir Y, Qiu J, Matsuoka N, Bolay H, Bermpohl D, Jin H, Wang X, Rosenberg GA, Lo EH, Moskowitz MA. Cortical spreading depression activates and upregulates MMP-9. J Clin Invest. 2004;113:1447–1455. doi: 10.1172/JCI21227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann GF, Burggraf D, Martens HK, Liebetrau M, Jager G, Wunderlich N, DeGeorgia M, Krieger DW. Mild to moderate hypothermia prevents microvascular basal lamina antigen loss in experimental focal cerebral ischemia. Stroke. 2004;35:764–769. doi: 10.1161/01.STR.0000116866.60794.21. [DOI] [PubMed] [Google Scholar]
- Heo JH, Lucero J, Abumiya T, Koziol JA, Copeland BR, del Zoppo GJ. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19:624–633. doi: 10.1097/00004647-199906000-00005. [DOI] [PubMed] [Google Scholar]
- Holtz ML, Craddock SD, Pettigrew LC. Rapid expression of neuronal and inducible nitric oxide synthases during post-ischemic reperfusion in rat brain. Brain Res. 2001;898:49–60. doi: 10.1016/s0006-8993(01)02140-0. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Nagase H. Preferential inactivation of tissue inhibitor of metalloproteinases-1 that is bound to the precursor of matrix metalloproteinase 9 (progelatinase B) by human neutrophil elastase. J Biol Chem. 1995;270:16518–16521. doi: 10.1074/jbc.270.28.16518. [DOI] [PubMed] [Google Scholar]
- Jourquin J, Tremblay E, Decanis N, Charton G, Hanessian S, Chollet AM, Le Diguardher T, Khrestchatisky M, Rivera S. Neuronal activity-dependent increase of net matrix metalloproteinase activity is associated with MMP-9 neurotoxicity after kainate. Eur J Neurosci. 2003;18:1507–1517. doi: 10.1046/j.1460-9568.2003.02876.x. [DOI] [PubMed] [Google Scholar]
- Justicia C, Panes J, Sole S, Cervera A, Deulofeu R, Chamorro A, Planas AM. Neutrophil infiltration increases matrix metalloproteinase-9 in the ischemic brain after occlusion/reperfusion of the middle cerebral artery in rats. J Cereb Blood Flow Metab. 2003;23:1430–1440. doi: 10.1097/01.WCB.0000090680.07515.C8. [DOI] [PubMed] [Google Scholar]
- Kohyama T, Liu X, Zhu YK, Wen FQ, Wang HJ, Fang Q, Kobayashi T, Rennard SI. Phosphodiesterase 4 inhibitor cilomilast inhibits fibroblast-mediated collagen gel degradation induced by tumor necrosis factor-alpha and neutrophil elastase. Am J Respir Cell Mol Biol. 2002;27:487–494. doi: 10.1165/rcmb.4818. [DOI] [PubMed] [Google Scholar]
- Lin B, Ginsberg MD. Quantitative assessment of the normal cerebral microvasculature by endothelial barrier antigen (EBA) immunohistochemistry: application to focal cerebral ischemia. Brain Res. 2000;865:237–244. doi: 10.1016/s0006-8993(00)02228-9. [DOI] [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- Lo EH, Wang X, Cuzner ML. Extracellular proteolysis in brain injury and inflammation: role for plasminogen activators and matrix metalloproteinases. J Neurosci Res. 2002;69:1–9. doi: 10.1002/jnr.10270. [DOI] [PubMed] [Google Scholar]
- Lu A, Ran R, Parmentier-Batteur S, Nee A, Sharp FR. Geldanamycin induces heat shock proteins in brain and protects against focal cerebral ischemia. J Neurochem. 2002;81:355–364. doi: 10.1046/j.1471-4159.2002.00835.x. [DOI] [PubMed] [Google Scholar]
- Molina CA, Montaner J, Abilleira S, Ibarra B, Romero F, Arenillas JF, Alvarez-Sabin J. Timing of spontaneous recanalization and risk of hemorrhagic transformation in acute cardioembolic stroke. Stroke. 2001;32:1079–1084. doi: 10.1161/01.str.32.5.1079. [DOI] [PubMed] [Google Scholar]
- Nishigaya K, Yagi S, Sato T, Kanemaru K, Nukui H. Impairment and restoration of the endothelial blood-brain barrier in the rat cerebral infarction model assessed by expression of endothelial barrier antigen immunoreactivity. Acta Neuropathol (Berl) 2000;99:231–237. doi: 10.1007/pl00007432. [DOI] [PubMed] [Google Scholar]
- Nito C, Kamiya T, Ueda M, Arii T, Katayama Y. Mild hypothermia enhances the neuroprotective effects of FK506 and expands its therapeutic window following transient focal ischemia in rats. Brain Res. 2004;1008:179–185. doi: 10.1016/j.brainres.2004.02.031. [DOI] [PubMed] [Google Scholar]
- Okada Y, Watanabe S, Nakanishi I, Kishi J, Hayakawa T, Watorek W, Travis J, Nagase H. Inactivation of tissue inhibitor of metalloproteinases by neutrophil elastase and other serine proteinases. FEBS Lett. 1988;229:157–160. doi: 10.1016/0014-5793(88)80817-2. [DOI] [PubMed] [Google Scholar]
- Petty MA, Wettstein JG. Elements of cerebral microvascular ischaemia. Brain Res Brain Res Rev. 2001;36:23–34. doi: 10.1016/s0165-0173(01)00062-5. [DOI] [PubMed] [Google Scholar]
- Pfefferkorn T, Rosenberg GA. Closure of the blood-brain barrier by matrix metalloproteinase inhibition reduces rtPA-mediated mortality in cerebral ischemia with delayed reperfusion. Stroke. 2003;34:2025–2030. doi: 10.1161/01.STR.0000083051.93319.28. Epub 2003. [DOI] [PubMed] [Google Scholar]
- Pfefferkorn T, Staufer B, Liebetrau M, Bultemeier G, Vosko MR, Zimmermann C, Hamann GF. Plasminogen activation in focal cerebral ischemia and reperfusion. J Cereb Blood Flow Metab. 2000;20:337–342. doi: 10.1097/00004647-200002000-00015. [DOI] [PubMed] [Google Scholar]
- Planas AM, Sole S, Justicia C. Expression and activation of matrix metalloproteinase-2 and -9 in rat brain after transient focal cerebral ischemia. Neurobiol Dis. 2001;8:834–846. doi: 10.1006/nbdi.2001.0435. [DOI] [PubMed] [Google Scholar]
- Planas AM, Sole S, Justicia C, Farre ER. Estimation of gelatinase content in rat brain: effect of focal ischemia. Biochem Biophys Res Commun. 2000;278:803–807. doi: 10.1006/bbrc.2000.3881. [DOI] [PubMed] [Google Scholar]
- Rice A, Banda MJ. Neutrophil elastase processing of gelatinase A is mediated by extracellular matrix. Biochemistry. 1995;34:9249–9256. doi: 10.1021/bi00028a038. [DOI] [PubMed] [Google Scholar]
- Rosell A, Ortega-Aznar A, Alvarez-Sabin J, Fernandez-Cadenas I, Ribo M, Molina CA, Lo EH, Montaner J. Increased brain expression of matrix metalloproteinase-9 after ischemic and hemorrhagic human stroke. Stroke. 2006;37:1399–1406. doi: 10.1161/01.STR.0000223001.06264.af. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Estrada EY, Dencoff JE. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke. 1998;29:2189–2195. doi: 10.1161/01.str.29.10.2189. [DOI] [PubMed] [Google Scholar]
- Smith WS. Safety of mechanical thrombectomy and intravenous tissue plasminogen activator in acute ischemic stroke. Results of the multi Mechanical Embolus Removal in Cerebral Ischemia (MERCI) trial, part I. AJNR Am J Neuroradiol. 2006;27:1177–1182. [PMC free article] [PubMed] [Google Scholar]
- Smith WS, Sung G, Starkman S, Saver JL, Kidwell CS, Gobin YP, Lutsep HL, Nesbit GM, Grobelny T, Rymer MM, Silverman IE, Higashida RT, Budzik RF, Marks MP. Safety and efficacy of mechanical embolectomy in acute ischemic stroke: results of the MERCI trial. Stroke. 2005;36:1432–1438. doi: 10.1161/01.STR.0000171066.25248.1d. [DOI] [PubMed] [Google Scholar]
- Snoek-van Beurden PA, Von den Hoff JW. Zymographic techniques for the analysis of matrix metalloproteinases and their inhibitors. Biotechniques. 2005;38:73–83. doi: 10.2144/05381RV01. [DOI] [PubMed] [Google Scholar]
- Sorrenti V, Di Giacomo C, Campisi A, Perez-Polo JR, Vanella A. Nitric oxide synthetase activity in cerebral post-ischemic reperfusion and effects of L-N(G)-nitroarginine and 7-nitroindazole on the survival. Neurochem Res. 1999;24:861–866. doi: 10.1023/a:1020906030328. [DOI] [PubMed] [Google Scholar]
- Thiyagarajan M, Kaul CL, Sharma SS. Neuroprotective efficacy and therapeutic time window of peroxynitrite decomposition catalysts in focal cerebral ischemia in rats. Br J Pharmacol. 2004;142:899–911. doi: 10.1038/sj.bjp.0705811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S, Nagel S, Kluge B, Schwab S, Heiland S, Koziol J, Gardner H, Hacke W. Topographically graded postischemic presence of metalloproteinases is inhibited by hypothermia. Brain Res. 2003;984:63–75. doi: 10.1016/s0006-8993(03)03088-9. [DOI] [PubMed] [Google Scholar]
- Zhang JW, Gottschall PE. Zymographic measurement of gelatinase activity in brain tissue after detergent extraction and affinity-support purification. J Neurosci Methods. 1997;76:15–20. doi: 10.1016/s0165-0270(97)00065-4. [DOI] [PubMed] [Google Scholar]