

Abstract
Two natural products have been synthesized using a ZnCl2-mediated benzylic coupling reaction. Both are potent inhibitors of the GTPase activity of FtsZ, a protein that is essential for bacterial cytokinesis.
The emergence of bacterial strains that are resistant to current drugs has prompted a renewed effort to discover new methods for fighting infectious disease.1 One promising new target is FtsZ, the bacterial analog of tubulin, which mediates bacterial cell division. During bacterial cytokinesis, FtsZ monomers polymerize at mid-cell to form the Z-ring, which eventually constricts, leading to septation and formation of daughter cells (Figure 1). FtsZ consumes GTP during Z-ring assembly, much like its eukaryotic analog tubulin during mitosis. As such, FtsZ is susceptible to inactivation by compounds that interfere with the GTPase activity of this protein.2
Figure 1.

Bacterial cell division and the role of FtsZ
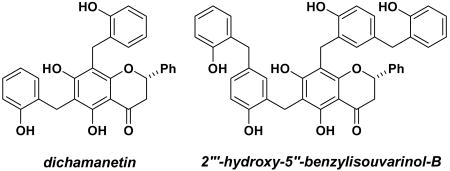
Recent studies have revealed several compounds that inhibit the GTPase activity of FtsZ and kill bacteria,3 and in one case several compounds were demonstrated to disrupt Z-ring formation.4 Zantrin Z1 (1, Figure 2), which was discovered in a high throughput in vitro screen for inhibition of GTPase activity,4 possesses a polyphenolic structure reminiscent of several natural products that exhibit potent antimicrobial activity. Dichamanetin (2) and 2″′-hydroxy-5″-benzyliso-uvarinol-B (3), isolated independently by Hufford and Anam from U. chamae and X. afticana respectively, exhibit comparable MIC values to zantrin Z1 when evaluated against a variety of bacterial strains.5 It is notable that these compounds show a high level of activity against gram positive bacteria (e.g. S. aureus and B. subtilis), and furthermore the MIC values are comparable to clinically relevant antibiotics.6
Figure 2.

Structures of synthetic (1) and naturally occurring (2 and 3) polyphenolic compounds
The structural similarity between polyphenolic compounds 1-3 suggested that they might all derive their antimicrobial activity by inhibiting the GTPase activity of FtsZ. In order to test this hypothesis, we undertook the syntheses of compounds 2 and 3. While naturally-occurring flavanones have attracted the attention of synthetic chemists and biologists alike, benzylated flavanones are quite rare, and as such no efficient syntheses of compounds related to 2 and 3 have been reported.7 A straightforward synthesis would allow us to evaluate the origin of their biological activity and prepare analogs that may be more potent.
The substituent symmetry of 2 and 3 suggested that a common core could be elaborated to provide both molecules. The formation of benzylic carbon-carbon bonds with electron-rich arenes is often achieved via ortho quinone methide (OQM) intermediates, which can be accessed by a variety of routes.8 Pinocembrin (4) could be converted to the OQM precursor by benzylic functionalization (Scheme 1, path A). We initially planned to explore halomethylation, hydroxymethylation, and aminomethylation, since all of these processes take place under neutral or acidic conditions. While all of these processes are well-established for phenols, the analogous transformations using resorcinols are almost unknown.9 Furthermore, the base sensitivity of the flavanone would limit the conditions that could be employed for the formation of the OQM intermediate. An alternate synthetic approach would involve functionalization of the incoming phenolic side-chain (Scheme 1, path B).
Scheme 1.

Retrosynthetic analysis of 2 and 3
Our synthesis began with the development of an efficient route to pinocembrin (Scheme 1). Flavanones related to pinocembrin have been prepared in high yield from the reaction of phenols with cinnamoyl chlorides through a Friedel-Crafts/cyclization sequence.10 Since this process is known to be low yielding for pinocembrin,11 we developed an aldol condensation/cyclization route that rapidly provides multi-gram quantities of pinocembrin.12 Trihydroxyacetophenone 5 is selectively bis-protected with methylchloromethyl ether, then converted to chalcone 6 under standard conditions. Cyclization with sodium acetate provided an equilibrium mixture of the cyclized product and chalcone starting material. Acidic hydrolysis of the MOM groups provided pinocembrin 4.
We explored several methods of benzylic functionalization of pinocembrin in an effort to prepare a suitable intermediate that would eventually lead to 2 and 3. We were able to produce both the morpholine (8, Scheme 3) and dimethylamine (9) Mannich bases from pinocembrin in high yield, though these reactions are not well established for complex resorcinol substrates.13 We made several unsuccessful attempts to convert diamines 8 and 9 directly to our desired product 2 using catalytic amounts of magnesium ethoxide, which has proven effective for dicarbonyl compounds.14 Attempts to convert Mannich bases 8 and 9 to the more reactive acetoxymethyl,15 hydroxymethyl,16 chloromethyl,17 and N-oxide18 derivatives were unsuccessful. Quaternization of the amino groups and subsequent displacement with phenol under Lewis acid-mediated conditions was unsuccessful.19 These experiments demonstrate that resorcinol-derived OQM intermediates are significantly more difficult to access than their phenol-derived counterparts, and are thus not suited to the synthesis of 2 and 3.
Scheme 3.

Aminomethylation of 4 and attempted displacement with phenol to produce 2
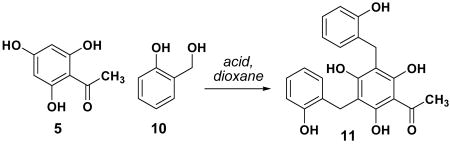
We next turned our attention to the use of benzylic side-chain substrates (Scheme 1, path B). This process was known to be extremely inefficient for the synthesis of dichamanetin.20 We employed trihydroxyacetophenone 5 as a model system to investigate reaction conditions for the aryl alkylation reaction with o-hydroxybenzyl alcohol (eq 1, Table 2). A survey of Lewis and protic acids revealed that ZnCl2 in dioxane afforded bisalkylated product 11 in 49% yield.21 Similar reaction conditions converted pinocembrin 4 to dichamanetin 2 in 59% yield to complete the first selective synthesis of this natural product (eq 2).
Table 2.
Optimization of benzylation conditions (Eq 1)
| |||||
|---|---|---|---|---|---|
| entry | temperature | acid (mol %) | yield | ||
| 1 | 130 °Ca | Sc(OTf)3 (100) | <10%b | ||
| 2 | 60 °C | Sc(OTf)3 (100) | <20%b | ||
| 3 | 100 °C | AlCl3 (100) | 21% | ||
| 4 | 100 °C | SnCl4 (25) | 30% | ||
| 5 | 100 °C | SnCl4 (200) | 33% | ||
| 6 | 130 °Ca | Mg(OTf)2 (100) | 18% | ||
| 7 | 100 °C | p-TsOH (25) | 24% | ||
| 8 | 100 °C | CSA (100) | 20% | ||
| 9 | 130 °Ca | ZnCl2 (100) | 49% | ||
| 10 | 130 °Ca | ZnBr2 (100) | 48% | ||
| 11 | 130 °Ca | ZnI2 (100) | 47% | ||
| 12 | 130 °Ca | ZnCl2 (200) | 49% | ||
| 13 | 60 °C | ZnCl2 (100) | <20%b | ||
| 14 | 23 °C | ZnCl2 (100) | - | ||
Unless otherwise noted, reactions were heated to reflux under argon for 24 h.
Heated for 1 h under microwave irradiation.
Based on LCMS (product not isolated)
 |
(2) |
We next addressed the synthesis of 2″′-hydroxy-5″-benzyl-isouvarinol-B (3, Scheme 4). Phenol 12 is made by selective mono-protection of commercially available 2,4′-dihydroxydiphenylmethane.22 Phenol 12 was hydroxyl-methylated using phenylboronic acid and paraformaldehyde23 to produce boronate ester 13. Hydrolysis of 13 with hydrogen peroxide provided the requisite benzylic alcohol 14 which was converted to 324 in high yield after benzylation and deprotection. The increased yield for the benzylation reaction relative to model compound 5 might be due to the decreased propensity of 4 to enolize and participate in undesirable side reactions.
Scheme 4.

Synthesis of compound 3
While the antimicrobial activity of these compounds has been documented, little is known about their mechanism of action. Both 2 and 3 are potent inhibitors of E. coli GTPase activity (Table 3) and they exhibit IC50 values similar to 1. These experiments indicate that the bacterial cell division protein FtsZ is a target of these compounds. Compound 11, which lacks the flavanone core structure, is much less potent.
Table 3.
Inhibition of E. coli FtsZ GTPase activity by 1, 2, 3, and 11
| compound | IC50 (μM) |
|---|---|
| 1 (zantrin Z1)a | 5.0 ± 0.5 |
| 2 (dichamanetin) | 12.5 ± 0.5 |
| 3 (2″′-hydroxy-5″-benzylisouvarinol-B) | 8.3 ± 0.5 |
| 11 | 60.4 ± 2.2 |
See reference 4a.
In summary, we have developed the first efficient route to hydroxybenzylated flavanone natural products. We synthesized dichamanetin and 2″′-hydroxy-5″-benzylisouvarinol-B from a common core structure using a new zinc chloride-mediated benzylic coupling reaction. The efficient synthesis described in this paper will allow the preparation of a panel of derivatives so that the mechanism of action can be studied in more detail.
Supplementary Material
Experimental procedures for the preparation of all new compounds and determination of IC50 values for 2, 3 and 11. This information is available free of charge via the internet at http://pubs.acs.org.
Scheme 2.

Synthesis of pinocembrin (4)
Table 1.
Antimicrobial activities (MICs, μM) of compounds 1-3
| compound | S. aureus | B. subtilis | M. smegmatis | E. coli | P. aeruginosa |
|---|---|---|---|---|---|
| 1 | 2.5 | 1.25 | -a | 20 | 40 |
| 2 | 1.7 | 1.7 | 3.4 | -b | -b |
| 3 | 10.7 | 2.6 | 3.8 | 2.3 | 15.4 |
not evaluated.
no significant activity.
Acknowledgments
J.T.S. thanks the National Institute for Allergy and Infectious Disease (NIAID) for funding of this research (R03 AI062905-01). J.T.S. acknowledges Amgen Pharmaceuticals for financial support and Harvard University Extension School for a Faculty Aide Award for I.M. D.C.R. acknowledges DARPA, the Charles A. Dana Foundation, and the National Institute of General Medical Sciences (NIGMS) for financial support. The authors thank Profs. Marc Kirschner and Tim Mitchison (Harvard Medical School) for insightful discussions.
Contributor Information
Sameer Urgaonkar, Broad Institute of Harvard and MIT, Chemical Biology Program, 320 Bent St, Cambridge, MA 0214.
Henry S. La Pierre, Broad Institute of Harvard and MIT, Chemical Biology Program, 320 Bent St, Cambridge, MA 0214
Israel Meir, Broad Institute of Harvard and MIT, Chemical Biology Program, 320 Bent St, Cambridge, MA 0214.
Henrik Lund, Department of Molecular Biology and Microbiology, Tufts University School of Medicine, 136 Harrison Avenue, Boston, MA 02111.
Debabrata RayChaudhuri, Department of Molecular Biology and Microbiology, Tufts University School of Medicine, 136 Harrison Avenue, Boston, MA 02111.
Jared T. Shaw, Broad Institute of Harvard and MIT, Chemical Biology Program, 320 Bent St, Cambridge, MA 0214.
References
- 1.(a) Walsh C. Nat Rev Microbiol. 2003;1:65–70. doi: 10.1038/nrmicro727. [DOI] [PubMed] [Google Scholar]; (b) Walsh C, Wright G. Chem Rev. 2005;105:391–393. doi: 10.1021/cr030100y. [DOI] [PubMed] [Google Scholar]
- 2.(a) Romberg L, Levin PA. Ann Rev Microbiol. 2003;57:125–154. doi: 10.1146/annurev.micro.57.012903.074300. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Margolin W. Curr Biol. 2003;13:R16–R18. doi: 10.1016/s0960-9822(02)01381-7. [DOI] [PubMed] [Google Scholar]; (c) Addinall SG, Holland B. J Mol Biol. 2002;318:219–236. doi: 10.1016/S0022-2836(02)00024-4. [DOI] [PubMed] [Google Scholar]; (d) Weiss DS. Mol Microbiol. 2004;54:588–597. doi: 10.1111/j.1365-2958.2004.04283.x. [DOI] [PubMed] [Google Scholar]; (e) Errington J, Daniel RA, Scheffers DJ. Microbiol Mol Biol Rev. 2003;67:52–65. doi: 10.1128/MMBR.67.1.52-65.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Wang J, Galgoci A, Kodali S, Herath KB, Jayasuriya H, Dorso K, Vicente F, Gonzalez A, Cully D, Bramhill D, Singh S. J Biol Chem. 2003;278:44424–44428. doi: 10.1074/jbc.M307625200. [DOI] [PubMed] [Google Scholar]; (b) Reynolds RC, Srivastava S, Ross LJ, Suling WJ, White EL. Bioorg Med Chem Lett. 2004;14:3161–3164. doi: 10.1016/j.bmcl.2004.04.012. [DOI] [PubMed] [Google Scholar]; (c) White EL, Suling WJ, Ross LJ, Seitz LE, Reynolds RC. J Antimicrob Chemother. 2002;50:111–114. doi: 10.1093/jac/dkf075. [DOI] [PubMed] [Google Scholar]
- 4.Margalit DN, Romberg L, Mets RB, Hebert AM, Mitchison TJ, Kirschner MW, RayChaudhuri D. Proc Nat'l Acad Sci USA. 2004;101:11821–11826. doi: 10.1073/pnas.0404439101.The antibacterial activity of this compound was first noted by Beaver: Beaver DJ, Shumard RS, Stoffel PJ. J Am Chem Soc. 1953;75:5579–5581.Subsequent to Beaver, Hakimelahi made similar observations with 1 and related analogs: Hakimelahi GH, Moshfegh AA. Helv Chim Acta. 1981;64:599–609.Moshfegh AA, Badri R, Hojjatie M, Kaviani M, Naderi B, Nazmi AH, Ramezanian M, Roozpeikar B, Hakimelahi GH. Helv Chim Acta. 1982;65:1221–1228.
- 5.(a) Hufford CD, Lasswell WL., Jr Lloydia. 1978;41:156–160. [PubMed] [Google Scholar]; (b) Anam EM. Ind J Chem, Sect B. 1994;33B:1009–1011. [Google Scholar]; See reference 4 for MIC values of 1
- 6.The National Committee for Clinical Laboratory standards (NCCLS) defines the effective MICs of methicillin, erythromycin, and vancomycin as 0.8, 0.4, and 5.8 μM respectively. Higher MICs indicate that a strain is becoming resistant to the drug. Leegaard TM, Caugant DA, Frøholm LO, Høiby EA. Clin Microbiol Infect. 2000;6:290–293. doi: 10.1046/j.1469-0691.2000.00086.x.
- 7.The synthesis of gericudranin, a related para-hydroxybenzylated 3-hydroxy flavanone has been reported: Choi YJ, Shim PJ, Ko KS, Kim HD. Heterocycles. 1996;43:1223–1228.
- 8.Van De Water RW, Pettus TRR. Tetrahedron. 2002;58:5367–5405. [Google Scholar]
- 9.(a) Tabatabai M, Vogt W, Boehmer V. Tetrahedron Lett. 1990;31:3295–3298. [Google Scholar]; (b) Tabatabai M, Vogt W, Baehmer V, Ferguson G, Paulus EF. Supramol Chem. 1994;4:147–152. [Google Scholar]
- 10.(a) Lee JM, Tseng TH, Lee YJ. Synthesis. 2001:2247–2254. [Google Scholar]; (b) Solladie G, Gehrold N, Maignan J. Eur J Org Chem. 1999:2309–2314. [Google Scholar]; (c) Talapatra B, Deb T, Talapatra S. Ind J Chem, Sect B. 1986;25B:1122–1125. [Google Scholar]; (d) Suresh RV, Iyer CSR, Iyer PR. Heterocycles. 1986;24:1925–1930. [Google Scholar]
- 11.Huzise SI, Tatsita H. Chem Ber. 1941;74B:275–278. [Google Scholar]
- 12.Huang C, Li S, Hou L, Li Y, Li Y. Synth Commun. 1999;29:1383–1392. [Google Scholar]; Zhao L, Li Y. Org Prep Proced Int. 1996;28:165–171. [Google Scholar]
- 13.(a) Ghantwal SR, Samant SD. J Indian Chem Soc. 2000;77:100–101. [Google Scholar]; (b) Jerzmanowska Z, Jurkowska-Kowalczyk E. Roczniki Chemii. 1970;44:1395–1401. [Google Scholar]
- 14.(a) Hellmann H, Pohlmann JLW. Liebigs Ann Chem. 1961;642:28–35. [Google Scholar]; (b) Hellmann H, Pohlmann JLW. Liebigs Ann Chem. 1961;643:38–42. [Google Scholar]
- 15.Mazzei M, Miele M, Nieddu E, Barbieri F, Bruzzo C, Alama A. Eur J Med Chem. 2001;36:915–923. [PubMed] [Google Scholar]
- 16.Crisp GT, Turner PD. Tetrahedron. 2000;56:407–415. [Google Scholar]
- 17.Sinhababu AK, Borchardt RT. Synth Commun. 1983;13:677–683. [Google Scholar]
- 18.McCamley K, Ripper JA, Singer RD, Scammells PJ. J Org Chem. 2003;68:9847–9850. doi: 10.1021/jo035243z. [DOI] [PubMed] [Google Scholar]; While this oxidation may have yielded appreciable amounts of the desired N-oxide, this compound was difficult to handle due to its apparent high aqueous solubility
- 19.Baik W, Lee HJ, Yoo CH, Jung JW, Kim BH. J Chem Soc, Perkin Trans 1. 1997;5:587–589. [Google Scholar]
- 20.Dichamanetin was produced in <2% yield using BF3-OEt2 for this reaction: Lasswell WL, Jr, Hufford CD. J Org Chem. 1977;42:1295–1302. doi: 10.1021/jo00428a006.
- 21.A related process has been reported using phenol-derived, but not resorcinol-derived, substrates. The reported reaction was conducted under conditions using a domestic microwave, and while the temperature was not monitored, the wattage applied suggests that the reaction takes place at >200 °C: Khalafi-Nezhad A, Rad MNS, Hakimelahi GH. Helv Chim Acta. 2003;86:2396–2403.
- 22.De Bruyn PJ, Foo LM, Lim ASC, Looney MG, Solomon DH. Tetrahedron. 1997;53:13915–13932. [Google Scholar]
- 23.Nagata W, Okada K, Aoki T. Synthesis. 1979:365–368. [Google Scholar]
- 24.The 1HNMR spectrum of synthetic 3 is consistent with the published data (ref. 5b), but no 13CNMR is reported and we were unable to obtain an authentic sample for comparison.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures for the preparation of all new compounds and determination of IC50 values for 2, 3 and 11. This information is available free of charge via the internet at http://pubs.acs.org.