

Abstract
CD36, a class-B scavenger receptor involved in multiple functions, including inflammatory signaling, may also contribute to ischemic brain injury through yet unidentified mechanisms. We investigated whether CD36 participates in the molecular events underlying the inflammatory reaction that accompanies cerebral ischemia and may contribute to the tissue damage. We found that activation of nuclear factor-κB, a transcription factor that coordinates postischemic gene expression, is attenuated in CD36-null mice subjected to middle cerebral artery occlusion. The infiltration of neutrophils and the glial reaction induced by cerebral ischemia were suppressed. Treatment with an inhibitor of inducible nitric oxide synthase, an enzyme that contributes to the tissue damage, reduced ischemic brain injury in wild-type mice, but not in CD36 nulls. In contrast to cerebral ischemia, the molecular and cellular inflammatory changes induced by intracerebroventricular injection of interleukin-1β were not attenuated in CD36-null mice. The findings unveil a novel role of CD36 in early molecular events leading to nuclear factor-κB activation and postischemic inflammation. Inhibition of CD36 signaling may be a valuable therapeutic approach to counteract the deleterious effects of postischemic inflammation.
Keywords: inflammation, Nox-2, COX-2, IL-1β, myeloperoxidase, iNOS
Introduction
Stroke is a leading cause of death and disability worldwide (American Heart Association, 2006). However, the only Food and Drug Administration-approved medical treatment for stroke is thrombolysis with tissue plasminogen activator (tPA) (for review, see Khaja and Grotta, 2007). Unfortunately, tPA is most effective and safe only when administered within 3 h after the onset of symptoms, a requirement met in <25% of cases (Barber et al., 2001; Reeves et al., 2006). In addition, because of exclusion criteria and concerns for hemorrhagic complications, <5% of stroke patients are treated with tPA (Heuschmann et al., 2003; Reeves et al., 2006). Therefore, there is a pressing need for therapies that can be administered at later times after ischemia.
Cerebral ischemia induces an inflammatory response in the injured brain (Wang et al., 2007). In animals as in humans, ischemia produced by occlusion of the middle cerebral artery (MCA) leads to adhesion of circulating leukocytes to cerebral endothelial cells, which then migrate through the vessel wall and infiltrate the damaged brain (Pozzilli et al., 1985; Barone et al., 1991; Akopov et al., 1996; Lindsberg et al., 1996; Forster et al., 1999). At the same time, astrocytes and microglia become activated (Ladeby et al., 2005; Pekny and Nilsson, 2005). These cellular events are driven by the expression of adhesion molecules and cytokines initiated by the transcription factor nuclear factor-κB (NF-κB) (Allan and Rothwell, 2001; Frijns and Kappelle, 2002; Schwaninger et al., 2006; Wang et al., 2007). Although a large number of studies have shown that suppression of postischemic inflammation is beneficial in experimental stroke (Mehta et al., 2007; Muir et al., 2007; Wang et al., 2007), initial therapeutic efforts targeting adhesion molecules were not successful (Sughrue et al., 2004). These failures have highlighted the complexities of the immune response induced by cerebral ischemia and the need to better understand the upstream pathways initiating inflammatory signaling (Wang et al., 2007).
The type-B scavenger receptor CD36 participates in multiple cellular functions (Febbraio and Silverstein, 2007). In particular, CD36 recognizes pathogen-associated molecular patterns and induces an inflammatory response through activation of NF-κB (Stuart et al., 2005; Triantafilou et al., 2006). CD36 is also involved in the mechanisms of cerebral ischemia. Thus, MCA occlusion upregulates CD36 in the ischemic brain, and CD36-null mice have smaller infarcts and better neurological outcome after focal ischemia (Cho et al., 2005). These observations raise the possibility that CD36 recognizes tissue injury signals after cerebral ischemia and initiates inflammatory gene expression.
Therefore, we examined whether CD36 contributes to the mechanisms of the inflammatory reaction associated with cerebral ischemia. We found that CD36 is required for the full expression of the NF-κB activation produced by cerebral ischemia and for the attendant inflammatory response. The participation of CD36 is restricted to the inflammation associated with cerebral ischemia and is not observed in the neuroinflammation induced by interleukin-1β (IL-1β). Thus, CD36 is involved in key molecular events underlying postischemic inflammation and could be a valuable therapeutic target for ischemic stroke.
Materials and Methods
Animals.
All experimental procedures were approved by the Institutional Animal Care and Use Committee of Weill Cornell Medical College. Experiments were performed in 2- to 3-month-old male CD36-null mice (Febbraio et al., 1999), mice lacking the Nox-2 subunit of NADPH oxidase (Pollock et al., 1995), and C57BL/6 wild-type controls, weighing 20–22 g. CD36 and Nox-2-null mice were obtained from in-house colonies (Park et al., 2004; Cho et al., 2005) and were congenic with the C57BL/6 strain. It has been previously demonstrated that the reduction in stroke volume in CD36 and Nox-2-null mice cannot be attributed to systemic effects or effects on cerebrovascular regulation (Walder et al., 1997; Park et al., 2004; Cho et al., 2005).
Transient middle cerebral artery occlusion.
Procedures for MCA occlusion have previously been published (Cho et al., 2005; Kawano et al., 2006; Kunz et al., 2007b), and are only summarized here. Mice were anesthetized with a mixture of isoflurane (1.5–2%), oxygen, and nitrogen. A fiber optic probe was glued to the parietal bone 2 mm posterior and 5 mm lateral to bregma, and connected to a laser-Doppler flowmeter (Periflux System 5000; Perimed, Järfälla, Sweden) for continuous monitoring of cerebral blood flow (CBF). For MCA occlusion, a heat-blunted monofilament surgical suture (6-0) was inserted into the exposed external carotid artery, advanced into the internal carotid artery, and wedged into the circle of Willis to obstruct the origin of the MCA. The filament was left in place for 30 min and then withdrawn. Only animals that exhibited a reduction in CBF >85% during MCA occlusion and a CBF recovery by >80% after 10 min of reperfusion were included in the study (Cho et al., 2005; Kunz et al., 2007b). The mice that did not meet these criteria were distributed equally among the groups studied and ranged from 0 to 2 per group. This procedure leads to reproducible infarcts similar in size and distribution to those reported by others using transient MCA occlusion of comparable duration (Plesnila et al., 2001; Borsello et al., 2003). Rectal temperature was monitored and kept constant (37.0 ± 0.5°C) during the surgical procedure and in the recovery period until the animals regained full consciousness.
Drug treatments.
Mice were randomly assigned to vehicle or treatment groups. The inducible nitric oxide synthase (iNOS) inhibitor aminoguanidine (AG) (100 mg/kg, i.p.; Sigma-Aldrich, St. Louis, MO) was administered 6 h after reperfusion and, thereafter, twice per day at days 1 and 2, and once at day 3. The cyclooxygenase-2 (COX-2) inhibitor NS398 (10 mg/kg in H2O, i.p.; Cayman Chemical, Ann Arbor, MI) was given 10 min and 6 h after reperfusion, and, thereafter, twice per day at days 1 and 2, and once at day 3. We have previously demonstrated that these administration protocols for AG or NS398 reduce postischemic iNOS or COX-2 activity, respectively, and attenuate infarct volume after focal cerebral ischemia without altering arterial pressure, blood gases, blood glucose, CBF, or body temperature (Iadecola et al., 1995; Zhang et al., 1996a; Nogawa et al., 1997; Zhang and Iadecola, 1998; Nagayama et al., 1999).
Infarct volume measurement.
As described in detail previously (Cho et al., 2005; Kawano et al., 2006; Kunz et al., 2007b), mice were killed 72 h after reperfusion, and their brains were removed, frozen, and sectioned (thickness 30 μm) in a cryostat. Brain sections were collected at 600 μm intervals throughout the ischemic lesion and stained with thionine. Infarct volume was determined using an image analyzer (MCID; Imaging Research, St. Catharines, Ontario, Canada). To eliminate the contribution of postischemic edema to the volume of injury, values were corrected for swelling according to the method of Lin et al. (1993) as previously described (Zhang and Iadecola, 1994).
Intracerebroventricular injection of interleukin-1β.
Mice were anesthetized and placed in a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA). After drilling a small hole in the left parietal bone (coordinates: 0.1 mm posterior to bregma and 0.9 mm lateral from midline), a Hamilton syringe (Hamilton, Reno, NV) was lowered 3.1 mm below the brain surface, and IL-1β (20 ng in 4 μl) (Cell Sciences, Canton, MA) or vehicle (saline) was injected. The dose of IL-1β was selected based on previous studies (Proescholdt et al., 2002; Ching et al., 2005), and its effectiveness was confirmed in preliminary experiments. After the procedure, mice were allowed to recover and returned to their cages.
Real-time PCR.
In some experiments, the mRNA for proinflammatory genes was examined after focal cerebral ischemia and intracerebroventricular injection of IL-1β using real-time PCR. The following genes were studied: CD36, iNOS, COX-2, endothelial-leukocyte adhesion molecule-1 (ELAM-1), intercellular adhesion molecule-1 (ICAM-1), Rac-2, and Nox-2. The proteins encoded by these genes are well known to be upregulated in the postischemic brain (Connolly et al., 1996; Zhang et al., 1996b; Iadecola et al., 1997, 2001; Kunz et al., 2007a). Mice were killed 24 and 72 h after reperfusion of MCA occlusion or 3, 6, and 12 h after intracerebroventricular injection of IL-1β, and their brains were removed. The forebrain was sectioned in half, and the right (ischemic) hemisphere after focal ischemia or the left hemisphere after intracerebroventricular injection was frozen in liquid nitrogen. Total RNA was prepared from the samples using Trizol reagent (Invitrogen, Carlsbad, CA). Quantitative determination of gene expression levels, using a two-step cycling protocol, was performed on Chromo 4 detector (Peltier Thermal Cycler; MJ Research, Watertown, MA). Primers for iNOS (forward, 5′-CAGCTGGGCTGTACAAACCTT-3′; reverse, 5′-CATTGGAAGTGAAGCGTTTCG-3′), COX-2 (forward, 5′-TGGTGCCTGGTCTGATGATG-3′; reverse, 5′-GTGGTAACCGCTCAGGTGTTG-3′), ELAM-1 (forward, 5′-CTCACTCCTGACATCGTCCTC-3′; reverse, 5′-ACGTTGTAAGAAGGCACATGG-3′), ICAM-1 (forward, 5′-GCCTTGGTAGAGGTGACTGAG-3′; reverse, 5′-GACCGGAGCTGAAAAGTTGTA-3′), Rac-2 (forward, 5′-GACACCATCGAGAAGCTGAAG-3′; reverse, 5′-GTGAGTGCAGAACATTCCAAGT-3′), Nox-2 (forward, 5′-CCAACTGGGATAACGAGTTCA-3′; reverse, 5′-GAGAGTTTCAGCCAAGGCTTC-3′), CD36 (forward, 5′-TTTCCTCTGACATTTGCAGGTCTA-3′; reverse, 5′-AAAGGCATTGGCTGGAAGAA-3′), and the mouse HPRT housekeeping gene (forward, 5′-AGTGTTGGATACAGGCCAGAC-3′; reverse, 5′-CGTGATTCAAATCCCTGAAGT-3′) were purchased from Invitrogen. Two microliters of diluted cDNA (1:10) were amplified by Platinum SYBR green qPCR supermix UDG (Invitrogen). The reactions were incubated at 50°C for 2 min and then at 95°C for 10 min. A PCR cycling protocol consisting of 15 s at 95°C and 1 min at 60°C for 45 cycles was required for quantification. Relative expression levels were calculated by according to Livak and Schmittgen (2001). Quantities of all targets in test samples were normalized to the mouse HPRT housekeeping gene, and values were normalized to respective control samples of sham-treated CD36+/+ mice.
Immunohistochemistry.
Immunoreactivity for the neutrophil marker myeloperoxidase (MPO), the astrocytic marker glial fibrillary acidic protein (GFAP), the macrophages/microglia marker F4/80, and CD36 were examined. Seventy-two hours after MCA occlusion or 12 h after intracerebroventricular injection, mice were anesthetized with sodium pentobarbital (120 mg/kg, i.p.) and perfused transcardially with saline. Brains were removed and frozen. Three consecutive brain sections (thickness, 14 μm) were cut and collected at the same rostrocaudal levels used for determination of infarct volume. Sections were fixed in methanol or Carnoy fixative, followed by permeabilization with Triton X-100. Sections processed for MPO immunohistochemistry were treated with H2O2 to inhibit endogenous peroxidases. Sections were incubated overnight with primary antibodies (MPO: 1:100, EMD Biosciences, La Jolla, CA; GFAP: 1:1000, Sigma-Aldrich; F4/80: 1:200, AbD Serotec, Raleigh, NC), followed by biotinylated secondary antibodies (MPO: 1:500, Santa Cruz Biotechnology, Santa Cruz, CA; GFAP and F4/80: 1:500, Jackson ImmunoResearch, West Grove, PA) at 25°C for 1 h. Sections were then incubated with Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA), and the immunoreactivity was visualized with diaminobenzidine (ImmPACT DAB peroxidase substrate; Vector Laboratories). Finally, nuclei were counterstained with methyl green. The specificity of the immunolabel was tested by omitting the primary antibody or by preadsorption with the antigen as described previously (Forster et al., 1999). For CD36 immunoreactivity, mice were perfused transcardially with heparinized saline followed by Histochoice Tissue Fixative (Sigma-Aldrich). Brains were removed, postfixed overnight, and placed in 30% sucrose (Sigma-Aldrich) for 24 h. Sections were incubated with monoclonal anti-CD36 antibody (1:500; Cayman Chemical) followed by a biotinylated secondary antibody (1:50; Sigma-Aldrich). The immunocomplex was visualized as described above.
Quantification of neutrophil infiltration.
Neutrophil infiltration is widely used as a marker of postischemic inflammation (Frijns and Kappelle, 2002). Mice were killed 72 h after reperfusion or 12 h after intracerebroventricular injection, respectively. Three adjacent brain sections were collected at 600 μm intervals, corresponding to the 15 brain levels used for infarct size determination (−3.7 to 3.5 from bregma), and were processed for MPO immunohistochemistry. Sections were examined under a microscope, and MPO-positive cells were counted in the ischemic hemisphere or in the hemisphere ipsilateral to the intracerebroventricular injection. The neutrophil counts for the three adjacent sections were averaged and displayed as number of neutrophils/brain section or total number of neutrophils/infarct. In infarcted brains, a regression analysis correlating the number of neutrophils at each rostrocaudal level with the area of the infarct was performed.
Electrophoretic mobility shift assay.
In NF-κB activation studies, mice were killed 4 h after ischemia-reperfusion or 1 h after intracerebroventricular injection of IL-1β, and their brains were removed. These time points were selected on the basis of previous studies on the time course of NF-κB activation (Gabriel et al., 1999; Stephenson et al., 2000; Proescholdt et al., 2002). The forebrain was sectioned in half, and the right (ischemic) hemisphere after focal ischemia or the left (injected) hemisphere after intracerebroventricular injection was sampled. The fresh brain samples were soaked and homogenized in hypotonic buffer containing 10 mm HEPES, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, and 0.5 mm DTT. Nuclei were collected by centrifugation and washed twice in hypotonic buffer, and nuclear proteins were extracted in high-salt buffer containing 20 mm HEPES, pH 7.9, 20% glycerol, 1.5 mm MgCl2, 800 mm NaCl, 0.1 mm EDTA, pH 8, and 1 mm DTT. Electrophoretic mobility shift assay (EMSA) was essentially performed as described previously (Anrather et al., 1999). Briefly, 6 μg of nuclear extracts were incubated for 30 min at room temperature with 60,000 cpm of double-stranded purified γ-32P ATP-labeled Ig κ light chain enhancer oligonucleotide (5′-AGTTGAGGGACTTTCCCAGGC-3′) for 30 min. For competition, either nonlabeled sense oligonucleotide or nonlabeled scrambled oligonucleotide (5′-ACAGTATCAAAGGCTCACATG-3′) was added. For the supershift assay, 2 μg of mouse monoclonal p65 antibody (clone F-6; Santa Cruz Biotechnology) was added, and reactions were incubated on ice for 1 h. Protein–DNA complexes were separated on 5% Tris/glycine/EDTA polyacrylamide gels, and bands were visualized by autoradiography.
Determination of reactive oxygen species
production. Reactive oxygen species (ROS) production was determined using in vivo hydroethidine microfluorography (Kondo et al., 1997), as previously described (Cho et al., 2005; Kunz et al., 2007a). Hydroethidine is a cell-permeable dye that is oxidized to ethidium by superoxide (Benov et al., 1998). Ethidium is trapped intracellularly by intercalating with DNA (Rothe and Valet, 1990). The fluorescence signal attributable to ethidium reflects cumulative ROS production during the period between administration of hydroethidine and killing of the animals. Hydroethidine (10 mg/kg) was injected into the jugular vein under isoflurane anesthesia 30 min after MCA occlusion, and mice were killed 3.5 h later. In experiments in which ROS production was assessed in IL-1β-treated mice, animals were killed 1 h after IL-1β injection. NF-κB-binding activity is increased at these times after ischemia or IL-1β injection (see Results). In some experiments, the ROS scavenger manganic(I–II)meso-tetrakis(4-benzoic acid) porphyrin (MnTBAP; 100 μg/4 μl, i.c.v.) was administered before MCA occlusion. Brains were removed, frozen, and cut in a cryostat (thickness, 20 μm), collected at 600 μm intervals. The sections were analyzed with a Nikon (Melville, NY) E800 fluorescence microscope equipped with a custom filter set (Chroma Technology, Rockingham, VT). Images were acquired by a computer-controlled digital monochrome camera (Coolsnap; Roper Scientific, Trenton, NJ) attached to the microscope. The analysis of ROS production was performed in a blinded manner using the IPLab software package (Scanalytics, Fairfax, VA) (Cho et al., 2005; Kunz et al., 2007a). After subtracting the camera dark current, pixel intensities of ethidium signals were assessed in the ischemic territory. Fluorescence intensities were measured in five serial sections per animal (rostrocaudal levels +1.6, +1.0, +0.4, −0.2, and −0.8 mm from bregma). The sum of the fluorescence intensity for each region was divided by the total number of pixels analyzed and expressed as relative fluorescence units (RFU) (Cho et al., 2005; Kunz et al., 2007a).
Statistical analysis.
Data are presented as mean ± SEM. Comparisons between two groups were statistically evaluated by the Student's t test. Multiple comparisons were evaluated by ANOVA followed by Newman–Keuls multiple comparison test. Differences were considered significant at p < 0.05.
Results
Postischemic inflammatory gene expression is attenuated in CD36−/− mice
First, we used CD36−/− mice to examine whether CD36 is needed for the upregulation of NF-κB-dependent transcripts after focal cerebral ischemia. These include iNOS, COX-2, ICAM-1, ELAM-1, and the NADPH oxidase subunit Nox-2 (Connolly et al., 1996; Zhang et al., 1996b; Iadecola et al., 1997, 2001; Kunz et al., 2007a). The neutrophil marker Rac-2 was also studied. In CD36+/+ mice, MCA occlusion upregulated iNOS, COX-2, ICAM-1, ELAM-1, Rac-2, and Nox-2 mRNA (n = 5 per group) (Fig. 1 A–F). Focal ischemia also increased CD36 expression (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In contrast to CD36+/+ mice, in CD36−/− mice (n = 5 per group), the expression of iNOS, ELAM-1, ICAM-1, Rac-2, and Nox-2 was markedly attenuated (Fig. 1A,C–F), whereas COX-2 expression was only slightly reduced at 72 h (Fig. 1B).
Figure 1.

Expression of mRNA for iNOS (A), COX-2 (B), ELAM-1 (C), ICAM-1 (D), Rac-2 (E), and Nox-2 (F) 24 and 72 h after MCA occlusion in CD36+/+ mice, CD36−/− mice, and CD36+/+ mice treated with the COX-2 inhibitor NS398. *p < 0.05 from CD36+/+; n = 5 per group; ANOVA and Newman–Keuls test.
The volume of the infarct produced by MCA occlusion is smaller in CD36−/− than in CD36+/+ mice (Cho et al., 2005) (Fig. 2A). To determine whether the attenuation in iNOS, ELAM-1, ICAM-1, Rac-2, and Nox-2 was a consequence of the reduction in infarct volume, we examined CD36+/+ mice treated with the COX-2 inhibitor NS398. NS398 reduced infarct volume to a value comparable with that of CD36−/− mice (Kunz et al., 2007a) (Fig. 2B). However, NS398 did not alter postischemic mRNA expression for COX-2, ELAM-1, ICAM-1, Rac-2, Nox-2, and CD36, although a small and transient reduction of iNOS was observed at 24 h (Fig. 1 A–F; supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Furthermore, the pattern of gene expression in the ischemic brain of Nox-2-null mice, which also have smaller infarcts (Kunz et al., 2007a), was different from that of CD36−/− mice (Fig. 3 A–E). Therefore, the attenuation in the expression of inflammation-related genes in CD36−/− mice cannot be attributed to the reduction in the size of the infarct.
Figure 2.

Effect of the iNOS inhibitor AG (A) or the COX-2 inhibitor NS398 (B) on infarct volume in CD36+/+ and CD36−/− mice. *p < 0.05 from vehicle; n = 6 per group; t test.
Figure 3.

Expression of mRNA for iNOS (A), COX-2 (B) ELAM-1 (C), ICAM-1 (D), and Rac-2 (E) 24 and 72 h after MCA occlusion in Nox-2+/+ or −/− mice. *p < 0.05 from Nox-2+/+; n = 5 per group; ANOVA and Newman–Keuls test.
The cellular inflammatory reaction associated with cerebral ischemia is attenuated in CD36−/− mice
In CD36+/+ mice, there were abundant GFAP-positive astrocytes surrounding the infarcted area 72 h after ischemia (Fig. 4Aa). At this time, there also was marked microglial activation as detected by the microglia/macrophage marker F4/80 (Fig. 4Ad). Abundant MPO-positive neutrophils were observed through the infarcted hemisphere (Fig. 4Ag), the neutrophil infiltration being greater at the rostrocaudal levels where the infarct was larger (Fig. 4B). In CD36−/− mice, the astrocytic and microglial activation were markedly attenuated (Fig. 4Ab,Ae) and the total number of infiltrating neutrophils was reduced by 86% (CD36+/+: 1938 ± 296; CD36−/−: 281 ± 92; p < 0.05; n = 5 per group) (Fig. 4Ah,B). In contrast, in CD36+/+ mice treated with NS398 the astrocytic response was not affected, and the microglial activation was only slightly attenuated compared with untreated mice (Fig. 4Ac,Af). The attenuation in neutrophil infiltration induced by NS398 was much less than that observed in CD36−/− mice (−38%; total neutrophils: 1199 ± 176; p < 0.05 from CD36−/− mice; n = 5 per group) (Fig. 4Ai,B). To determine whether the reduction in neutrophil infiltration in CD36−/− mice was commensurate with the reduction in infarct size, a linear regression analysis was performed between the number of neutrophils at each rostrocaudal level and the corresponding infarct area. There was a linear relationship between number of neutrophils and infarct area (Fig. 4C) (R2 = 0.95; p < 0.05; n = 14). However, the slope of the regression line in CD36−/− mice (6.9 ± 0.8) was lower than that of CD36+/+ mice (11.4 ± 0.8; p < 0.05), indicating that the reduction in neutrophils was greater than anticipated from the smaller size of the infarct. In contrast, in CD36+/+ mice treated with NS398, the slope of the regression line overlapped with that of untreated mice (11.5 ± 0.9; p > 0.05) (Fig. 4C), indicating that the reduction in the number of neutrophils was proportional to the reduction in infarct size.
Figure 4.
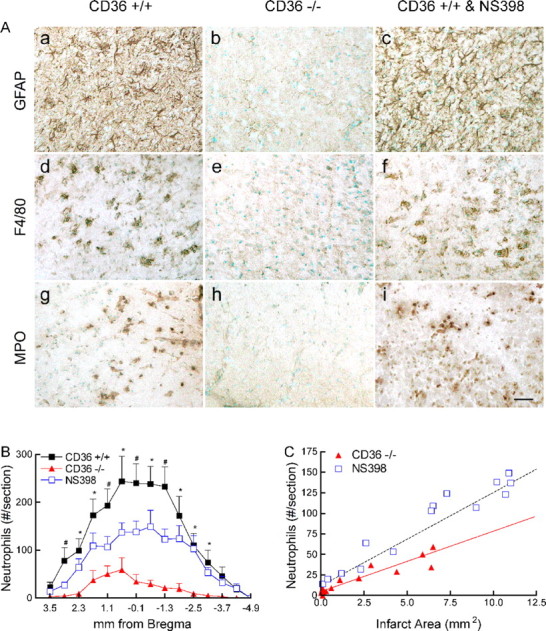
Cellular inflammatory reaction 72 h after MCA occlusion. A, GFAP (a–c), F4/80 (d–f), and MPO (g–i) in CD36+/+ (a, d, g), CD36−/− (b, e, h), and CD36+/+ mice treated with the COX-2 inhibitor NS398 (c, f, i). Images were taken at the medial border zone of the cortical infarct. Scale bar, 50 μm. B, Rostrocaudal distribution of neutrophils throughout the infarct. *p < 0.05 from CD36−/− mice; #p < 0.05 from CD36−/− and CD36+/+ mice treated with NS398; n = 5 per group; ANOVA and Newman–Keuls test. C, Linear regression analysis between the infarct area at each rostrocaudal level depicted in B and the number of neutrophils at that level. The dashed line represents the correlation between infarct area and neutrophils in CD36+/+ mice. The red line represents the regression line correlating infarct area and neutrophils in CD36−/− mice. Note that the points pertaining to CD36+/+ mice treated with NS398 overlap with the line of CD36+/+ mice.
iNOS inhibition does not reduce infarct volume in CD36−/− mice
We then sought to provide evidence that the attenuation in inflammation was responsible for the reduced injury volume in CD36−/− mice. We reasoned that, if postischemic inflammation is attenuated in CD36−/− mice, then agents that reduce infarct volume by counteracting the destructive actions of inflammation should not be effective in CD36−/− mice. iNOS is a major effector of the tissue damage produced by inflammation (Iadecola et al., 1995, 1997; Loihl et al., 1999; Parmentier et al., 1999), and its expression is suppressed in CD36−/− mice (Fig. 1A). Therefore, we examined the effects of the iNOS inhibitor AG in CD36−/− mice. AG attenuated the volume of the infarct in CD36+/+ (p < 0.05; n = 6 per group), but not in CD36−/− mice (p > 0.05) (Fig. 2A). In contrast, the COX-2 inhibitor NS398 attenuated the lesion in CD36−/− mice (p < 0.05; n = 6 per group) (Fig. 2B). Therefore, COX-2 inhibition, but not iNOS inhibition, attenuates infarct volume in CD36-null mice. The observation that COX-2 inhibition reduced infarct volume in CD36-null mice attests to the fact that CD36 and COX-2 contribute to ischemic injury through different pathogenic mechanisms.
Intracerebroventricular injection of IL-1β induces neuroinflammation in CD36−/− mice
The attenuated inflammatory reaction after cerebral ischemia in CD36−/− mice could reflect a generalized anti-inflammatory phenotype conferred by inactivation of this scavenger receptor. To address this issue, we used a model of neuroinflammation produced by intracerebroventricular injection of IL-1β (Proescholdt et al., 2002; Ching et al., 2005). In CD36+/+ mice, IL-1β upregulated iNOS, COX-2, ELAM-1, ICAM-1, Rac-2, and Nox-2 mRNA (p < 0.05; n = 5 per group) (Fig. 5 A–F). In addition, IL-1β induced astrocytic activation, neutrophil infiltration, and a modest activation of microglia (Fig. 6A). These changes were comparable with those reported previously (Proescholdt et al., 2002), and were observed in the periventricular regions of the forebrain and along the injection needle track (Fig. 6B). The upregulation of inflammatory genes induced by IL-1β did not differ between CD36+/+ and −/− mice (p > 0.05), with the exception of Nox-2, which was reduced in the nulls (Fig. 5F). The astrocytic and microglial activation did not differ between CD36+/+ and −/− mice, but the neutrophil infiltration was more marked in CD36−/− mice, although the increase reached statistical significance only at one rostrocaudal level (Fig. 6 A–C) (n = 5 per group). Therefore, IL-1β is able to induce a robust neuroinflammatory response in CD36-null mice.
Figure 5.

Expression of mRNA for iNOS (A), COX-2 (B), ELAM-1 (C), ICAM-1 (D), Rac-2 (E), and Nox-2 (F) 3, 6, and 12 h after intracerebroventricular injection of IL-1β. Sham mice were prepared for injection of IL-1β, but the skull was not drilled and the needle was not inserted. Saline mice received an injection of vehicle (saline). *p < 0.05 from CD36+/+; n = 5 per group; ANOVA and Newman–Keuls test.
Figure 6.
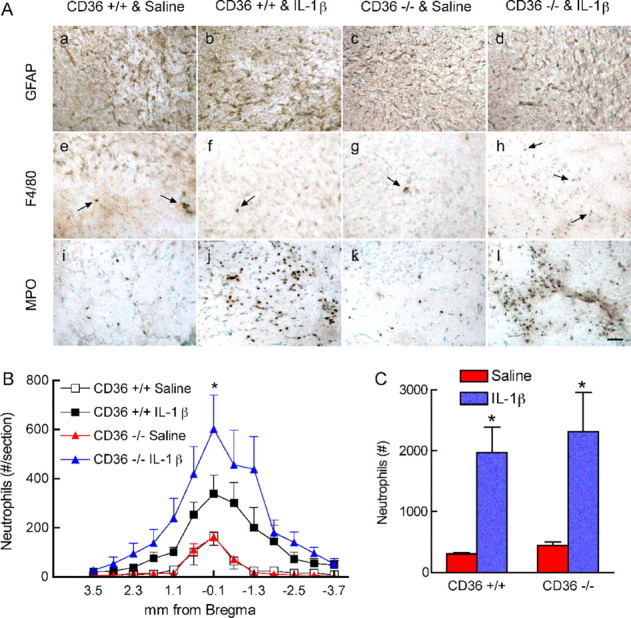
Cellular inflammatory reaction 24 h after intracerebroventricular injection of IL-1β. A, GFAP (a–d), F4/80 (e–h), and MPO (i–l) in CD36+/+ mice treated with saline (a, e, i), CD36+/+ mice treated with IL-1β (b, f, j), CD36−/− mice treated with saline (c, g, k), and CD36−/− mice treated with IL-1β (d, h, l). Images were taken in the periventricular region of the striatum. Arrows in e–h indicate F4/80-positive cells. Scale bar, 50 μm. B, Rostrocaudal distribution of neutrophils throughout the periventricular regions of the forebrain. *p < 0.05 from CD36+/+ mice treated with IL-1β; n = 5 per group; ANOVA and Newman–Keuls test. C, Total number of neutrophils in the brain of mice treated with IL-1β. *p < 0.05 from saline; t test.
NF-κB activation is suppressed in CD36−/− mice after stroke, but not IL-1β injection
NF-κB is the major transcription factor driving the postischemic expression of inflammatory genes (Schwaninger et al., 2006; Wang et al., 2007). Therefore, we examined the in vitro binding activity of this transcription factor in CD36+/+ and −/− mice using EMSA. In agreement with previous reports (Gabriel et al., 1999; Stephenson et al., 2000; Proescholdt et al., 2002), in wild-type mice MCA occlusion or intracerebroventricular injection of IL-1β increased NF-κB DNA binding activity in the affected brain (Fig. 7A,B). Specificity of the reaction was ensured by competition with a nonlabeled sense Ig κ light chain enhancer oligonucleotide, which led to complete loss of the signal, whereas addition of unlabeled scrambled probe had no effect. Supershift assays with a p65-specific antibody demonstrated presence of p65 in the NF-κB binding complex (Fig. 7C). In CD36−/− mice, postischemic NF-κB binding activity was attenuated (Fig. 7A). However, a comparable attenuation was not observed in CD36+/+ mice in which the infarct was reduced by treatment with NS398 (Fig. 7A). Moreover, in contrast with MCA occlusion, the NF-κB activation produced by intracerebroventricular injection of IL-1β did not differ between CD36+/+ and −/− mice (Fig. 7B).
Figure 7.

EMSA in CD36+/+ and −/− mice. A, NF-κB binding activity in CD36+/+ (wt) and −/− (ko) mice with and without MCA occlusion (MCAO) and treatment with NS398. B, NF-κB binding activity in wt and ko with and without intracerebroventricular injection of IL-1β. C, NF-κB binding activity in CD36+/+ mice with MCAO or intracerebroventricular injection of IL-1β. The signal is eliminated by addition of unlabeled sense oligonucleotide (co Ig κB) but not by unlabeled scrambled oligonucleotide (co sc Ig κB), indicating specificity of the reaction. Treatment with p65 antibodies (p65 Ab) leads to a band shift, demonstrating the presence of p65 in the NF-κB complex. The asterisks indicate nonspecific binding activity. Data are representative of four independent experiments. D, ROS production assessed using hydroethidine fluoromicrography 4 h after MCAO. The ROS scavenger MnTBAP blocks postischemic ROS production (p > 0.05 from sham; p < 0.05 from vehicle). *p < 0.05 from sham; ANOVA and Newman–Keuls test; n = 5 per group. E, NF-κB binding activity in wt mice, with or without MCAO, treated with vehicle or MnTBAP intracerebroventricularly. Data are representative of three independent experiments.
Role of reactive oxygen species in NF-κB activation
CD36-null mice have a reduced ROS production after MCA occlusion (Cho et al., 2005), which could play a role in the postischemic attenuation of NF-κB binding activity, a redox-sensitive transcription factor (Hayden and Ghosh, 2004). Therefore, we explored whether ROS are required for the NF-κB activation induced by cerebral ischemia or IL-1β injection. Administration of the ROS scavenger MnTBAP blocked postischemic ROS production (Fig. 7D) (p > 0.05 from sham; p < 0.05 from vehicle; n = 5 per group), but failed to reduce NF-κB binding activity to a level comparable with that observed in CD36-null mice (Fig. 7E). On the other hand, intracerebroventricular injection of IL-1β did not increase ROS production (vehicle: 18.7 ± 1.7 RFU; IL-1β: 18.9 ± 1.1; p > 0.05; n = 4 per group), but it markedly increased NF-κB binding activity (Fig. 7B).
Discussion
We have demonstrated that CD36-null mice have a reduced inflammatory response to focal cerebral ischemia. Thus, the expression of iNOS, ICAM-1, ELAM-1, Rac-2, and Nox-2 was markedly attenuated in CD36−/− mice after MCA occlusion, but not after IL-1β-induced neuroinflammation. These molecular changes were associated with a substantial attenuation of the glial activation and neutrophil infiltration evoked by cerebral ischemia, well established cellular markers of postischemic inflammation. Administration of the iNOS inhibitor AG did not reduce infarct volume in CD36-null mice, suggesting that the protective effect conferred by CD36 deficiency is partly attributable to suppression of iNOS expression. In contrast, inhibition of COX-2, an enzyme whose expression was not attenuated by CD36 deletion, reduced the damage in CD36-null mice. Next, we investigated the molecular substrates of the reduced inflammatory response to cerebral ischemia in CD36-null mice. We found that the postischemic activation of NF-κB, a key transcription factor coordinating gene expression after ischemia (Schwaninger et al., 2006), is reduced in CD36-null mice. In contrast, brain NF-κB activation was not attenuated after intracerebroventricular injection of IL-1β. These observations, collectively, demonstrate that CD36 is specifically required for the full expression of the NF-κB activation induced by cerebral ischemia and for the attendant inflammatory response.
The intensity of the inflammatory reaction is related to the size of the ischemic lesion, so that small infarcts have less inflammation than large infarcts (Akopov et al., 1996). Therefore, the attenuation in brain inflammation in CD36-null mice could be a consequence of the reduced infarct size. However, this is not the case, because NS398 produced a reduction in infarct volume comparable with that observed in CD36-null mice, but did not mimic the attenuation in inflammatory gene expression observed in CD36 nulls. Similarly, the attenuation in infiltrating neutrophil observed with NS398 was commensurate with the reduction in infarct size, whereas in CD36 nulls neutrophil infiltration was suppressed more than expected on the basis of the reduction in infarct size. Such attenuation in neutrophil infiltration is not attributable to a global failure of neutrophil migration across the blood–brain barrier, because intracerebroventricular injection of IL-1β induced a strong neutrophil response in CD36-null mice. These observations demonstrate that the attenuation in the inflammatory response in CD36-null mice cannot be attributed to secondary effects of the reduction in infarct size or to a generalized inability to mount an inflammatory response. Rather, the findings reinforce the conclusion that CD36 is specifically involved in the molecular events leading to NF-κB activation and inflammation after focal cerebral ischemia.
Increasing evidence suggests that NF-κB plays a crucial role in postischemic gene expression and ischemic brain injury (Schwaninger et al., 2006). NF-κB is central to the expression of critical adaptive responses in multiple cell types, especially those of the immune system (Hayden and Ghosh, 2004). Cerebral ischemia leads to NF-κB activation in neurons, astrocytes, microglia, and infiltrating inflammatory cells (Gabriel et al., 1999; Schneider et al., 1999; Stephenson et al., 2000; Nurmi et al., 2004). Mice with a null mutation of the NF-κB subunit p50 have a smaller infarct after MCA occlusion (Schneider et al., 1999). Furthermore, mice deficient in IKK2, a kinase required for NF-κB activation, are less susceptible to focal cerebral ischemia (Herrmann et al., 2005). NF-κB activation in the early stages of cerebral ischemia triggers a more protracted series of molecular events that leads to a cellular inflammatory response lasting several days. Although several global stimuli could potentially activate NF-κB after cerebral ischemia, including the neurotransmitter glutamate, free radicals, or hypoxia (Schwaninger et al., 2006), no specific signaling mechanism has been identified. Thus, CD36 is the first receptor demonstrated to be necessary for the full expression of postischemic NF-κB activation. The involvement of CD36 is remarkably specific for the NF-κB activation induced by cerebral ischemia, because CD36 is not required for the activation of NF-κB induced by IL-1β.
What are the mechanisms triggering CD36 activation in cerebral ischemia? CD36 binds a wide variety of molecules, including modified lipids, amyloid-β, thrombospondin-1, and advanced glycation endproducts (Febbraio and Silverstein, 2007). In brain, CD36 is expressed mainly in microglia and endothelial cells and is upregulated after cerebral ischemia (Cho et al., 2005). Therefore, ligands generated in blood or brain could have access to cells expressing CD36. The ligands mediating CD36 activation in ischemia have not been identified. Thrombospondin-1 is unlikely to be involved, because this protein is upregulated relatively late after focal cerebral ischemia (Hayashi et al., 2003). On the other hand, cerebral ischemia is well known to produce numerous species of modified lipids (Adibhatla et al., 2006). For example, modified low-density lipoprotein (LDL), such as oxidized LDL, is formed after cerebral ischemia and could activate CD36 (Uno et al., 2003; Shie et al., 2004; Vibo et al., 2007). In addition, hypoxia has been reported to produce diacylglyceride, a CD36 ligand that is involved in inflammatory signaling via toll-like receptors 2/6 (TLR2/6; see below) (Bruder et al., 2005; Hoebe et al., 2005). Therefore, ischemia has the potential of generating several CD36 ligands that could play a role in postischemic CD36 signaling.
Another question concerns the molecular pathways linking CD36 to NF-κB activation. The free radical production induced by cerebral ischemia is attenuated in CD36−/− mice (Cho et al., 2005). Because NF-κB is a redox-sensitive transcription factor (Hayden and Ghosh, 2004), it is possible that the attenuation in NF-κB observed in CD36−/− mice is related to the reduced oxidative stress in these mice. However, this possibility seems unlikely, because we have shown here that the antioxidant MnTBAP suppresses ROS production after cerebral ischemia, but it does not attenuate NF-κB binding activity. In contrast, in CD36-null mice, in which ROS production is also attenuated (Cho et al., 2005), NF-κB binding activity is markedly suppressed. Therefore, the reduced NF-κB activation observed in CD36-null mice cannot be attributed solely to reduced oxidative stress. Conversely, IL-1β induces NF-κB activation without increasing ROSs. These observations suggest that in ischemia or IL-1β-induced neuroinflammation, ROSs are not required for the activation of NF-κB. On the other hand, recent data suggest that CD36 forms a signaling complex with TLR2/6 that activates NF-κB in response to bacterial wall components (Hoebe et al., 2005; Stuart et al., 2005; Triantafilou et al., 2006). Therefore, it is conceivable that also in cerebral ischemia CD36 could form a receptor complex with TLR2/6, and activate NF-κB through the canonical pathway. This possibility is reinforced by the observation that mice deficient in TLR2 signaling are protected from cerebral ischemia (Ziegler et al., 2007; Tang et al., 2007). Future studies will have to address the role of TLR2/6 and their association with CD36 in the setting of ischemic brain injury and, possibly, other brain pathologies as well.
In conclusion, we have demonstrated that CD36-null mice have a profound attenuation in the molecular and cellular inflammatory response induced by focal cerebral ischemia. This effect is attributable to a role of CD36 in NF-κB activation in the setting of cerebral ischemia, but not in the neuroinflammation induced by intracerebroventricular injection of IL-1β. Attenuation of the damage produced by postischemic inflammation contributes to the neuroprotection observed in CD36-null mice. The present study raises the possibility that CD36 acts as a sensor for specific tissue injury signals generated during the early stages of cerebral ischemia. The CD36-dependent NF-κB activation triggers proinflammatory signaling that contributes to the inflammatory response associated with cerebral ischemia. Therefore, the findings unveil a previously unrecognized role of CD36 in the NF-κB activation and inflammatory response associated with cerebral ischemia. CD36, alone or in a complex with TLR2/6, may be a promising target for suppressing the tissue damage initiated by postischemic inflammation.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants NS34179 and NS35806 (C.I.) and by Deutsche Forschungsgemeinschaft Grant KU 1990/1-1 (A.K.). C.I. is the recipient of a Javits Award from NIH–National Institute of Neurological Disorders and Stroke.
References
- Adibhatla RM, Hatcher JF, Dempsey RJ. Lipids and lipidomics in brain injury and diseases. AAPS J. 2006;8:E314–E321. doi: 10.1007/BF02854902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akopov SE, Simonian NA, Grigorian GS. Dynamics of polymorphonuclear accumulation in acute cerebral infarction and their correlation with brain tissue damage. Stroke. 1996;27:1739–1743. doi: 10.1161/01.str.27.10.1739. [DOI] [PubMed] [Google Scholar]
- Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci. 2001;2:734–744. doi: 10.1038/35094583. [DOI] [PubMed] [Google Scholar]
- American Heart Association. Heart disease and stroke statistics—2006 update. Dallas, TX: 2006. [Google Scholar]
- Anrather J, Csizmadia V, Soares MP, Winkler H. Regulation of NF-kappaB RelA phosphorylation and transcriptional activity by p21(ras) and protein kinase Czeta in primary endothelial cells. J Biol Chem. 1999;274:13594–13603. doi: 10.1074/jbc.274.19.13594. [DOI] [PubMed] [Google Scholar]
- Barber PA, Zhang J, Demchuk AM, Hill MD, Buchan AM. Why are stroke patients excluded from TPA therapy? An analysis of patient eligibility. Neurology. 2001;56:1015–1020. doi: 10.1212/wnl.56.8.1015. [DOI] [PubMed] [Google Scholar]
- Barone FC, Hillegass LM, Price WJ, White RF, Lee EV, Feuerstein GZ, Sarau HM, Clark RK, Griswold DE. Polymorphonuclear leukocyte infiltration into cerebral focal ischemic tissue: myeloperoxidase activity assay and histologic verification. J Neurosci Res. 1991;29:336–345. doi: 10.1002/jnr.490290309. [DOI] [PubMed] [Google Scholar]
- Benov L, Sztejnberg L, Fridovich I. Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radic Biol Med. 1998;25:826–831. doi: 10.1016/s0891-5849(98)00163-4. [DOI] [PubMed] [Google Scholar]
- Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, Bogousslavsky J, Bonny C. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. 2003;9:1180–1186. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- Bruder ED, Lee PC, Raff H. Lipid and fatty acid profiles in the brain, liver, and stomach contents of neonatal rats: effects of hypoxia. Am J Physiol Endocrinol Metab. 2005;288:E314–E320. doi: 10.1152/ajpendo.00362.2004. [DOI] [PubMed] [Google Scholar]
- Ching S, He L, Lai W, Quan N. IL-1 type I receptor plays a key role in mediating the recruitment of leukocytes into the central nervous system. Brain Behav Immun. 2005;19:127–137. doi: 10.1016/j.bbi.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Cho S, Park EM, Febbraio M, Anrather J, Park L, Racchumi G, Silverstein RL, Iadecola C. The class B scavenger receptor CD36 mediates free radical production and tissue injury in cerebral ischemia. J Neurosci. 2005;25:2504–2512. doi: 10.1523/JNEUROSCI.0035-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly ES, Jr, Winfree CJ, Springer TA, Naka Y, Liao H, Yan SD, Stern DM, Solomon RA, Gutierrez-Ramos JC, Pinsky DJ. Cerebral protection in homozygous null ICAM-1 mice after middle cerebral artery occlusion. Role of neutrophil adhesion in the pathogenesis of stroke. J Clin Invest. 1996;97:209–216. doi: 10.1172/JCI118392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Febbraio M, Silverstein RL. CD36: implications in cardiovascular disease. Int J Biochem Cell Biol. 2007;39:2012–2030. doi: 10.1016/j.biocel.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Febbraio M, Abumrad NA, Hajjar DP, Sharma K, Cheng W, Pearce SF, Silverstein RL. A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J Biol Chem. 1999;274:19055–19062. doi: 10.1074/jbc.274.27.19055. [DOI] [PubMed] [Google Scholar]
- Forster C, Clark HB, Ross ME, Iadecola C. Inducible nitric oxide synthase expression in human cerebral infarcts. Acta Neuropathol (Berl) 1999;97:215–220. doi: 10.1007/s004010050977. [DOI] [PubMed] [Google Scholar]
- Frijns CJ, Kappelle LJ. Inflammatory cell adhesion molecules in ischemic cerebrovascular disease. Stroke. 2002;33:2115–2122. doi: 10.1161/01.str.0000021902.33129.69. [DOI] [PubMed] [Google Scholar]
- Gabriel C, Justicia C, Camins A, Planas AM. Activation of nuclear factor-kappaB in the rat brain after transient focal ischemia. Brain Res Mol Brain Res. 1999;65:61–69. doi: 10.1016/s0169-328x(98)00330-1. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Noshita N, Sugawara T, Chan PH. Temporal profile of angiogenesis and expression of related genes in the brain after ischemia. J Cereb Blood Flow Metab. 2003;23:166–180. doi: 10.1097/01.WCB.0000041283.53351.CB. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Herrmann O, Baumann B, de Lorenzi R, Muhammad S, Zhang W, Kleesiek J, Malfertheiner M, Kohrmann M, Potrovita I, Maegele I, Beyer C, Burke JR, Hasan MT, Bujard H, Wirth T, Pasparakis M, Schwaninger M. IKK mediates ischemia-induced neuronal death. Nat Med. 2005;11:1322–1329. doi: 10.1038/nm1323. [DOI] [PubMed] [Google Scholar]
- Heuschmann PU, Berger K, Misselwitz B, Hermanek P, Leffmann C, Adelmann M, Buecker-Nott HJ, Rother J, Neundoerfer B, Kolominsky-Rabas PL. Frequency of thrombolytic therapy in patients with acute ischemic stroke and the risk of in-hospital mortality: the German Stroke Registers Study Group. Stroke. 2003;34:1106–1113. doi: 10.1161/01.STR.0000065198.80347.C5. [DOI] [PubMed] [Google Scholar]
- Hoebe K, Georgel P, Rutschmann S, Du X, Mudd S, Crozat K, Sovath S, Shamel L, Hartung T, Zahringer U, Beutler B. CD36 is a sensor of diacylglycerides. Nature. 2005;433:523–527. doi: 10.1038/nature03253. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Zhang F, Xu X. Inhibition of inducible nitric oxide synthase ameliorates cerebral ischemic damage. Am J Physiol. 1995;268:R286–R292. doi: 10.1152/ajpregu.1995.268.1.R286. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Zhang F, Casey R, Nagayama M, Ross ME. Delayed reduction in ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J Neurosci. 1997;17:9157–9164. doi: 10.1523/JNEUROSCI.17-23-09157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Niwa K, Nogawa S, Zhao X, Nagayama M, Araki E, Morham S, Ross ME. Reduced susceptibility to ischemic brain injury and NMDA-mediated neurotoxicity in cyclooxygenase-2 deficient mice. Proc Natl Acad Sci USA. 2001;98:1294–1299. doi: 10.1073/pnas.98.3.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano T, Anrather J, Zhou P, Park L, Wang G, Frys KA, Kunz A, Cho S, Orio M, Iadecola C. Prostaglandin E(2) EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat Med. 2006;12:225–229. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]
- Khaja AM, Grotta JC. Established treatments for acute ischaemic stroke. Lancet. 2007;369:319–330. doi: 10.1016/S0140-6736(07)60154-8. [DOI] [PubMed] [Google Scholar]
- Kondo T, Reaume AG, Huang T-T, Carlson E, Murakami K, Chen SF, Hoffman EK, Scott RW, Epstein CJ, Chan PH. Reduction of CuZn-superoxide dismutase activity exacerbates neuronal cell injury and edema formation after transient focal cerebral ischemia. J Neurosci. 1997;17:4180–4189. doi: 10.1523/JNEUROSCI.17-11-04180.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz A, Anrather J, Zhou P, Orio M, Iadecola C. Cyclooxygenase-2 does not contribute to postischemic production of reactive oxygen species. J Cereb Blood Flow Metab. 2007a;27:545–551. doi: 10.1038/sj.jcbfm.9600369. [DOI] [PubMed] [Google Scholar]
- Kunz A, Park L, Abe T, Gallo EF, Anrather J, Zhou P, Iadecola C. Neurovascular protection by ischemic tolerance: role of nitric oxide and reactive oxygen species. J Neurosci. 2007b;27:7083–7093. doi: 10.1523/JNEUROSCI.1645-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladeby R, Wirenfeldt M, Garcia-Ovejero D, Fenger C, Dissing-Olesen L, Dalmau I, Finsen B. Microglial cell population dynamics in the injured adult central nervous system. Brain Res Brain Res Rev. 2005;48:196–206. doi: 10.1016/j.brainresrev.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Lin T-N, He YY, Wu G, Khan M, Hsu CY. Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke. 1993;24:117–121. doi: 10.1161/01.str.24.1.117. [DOI] [PubMed] [Google Scholar]
- Lindsberg PJ, Carpen O, Paetau A, Karjalainen-Lindsberg ML, Kaste M. Endothelial ICAM-1 expression associated with inflammatory cell response in human ischemic stroke. Circulation. 1996;94:939–945. doi: 10.1161/01.cir.94.5.939. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Loihl AK, Asensio V, Campbell IL, Murphy S. Expression of nitric oxide synthase (NOS)-2 following permanent focal ischemia and the role of nitric oxide in infarct generation in male, female and NOS-2 gene-deficient mice. Brain Res. 1999;830:155–164. doi: 10.1016/s0006-8993(99)01388-8. [DOI] [PubMed] [Google Scholar]
- Mehta SL, Manhas N, Raghubir R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev. 2007;54:34–66. doi: 10.1016/j.brainresrev.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Muir KW, Tyrrell P, Sattar N, Warburton E. Inflammation and ischaemic stroke. Curr Opin Neurol. 2007;20:334–342. doi: 10.1097/WCO.0b013e32813ba151. [DOI] [PubMed] [Google Scholar]
- Nagayama M, Niwa K, Nagayama T, Ross ME, Iadecola C. The cyclooxygenase-2 inhibitor NS-398 ameliorates cerebral ischemic injury in wild-type mice but not in mice with deletion of the inducible nitric oxide synthase gene. J Cereb Blood Flow Metab. 1999;19:1213–1219. doi: 10.1097/00004647-199911000-00005. [DOI] [PubMed] [Google Scholar]
- Nogawa S, Zhang F, Ross ME, Iadecola C. Cyclo-oxygenase-2 gene expression in neurons contributes to ischemic brain damage. J Neurosci. 1997;17:2746–2755. doi: 10.1523/JNEUROSCI.17-08-02746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurmi A, Lindsberg PJ, Koistinaho M, Zhang W, Juettler E, Karjalainen-Lindsberg ML, Weih F, Frank N, Schwaninger M, Koistinaho J. Nuclear factor-kappaB contributes to infarction after permanent focal ischemia. Stroke. 2004;35:987–991. doi: 10.1161/01.STR.0000120732.45951.26. [DOI] [PubMed] [Google Scholar]
- Park L, Anrather J, Zhou P, Frys K, Wang G, Iadecola C. Exogenous NADPH increases cerebral blood flow through NADPH oxidase-dependent and -independent mechanisms. Arterioscler Thromb Vasc Biol. 2004;24:1860–1865. doi: 10.1161/01.ATV.0000142446.75898.44. [DOI] [PubMed] [Google Scholar]
- Parmentier S, Bohme GA, Lerouet D, Damour D, Stutzmann JM, Margaill I, Plotkine M. Selective inhibition of inducible nitric oxide synthase prevents ischaemic brain injury. Br J Pharmacol. 1999;127:546–552. doi: 10.1038/sj.bjp.0702549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- Plesnila N, Zinkel S, Le DA, Amin-Hanjani S, Wu Y, Qiu J, Chiarugi A, Thomas SS, Kohane DS, Korsmeyer SJ, Moskowitz MA. BID mediates neuronal cell death after oxygen/glucose deprivation and focal cerebral ischemia. Proc Natl Acad Sci USA. 2001;98:15318–15323. doi: 10.1073/pnas.261323298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, Orkin SH, Doerschuk CM, Dinauer MC. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet. 1995;9:202–209. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- Pozzilli C, Lenzi GL, Argentino C, Carolei A, Rasura M, Signore A, Bozzao L, Pozzilli P. Imaging of leukocytic infiltration in human cerebral infarcts. Stroke. 1985;16:251–255. doi: 10.1161/01.str.16.2.251. [DOI] [PubMed] [Google Scholar]
- Proescholdt MG, Chakravarty S, Foster JA, Foti SB, Briley EM, Herkenham M. Intracerebroventricular but not intravenous interleukin-1beta induces widespread vascular-mediated leukocyte infiltration and immune signal mRNA expression followed by brain-wide glial activation. Neuroscience. 2002;112:731–749. doi: 10.1016/s0306-4522(02)00048-9. [DOI] [PubMed] [Google Scholar]
- Reeves MJ, Broderick JP, Frankel M, LaBresh KA, Schwamm L, Moomaw CJ, Weiss P, Katzan I, Arora S, Heinrich JP, Hickenbottom S, Karp H, Malarcher A, Mensah G. The Paul Coverdell National Acute Stroke Registry: initial results from four prototypes. Am J Prev Med. 2006;31:S202–S209. doi: 10.1016/j.amepre.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Rothe G, Valet G. Flow cytometric analysis of respiratory burst activity in phagocytes with hydroethidine and 2′,7′-dichlorofluorescin. J Leukoc Biol. 1990;47:440–448. [PubMed] [Google Scholar]
- Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M. NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med. 1999;5:554–559. doi: 10.1038/8432. [DOI] [PubMed] [Google Scholar]
- Schwaninger M, Inta I, Herrmann O. NF-kappaB signalling in cerebral ischaemia. Biochem Soc Trans. 2006;34:1291–1294. doi: 10.1042/BST0341291. [DOI] [PubMed] [Google Scholar]
- Shie FS, Neely MD, Maezawa I, Wu H, Olson SJ, Jurgens G, Montine KS, Montine TJ. Oxidized low-density lipoprotein is present in astrocytes surrounding cerebral infarcts and stimulates astrocyte interleukin-6 secretion. Am J Pathol. 2004;164:1173–1181. doi: 10.1016/S0002-9440(10)63205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson D, Yin T, Smalstig EB, Hsu MA, Panetta J, Little S, Clemens J. Transcription factor nuclear factor-kappa B is activated in neurons after focal cerebral ischemia. J Cereb Blood Flow Metab. 2000;20:592–603. doi: 10.1097/00004647-200003000-00017. [DOI] [PubMed] [Google Scholar]
- Stuart LM, Deng J, Silver JM, Takahashi K, Tseng AA, Hennessy EJ, Ezekowitz RA, Moore KJ. Response to Staphylococcus aureus requires CD36-mediated phagocytosis triggered by the COOH-terminal cytoplasmic domain. J Cell Biol. 2005;170:477–485. doi: 10.1083/jcb.200501113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sughrue ME, Mehra A, Connolly ES, Jr, D'Ambrosio AL. Anti-adhesion molecule strategies as potential neuroprotective agents in cerebral ischemia: a critical review of the literature. Inflamm Res. 2004;53:497–508. doi: 10.1007/s00011-004-1282-0. [DOI] [PubMed] [Google Scholar]
- Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, Lathia JD, Siler DA, Chigurupati S, Ouyang X, Magnus T, Camandola S, Mattson MP. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci USA. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triantafilou M, Gamper FG, Haston RM, Mouratis MA, Morath S, Hartung T, Triantafilou K. Membrane sorting of toll-like receptor (TLR)-2/6 and TLR2/1 heterodimers at the cell surface determines heterotypic associations with CD36 and intracellular targeting. J Biol Chem. 2006;281:31002–31011. doi: 10.1074/jbc.M602794200. [DOI] [PubMed] [Google Scholar]
- Uno M, Kitazato KT, Nishi K, Itabe H, Nagahiro S. Raised plasma oxidised LDL in acute cerebral infarction. J Neurol Neurosurg Psychiatry. 2003;74:312–316. doi: 10.1136/jnnp.74.3.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vibo R, Korv J, Roose M, Kampus P, Muda P, Zilmer K, Zilmer M. Acute phase proteins and oxidised low-density lipoprotein in association with ischemic stroke subtype, severity and outcome. Free Radic Res. 2007;41:282–287. doi: 10.1080/10715760601083235. [DOI] [PubMed] [Google Scholar]
- Walder CE, Green SP, Darbonne WC, Mathias J, Rae J, Dinauer MC, Curnutte JT, Thomas GR. Ischemic stroke injury is reduced in mice lacking a functional NADPH oxidase. Stroke. 1997;28:2252–2258. doi: 10.1161/01.str.28.11.2252. [DOI] [PubMed] [Google Scholar]
- Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol. 2007;184:53–68. doi: 10.1016/j.jneuroim.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Iadecola C. Infarct measurement methodology. J Cereb Blood Flow Metab. 1994;14:697–698. doi: 10.1038/jcbfm.1994.88. [DOI] [PubMed] [Google Scholar]
- Zhang F, Iadecola C. Temporal characteristics of the protective effect of aminoguanidine on cerebral ischemic damage. Brain Res. 1998;802:104–110. doi: 10.1016/s0006-8993(98)00557-5. [DOI] [PubMed] [Google Scholar]
- Zhang F, Casey RM, Ross ME, Iadecola C. Aminoguanidine ameliorates and L-arginine worsens brain damage from intraluminal middle cerebral artery occlusion. Stroke. 1996a;27:317–323. doi: 10.1161/01.str.27.2.317. [DOI] [PubMed] [Google Scholar]
- Zhang RL, Chopp M, Zhang ZG, Phillips ML, Rosenbloom CL, Cruz R, Manning A. E-selectin in focal cerebral ischemia and reperfusion in the rat. J Cereb Blood Flow Metab. 1996b;16:1126–1136. doi: 10.1097/00004647-199611000-00006. [DOI] [PubMed] [Google Scholar]
- Ziegler G, Harhausen D, Schepers C, Hoffmann O, Rohr C, Prinz V, Konig J, Lehrach H, Nietfeld W, Trendelenburg G. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochem Biophys Res Commun. 2007;359:574–579. doi: 10.1016/j.bbrc.2007.05.157. [DOI] [PubMed] [Google Scholar]