

Abstract
Background
Extracellular acidosis (EA) regulates Heme Oxygenase-1 (HO-1) expression in vascular smooth muscle cells via transcriptional and posttranscriptional mechanisms but the signaling pathways involved are not known. We examined the role of Mitogen-Activated Protein Kinase (MAPK) pathways in HO-1 regulation by EA.
Methods
Primary rat aortic smooth muscle cells were exposed to EA or physiologic pH. Levels of the total and phosphorylated forms of p38, extracellular signal-regulated protein kinases1/2 (ERK1/2), c-Jun N-terminal kinases/stress-activated protein kinases (JNK1/2), and HO-1 protein were assessed by Western analysis and HO-1 mRNA levels were assessed by quantitative PCR. Inhibition of p38 MAPK was achieved with the chemical inhibitor SB203580, or adenoviral infection of a dominant-negative form of p38α. Phospho p38 MAPK activity was evaluated with an in vitro kinase activity assay. Binding of Activator Protein-1(AP-1), a known target of MAPK pathways, was assessed by Electromobility shift assay (EMSA)
Results
EA induced phosphorylation of p38 MAPK in a biphasic manner while total p38 was unchanged. EA did not alter levels of phospho ERK 1/2 and phospho JNK 1/2. There was increased phospho p38 MAPK activity in the setting of EA which preceded the induction of HO-1. Inhibition of phospho p38 activity with either SB20358 or a dominant negative p38α oligonucleotide abrogated the induction of HO-1 by EA. Increased specific binding of AP-1 in the setting of EA was shown by EMSA.
Conclusion
Increased phospho p38 activity precedes and likely mediates HO-1 induction by EA. Increased AP-1 binding may underlie the transcriptional regulation of HO-1 by EA.
INTRODUCTION
Extracellular acidosis is a clinically relevant cellular stressor with profound effects on cardiovascular homeostasis but the molecular mechanisms underlying vascular cell responses to acidosis are incompletely understood. The pH of extracellular fluid is normally maintained within a narrow range (7.37–7.42) which is essential to normal metabolic function. When the well orchestrated, multi-system process that preserves the normal alkalinity of the extracellular fluid fails, acute or chronic acid-base disturbances ensue. The extreme range of acute plasma pH that is compatible with life is 6.9–7.8. Although the clinical significance of acidosis is widely appreciated, significant gaps in our understanding of vascular cell responses to acidosis exist. Specifically, potential adaptive responses to acidosis have not been adequately explored in vascular cells.
We previously reported that extracellular acidosis (EA), induces the expression of Heme Oxygenase-1 (HO-1) in primary vascular smooth muscle cells (VSMCs) derived from the systemic as well as the pulmonary circulation 1. HO-1 is a cytoprotective molecule known to play an important role in vascular homeostasis through several mechanisms including regulation of VSMC proliferation 2–4 and apoptosis5, 6 and its importance in diseases of the vessel wall is the subject of intense investigation. Since HO-1 is an important regulator of VSMC homeostasis, we reasoned that its regulation by EA may represent an adaptive response and we sought to define the mechanisms of this regulation. We previously reported that both transcriptional and posttranscriptional mechanisms underlie the induction of HO-1 by EA in VSMCs, and that Nitric oxide did not appear to mediate it 1. In this study we further define the molecular mechanisms underlying HO-1 regulation by EA by identifying the role of the Mitogen-Activated Protein Kinase (MAPK) pathways.
Activation of the MAPK pathways (p38, ERK1/2 and JNK 1/2) plays a central role in cellular responses to a multitude of extracellular signals 7 including EA 8, 9. Although both ERK1/2 and p38 MAPKs have been implicated in cellular responses to acidosis in a variety of cell types 8, 9, the effect of EA on MAPK pathways has not been studied in VSMCs. In addition, although MAPK pathways are involved in the regulation of HO-1 by various inducers, this process appears to be cell- and stimulus- specific 10–14. The present study supports that the p38 MAPK pathway mediates the regulation of HO-1 by EA in VSMCs.
METHODS
Cell culture and exposure to EA
The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996). Primary VSMCs were isolated from adult rat aortas and their state of differentiation was characterized with VSMC markers as previously described 1. These primary rat aortic smooth muscle cells (RASMCs) were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS) and 2mM Glutamine and passaged every 3 to 4 days (used between passages 4–10). When they reached approximately 80% confluence, cells were subjected to 48 hours of serum deprivation (0.5% FCS) prior to exposure to media of different pH values (7.4 or 6.8). We used bicarbonate-free DMEM/F12 (1:1)/0.5% FCS media, pH-adjusted to the desired value with 100mM Hepes buffer. The cells were incubated in a CO2-free 37 °C incubator for different time periods and they were then harvested for extraction of RNA or protein. Because subtle cellular stimuli such as media replenishment may lead to changes in MAPK phosphorylation and/or HO-1 expression, our time course experiments were done in media of physiologic pH (7.4) in parallel to exposure to acidic media (pH 6.8) and normalized.
Kinase inhibitor treatments
The p38 MAPK inhibitor SB203580 and the ERK1/2 inhibitor PD098059 were purchased from Calbiochem. The cells were treated with 50nM to 5μM SB203580, or 50nm to 10 μM PD098059 for one hour prior to and during exposure to EA (pH 6.8) or media of physiologic pH (7.4). Control cells were treated with vehicle alone (dimethylsulfoxide- DMSO)
Western analysis
This was done using standard methods 15. We used a polyclonal anti-rat HO-1 antibody (Stressgen, SPA 896) at 1:1000 dilution for Western analysis. Activation of p38 MAPK was assayed by Western blot analysis using an antibody that specifically recognizes the dually phosphorylated active form of the enzyme (on residues Thr180 and Tyr182). Phospho-p44/42 (Thr202/Tyr204) and phospho-SAPK/JNK(Thr183/Tyr185) were also used for detection of the phosphorylated forms of p44/42 (ERK 1/2) and JNK 1/2. All MAPK antibodies were from Cell signaling Technology (Bevery, MA). Alpha tubulin and/or total p38 levels were used for normalization of the Western blots. Quantification of signals was done using NIH image software.
Real time RT-PCR
Total cellular RNA was extracted with the Trizol reagent (Invitrogen) after exposure to the experimental conditions and 1 μg was DNase-treated (Promega), and reverse transcribed (Qiagen, Omniscript Reverse Transcriptase) using random primers. Quantitative PCR analysis was performed in triplicate for each sample using the M× 40000 machine (Stratagene). Primers and FAM-labeled Taqman probes were obtained from Applied Biosystems. Ct values for HO-1 (a 132 bp amplicon spanning exon 2 and 3 of the rat HO-1 gene) were standardized against the internal ribosomal RNA (18s) control probe to calculate ΔCt values. ΔΔCt values were calculated by subtracting the ΔCt values of the samples from cells exposed to pH 7.4 from the mean ΔCt value of all other experimental conditions. Fold change was calculated using the formula 2−ΔΔCt.
p38 MAP Kinase activity assay
Kinase activity was performed using an assay kit according to the manufacturer’s instructions (Cell Signaling, Beverly, MA). After exposure to the experimental conditions, 200 μg of cell lysates were immunoprecipitated with immobilized phospho p38 MAPK antibody (Thr180 and Tyr182). An in vitro kinase assay was performed using ATF-2 fusion protein as a substrate and phosphorylation of ATF-2 (Thr71) was detected by Western blotting with phospho-ATF-2 (pATF2) antibody.
Nuclear protein extraction and electromobility shift assay (EMSA)
Cell nuclear extracts were prepared according to the method of Schreiber et al 16 and total protein was quantified with the Bradford assay. For EMSA, 5 ug nuclear proteins were incubated for 10 minutes at room temperature in a 20uL binding mixture containing 0mmol/L HEPES, pH 7.9, 0.5 mmol/L MgCl2, 0.1 mmol/L EDTA, 5% glycerol, 100 mmol/L KCl, and 1ug polydeoxyinosinic-deoxycytidylic acid. Radiolabeled oligonucleotide probes (22000 cpm) designed to bind to AP-1, mutant AP-1 and SP-1 were then added, and the incubation was continued for 20 minutes. Binding in the presence of c-Jun or c-Fos antibodies was assessed by including these antibodies in the binding mixture. The binding mixture was fractionated on a 4% polyacrylamide gel in 0.5x TBE (1XTBE consists of 89mmol/L Tris Base, 89mmol/L Boric Acid and 2mmol/L EDTA) at 4 °C and autoradiography was performed.
Plasmid constructs, generation of adenoviral vectors containing p38 Dominant negative oligonucleotide, and adenoviral infection
The dominant-negative mutant mammalian p38α expression plasmid pCMV-Flag-p38(agf) was generously provided by Dr. Roger Davis (University of Massachusetts, Worcester, MA) and was previously described 7. Briefly, the dominant negative form of p38 has an Alanine instead of a Threonine in position 180 and a Phenylalanine instead of a Tyrosine in position 182 (AGF instead of TGY) therefore cannot be activated by dual phosphorylation on Threonine 180 and Tyrosine 182. The mutant p38 fragment was subcloned into the adenovirus vector pDC316 (Microbix Biosystems Inc, Toronto, Canada) at HindIII/XbaI sites. Following the instructions of the AdMax™ system (Microbix Biosystems Inc), the p38 Dominant Negative Mutant plasmid was co-transfected with the viral genomic plasmid (pBHGloxE1, 3Cre) into QBI-293A cells (Q.Biogene, Montreal, Canada). The viral plaques were isolated and screened with an anti-Flag antibody to determine the transfected p38 module. Multiplicity of infection (MOI) of positive virus was titrated. An adenovirus expressing β-gal was used as control for viral infection as previously described 17.
Assessment of RASMC proliferation
After exposure to the experimental conditions, cell proliferation was assessed by cell number and DNA synthesis was measured by 5-bromo-2′-deoxyuridine (BrdU) incorporation. Quadruplicate cultures of primary RASMCs were plated at equal numbers and when they reached ~ 40% confluence, they were serum deprived (0.5% FBS) for 48 hours. They were then stimulated to grow in serum and/or Platelet-Derived Growth Factor –BB (PDGF-BB, 25ng/ml) in media of pH 6.8 or pH 7.4. Viable cells were counted at serial time points from parallel plates using the Trypan blue exclusion method and the number of dead cells per condition was noted. For assessment of DNA synthesis, RASMCs were plated at equal numbers in a 96-well plate, and after 48 hours of serum deprivation (0.5% FBS), they were stimulated to grow in serum or PDGF-BB in media of pH 6.8 or 7.4 overnight. A 5-bromo-2′-deoxyuridine (BrdU) chemiluminescence immunoassay (Roche) was then used to compare DNA synthesis between the two experimental conditions.
Migration assays
Equal number of RASMCs were plated on the top chamber of Transwell plates (pore size 8.0 mM) and 50 ng/ml of PDGF-BB was added in the lower chamber. After exposure to the experimental conditions, the top chamber was scraped and live cells were quantified in the lower chamber by an acid phosphatase enzymatic assay as previously described 17, 18.
Statistical methods
The nonparametric ANOVA (Kruskal Wallis) test was used to compare median values among the experimental groups and differences were considered significant if p < 0.05.
RESULTS
EA increases phospho p38 (pp38) and HO-1 protein levels and increases pp38 MAPK activity in primary vascular smooth muscle cells
As we had previously shown there is a time-dependent induction of HO-1 protein upon exposure of primary RASMCs to acidic media which begins after 8 hours of exposure and peaks at 18 hours 1 (Figures 1A and 1C). To examine the role of MAPKs in the induction of HO-1 by EA we performed time course experiments of the phosphorylated forms of p38, ERK1/2 and JNK1/2 proteins in RASMCs exposed to media of pH 7.4 or 6.8. As shown in Figures 1A, 1B and 1D, although there was an acute increase in the phosphorylated forms of all three MAPKs following exposure of the cells to fresh media of either pH, this transient increase was not followed by changes in HO-1 protein levels. However, in the setting of EA, there was a biphasic induction of phospho p38 (pp38), with a second peak at 8 hours which preceded the induction of HO-1 protein by EA (Figures 1A, 1B and IC). This correlated with increased p38 MAPK activity as shown in Figure 2: following exposure to EA for 8 hours, p38 MAPK was immunoprecipitated and its activity was assessed using recombinant ATF-2 as a substrate. As shown in Figure 2, pp38-mediated ATF-2 phosphorylation in vitro was significantly increased in the setting of EA and treatment with the p38 MAPK inhibitor, SB203580, led to significant reduction of pp38 activity.
Figure 1. Time course of phospho p38, phospho Erk1/2, phospho JNK1/2 and HO-1 protein after exposure to EA in RASMCs.
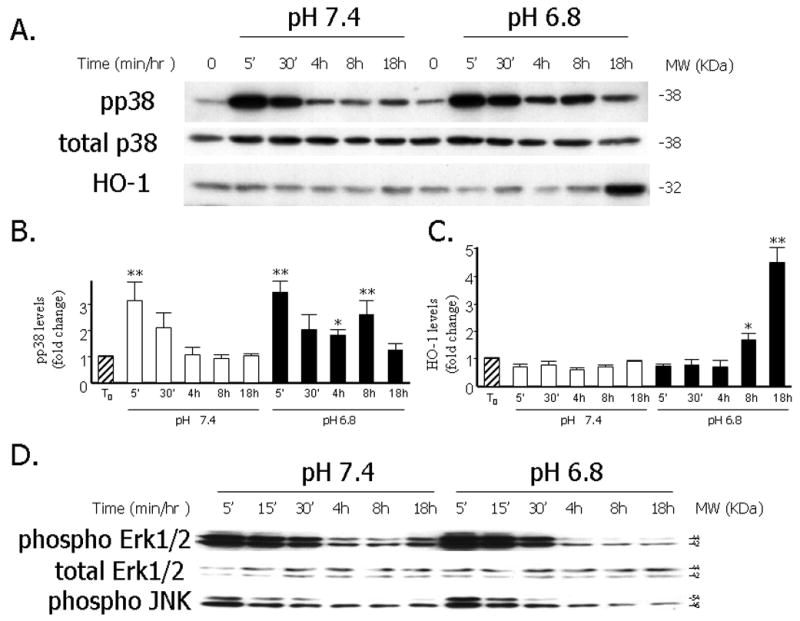
A. Representative Western blot depicting levels of pp38, total p38 and HO-1 protein after exposure to EA (pH 6.8) or media of physiologic pH (7.4) for different time periods. B. Quantitative analysis of levels of pp38 (normalized to total p38) following exposure to the experimental conditions. Data are expressed as the mean and standard error of the mean (SEM). Eight independent experiments are represented. *: p< 0.05, **: p< 0.001, statistically significant differences as compared to T0. C. Quantitative analysis of levels of HO-1 (normalized to α-tubulin) following exposure to the experimental conditions. Data are expressed as the mean and standard error of the mean (SEM). Eight independent experiments are represented. *: p< 0.05, **: p< 0.001, statistically significant differences as compared to T0. D. Representative Western blot depicting pErk/1/2, total Erk1/2 and pJNK1/2 protein levels after exposure to EA or media of physiologic pH for different time periods.
Figure 2. Increased pp38 activity in RASMCs exposed to EA and inhibition of pp38 activity after treatment with the p38 MAPK inhibitor SB203580.
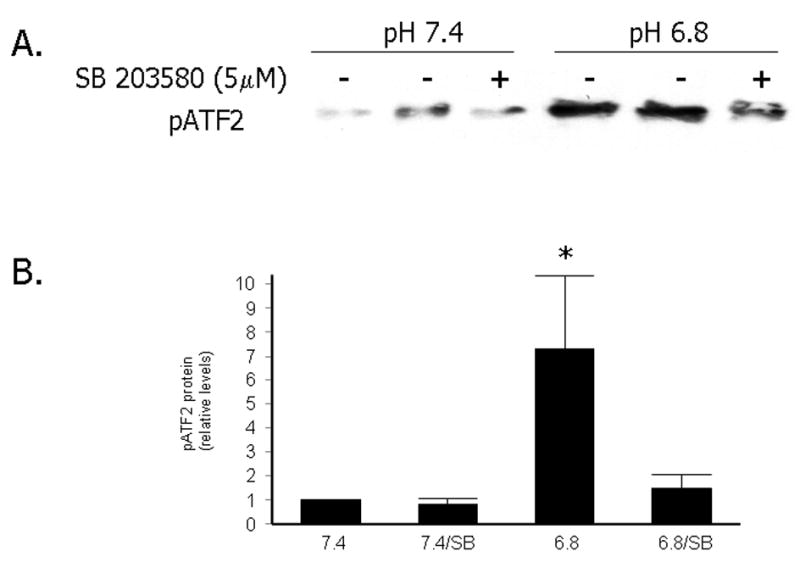
A. Representative immunoprecipitation with pp38 antibody, followed by in vitro kinase reaction with ATF-2 GST fusion protein and detection of the phosphorylated ATF-2 by Western blotting. Cells were exposed to pH 7.4 or 6.8 for 8 hours after pretreatment with DMSO or SB203580 (5 μM) for one hour. B. Quantitative analysis of levels of pATF2 following exposure to the experimental conditions and in vitro kinase assay. Data are expressed as the mean and standard error of the mean (SEM). Five independent experiments are represented *: p< 0.01, statistically significant difference as compared to pp38 activity at pH 7.4, 7.4/SB and 6.8/SB.
Pharmacologic inhibition of p38 MAPK with SB203580, attenuates the induction of HO-1 expression by EA
In order to determine the potential role of the p38 MAPK signaling pathway in the induction of HO-1 by EA we pretreated primary RASMCs with the p38 MAPK inhibitor, SB230580, for one hour prior to exposure to media of pH 7.4 or 6.8. RNA and protein were then isolated and quantitative PCR and Western blots were performed respectively. As shown in Figure 3A, and as we previously reported, in the vehicle-treated cells, there was a significant induction of HO-1 mRNA after exposure to acidic media for 8 hours. This induction was reduced significantly following pretreatment with 5uM SB203580. At the protein level, HO-1 induction by EA is maximal at 18 hours as we previously reported. As shown in Figure 3B, treatment with the p38 MAPK inhibitor, SB203580, was effective in preventing the induction of HO-1 protein by EA while it had no effect on HO-1 protein levels at pH 7.4. Pretreatment with the ERK1/2 MAPK inhibitor PD098059 (10uM) had no effect on HO-1 protein induction by EA. All doses of SB203580, from 50nM to 5 uM, as well as treatment with another p38 MAPK inhibitor, SB202190 were effective in inhibiting HO-1 protein upregulation by EA (data not shown).
Figure 3. Inhibitory effect of the p38 MAPK inhibitor SB203580 on HO-1 mRNA and protein induction after exposure of RASMCs to acidic media.
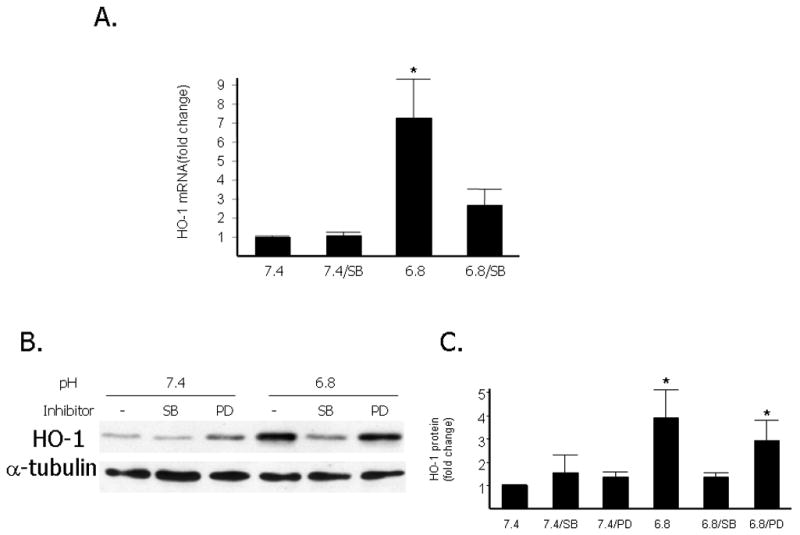
A. Quantitative analysis of HO-1 mRNA levels (normalized to 18s mRNA by Q-PCR) after exposure of RASMCs to acidic media (pH 6.8) compared to exposure to media of pH 7.4 with or without treatment with 5μM of SB203580. Cells were pretreated with 5μM SB203580 for one hour or DMSO (vehicle) and exposed to an extracellular pH of 6.8 or 7.4 for 8 hours. Data are expressed as the mean and standard error of the mean (SEM). Six independent experiments are represented. *p<0.05 compared to HO-1 mRNA at pH 7.4. B. Representative Western blot depicting HO-1 protein levels compared to levels of α-tubulin used as loading control. Cells were pretreated for one hour with DMSO (vehicle), the p38 inhibitor SB203580 (5μM-SB), or the Erk1/2 inhibitor PD098059 (10μM-PD) and were then exposed to an extracellular pH of 6.8 or 7.4 for 18 hours. C: Quantitative analysis of HO-1 protein levels after exposure of RASMCs to acidic media compared to exposure to media of pH 7.4 with or without treatment with 5μM of SB203580 or 10 μM of PD 098059. Data are expressed as the mean and standard error of the mean (SEM). *: p< 0.05, statistically significant difference as compared to pH 7.4, 7.4/SB, 7.4/PD and 6.8/SB. Six independent experiments are represented and quantitative analysis was done with NIH Image software and normalized to α-tubulin levels.
Adenoviral infection with a dominant-negative form of p38α attenuates the induction of HO-1 expression by EA
To account for possible non-specific effects of the chemical inhibitors of p38 MAPK signaling on HO-1 induction by EA, we performed adenoviral infection of primary RASMCs with a dominant-negative form of p38α. Figure 4A shows the presence of Flag signal and increased total p38 levels after successful transfection of QBI-293A cells with the original Flag-tagged plasmid pCMV-Flag-p38(agf) as well as the new plasmid we created by subcloning the dominant negative p38 fragment into the adenoviral vector pDC316, denoted pDC316-Flag-p38(agf). Untransfected or transfected with a β-gal plasmid (pDC316-β-gal) QBI-293A cells were used as negative controls. Figure 4B shows the presence of Flag signal after infection of primary RASMCs with the p38-dominant negative-expressing adenovirus at different multiplicities of infection (MOIs). Uninfected or infected with a β-gal-expressing adenovirus RASMCs were used as controls. As shown in Figures 4C, 4D and 4E there was a significant decrease in the magnitude of acid-induction of HO-1 protein and RNA levels in RASMCs infected with the adenoviral vector expressing p38 dominant-negative compared to cells infected with adenoviral vector expressing β-gal. Levels of HO-1 mRNA and protein were significantly higher in uninfected cells exposed to EA compared to uninfected cells kept at pH 7.4. Adenoviral infection had no effect on HO-1 expression at pH 7.4 but we observed significantly higher levels of HO-1 mRNA in RASMCs infected with β-gal-expressing adenovirus in the setting of EA. We interpreted this as evidence of a non-specific effect of the combination of adenoviral infection and exposure to acidosis that lead to increased levels of HO-1. Despite this apparent non specific effect of adenoviral infection on HO-1 at pH 6.8, infection with adenovirus expressing p38α dominant-negative significantly reduced HO-1 mRNA and protein levels in the setting of EA.
Figure 4. Effect of adenoviral infection with a dominant-negative p38 mutant on HO-1 induction by EA.
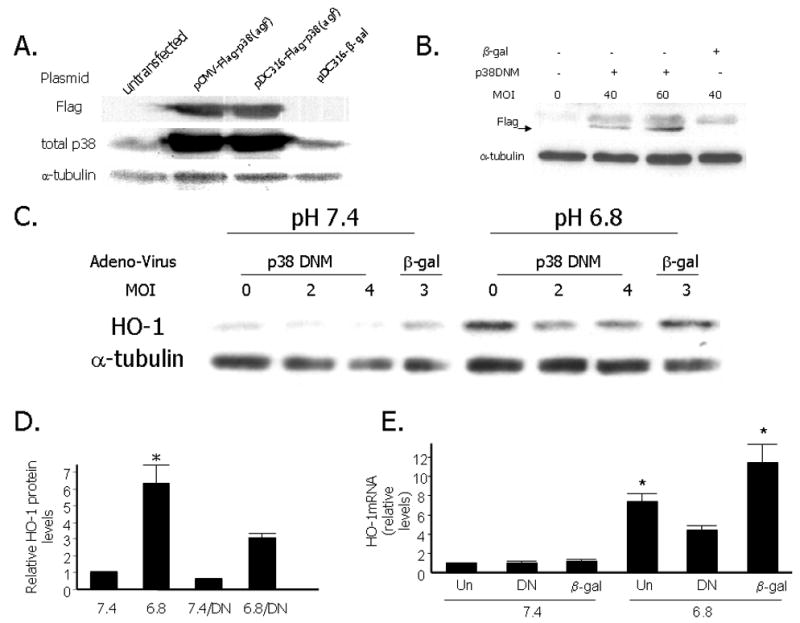
A. Western analysis depicting Flag, total p38 and α-tubulin levels in QBI293A cells that were either untransfected, transfected with the original plasmid pCMV-Flag-p38(agf), or the adenoviral vector containing the dominant form of p38, denoted pDC316-Flag-p38 (agf), or the β-gal plasmid pDC316-β-gal. B. Representative Western analysis showing Flag and α-tubulin levels in RASMCs that were either uninfected, or infected with p38 dominant negative-expressing adenovirus (at MOI 4O or 60), or infected with a β-gal expressing adenovirus. C. Representative Western analysis depicting HO-1 levels in RASMCs exposed to media of pH 6.8 or 7.4 for 8 hours with or without adenoviral infection with a p38 dominant negative mutant (DN) or β-galactosidase (β-gal) at the indicated multiplicity of infection (MOI). D. Quantitative analysis of HO-1 protein levels after exposure of RASMCs to acidic media (pH 6.8) compared to exposure to media of pH 7.4 with or without adenoviral infection with p38 dominant negative (DN) mutant or β-gal. Data are expressed as the mean and standard error of the mean (SEM). Six independent experiments are represented. * p<0.05 compared to 7.4,7.4/DN and 6.8/DN. E. Quantitative analysis of HO-1 mRNA levels (normalized to 18s mRNA by Q-PCR) after exposure of RASMCs to acidic media (pH 6.8) compared to exposure to media of pH 7.4 following adenoviral infection with p38 DN or β-gal. Fourty eight hours after adenoviral infection with DN or β-gal (MOI 2), RASMCs were subjected to 48 hours of serum deprivation and exposed to an extracellular pH of 6.8 or 7.4 for 8 hours. Uninfected RASMCs (Un) were also subjected to 48 hours of serum deprivation and exposed to the same experimental conditions. Data are expressed as the mean and standard error of the mean (SEM). Five independent experiments are represented. *p<0.05 compared to HO-1 mRNA at pH 7.4.
Increased AP-1/DNA specific binding after exposure of RASMCs to EA
Given that the 5′ region upstream of the HO-1 promoter contains multiple Activator Protein-1 (AP-1) sites, which is a known downstream target of MAPK pathways, in the next set of experiments we examined by electromobility shift assays whether EA enhances the binding of nuclear proteins to the AP-1 sequence. Radioactively labeled oligonucleotide probes for wild type AP-1 (Ap-1 W), mutant AP-1 (AP-1 M) or the unrelated oligonucleotide SP-1 were incubated with nuclear extracts from RASMCs exposed to media of pH 6.8 or 7.4 for 8 hours. Compared to nuclear extracts from cells exposed to pH 7.4, nuclear extracts of cells exposed to EA showed increased specific AP-1 binding (Figure 5B). The specifity of this binding was shown by competition with cold AP-1 probe and lack of recognition by a mutant AP-1 probe (Figure 5B). No change in binding of nuclear proteins to the SP-1 site was observed between the two experimental conditions (Figure 5C). Treatment with the p38 MAPK inhibitor, SB203580, attenuated AP-1 binding thus supporting that the p38 MAPK pathway leads to enhanced AP-1 binding. In vitro binding in the presence of c-Jun or c-Fos antibodies showed either attenuated or absent binding, however this effect was similar under both physiologic and acidic conditions and no supershifted bands were detected. Thus, although our data support that nuclear extracts from cells exposed to EA exhibit increased AP-1 binding in vitro, we cannot determine whether the transcription factors c-Fos and c-Jun are involved in the increased binding at the AP-1 site in the setting of EA and we cannot exclude the role of other regulatory sites in the transcriptional regulation of HO-1 by EA and/or p38MAPK.
Figure 5. Increased AP-1/DNA specific binding of nuclear proteins after exposure of RASMCs to EA.
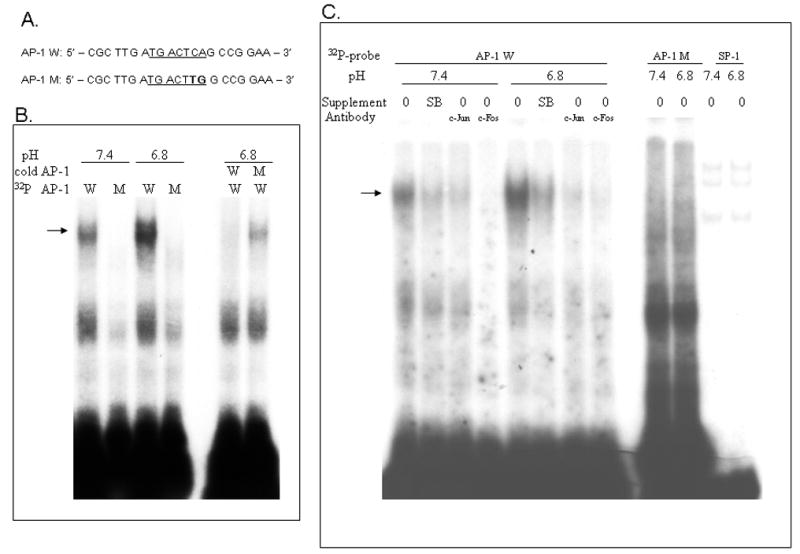
A. Oligonucleotide sequences of wild type (W) and mutant (M) AP-1 probes used for Electromobility shift assays (EMSAs). B. Representative EMSA showing increased specific binding of nuclear proteins from RASMCs exposed to acidic media for 8 hours to a wild type AP-1 DNA probe compared to nuclear proteins from RASMCS exposed to media of pH 7.4. The arrow indicates the specific protein/DNA binding band that is absent in the presence of the mutant AP-1 probe and is competed away by inclusion of cold wild type AP-1 probe but not mutant AP-1 probe in the binding mixture. C. Pretreatment of cells with the p38 MAPK inhibitor SB203580 led to decreased binding to the AP-1 site. Binding in the presence of c-Jun or c-Fos was assessed by including these antibodies in the binding mixture. Binding to an SP-1 probe showed no difference between nuclear extracts from cells exposed to acidic media or pH 7.4. The presented data are representative of experiments performed in triplicate.
EA inhibits RASMC proliferation and migration
To begin to define the functional implications of altered intracellular signaling and HO-1 gene regulation by EA, we examined whether exposure of RASMCs to EA alters their proliferative and migratory responses to growth stimuli. We found that RASMC proliferation in response to serum or Platelet-Derived Growth Factor-BB (PDGF-BB) stimulation was inhibited in the setting of EA (Figure 6A) while the number of dead cells under the same experimental conditions was less than 10% and not altered by exposure to EA. This finding corresponded to significantly lower BrdU incorporation in the setting of EA thus supporting that EA exerts an inhibitory effect in DNA synthesis in RASMCs (Figure 6B). RASMC migration in response to PDGF-BB was also decreased in the setting of EA (Figure 6C).
Figure 6. Inhibition of RASMC proliferation and migration by EA.
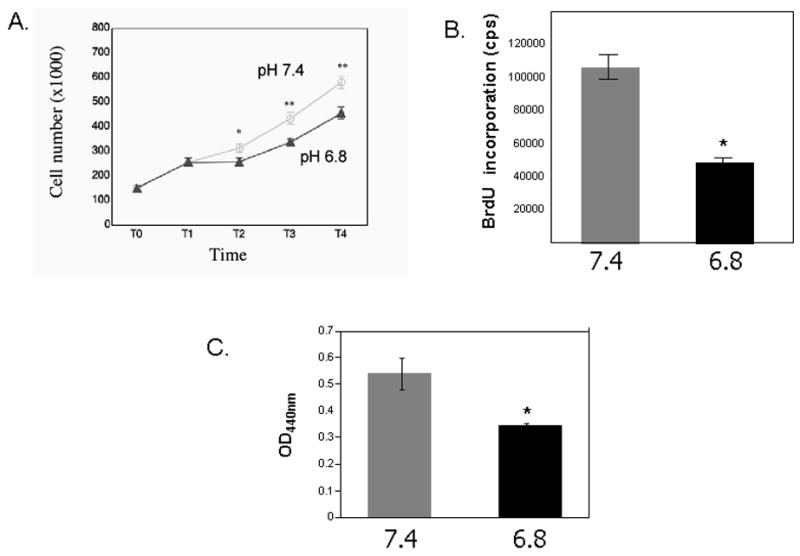
A. Cell number in response to serum and Platelet-derived Growth Factor-BB (PDGF-BB). Quadruplicate cultures of primary rat VSMCs were plated at equal numbers and grown to 40% confluence (T0). They were serum deprived (0.5% FBS) for 48 hours (T1) and then stimulated to proliferate in serum in media of pH 6.8 or pH 7.4 overnight (T2). A second growth stimulus (PDGF-BB, 25ng/ml) was then added and cell counts were assessed 18 hours (T3) and 48 hours (T4) after the addition of PDGF-BB. The mean and SEM values are shown * p< 0.05, ** p<0.001(comparisons were made between the two experimental conditions). B. BrdU incorporation in primary RASMCs in response to PDGF-BB in the setting of extracellular acidosis (pH6.8) or physiologic pH (7.4). RASMCs were plated at equal numbers (15,000 cells/well in a 96-well plate) and incubated at 37° C overnight to ensure attachment. Plating efficiency was assessed by counting cell numbers in parallel cultures 24 hours later. Cells were serum deprived for 48 hours then stimulated to proliferate with PDGF-BB (25ng/ml) in media of pH 6.8 or pH 7.4 overnight. A 5-bromo-2′-deoxyuridine (BrdU) chemiluminescence immunoassay (Roche) was used to measure DNA synthesis. Mean and SEM are shown. *p<0.0001. C. Cell migration assays were performed using Transwell plates (Transwell-Costar corp). Subconfluent cells of primary RASMCs were trypsinized, neutralized and resuspended in DMEM, 0.1% BSA. The same number of cells (5×105) were added to the top of each migration chamber in the setting of extracellular acidosis (pH 6.8) or physiologic pH (7.4) and were allowed to migrate to the underside of the chamber overnight in the presence of 50ng/ml PDGF-BB in the lower chamber. The upper side of the filter was then scraped off and the filter was removed. VSMC that had migrated to the lower side of the filter were quantified with an acid phosphatase enzymatic assay. Mean optical density at 440nm (OD440nm) and SEM are shown* p<0.05
DISCUSSION
In this study, using pharmacologic inhibition and a dominant negative approach, we found that the p38 MAPK pathway is involved in the regulation of HO-1 by EA in primary rat aortic smooth muscle cells. We found a biphasic increase in phospho-p38 MAPK protein levels and increased p38 MAPK activity after exposure of RASMCs to EA which preceded the induction of HO-1. This response to EA was not seen in the ERK1/2 and JNK pathways. Inhibition of p38 activity with the chemical inhibitor SB203580, or with adenoviral infection with a p38 dominant negative-expressing virus, inhibited the induction of HO-1 by EA. Since we also found increased specific binding for AP-1 in the setting of EA, which was inhibited after treatment with the p38 inhibitor SB203580, we speculate that increased AP-1 binding resulting from p38 MAPK signaling may underlie the transcriptional activation of HO-1 in the setting of EA. However, the role of the p38 MAPK pathway in the posttranscriptional regulation of HO-1 by acidosis should also be explored. In addition, we found that EA inhibited RASMC proliferation and migration.
Extracellular acidosis, a clinically relevant cellular stressor, alters intracellular signaling pathways and gene expression in various cell types but regulation of gene expression by EA in RASMCs had not been previously reported. In addition, the sensing and signaling events linking EA to gene expression have not been studied in VSMCs. We previously reported that EA increases the transcriptional rate and mRNA stability of HO-1 in rat aortic and pulmonary artery smooth muscle cells and that this regulation was not mediated by nitric oxide 1. We have now identified p38MAPK as the signaling pathway mediating, at least in part, the acid induction of HO-1 in RASMCs. Since we also found inhibition of RASMC proliferation and migration in the setting of EA, future studies should address the role of HO-1 in mediating the effects of acidosis in vascular homeostasis and the potential role of EA in the pathogenesis and reversal of vascular disorders.
MAPKs are a family of serine/threonine kinases that respond to a variety of cellular stimuli such as growth factor stimulation, osmotic stress, cytokines, extracellular matrix components and extracellular acidosis 7–9. Protein phosphorylation events are involved in the regulation of gene expression by activating transcription factors and through post-transcriptional mechanisms. Phosphorylation of p38 MAPK was reported to mediate the acid-induction of Phosphoenolpyruvate carboxykinase (PEPCK) transcription in proximal tubular epithelial cells by Feifel et al 9. Similar to our finding of a biphasic induction of phospho p38 by EA in RASMCs, this study also showed that exposure of tubular epithelial cells to EA resulted in a biphasic phosphorylation of p38 MAPK with peaks occurring at 0.5–1 hour and at 9 hours. In contrast, Xu et al reported that EA led to induction of phospho ERK1/2 within 10 minutes of exposure to acidic media and this was followed by induction of Vascular Endothelial Growth Factor (VEGF) expression within an hour in human glioblastoma cells 8. Regulation of the p38 MAPK pathway by EA in RASMCs has not been previously reported. Not surprisingly, we found an acute but transient increase in the phosphorylated forms of all three MAPKs (p38, ERK 1/2 and JNK1/2) upon exposure to fresh media regardless of the extracellular pH. This transient increase was most likely triggered by exposure to fresh media (containing 0.5% serum) and, although of significant magnitude, did not lead to induction of HO-1. The biphasic induction of phosphorylation by EA was unique to p38 MAPK, as was previously reported in renal epithelial cells 9.
The mechanisms of gene regulation in response to activation of the MAPK pathways include transcriptional activation and posttranscriptional mRNA stabilization. As we previously reported by nuclear run on and RNA stability experiments both mechanisms contribute to HO-1 regulation by EA in VSMCs 1. A well characterized mechanism of transcriptional activation in response to MAPK activation is enhanced binding of various transcription factors to the AP-1 site. Although we found enhanced AP-1 binding in the setting of EA in RASMCs, which was decreased in the presence of the p38 MAPK inhibitor SB203580, we have no definitive evidence that p38 MAPK is involved in the transcriptional activation of HO-1 by EA. Systematic evaluation of the several regulatory elements (including several AP-1 binding sites) found in the 5′ region upstream of the HO-1 promoter will likely define their importance in mediating the pH response. Alternatively, activation of the p38 MAPK pathway may mediate HO-1 mRNA stabilization in the setting of EA. The mechanisms of HO-1 mRNA stabilization in the setting of EA may involve inhibition or delay of deadenylation of the poly A tail, inhibition of the 5′ decapping or a subsequent step in the mRNA decay 19. Interestingly, stabilization of HO-1 mRNA has previously been reported in response to nitric oxide in human fibroblasts and this was associated with delay in the deadenylation process 19. Stabilization of other mRNAs in response to p38 MAPK signaling was also reported by Wang et al who found that both AU-rich element-dependent and independent mechanisms underlie the stabilization process 20.
Involvement of the MAPK pathways in the regulation of HO-1 by other inducers has been previously reported although this process appears to be cell- and stimulus-specific. Kocanova et al showed that induction of HO-1 by photodynamic therapy in cancer cells required the p38MAPK and PI3K pathways 12. Elbirt et al showed that both ERK and p38 MAPKs were involved in the transcriptional regulation of HO-1 by sodium arsenite in hepatoma cells and that at least one AP-1 element was involved in this response 10. In contrast, Kietzmann et al reported that the JNK MAPK pathway mediated the regulation of HO-1 by arsenite in primary rat hepatocytes whereas the ERK and p38 MAPK pathways were not involved in this regulation 11. These authors found that the JNK pathway increased HO-1 expression through interaction of c-Jun with AP-1, whereas the p38 MAPK pathway inhibited HO-1 promoter activity through interaction with an E-box. Specifically in vascular smooth muscle cells, p38 MAPK was found to be involved in HO-1 induction by both the polyphenolic compound quercetin 14 and by Bone Morphogenetic Protein 4 (BMP4) 13 but the molecular mechanisms underlying this regulation were not studied.
In the clinical setting, acidosis is known to alter vascular tone 21, 22 in a vascular bed-specific manner: in the systemic circulation, it is associated with vasodilation whereas in the pulmonary circulation it causes vasoconstriction. This vascular bed specificity is most likely due to distinct responses of specific ion channels in pulmonary Vs systemic VSMCs to acidosis 23: in systemic VSMCs, activation of the ATP-sensitive K+ channel Kir 6.1/SUR 2B is critical in inducing vasodilation during hypercapnic acidosis 22, 24. In contrast, impaired activity of distinct subtypes of voltage gated potassium channels (Kvs) was found in pulmonary VSMCs exposed to normocapnic acidosis 23, 25 and may mediate the pulmonary vasoconstrictive effect of acidosis. Beyond the effects of acidosis on vascular tone, no previous studies have addressed the potential role of acidosis in regulation of VSMC gene expression, proliferation and migration. We found that EA increased the expression of HO-1 in RASMCs via the p38 MAPK pathway and inhibited cellular proliferation and migration. The underlying molecular mechanisms as well as the functional implications of this regulation need to be examined in future studies.
Acknowledgments
Funding: National Institutes of Health (K08-HL03917 to H.C.); Charles H. Hood Foundation Research Grant (to H.C.).
We gratefully acknowledge Dr. Roger Davis for providing us with the p38 dominant-negative plasmid and we thank Dr. Stella Kourembanas, Dr. Mark Perrella and Dr. S. Alex Mitsialis for useful suggestions.
Footnotes
Conflict of Interest: none declared.
References
- 1.Christou H, Bailey N, Kluger MS, Mitsialis SA, Kourembanas S. Extracellular acidosis induces heme oxygenase-1 expression in vascular smooth muscle cells. American journal of physiology. 2005 Jun;288(6):H2647–2652. doi: 10.1152/ajpheart.00937.2004. [DOI] [PubMed] [Google Scholar]
- 2.Morita T, Kourembanas S. Endothelial cell expression of vasoconstrictors and growth factors is regulated by smooth muscle cell-derived carbon monoxide. The Journal of clinical investigation. 1995 Dec;96(6):2676–2682. doi: 10.1172/JCI118334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morita T, Mitsialis SA, Koike H, Liu Y, Kourembanas S. Carbon monoxide controls the proliferation of hypoxic vascular smooth muscle cells. The Journal of biological chemistry. 1997 Dec 26;272(52):32804–32809. doi: 10.1074/jbc.272.52.32804. [DOI] [PubMed] [Google Scholar]
- 4.Ollinger R, Bilban M, Erat A, Froio A, McDaid J, Tyagi S, Csizmadia E, Graca-Souza AV, Liloia A, Soares MP, Otterbein LE, Usheva A, Yamashita K, Bach FH. Bilirubin: a natural inhibitor of vascular smooth muscle cell proliferation. Circulation. 2005 Aug 16;112(7):1030–1039. doi: 10.1161/CIRCULATIONAHA.104.528802. [DOI] [PubMed] [Google Scholar]
- 5.Liu XM, Chapman GB, Wang H, Durante W. Adenovirus-mediated heme oxygenase-1 gene expression stimulates apoptosis in vascular smooth muscle cells. Circulation. 2002 Jan 1;105(1):79–84. doi: 10.1161/hc0102.101369. [DOI] [PubMed] [Google Scholar]
- 6.Liu XM, Chapman GB, Peyton KJ, Schafer AI, Durante W. Carbon monoxide inhibits apoptosis in vascular smooth muscle cells. Cardiovascular research. 2002 Aug 1;55(2):396–405. doi: 10.1016/s0008-6363(02)00410-8. [DOI] [PubMed] [Google Scholar]
- 7.Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. The Journal of biological chemistry. 1995 Mar 31;270(13):7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 8.Xu L, Fukumura D, Jain RK. Acidic extracellular pH induces vascular endothelial growth factor (VEGF) in human glioblastoma cells via ERK1/2 MAPK signaling pathway: mechanism of low pH-induced VEGF. The Journal of biological chemistry. 2002 Mar 29;277(13):11368–11374. doi: 10.1074/jbc.M108347200. [DOI] [PubMed] [Google Scholar]
- 9.Feifel E, Obexer P, Andratsch M, Euler S, Taylor L, Tang A, Wei Y, Schramek H, Curthoys NP, Gstraunthaler G. p38 MAPK mediates acid-induced transcription of PEPCK in LLC-PK(1)-FBPase(+) cells. Am J Physiol Renal Physiol. 2002 Oct;283(4):F678–688. doi: 10.1152/ajprenal.00097.2002. [DOI] [PubMed] [Google Scholar]
- 10.Elbirt KK, Whitmarsh AJ, Davis RJ, Bonkovsky HL. Mechanism of sodium arsenite-mediated induction of heme oxygenase-1 in hepatoma cells. Role of mitogen-activated protein kinases. The Journal of biological chemistry. 1998 Apr 10;273(15):8922–8931. doi: 10.1074/jbc.273.15.8922. [DOI] [PubMed] [Google Scholar]
- 11.Kietzmann T, Samoylenko A, Immenschuh S. Transcriptional regulation of heme oxygenase-1 gene expression by MAP kinases of the JNK and p38 pathways in primary cultures of rat hepatocytes. The Journal of biological chemistry. 2003 May 16;278(20):17927–17936. doi: 10.1074/jbc.M203929200. [DOI] [PubMed] [Google Scholar]
- 12.Kocanova S, Buytaert E, Matroule JY, Piette J, Golab J, de Witte P, Agostinis P. Induction of heme-oxygenase 1 requires the p38(MAPK) and PI3K pathways and suppresses apoptotic cell death following hypericin-mediated photodynamic therapy. Apoptosis. 2007 Apr;12(4):731–741. doi: 10.1007/s10495-006-0016-x. [DOI] [PubMed] [Google Scholar]
- 13.Yang X, Lee PJ, Long L, Trembath RC, Morrell NW. BMP4 Induces HO-1 via a Smad Independent, p38MAPK Dependent Pathway in Pulmonary Artery Myocytes. Am J Respir Cell Mol Biol. 2007 Jun 28; doi: 10.1165/rcmb.2006-0360OC. [DOI] [PubMed] [Google Scholar]
- 14.Lin HC, Cheng TH, Chen YC, Juan SH. Mechanism of heme oxygenase-1 gene induction by quercetin in rat aortic smooth muscle cells. Pharmacology. 2004 Jun;71(2):107–112. doi: 10.1159/000076947. [DOI] [PubMed] [Google Scholar]
- 15.Minamino T, Christou H, Hsieh CM, Liu Y, Dhawan V, Abraham NG, Perrella MA, Mitsialis SA, Kourembanas S. Targeted expression of heme oxygenase-1 prevents the pulmonary inflammatory and vascular responses to hypoxia. Proceedings of the National Academy of Sciences of the United States of America. 2001 Jul 17;98(15):8798–8803. doi: 10.1073/pnas.161272598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res. 1989 Aug 11;17(15):6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu X, Chang MS, Mitsialis SA, Kourembanas S. Hypoxia regulates bone morphogenetic protein signaling through C-terminal-binding protein 1. Circ Res. 2006 Aug 4;99(3):240–247. doi: 10.1161/01.RES.0000237021.65103.24. [DOI] [PubMed] [Google Scholar]
- 18.Connolly DT, Knight MB, Harakas NK, Wittwer AJ, Feder J. Determination of the number of endothelial cells in culture using an acid phosphatase assay. Anal Biochemistry. 1986;152(1):136–140. doi: 10.1016/0003-2697(86)90131-4. [DOI] [PubMed] [Google Scholar]
- 19.Leautaud V, Demple B. Regulation of heme oxygenase-1 mRNA deadenylation and turnover in NIH3T3 cells by nitrosative or alkylation stress. BMC molecular biology. 2007;8:116. doi: 10.1186/1471-2199-8-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang S, Zhang J, Zhang Y, Kern S, Danner RL. Nitric oxide-p38 MAPK signaling stabilizes mRNA through AU-rich element-dependent and -independent mechanisms. Journal of leukocyte biology. 2008 Apr;83(4):982–990. doi: 10.1189/jlb.0907641. [DOI] [PubMed] [Google Scholar]
- 21.Austin C, Wray S. Interactions between Ca(2+) and H(+) and functional consequences in vascular smooth muscle. Circ Res. 2000 Feb 18;86(3):355–363. doi: 10.1161/01.res.86.3.355. [DOI] [PubMed] [Google Scholar]
- 22.Wang X, Wu J, Li L, Chen F, Wang R, Jiang C. Hypercapnic acidosis activates KATP channels in vascular smooth muscles. Circ Res. 2003 Jun 13;92(11):1225–1232. doi: 10.1161/01.RES.0000075601.95738.6D. [DOI] [PubMed] [Google Scholar]
- 23.Berger MG, Vandier C, Bonnet P, Jackson WF, Rusch NJ. Intracellular acidosis differentially regulates KV channels in coronary and pulmonary vascular muscle. The American journal of physiology. 1998 Oct;275(4 Pt 2):H1351–1359. doi: 10.1152/ajpheart.1998.275.4.H1351. [DOI] [PubMed] [Google Scholar]
- 24.Santa N, Kitazono T, Ago T, Ooboshi H, Kamouchi M, Wakisaka M, Ibayashi S, Iida M. ATP-sensitive potassium channels mediate dilatation of basilar artery in response to intracellular acidification in vivo. Stroke. 2003 May;34(5):1276–1280. doi: 10.1161/01.STR.0000068171.01248.97. [DOI] [PubMed] [Google Scholar]
- 25.Steidl JV, Yool AJ. Differential sensitivity of voltage-gated potassium channels Kv1.5 and Kv1.2 to acidic pH and molecular identification of pH sensor. Mol Pharmacol. 1999 May;55(5):812–820. [PubMed] [Google Scholar]