

Abstract
Feline infectious peritonitis (FIP) is a terminal disease of cats caused by systemic infection with a feline coronavirus (FCoV). FCoV biotypes that cause FIP are designated feline infectious peritonitis virus (FIPV), and are distinguished by their ability to infect macrophages and monocytes. Antigenically similar to their virulent counterparts are FCoV biotypes designated feline enteric coronavirus (FECV), which usually cause only mild enteritis and are unable to efficiently infect macrophages and monocytes. The FCoV spike protein mediates viral entry into the host cell and has previously been shown to determine the distinct tropism exhibited by certain isolates of FIPV and FECV, however, the molecular mechanism underlying viral pathogenesis has yet to be determined. Here we show that the FECV strain WSU 79-1683 (FECV-1683) is highly dependent on host cell cathepsin B and cathepsin L activity for entry into the host cell, as well as on the low pH of endocytic compartments. In addition, both cathepsin B and cathepsin L are able to induce a specific cleavage event in the FECV-1683 spike protein. In contrast, host cell entry by the FIPV strains WSU 79-1146 (FIPV-1146) and FIPV-DF2 proceeds independently of cathepsin L activity and low pH, but is still highly dependent on cathepsin B activity. In the case of FIPV-1146 and FIPV-DF2, infection of primary feline monocytes was also dependent on host cell cathepsin B activity, indicating that host cell cathepsins may play a role in the distinct tropisms displayed by different feline coronavirus biotypes.
Keywords: Feline coronavirus, Feline infectious peritonitis, Virus entry, Cathepsin, Endosome, Spike protein
1. Introduction
Coronaviruses are enveloped positive-stranded RNA viruses that replicate in the cytoplasm (Lai and Holmes, 2001, Perlman et al., 2008). They have a distinctive set of club-shaped spikes on their envelope, comprised of the viral spike protein (S). The coronavirus S protein is a primary determinant of cell tropism and pathogenesis, being responsible (and apparently sufficient) for receptor binding and fusion (Gallagher and Buchmeier, 2001). Overall, the coronavirus S protein is categorized as a class I fusion protein (Earp et al., 2005) based on the presence of characteristic heptad repeats (Bosch et al., 2003, Chambers et al., 1990, Kliger and Levanon, 2003); as such it shows characteristic features of fusion proteins of influenza virus (HA), retroviruses (Env) and paramyxoviruses (F) for which there is extensive characterization at a structural and biophysical level (Colman and Lawrence, 2003). Many viral fusion proteins are known to be activated following cleavage by host cell proteases (Klenk and Garten, 1994). This has been most extensively documented for orthomyxo- and paramyxoviruses. In the case of high pathogenicity avian influenza strains, mutations in the region of the cleavage site that modify it from a trypsin-like (monobasic) to a furin-like (polybasic) cleavage site allow systemic spread within the host and hence show drastically increased virulence (Klenk et al., 2008). Although coronaviruses do show structural similarities to canonical class I fusion proteins, they appear to have significant differences in regard to the role of proteolytic cleavage for fusion activation (Bosch and Rottier, 2008).
Coronavirus S proteins consist of two principal domains (S1 and S2), with S1 responsible for receptor binding, and S2 mediating membrane fusion. In group 3 coronaviruses (e.g., infectious bronchitis virus; IBV) there is ubiquitous cleavage of the S precursor protein to form the S1 and S2 subunits (Cavanagh et al., 1986), with cleavage apparently carried out by furin. In group 2 viruses (e.g., for mouse hepatitis virus; MHV), S1/S2 cleavage is variable (Frana et al., 1985). The ability to be cleaved at the S1/S2 boundary has been studied quite extensively for MHV-1 and 4 (Wentworth and Holmes, 2007). While there is some correlation between S1/S2 cleavage and increased cell–cell fusion, in general there appears to be no direct link between syncytia formation (and by analogy S1/S2 protein cleavage) and pathogenicity (Hingley et al., 2002). For the group 2 MHV-2, the S protein remains uncleaved in the virus particle (Qiu et al., 2006). For group 1 viruses (e.g., human coronavirus NL63 and feline coronavirus; FCoV), S1/S2 cleavage is not considered to occur (Vennema et al., 1990), however, recent identification of a group 1 coronavirus with a furin-like motif at S1/S2 (Pratelli et al., 2003) suggests this may not be the case with all group 1 members. Infection of cells by several coronaviruses has been shown to be sensitive to the presence of lysosomotropic agents, such as NH4Cl, which raise the pH of the endosome (Blau and Holmes, 2001, Gallagher et al., 1991, Hansen et al., 1998, Li and Cavanagh, 1990, Li and Cavanagh, 1992, Mizzen et al., 1985). These data suggest a pH-dependent route of coronavirus entry through endosomes. In some cases (e.g., IBV and MHV-4), the coronavirus fusion reaction has been clearly shown to be triggered by low pH (Chu et al., 2006, Eifart et al., 2007), whereas in other cases, e.g., SARS-CoV, it is suggested that the low pH of the endosome is important for activation of cathepsin proteases that prime the fusion event (Simmons et al., 2005), with fusion activation itself considered to be pH-independent.
The classical cathepsins are cysteine proteases typically present within the endosome/lysosome system of host cells (Barrett et al., 2004). While a role for cathepsins during entry of the non-enveloped reoviruses has been known for some time (Ebert et al., 2002, Golden et al., 2004), it has recently become apparent that the fusion proteins from several families of enveloped viruses can be cleaved by these proteases. Such examples are the recently identified paramyxoviruses Hendra and Nipah (Diederich et al., 2005, Pager et al., 2006, Pager and Dutch, 2005), as well as Ebola virus (Chandran et al., 2005, Sanchez, 2007, Schornberg et al., 2006) and Moloney murine leukemia virus (Kumar et al., 2007). In the case of coronaviruses, SARS-CoV and MHV-2 are thought to require cathepsin activation (Huang et al., 2005, Qiu et al., 2006, Simmons et al., 2004, Simmons et al., 2005), with cathepsin L predominantly involved in SARS-CoV entry, and cathepsin B mediating MHV-2 cleavage during virus entry. For SARS-CoV, neither the site of cathepsin L-mediated proteolysis nor the cleaved products produced are known, although for MHV-2 cathepsin B may cleave at the S1/S2 junction (Qiu et al., 2006). Other coronaviruses such as MHV-4, HCoV-NL63 and IBV are not considered to be dependent on cathepsins (Chu et al., 2006, Huang et al., 2005, Qiu et al., 2006).
Feline infectious peritonitis (FIP) is a highly lethal systemic infection of cats caused by a group I feline coronavirus (FCoV) of the FIPV biotype (Haijema et al., 2007, Olsen, 1993). However, under normal circumstances FCoVs cause only mild and often inapparent enteritis; in this case the virus is classified as the feline enteric coronavirus (FECV) biotype (de Groot-Mijnes et al., 2005, Haijema et al., 2007). FCoVs can exist as either serotype I, or less commonly as serotype II, in which case the spike protein share extensive similarity with canine coronavirus (CCoV) and transmissible gastroenteritis virus (TGEV). Due to the relative ease of growth in cell culture, the serotype II FCoVs have been subject to some degree of molecular characterization. In particular, two strains of serotype II FCoV, WSU 79-1683 and WSU 79-1146, have served as model viruses that cause either mild enteritis or FIP, respectively (Pedersen et al., 1984a, Pedersen et al., 1984b). The viruses are thus referred to as FECV-1683 and FIPV-1146. In the “internal mutation” model of FIP it is believed that a process of mutation within an individual cat confers the ability of a FECV to infect macrophages and monocytes, and so allow systemic spread and become a virus that causes FIP (Vennema et al., 1998). Despite extensive study, the genomic differences that determine whether an FCoV will behave as an FECV or an FIPV are still largely unknown (Haijema et al., 2007). However, in the case of the serotype II FCoVs, mapping studies between the FECV-1683 and FIPV-1146 spike proteins have determined critical domains involved in the switch to FIPV. Surprisingly, the S1 domain (receptor-binding) domain is not important, and the critical region was mapped to the C-terminal part of the S2 (fusion) domain (Rottier et al., 2005).
In light of the recently determined involvement of cathepsins in activation of other coronavirus S proteins, we reasoned that the FCoV spike protein might be a target of cathepsins during virus entry, which might explain some of the unusual pathogenic properties of FIPV. Here we examined the entry pathway of feline coronaviruses and show a differential role for low pH and spike protein cleavage by cathepsin B and cathepsin L.
2. Materials and methods
2.1. Cell lines and primary feline monocytes
Crandell–Rees feline kidney (CRFK) cells and feline fetal lung cells (AK-D) were obtained from the American Type Culture Collection (ATCC). A-72 cells were provided by Dr. Colin Parrish (Cornell University, Baker Institute for Animal Health). Fc2Lu cells were provided by Dr. Ed Dubovi (Animal Health Diagnostic Center, New York State College of Veterinary Medicine, Cornell University). Primary feline monocytes were individually purified from the blood of three SPF cats (Liberty Research, Inc., Waverly, NY) using a standard Ficoll-paque gradient (GE Healthcare) as specified by the manufacturer. Monocytes were seeded in 24-well plates and allowed to attach to culture-treated glass coverslips overnight. After washing, the purity of monocyte preparations was checked by immunofluorescence microscopy using the monocyte marker DH59B. CRFK, Fc2Lu, AK-D and A-72 cells were grown in the presence of 5% CO2 at 37 °C in RPMI-1640 media pH 7.4 supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, 100 U/ml penicillin and 10 μg/ml streptomycin. Monocytes were cultured under the same conditions except the media was supplemented with 20% FBS.
2.2. Viruses and antibodies
FIPV WSU 79-1146 (FIPV-1146) was obtained from the ATCC. FECV WSU 79-1683 (FECV-1683) and FIPV-DF2 were provided by Dr. Ed Dubovi (Animal Health Diagnostic Center, New York State College of Veterinary Medicine, Cornell University). FIPV-1146 and FECV-1683 were grown in A-72 cells and FIPV-DF2 was grown in CRFK cells, and supernatant collected after CPE was observed in 80% of cells. Supernatant was clarified by a low speed centrifugation step (1250 × g for 10 min) and viral particles were then pelleted by centrifugation at 28,000 rpm in a SW28 rotor (Sorvall) for 60 min. Pellets were resuspended in either phosphate-buffered saline pH 7.4 for infection assays, or acetate buffer pH 5.0 for cathepsin cleavage assays. Virus titers were determined with TCID50 assays on CRFK cells, using standard techniques (Gray, 1999). Anti-FCoV nucleocapsid (17B7.1) (Olsen et al., 1992) and spike (22G6.4) monoclonal antibodies were provided by Dr. Ed Dubovi (Animal Health Diagnostic Center, New York State College of Veterinary Medicine, Cornell University). The monocyte marker antibody (anti-CD127a mAb DH59B) was obtained from Veterinary Medical Research & Development, Inc. (Pullman, WA).
2.3. Inhibitors
The cysteine protease inhibitor (2S,3S)-trans-epoxysuccinyl-l-leucylamido-3-methylbutane ethyl ester loxistatin (E-64d), the cathepsin B inhibitor [l-3-trans-(propylcarbamoyl)oxirane-2-carbonyl]-l-isoleucyl-l-proline methyl ester (CA074-Me), the cathepsin K inhibitor 1-(N-benzyloxycarbonyl-leucyl)-5-(N-Boc-phenylalanyl-leucyl)carbohydrazide (Boc-l) and bafilomycin A1 were purchased Calbiochem (San Diego, CA). The cathepsin L inhibitor Z-Phe-Tyr(t-Bu)-diazomethylketone (Z-FY-(t-Bu)-DMK) was purchased from Axxora (San Diego, CA).
2.4. Growth and infection assays
For growth assays, A-72 cells were seeded to 75% confluency on 24-well tissue culture-treated plates. Cells were pre-incubated with specified treatments in serum-free media for 60 min before addition of virus to a final concentration of 106 TCID50/ml. After 3 h, cells were washed and media replaced with fresh serum-free media. After 9 h, supernatant was collected and frozen at −80 °C until later determination of viral titer by TCID50 assay. For infection assays, cells were seeded to 75% confluency on 24-well plates with tissue culture-treated glass coverslips. Cells were treated with inhibitors as specified, either 60 min before, or 60 min after the addition of virus. Virus was added to a final concentration of 106 TCID50/ml, which infected 20–50% of control cells under the conditions tested. 6 h post-infection, cells were fixed in 3% paraformaldehyde and stained with the anti-FCoV nucleocapsid mAb 17B7.1, essentially as described previously (Chu et al., 2006). Influenza virus and Sendai virus infection assays were performed as previously described (Chu et al., 2006). All assays were repeated at least three times and 8–15 widefield images were captured and quantified per experimental replicate. Cells were viewed on a Nikon Eclipse E600 fluorescence microscope, and images were captured with a Sensicam EM camera and IPLab software before transfer into ImageJ software (http://rsb.info.nih.gov/ij/) for determination of infection frequency.
2.5. Cleavage assays and Western blot analysis
Concentrated viral particles (1012 TCID50/ml) were suspended in acetate buffer pH 5.0. 50 μl of virus preparation was incubated with either purified cathepsin L (Calbiochem, San Diego, CA) or purified cathepsin B (Calbiochem, San Diego, CA) at a final concentration of 1 μM for 1 h at 37 °C, followed by the addition of 500 U PNGase F (New England Biolabs, Ipswich, MA) to allow differentiation of any multiply glycosylated species. SDS sample buffer was added and the reaction was heated at 95 °C before separation using a 4–20% SDS-PAGE gel at 200 V for 2 h. Gels were electroblotted to PVDF membrane, blocked with 5% bovine serum albumin and probed with the anti-FCoV spike protein mAb 22G6.4. Membranes were developed using anti-mouse antibody linked to horseradish peroxidase (SouthernBiotech, Birmingham, AL) and ECL substrate (Pierce, Rockford, IL) and images captured using a Fujifilm LAS-3000 CCD camera. Western blot densitometry analysis of spike protein cleavage was performed using ImageJ software. Percent cleavage was determined by dividing pixel intensity of the cleaved spike protein band by total pixel intensity of the cleaved and uncleaved spike protein bands combined.
3. Results
3.1. Effect of cysteine proteases on the propagation of FIPV-1146 and FECV-1683 in vitro
To determine the role of cysteine proteases in the life cycle of FIPV-1146 and FECV-1683, we first pre-treated A-72 cells with the cell-permeable irreversible cysteine protease inhibitor E-64d, which non-selectively inhibits a range of cysteine proteases. Cells were then infected with either FIPV-1146 or FECV-1683. As a control, cells were infected with influenza virus, which is not believed to require proteolytic activation by cysteine proteases during entry. We collected cell supernatants at 9 h post-infection and assayed virus production by TCID50 assay. Pre-treatment with E-64d significantly reduced the production of both FIPV-1146 and FECV-1683 viral particles, as compared to control cells pre-treated with DMSO alone (Fig. 1 ). As expected, E-64d had no effect on influenza virus infection.
Fig. 1.

The effect of cysteine protease inhibitors on the growth of FIPV-1146 and FECV-1683. A-72 cells were pre-treated with specified inhibitors at a concentration of 10 μM and then infected with virus. 3 h post-infection, fresh media without inhibitor was added to cells and 9 h post-infection media was collected. Production of extracellular virus from at least three independent experiments was determined by TCID50 assay. Error bars represent the standard deviation of the mean.
To further investigate the potential roles of cathepsins in the growth of FIPV-1146 and FECV-1683, cells were then pre-treated with specific inhibitors of cathepsin B (CA074-Me), cathepsin L (Z-FY-(t-Bu)-DMK) and cathepsin K (Boc1). As shown in Fig. 1, the cathepsin B inhibitor considerably limited the production of both FIPV-1146 and FECV-1683. In contrast, the cathepsin L inhibitor had only a negligible effect on FIPV-1146; however, it greatly diminished the propagation of FECV-1683. Cathepsin K inhibitor had no significant effect on either virus (Fig. 1). The specific cathepsin inhibitors also had no effect on influenza virus infection. These data indicate that both FIPV-1146 and FECV-1683 require cathepsin B activity for propagation, whereas FECV-1683 selectively requires cathepsin L activity.
3.2. Role of cathepsin B and L during entry of FIPV-1146 and FECV-1683 into host cells
To establish if the effects of the cathepsin B and L inhibitors occurred during virus entry, A-72 cells were treated with five log dilutions of inhibitor either 60 min pre-infection, or 60 min post-infection, and infected with FIPV-1146 or FECV-1683. Cells were fixed 6 h post-infection, and infected cells were detected by immunofluorescence microscopy using the anti-FCoV nucleocapsid monoclonal antibody 17B7.1, and quantified (Fig. 2 ). Pre-incubation with the specific cathepsin B inhibitor almost completely blocked infection by FIPV-1146, however, treatment after entry had no effect (Fig. 2). Consistent with data from viral growth assays, the cathepsin L inhibitor had no effect on FIPV-1146 entry, except for some limited effect at the highest concentration tested (Fig. 2B). Pre-incubation with either cathepsin B or cathepsin L inhibitor both potently blocked infection by FECV-1683, and again treatment after entry with either inhibitor had no effect. As a control, cells were treated with inhibitors and infected with influenza virus. Infected cells were fixed 6 h post-infection and detected by immunofluorescence microscopy with the anti-influenza nucleoprotein monoclonal antibody H19 (Fig. 2). Neither the cathepsin B nor the cathepsin L inhibitor had an effect on influenza virus entry. Pre-treatment with the cathepsin K inhibitor had no effect on the cellular entry of either FIPV-1146, FECV-1683 or influenza virus (data not shown). These data show that the inhibitory effects of the cathepsin inhibitors are manifested during virus entry.
Fig. 2.
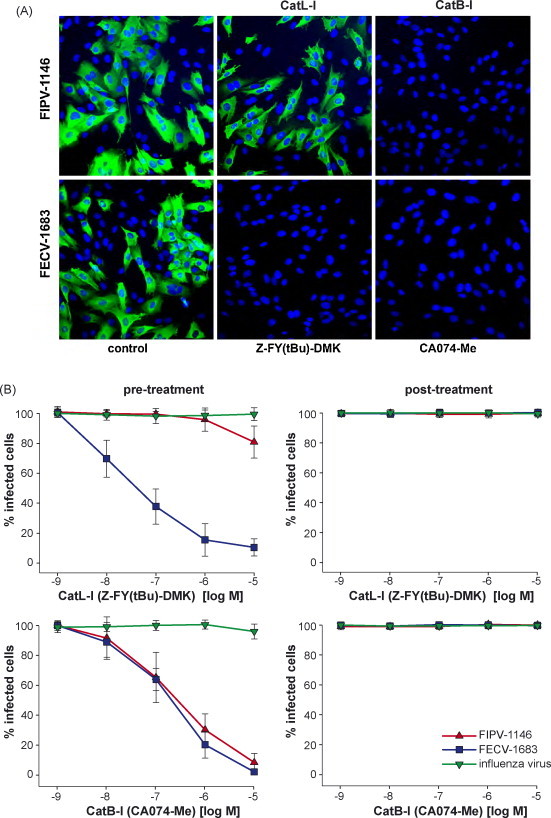
The effect of cathepsin L and cathepsin B inhibitors on the entry of FIPV-1146 and FECV-1683. A-72 cells were pre-treated with the cathepsin L inhibitor Z-FY-(t-Bu)-DMK (CatL-I) or the cathepsin B inhibitor CA074-Me (CatB-I) at the concentration specified, and then infected with virus. 6 h post-infection cells were fixed and stained for immunofluorescence microscopy with the anti-FCoV nucleocapsid mAb 17B7.1 (A). Images from at least three independent experiments were processed and quantified (B). To control for non-specific effects on replication, cells were also treated with inhibitors 1 h post-infection. For quantification, >1000 cells were scored from three independent replicates of each experimental condition. Error bars represent the standard deviation of the mean.
3.3. Effect of lysosomotropic agents on the cellular entry of FIPV-1146 and FECV-1683
Lysosomotropic agents such as NH4Cl and bafilomycin A are well established to disrupt the acidic environment of endocytic compartments. To assess the role of low pH during entry of FIPV-1146 and FECV-1683, A-72 cells were pre-treated with either NH4Cl or bafilomycin A1. We first assessed the effect of NH4Cl, a weak base that acts to neutralize the low pH of the endosome. Pre-treatment with 10 mM NH4Cl only partially inhibited entry of FIPV-1146 (approximately 20% inhibition), however, the same concentration almost completely blocked entry by FECV-1683 (Fig. 3 ). Even following pre-treatment with 30 mM NH4Cl, FIPV-1146 entry was only partially blocked as compared to FECV-1683. As controls for these experiments, we used influenza virus and Sendai virus. Influenza virus is well established to require low pH to trigger its fusion mechanism and was completely inhibited by pre-treatment with 10 mM NH4Cl, analogous to the results obtained with FECV-1683 (Fig. 3). Sendai virus is known to fuse at a neutral pH and accordingly showed no reduction in entry with cell pre-treated with NH4Cl (Fig. 3).
Fig. 3.
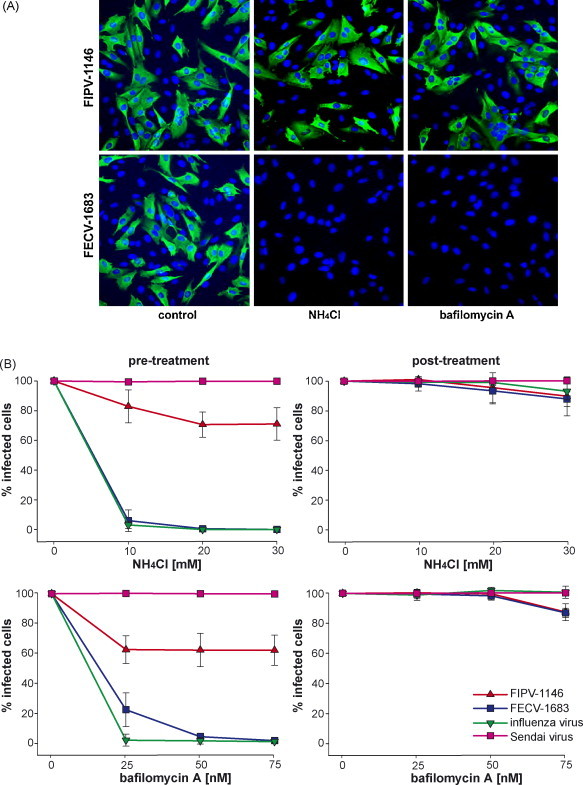
The effect of lysosomotropic agents on the entry of FIPV-1146 and FECV-1683. A-72 cells were pre-treated with NH4Cl or bafilomycin A at the specified concentration and then infected with virus. 6 h post-infection cells were fixed and stained for immunofluorescence microscopy with the anti-FCoV nucleocapsid mAb 17B7.1 (A). Images from at least three independent experiments were processed and quantified (B). To control for non-specific effects on replication, cells were also treated with lysomotrophic agents 1 h post-infection. For quantification, >1000 cells were scored from three independent replicates of each experimental condition. Error bars represent the standard deviation of the mean.
We next assessed the effect of bafilomycin A1, an inhibitor of the vacuolar-type H+-ATPase. Pre-treatment of cells with 50 nM bafilomycin A1 completely blocked entry of both FECV-1683 and influenza virus, but only partially blocked entry by FIPV-1146 (approximately 40% inhibition). Sendai virus entry was unaffected by bafilomycin A1 at the concentrations tested. Treatment after entry with both NH4Cl and bafilomycin A1 had a minimal effect on FIPV-1146 and FECV-1683 at the concentrations shown in Fig. 3, however, higher concentrations (>100 nM bafilomycin A1 and >30 mM NH4Cl) were able to significantly reduce infectivity of both viruses (data not shown). Post-treatment with bafilomycin had no effect on influenza or Sendai virus infection at the concentrations shown in Fig. 3. Overall these data show that FECV-1683 entry is highly dependent on low endosomal pH, whereas FIPV-1146 is much less dependent on low pH during entry.
3.4. Cathepsins and low pH play a general role for serotype II feline coronavirus infection of multiple cell types
To determine whether the effects of low pH and cathepsin inhibitors were more generally applicable to serotype II coronavirus infections, we repeated key experiments using an additional feline coronavirus strain (FIPV-DF2), and also tested a variety of established feline cell lines (CRFK, AK-D and Fc2Lu). As shown in Fig. 4 , the results were consistent with our previous data (see Fig. 1, Fig. 2). The cathepsin B inhibitor CA074-Me almost completely prevented infection of all the feline coronaviruses tested, whereas the cathepsin L inhibitor Z-FY-(t-Bu)-DMK, selectively inhibited FECV-1683, but showed little or no effect on infection by FIPV-1146 or FIPV-DF2. Likewise, the weak base NH4Cl selectively inhibited FECV-1683 in all the cell lines tested, but showed only a marginal effect on the FIPV biotypes.
Fig. 4.
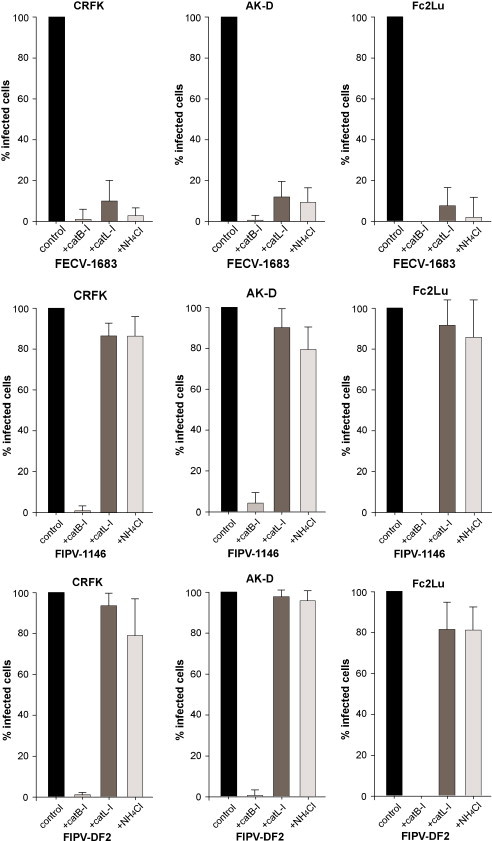
The effects of cathepsin L and cathepsin B inhibitors, and low pH, on the infection of feline cells by serotype II FCoVs. CRFK, AK-D or Fc2Lu cells were pre-treated with either 10 μM cathepsin B inhibitor CA074-Me (CatB-I), 10 μM cathepsin L inhibitor Z-FY-(t-Bu)-DMK (CatL-I), or 10 mM NH4Cl, and then infected with virus. 6 h post-infection cells were fixed and stained for immunfluorescence microscopy with the anti-FCoV nucleocapsid mAb 17B7.1. Images from at least three independent experiments were processed and quantified. For quantification >500 cells were scored from three independent replicates. Error bars represent the standard deviation of the mean.
3.5. Cleavage of the FIPV-1146 and FECV-1683 spike protein by cathepsin B and cathepsin L
To investigate whether the effect of cathepsins B and L occurred at the level of spike protein cleavage, concentrated FIPV-1146 and FECV-1683 viral particles were incubated with purified, activated cathepsin B and cathepsin L for 1 h at 37 °C, and then analyzed by Western blot with the anti-FCoV monoclonal antibody 22G6.4, specific for the S protein. As shown in Fig. 5 , cathepsin L was unable to cleave the spike protein of FIPV-1146 under the conditions tested, but was clearly able to cleave the FECV-1683 spike protein, with an obvious cleavage product of approximately 150–160 kDa. In contrast, cathepsin B was able to cleave both FIPV-1146 and FECV-1683 spike proteins, also producing a cleavage product of approximately 150–160 kDa (Fig. 5). The samples shown in Fig. 5 were treated with the endoglycosidase PNGase F, which resulted in a clearer representation of the cleaved product by Western blot, compared to samples without PNGaseF pre-treatment, most likely due to heterogenous glycosylation of the spike protein in samples without the glycosidase (data not shown). Overall, these data indicate that cathepsin L selectively cleaves the S protein of FECV-1683, whereas cathepsin B can cleave the S protein of both FECV-1683 and FIPV-1146.
Fig. 5.
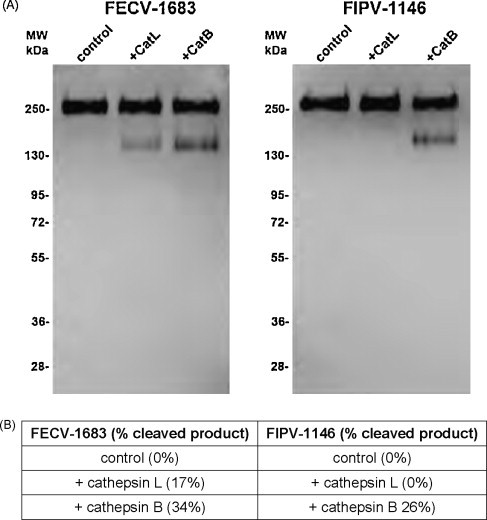
Cleavage of FIPV-1146 and FECV-1683 by purified cathepsins. Concentrated virus preparations were incubated with purified cathepsin B, cathepsin L, or buffer alone for 1 h at 37 °C, followed by treatment with PNGase F. (A) Samples were then analyzed by western blot with the anti-FCoV spike mAb 22G6.4. (B) Quantification of western blot to show degree of cleavage.
3.6. Effect of cathepsins B and L on the infection of serotype II feline coronavirus into primary feline blood monocytes
The in vivo targets of the highly virulent coronaviruses FIPV-1146 and FIPV-DF2 include feline monocytes and macrophages. In order to assess whether cathepsins B and L might play a role in the entry of FIPV-1146 and FIPV-DF2 into its in vivo target, we isolated CD172a-positive, primary feline blood monocytes, pre-treated the cells with the cathepsin B inhibitor CA074-Me, or the cathepsin L inhibitor Z-FY-(t-Bu)-DMK, and then infected them with FIPV-1146 or FIPV-DF2. As shown in Fig. 6 , the cathepsin B inhibitor significantly reduced infection by FIPV-1146 and FIPV-DF2 (approximately 85–95% inhibition), however, the block was not as complete as observed in cell lines (see Fig. 2, Fig. 4). In line with our data using cell lines, the cathepsin L inhibitor showed no significant effect on FIPV infection of primary feline monocytes. As expected (Dewerchin et al., 2005), primary feline blood monocytes were only negligibly susceptible to infection with FECV-1683, hence a role for cathepsin B or L cannot be determined in this case (Fig. 6). These data indicate that cathepsin B-mediated cleavage of FIPV-1146 and FIPV-DF2 may play an important role in cells infected in vivo during the course of FIP.
Fig. 6.

The effect of cathepsin inhibitors on the infection of primary feline blood monocytes. Monocytes were purified from the blood of SPF cats, pre-treated with the cathepsin B inhibitor CA074-Me (CatB-I) or the cathepsin L inhibitor Z-FY-(t-Bu)-DMK (CatL-I) at a concentration 10 μM, and infected with virus. 12 h post-infection, cells were fixed and stained for immunofluorescence microscopy with the anti-FCoV nucleocapsid mAb 17B7.1 (A). Images from at least three independent experiments were processed and quantified (B). For quantification, >200 cells were scored from three independent replicates of each experimental condition. Error bars represent the standard deviation of the mean.
4. Discussion
We show here the effects of low pH and cathepsin cleavage of the serotype II feline coronavirus spike protein, with a differential effect of low pH, and cathepsin B versus cathepsin L cleavage for the FECV and FIPV biotypes. The two biotypes are genotypically very similar and the strains FECV-1683 and FIPV-1146, in particular, are known to share a high degree of amino acid identity in their spike protein (>95%) yet cause radically different clinical outcomes—lethal vasculitis for FIPV-1146, compared to mild enteritis for FECV-1683 (Haijema et al., 2007). Previous studies have shown that the S2 domain of the viral spike protein is a critical discriminating factor in FECV-1683 and FIPV-1146 pathogenesis (Rottier et al., 2005). The studies presented here raise the possibility that differential proteolysis of the spike (S2) protein might be an important factor in explaining the different pathogenic properties of these viruses.
At present, we cannot definitely determine the exact point in the FCoV infectious cycle that cathepsin cleavage would occur. However, the in vivo localization of cathepsins to the endosome/lysosome system, together with absence of any obvious cleaved product in purified extracellular FCoV particles unless exogenous activated cathepsin is added, would strongly suggest that cleavage occurs in the endosome during virus entry. In support of this, we find that addition of cathepsin inhibitors after virus entry has occurred has no detectable effect on viral replication. While the use of feline aminopeptidase N (fAPN) as a receptor for serotype II feline coronaviruses is well established (Tresnan et al., 1996, Tusell et al., 2007, Van Hamme et al., 2007), many other aspects of the entry pathway have not been explored in great detail for these viruses. Based on the effects of lysosomotropic agents shown here, we suggest that all serotype II feline coronaviruses enter the endocytic pathway for virus entry, where they are primed for fusion activation by cathepsin cleavage, but that FECV-1683 is much more dependent on endosomal function, suggesting that FIPV-1146 and FIPV-DF2 may escape into the cytosol earlier in the endocytic pathway. Possibilities that might account for this are the differential cleavage of the FIPV spike protein by cathepsin L, or alternatively a lower threshold for any necessary low pH-dependent conformational changes for FIPV spike. It remains to be determined whether a similar situation with cathepsin cleavage exists for the serotype I FCoVs, which are not considered to share the same receptor as FECV-1683 and FIPV-1146 (Dye et al., 2007).
In general, the cleavage site of cathepsins is difficult to predict based on sequence specificity, and so we cannot at present determine specifically where in the spike protein cathepsin B or L might cleave. However, based on the single 150–160 kDa cleaved product seen in Fig. 5 for both cathepsin B and L, it does appear that the spike protein is subject to single, distinct cleavage event for both proteases, most likely occurring at the same approximate position in S. We used the monoclonal antibody 22G6.4 to show this specific cleavage event, and while the epitope recognized by 22G6.4 is not known, by analogy to the many other S-specific FCoV monoclonal antibodies that have epitopes in S1 (Corapi et al., 1995, Olsen et al., 1993), we tentatively suggest that the 22G6.4-reactive epitope is also most likely to be within the S1 domain. Based on the cleavage pattern shown in Fig. 5., it does not appear that cleavage occurs at the S1/S2 boundary, but rather, the 150–160 kDa cleavage products observed may represent cleavage within the C-terminal part of S2. As the C-terminal part of FCoV S2 has been previously considered to specify the pathogenic properties of FIPV (Rottier et al., 2005), it will be interesting to determine if any of the specific mutations in the FIPV-1146 spike can account for the differential cathepsin-sensitivity observed here.
Interestingly, treatment with cathepsins L or B resulted in only a fraction of the spike protein being cleaved (approximately 17–34% cleaved depending on the cathepsin/virus combination; see Fig. 5B). This may indicate that the cleavage site is relatively inaccessible, and that only a small number of spike proteins are cleaved during virus entry. In this regard, it is noteworthy that in many cases cleavage activation of a viral fusion protein does not necessarily occur on all copies of the protein; for example see Michalski et al. (2000). Our data also indicate that for FECV-1683 (which is cleaved by both cathepsin B and L), cleavage occurs to a greater degree than with cathepsin B (see Fig. 5). These data are consistent with inhibitor treatments where cathepsin B inhibitor had a greater effect on virus infection than cathepsin L inhibitor. Thus we consider that cathepsin B may be the more important protease mediating FCoV entry, with cathepsin L possibly having a more secondary role.
Cysteine proteases, such as cathepsins B, L, K and S, are emerging as therapeutic targets for a range of diseases, including cancer, osteoarthritis and autoimmune disorders, with some candidate drugs currently in clinical trials (Turk and Guncar, 2003, Vasiljeva et al., 2007). Cathepsin inhibitors also show promise as drugs targeting virus entry (Vasiljeva et al., 2007). Our results show a clear role for cathepsin B during entry of serotype II feline coronaviruses, raising the possibility that cathepsin B inhibitors may be effective therapeutics to treat this incurable and lethal affliction of cats.
Acknowledgements
We thank Fred Scott, Joel Baines and Sandrine Belouzard for helpful advice and discussions during the course of this work, and Ed Dubovi for kind provision of reagents. We also thank A. Damon Ferguson for technical assistance. A.D.R. was supported by grant T32AI007618 (Training in Molecular Virology and Pathogenesis) from the National Institutes of Health. This work was supported by a research grant from the Winn Feline Foundation.
References
- Barrett A.J., Rawlings N.D., Woessner J.F. Elsevier Academic Press; London: 2004. Handbook of Proteolytic Enzymes. [Google Scholar]
- Blau D.M., Holmes K.V. Human coronavirus HCoV-229E enters susceptible cells via the endocytic pathway. Adv. Exp. Med. Biol. 2001;494:193–198. doi: 10.1007/978-1-4615-1325-4_31. [DOI] [PubMed] [Google Scholar]
- Bosch B.J., Rottier P.J. Nidovirus entry into cells. In: Perlman S., Gallagher T., Snijder E.J., editors. Nidoviruses. ASM Press; Washington, DC: 2008. pp. 157–178. [Google Scholar]
- Bosch B.J., van der Zee R., de Haan C.A., Rottier P.J. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J. Virol. 2003;77:8801–8811. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D., Davis P.J., Pappin D.J., Binns M.M., Boursnell M.E., Brown T.D. Coronavirus IBV: partial amino terminal sequencing of spike polypeptide S2 identifies the sequence Arg-Arg-Phe-Arg-Arg at the cleavage site of the spike precursor propolypeptide of IBV strains Beaudette and M41. Virus Res. 1986;4:133–143. doi: 10.1016/0168-1702(86)90037-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers P., Pringle C.R., Easton A.J. Heptad repeat sequences are located adjacent to hydrophobic regions in several types of virus fusion glycoproteins. J. Gen. Virol. 1990;71(Pt 12):3075–3080. doi: 10.1099/0022-1317-71-12-3075. [DOI] [PubMed] [Google Scholar]
- Chandran K., Sullivan N.J., Felbor U., Whelan S.P., Cunningham J.M. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science. 2005;308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu V.C., McElroy L.J., Chu V., Bauman B.E., Whittaker G.R. The avian coronavirus infectious bronchitis virus undergoes direct low-pH-dependent fusion activation during entry into host cells. J. Virol. 2006;80:3180–3188. doi: 10.1128/JVI.80.7.3180-3188.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman P.M., Lawrence M.C. The structural biology of type I viral membrane fusion. Nat. Rev. Mol. Cell Biol. 2003;4:309–319. doi: 10.1038/nrm1076. [DOI] [PubMed] [Google Scholar]
- Corapi W.V., Darteil R.J., Audonnet J.C., Chappuis G.E. Localization of antigenic sites of the S glycoprotein of feline infectious peritonitis virus involved in neutralization and antibody-dependent enhancement. J. Virol. 1995;69:2858–2862. doi: 10.1128/jvi.69.5.2858-2862.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot-Mijnes J.D., van Dun J.M., van der Most R.G., de Groot R.J. Natural history of a recurrent feline coronavirus infection and the role of cellular immunity in survival and disease. J. Virol. 2005;79:1036–1044. doi: 10.1128/JVI.79.2.1036-1044.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewerchin H.L., Cornelissen E., Nauwynck H.J. Replication of feline coronaviruses in peripheral blood monocytes. Arch. Virol. 2005;150:2483–2500. doi: 10.1007/s00705-005-0598-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederich S., Moll M., Klenk H.D., Maisner A. The nipah virus fusion protein is cleaved within the endosomal compartment. J. Biol. Chem. 2005;280:29899–29903. doi: 10.1074/jbc.M504598200. [DOI] [PubMed] [Google Scholar]
- Dye C., Temperton N., Siddell S.G. Type I feline coronavirus spike glycoprotein fails to recognize aminopeptidase N as a functional receptor on feline cell lines. J. Gen. Virol. 2007;88:1753–1760. doi: 10.1099/vir.0.82666-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earp L.J., Delos S.E., Park H.E., White J.M. The many mechanisms of viral membrane fusion proteins. Curr. Top. Microbiol. Immunol. 2005;285:25–66. doi: 10.1007/3-540-26764-6_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert D.H., Deussing J., Peters C., Dermody T.S. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J. Biol. Chem. 2002;277:24609–24617. doi: 10.1074/jbc.M201107200. [DOI] [PubMed] [Google Scholar]
- Eifart P., Ludwig K., Bottcher C., de Haan C.A., Rottier P.J., Korte T., Herrmann A. Role of endocytosis and low pH in murine hepatitis virus strain A59 cell entry. J. Virol. 2007;81:10758–10768. doi: 10.1128/JVI.00725-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frana M.F., Behnke J.N., Sturman L.S., Holmes K.V. Proteolytic cleavage of the E2 glycoprotein of murine coronavirus: host-dependent differences in proteolytic cleavage and cell fusion. J. Virol. 1985;56:912–920. doi: 10.1128/jvi.56.3.912-920.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher T.M., Buchmeier M.J. Coronavirus spike proteins in viral entry and pathogenesis. Virology. 2001;279:371–374. doi: 10.1006/viro.2000.0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher T.M., Escarmis C., Buchmeier M.J. Alteration of the pH dependence of coronavirus-induced cell fusion: effect of mutations in the spike glycoprotein. J. Virol. 1991;65:1916–1928. doi: 10.1128/jvi.65.4.1916-1928.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden J.W., Bahe J.A., Lucas W.T., Nibert M.L., Schiff L.A. Cathepsin S supports acid-independent infection by some reoviruses. J. Biol. Chem. 2004;279:8547–8557. doi: 10.1074/jbc.M309758200. [DOI] [PubMed] [Google Scholar]
- Gray J. Assays for virus infection. In: Cann A.J., editor. Virus Culture. Oxford University Press; Oxford: 1999. pp. 81–109. [Google Scholar]
- Haijema B.J., Rottier P.J., de Groot R.J. Feline coronaviruses: a tale of two-faced types. In: Thiel V., editor. Coronaviruses. Molecular and Cellular Biology. Caister Academic Press; Norfolk, UK: 2007. pp. 183–203. [Google Scholar]
- Hansen G.H., Delmas B., Besnardeau L., Vogel L.K., Laude H., Sjostrom H., Noren O. The coronavirus transmissible gastroenteritis virus causes infection after receptor-mediated endocytosis and acid-dependent fusion with an intracellular compartment. J. Virol. 1998;72:527–534. doi: 10.1128/jvi.72.1.527-534.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingley S.T., Leparc-Goffart I., Seo S.H., Tsai J.C., Weiss S.R. The virulence of mouse hepatitis virus strain A59 is not dependent on efficient spike protein cleavage and cell-to-cell fusion. J. Neurovirol. 2002;8:400–410. doi: 10.1080/13550280260422703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang I.C., Bosch B.J., Li F., Li W., Lee K.H., Ghiran S., Vasilieva N., Dermody T.S., Harrison S.C., Dormitzer P.R., Farzan M., Rottier P.J., Choe H. SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J. Biol. Chem. 2005;10:3198–3203. doi: 10.1074/jbc.M508381200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenk H.-D., Garten W. Activation cleavage of viral spike proteins by host proteases. In: Wimmer E., editor. Cellular Receptors for Animal Viruses. Cold Spring Harbor Press; Cold Spring Harbor, NY: 1994. pp. 241–280. [Google Scholar]
- Klenk H.-D., Matrosovich M., Stech J. Avian influenza: molecular mechansims of pathogenesis. In: Mettenleiter T.C., Sobrino F., editors. Animal Viruses: Molecular Biology. Caister Academic Press; Norfolk, UK: 2008. pp. 253–303. [Google Scholar]
- Kliger Y., Levanon E.Y. Cloaked similarity between HIV-1 and SARS-CoV suggests an anti-SARS strategy. BMC Microbiol. 2003;3:20. doi: 10.1186/1471-2180-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P., Nachagari D., Fields C., Franks J., Albritton L.M. Host cell cathepsins potentiate Moloney murine leukemia virus infection. J. Virol. 2007;81:10506–10514. doi: 10.1128/JVI.02853-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M.C., Holmes K.V. Coronaviridae: the viruses and their replication. In: Knipe D.M., Howely P.M., editors. Fields Virology. Lippincott Wilkins and Williams; Philadelphia: 2001. [Google Scholar]
- Li D., Cavanagh D. Role of pH in syncytium induction and genome uncoating of avian infectious bronchitis coronavirus (IBV) Adv. Exp. Med. Biol. 1990;276:33–36. doi: 10.1007/978-1-4684-5823-7_5. [DOI] [PubMed] [Google Scholar]
- Li D., Cavanagh D. Coronavirus IBV-induced membrane fusion occurs at near-neutral pH. Arch. Virol. 1992;122:307–316. doi: 10.1007/BF01317192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski W.P., Crameri G., Wang L., Shiell B.J., Eaton B. The cleavage activation and sites of glycosylation in the fusion protein of Hendra virus. Virus Res. 2000;69:83–93. doi: 10.1016/s0168-1702(00)00169-6. [DOI] [PubMed] [Google Scholar]
- Mizzen L., Hilton A., Cheley S., Anderson R. Attenuation of murine coronavirus infection by ammonium chloride. Virology. 1985;142:378–388. doi: 10.1016/0042-6822(85)90345-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen C.W. A review of feline infectious peritonitis virus: molecular biology, immunopathogenesis, clinical aspects, and vaccination. Vet. Microbiol. 1993;36:1–37. doi: 10.1016/0378-1135(93)90126-R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen C.W., Corapi W.V., Ngichabe C.K., Baines J.D., Scott F.W. Monoclonal antibodies to the spike protein of feline infectious peritonitis virus mediate antibody-dependent enhancement of infection of feline macrophages. J. Virol. 1992;66:956–965. doi: 10.1128/jvi.66.2.956-965.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen C.W., Corapi W.V., Jacobson R.H., Simkins R.A., Saif L.J., Scott F.W. Identification of antigenic sites mediating antibody-dependent enhancement of feline infectious peritonitis virus infectivity. J. Gen. Virol. 1993;74(Pt 4):745–749. doi: 10.1099/0022-1317-74-4-745. [DOI] [PubMed] [Google Scholar]
- Pager C.T., Dutch R.E. Cathepsin L is involved in proteolytic processing of the Hendra virus fusion protein. J. Virol. 2005;79:12714–12720. doi: 10.1128/JVI.79.20.12714-12720.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pager C.T., Craft W.W., Jr., Patch J., Dutch R.E. A mature and fusogenic form of the Nipah virus fusion protein requires proteolytic processing by cathepsin L. Virology. 2006;346:251–257. doi: 10.1016/j.virol.2006.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen N.C., Black J.W., Boyle J.F., Evermann J.F., McKeirnan A.J., Ott R.L. Pathogenic differences between various feline coronavirus isolates. Adv. Exp. Med. Biol. 1984;173:365–380. doi: 10.1007/978-1-4615-9373-7_36. [DOI] [PubMed] [Google Scholar]
- Pedersen N.C., Evermann J.F., McKeirnan A.J., Ott R.L. Pathogenicity studies of feline coronavirus isolates 79-1146 and 79-1683. Am. J. Vet. Res. 1984;45:2580–2585. [PubMed] [Google Scholar]
- Perlman S., Gallagher T., Snijder E.J. ASM Press; Washington, DC: 2008. Nidoviruses. [Google Scholar]
- Pratelli A., Martella V., Decaro N., Tinelli A., Camero M., Cirone F., Elia G., Cavalli A., Corrente M., Greco G., Buonavoglia D., Gentile M., Tempesta M., Buonavoglia C. Genetic diversity of a canine coronavirus detected in pups with diarrhoea in Italy. J. Virol. Methods. 2003;110:9–17. doi: 10.1016/S0166-0934(03)00081-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z., Hingley S.T., Simmons G., Yu C., Das Sarma J., Bates P., Weiss S.R. Endosomal proteolysis by cathepsins is necessary for murine coronavirus mouse hepatitis virus type 2 spike-mediated entry. J. Virol. 2006;80:5768–5776. doi: 10.1128/JVI.00442-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottier P.J., Nakamura K., Schellen P., Volders H., Haijema B.J. Acquisition of macrophage tropism during the pathogenesis of feline infectious peritonitis is determined by mutations in the feline coronavirus spike protein. J. Virol. 2005;79:14122–14130. doi: 10.1128/JVI.79.22.14122-14130.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez A. Analysis of filovirus entry into vero e6 cells, using inhibitors of endocytosis, endosomal acidification, structural integrity, and cathepsin (B and L) activity. J. Infect. Dis. 2007;196(Suppl. 2):S251–258. doi: 10.1086/520597. [DOI] [PubMed] [Google Scholar]
- Schornberg K., Matsuyama S., Kabsch K., Delos S., Bouton A., White J. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J. Virol. 2006;80:4174–4178. doi: 10.1128/JVI.80.8.4174-4178.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G., Gosalia D.N., Rennekamp A.J., Reeves J.D., Diamond S.L., Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. U.S.A. 2005;102:11876–11881. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G., Reeves J.D., Rennekamp A.J., Amberg S.M., Piefer A.J., Bates P. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc. Natl. Acad. Sci. U.S.A. 2004;101:4240–4245. doi: 10.1073/pnas.0306446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tresnan D.B., Levis R., Holmes K.V. Feline aminopeptidase N serves as a receptor for feline, canine, porcine, and human coronaviruses in serogroup I. J. Virol. 1996;70:8669–8674. doi: 10.1128/jvi.70.12.8669-8674.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk D., Guncar G. Lysosomal cysteine proteases (cathepsins): promising drug targets. Acta Crystallogr. D: Biol. Crystallogr. 2003;59:203–213. doi: 10.1107/s0907444902021479. [DOI] [PubMed] [Google Scholar]
- Tusell S.M., Schittone S.A., Holmes K.V. Mutational analysis of aminopeptidase N, a receptor for several group 1 coronaviruses, identifies key determinants of viral host range. J. Virol. 2007;81:1261–1273. doi: 10.1128/JVI.01510-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hamme E., Dewerchin H.L., Cornelissen E., Nauwynck H.J. Attachment and internalization of feline infectious peritonitis virus in feline blood monocytes and Crandell feline kidney cells. J. Gen. Virol. 2007;88:2527–2532. doi: 10.1099/vir.0.82991-0. [DOI] [PubMed] [Google Scholar]
- Vasiljeva O., Reinheckel T., Peters C., Turk D., Turk V., Turk B. Emerging roles of cysteine cathepsins in disease and their potential as drug targets. Curr. Pharm. Des. 2007;13:387–403. doi: 10.2174/138161207780162962. [DOI] [PubMed] [Google Scholar]
- Vennema H., Heijnen L., Zijderveld A., Horzinek M.C., Spaan W.J. Intracellular transport of recombinant coronavirus spike proteins: implications for virus assembly. J. Virol. 1990;64:339–346. doi: 10.1128/jvi.64.1.339-346.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennema H., Poland A., Foley J., Pedersen N.C. Feline infectious peritonitis viruses arise by mutation from endemic feline enteric coronaviruses. Virology. 1998;243:150–157. doi: 10.1006/viro.1998.9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentworth D.E., Holmes K.V. Coronavirus binding and entry. In: Thiel V., editor. Coronaviruses. Molecular and Cellular Biology. Caister Academic Press; Norfolk, UK: 2007. pp. 3–31. [Google Scholar]