

Abstract
Prior studies by many laboratories have illustrated that ethanol can elicit a cascade of caspase-dependent apoptotic events in cultured neurons. Studies in our laboratory have connected this to oxidative stress and effects on fetal cortical neuron glutathione homeostasis.
Aims
The intent of the following studies is to address mechanisms underlying ethanol-associated DNA damage that may be connected to apoptotic death of neurons.
Methods
Cultures of fetal rat cerebral cortical neurons were utilized. Estimates of DNA damage was determined by Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining and nuclear condensation; Poly(ADP-ribose) polymerase-1 (PARP-1) expression was determined by immunostaining and Western blotting; and occurrence of parylation and AIF translocations were assessed by Western blotting.
Results
Ethanol treatment of the neurons generated increases in DNA damage by 4 hours while nuclear condensation was low at the short exposure period but increased markedly by 24 hours. This was temporally related to a marked up-regulation of PARP-1 expression. Activity of PARP-1, as assessed by PolyADP-ribose (PAR) formation, occurred within 15 minutes and peaked by 6 to 8 hours of ethanol treatment. An almost complete translocation of apoptosis inducing factor (AIF) from mitochondria to the nucleus occurred by 24 hours of ethanol treatment (4.0 mg/ml). Ethanol treatment for 4, 12 and 24 hours elicited an increasing caspase-mediated cleavage of PARP-1 to its 24kDa fragment.
Conclusions
These data illustrate the rapid occurrence of DNA damage following ethanol exposure and that PARP-1 pathways may play a role in the subsequent apoptotic death of these neurons.
Keywords: ethanol, PARP-1, AIF, DNA damage, neuron, apoptosis, oxidative stress
INTRODUCTION
Maternal alcohol consumption can impair offspring brain development in humans and in animal models. One mechanism underlying this may be an ethanol-related reduction in the number of neurons in various brain regions such as cerebral cortex, hippocampus, cerebellum and olfactory bulb. This response likely reflects altered neuron migration and enhanced cell death (Akbar et al., 2006; Barnes and Walker, 1981; Bhave et al., 2000; Chandler et al., 1997; Cheema et al., 2000; Crews et al., 1999; de la Monte et al., 2002; Dikranian et al., 2005; Goodlett et al., 1991; Jacobs and Miller, 2001; Luo et al., 1997; McAlhany et al., 2000; Miller and Potempa, 1990; Miller, 1996; Nowoslawski et al., 2005; Valles et al., 1997; Vangipuram et al., 2008; Vaudry et al., 2002; West et al., 1984, 1990; Xu et al., 2005; Young et al., 2003). There is compelling evidence in vivo and in cultured neurons that ethanol-related neuron death is by apoptotic processes which can be mechanistically connected to ethanol-initiated oxidative stress (Ramachandran et al., 2003; Rathinam et al., 2006; Watts et al., 2005). There appear to be multiple apoptotic pathways activated in neurons by ethanol, including the well documented intrinsic components associated with effector caspase (caspase-3) activation and DNA damage (Ramachandran et al., 2001, 2003). The latter is reflected in end-point DNA fragmentation in neurons committed to apoptotic death (Ramachandran et al., 2003; Watts et al., 2005). However, increases in TUNEL positive cells have also been documented in neonatal brains following in vivo ethanol exposure and in ethanol-exposed cultured fetal cerebral cortical neurons (Maffi et al., 2008). This suggests lesser DNA damage such as nicks which are produced by reactive oxygen species. Other than an ethanol-related AIF release by brain mitochondria (Ramachandran et al., 2001), little is known about mechanisms underlying ethanol induction of DNA damage in neurons. However, the occurrence of oxidative stress in this setting supports the concept that DNA strand nicks occur, which would trigger PARP-1 activation.
PARP-1 is a Zn-finger nuclear protein activated by DNA strand breaks and utilizes β-NAD as a substrate to catalyze the synthesis of ADP-ribose polymers on nuclear proteins, including PARP-1 itself (Kameshita et al., 1984). Kaufman et al (1993) first described the cleavage of PARP-1 during apoptosis in cells treated with different drugs. Full length PARP-1 is 113kDa which, during apoptosis is cleaved at the DEVD site to generate an 89- and a 24kDa fragment (Kaufmann et al., 1993; Lazebenik et al., 1994; Los et al., 1997, 1999; Nicholson et al., 1995). Both oxidative and nitrosative stress can trigger DNA strand breakage thereby activating PARP-1 in turn affording protection against cellular injury in various types of oxidant-induced cell death in vitro (Szabo and Dawson, 1998). Yet, excessive and possibly prolonged DNA damage causes hyperactivation of PARP-1 and the related massive NAD+ usage can affect cellular energetics and function (Sims et al., 1983; Yamamoto et al., 1981). PARP-1 plays a key role in neuronal death and survival under multiple stress conditions (Virag and Szabo, 2002). Its activation is a mediator of neuron death during excitotoxicity, ischemia, and oxidative stress, such that PARP-1 gene deletion or pharmacological inhibition can markedly improve neuronal survival in these settings (Eliasson et al., 1997; Endres et al., 1997; Mandir et al., 2000; Zhang et al., 1994).
Finally, there are functional associations between AIF and PARP-1. Yu et al (2002 and 2006) demonstrated that PARP-1 activation signals translocation of AIF, a 67 kDa flavoprotein, from the mitochondria to the nucleus, resulting in a “caspase-independent” pathway of programmed cell death. The following studies address the activation of these apoptosis pathways in cultured fetal cortical neurons exposed to ethanol.
MATERIALS AND METHODS
Minimal essential medium (MEM), fetal bovine serum, horse serum, and tissue culture plastics were from Invitrogen Corp (Carlsbad, CA). Apotag Fluorescein kits were from Millipore (Billerica, MA) Inh2BP, Benzamide, polylysine, uridine and 5-fluoro-2’-deoxyuridine were from Sigma Chemicals (St Louis, MO). Fluorescence–tagged secondary antibodies were purchased from Jackson Immunoresearch Laboratories (West Grove, PA). Alkaline phosphatase conjugated secondary antibodies were purchased from Vector Laboratories (Burlingame, CA). Antibodies towards AIF and PARP-1 were from Santa Cruz Biotechnologies (Santa Cruz, CA). The kit for protein estimation was from Bio-Rad (Hercules, CA). Nitrocellulose membrane was from Schleicher & Schuell (Keene, NH); chemiluminescence reagent kit from Pierce (Milwaukee, WI) X-OMAT AR film was from Eastman Kodak (Rochester, NY); and all other reagents were purchased from Sigma-Aldrich (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA).
Cell Culture
Primary cortical neuron cultures from embryonic day 16-17 rats were prepared as described by Dutton (1990). The dorsolateral cerebral cortex was carefully dissected from the fetal brain and finely minced and dissociated in Hank’s balanced salt solution (HBSS). Cells were then titrated in minimal essential medium (MEM) supplemented with 10% fetal bovine serum and 10% horse serum and were counted and seeded on dishes. All dishes and coverslips are coated with poly-D-lysine overnight. Neurons were maintained in the above-described media at 37°C in a humidified atmosphere of 95% air and 5% CO2. After 24 hr, the media was replaced with MEM containing 10% horse serum and uridine (10 mg/ml) and 5-fluoro-2’-deoxyuridine (4 mg/ml) to prevent the growth of astrocytes. Media were again changed 48 hr later (MEM with 10% horse serum). On the fifth day of culture, the cells were treated with ethanol for various time points. This is a well-documented culture system, largely free of glia, in which viability of neurons is maintained throughout the 6 days of culture. We confirmed that there was greater than a 95% purity of neuron populations by staining with an antibody specific for MAP2 (data not shown). For all experiments, controls were included for each measure and time point to compensate for any culture-related changes, and the numbers of replicates are noted in the figure legends.
Treatment
On the fifth day of culture, 5.1 μls of ethanol (200 proof) were added directly per 1 ml of media, yielding an initial concentration 4.0 mg/ml; thus neither ethanol-treated cells nor control cells were exposed to a medium change on the day of treatment. For ethanol-exposed cells, a beaker filled with ethanol was left overnight in the incubator to minimize evaporation from the medium. This is a system that effectively maintains ethanol concentrations in the medium (Devi et al., 1993). The ethanol concentration 4.0 mg/ml will decline to not less than about 3.8 mg/ml, within 24 hours. This was confirmed by assays of ethanol concentration by the alcohol dehydrogenase method. For all treatments, controls were included that were matched for time in culture. Justifications for the models: The 4 mg/ml concentration is not commonly seen clinically, but in select experiments e.g. AIF release, both 2.5 and 4 mg/ml ethanol concentrations were utilized to extend interpretation of the data past a mechanistic proof of concept. We have observed a comparable permeabilization of fetal brain mitochondria that had been exposed to ethanol in in vivo binge settings where maternal blood ethanol levels peaked at about 2.5 mg/ml (Ramachandran et al., 2001). Additionally the in vitro studies below were short-term whereas alcohol abuse is usually prolonged, and our intent was to reflect the relatively high ethanol levels that have been shown to elicit maximal damage to the developing brain. The use of cerebral cortical neurons reflects an extensive literature illustrating ethanol effects on the cerebral cortex in vivo and on cultured fetal cortical neurons (see the Introduction).
Subcellular Fractionation
Subcellular fractionations were modified from the protocol described by (Nijhawan et al., 2003). Cortical neurons that were seeded at a density of 8.5×106 cells/ml were harvested in PBS. After centrifugation at 1000g, the cell pellet was resuspended in Buffer (20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 250 mM Sucrose and protease inhibitors) and neurons lysed by passage through a 23-gauge needle 20 times. The lysate was centrifuged at 1000g, 10,000g, and 100,000g yielding the nuclear, mitochondrial and microsomal membrane pellets, respectively. The 100,000g supernatant was the cytosolic fraction. Nuclear proteins were extracted from the 1000g pellet in extraction lysis buffer (20 mM Tris, 125 mM NaCl, 5 mM MgCl2, 0.2 mM EDTA, 12% glycerol and 0.1% NP40 (with protease inhibitors) and the nuclei sonicated. The suspension was then spun at 14,000 rpm. Prior to processing samples by the subcellular fractionation method, parts of the whole protein extracts were set aside and assessed for equal starting concentration of protein using β-tubulin. After fractionation, mitochondrial and nuclear fractions were checked for purity using anti-cytochrome C oxidase subunit II and anti- H2B respectively.
Immunostaining
Neurons cultured on cover slips coated with poly-D-lysine overnight were washed with PBS, this was followed by fixation in 2% paraformaldehyde in PBS at room temperature, followed by incubation in blocking solution (2% normal goat serum, 2% fish skin gelatin, 0.25% Triton ×100 and 1% bovine serum albumin in PBS) and incubation overnight with primary antibody. Cells were washed with PBS and then incubated with secondary antibody (FITC or CY3 conjugated antibody) in blocking solution. Cells were washed with PBS and DAPI was added to stain for nuclei. Fluorescence microscopy was performed using an Olympus B-MAX microscope (Olympus, Tokyo, Japan) and recorded on a “Spot II” digital camera (Diagnostic Instruments, Tokyo, Japan). The exposure time used to image controls was kept constant for imaging experimental samples. Image J software provided by NIH was used to quantify signal intensity and the threshold for fluorescent measurements was kept constant while measuring across images to eliminate errors in quantifying signal intensities.
Western Blotting
The protein concentration of extracts of neurons was determined using the Biorad assay according to the manufacturer’s instructions. Western blotting was performed and membranes were incubated with a 1:250 dilution of primary antibody. The primary antibody was detected using HRP-conjugated secondary anti-rabbit or anti-goat or anti-mouse antiserum followed by use of a chemiluminescence reagent kit according to the manufacturer’s instructions. The blots were subject to autoradiography. In some cases, AP conjugated secondary antibodies were used followed by color development with NBT/BCIP to visualize protein bands. Quantifications of bands were done by scanning autoradiographs and saving files as TIFF images and then using Scion Image software (NIH) to quantify bands.
TUNEL Staining
Staining protocol provided with the apoptag kit was followed. Cells were grown on coverslips coated with poly-D-lysine overnight, washed with PBS, and fixed in 1% paraformaldehyde. Neurons were post fixed in pre-cooled Ethanol: Acetic acid (2:1) at -20°C. TdT enzyme was pipetted onto the section and incubated in a humidified chamber. The cells were washed with three changes of PBS, anti-dioxigenin fluorescein conjugate was added on the coverslip, and incubated in a humidified dark chamber. Mounting was carried out by applying a mounting medium containing DAPI. Fluorescence microscopy was performed using an Olympus B-MAX microscope (Olympus, Tokyo, Japan) and recorded on a “Spot II” digital camera (Diagnostic Instruments, Tokyo, Japan).
Statistical Analysis
One-way ANOVA was used to determine statistical significance of the data. Significant differences among control and ethanol-treated samples were calculated using the Bonferroni’s multiple comparison test with the Prism Biostatic Program (GraphPad Software, San Diego, CA). A P value of <0.05 was considered statistically significant.
RESULTS
Ethanol-induces DNA damage
DNA damage is one of the hallmarks of apoptosis, the visualization of which is possible by TUNEL staining, an assay for DNA breaks by enzymatic labeling of free 3’-OH termini. In Fig.1, the control panels were obtained prior to ethanol (4.0 mg/ml) treatment and most cell nuclei appear normal and uncondensed as seen by DAPI (blue) stain. However, by 4 hours of ethanol exposure (4.0 mg/ml), there were increases in DNA nicks localized in the nuclei as seen by FITC (green) stain and some cells exhibit chromosomal condensation. Ethanol treatment for 24 hours elicited much more extensive chromosomal condensation, yet at this time point there are cells that are resistant to ethanol treatment. We have recently experimentally addressed this question of selective vulnerability to ethanol (Maffi et al., 2008). The initial DNA damage e.g. nicks, illustrated in Figure 1 occurred much earlier after ethanol exposure than late-stage DNA fragmentation which does not become statistically significant until 12 hours of ethanol exposure (Ramachandran et al., 2003; Watts et al., 2005). Five different fields were randomly photographed for each treatment. A representative photograph for each treatment is featured in Fig 1.
Figure 1. Ethanol increases DNA damage and nuclear condensation.
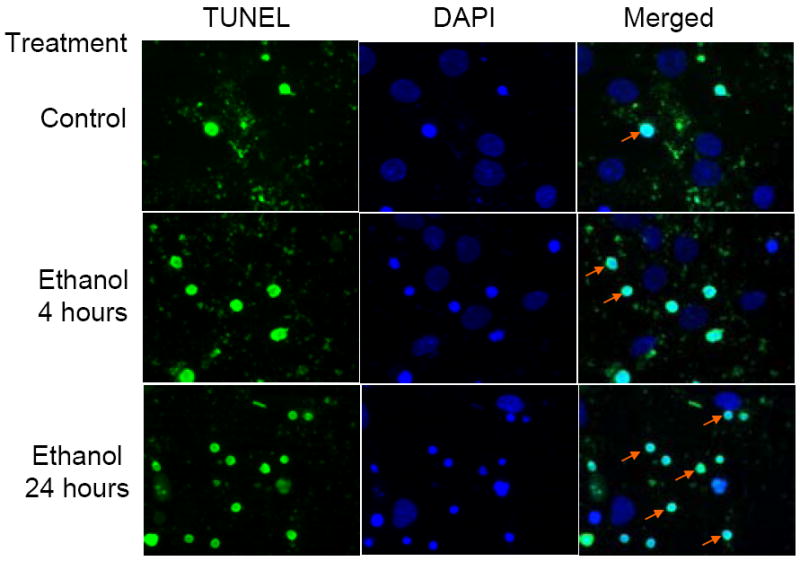
This panel illustrates the effects of ethanol on neuronal DNA damage. Cells were treated with 4mg/ml ethanol for 4 and 24 hours and assessed for DNA damage and chromatin condensation using the TUNEL assay. DNA damage as assessed by TUNEL can be visualized by the green FITC stain and the nuclei are stained blue with DAPI. The first column shows increases in the number of cells that demonstrate DNA damage. The middle column shows all the nuclei of cells present in the field, the number of condensed nuclei being far greater in the 24 hours ethanol treated row. The right most column is a merge of the TUNEL and DAPI stained pictures. Arrows point to examples of condensed nuclei.
Ethanol increases expression of PARP-1, its activation and inactivation
Ethanol elicits in cultured fetal cortical neurons, a rapid increase in ROS which is ultimately followed by increased measures of DNA fragmentation and apoptotic death (Ramachandran et al., 2003; Rathinam et al., 2006; Watts et al., 2005). Figure 2 illustrates that ethanol (4 mg/ml, 24 hours) produced enhanced PARP-1 expression which was heightened about 10 fold as compared to the control (Panel A). This increase is further illustrated in the accompanying bar graph (Panel B). The antibody used for these studies detected the full length protein as well as the 24kDa fragment. Three random fields were photographed and individual cells (n=5) were quantified from control and ethanol treated samples.
Fig 2. PARP-1 expression is enhanced with ethanol treatment.
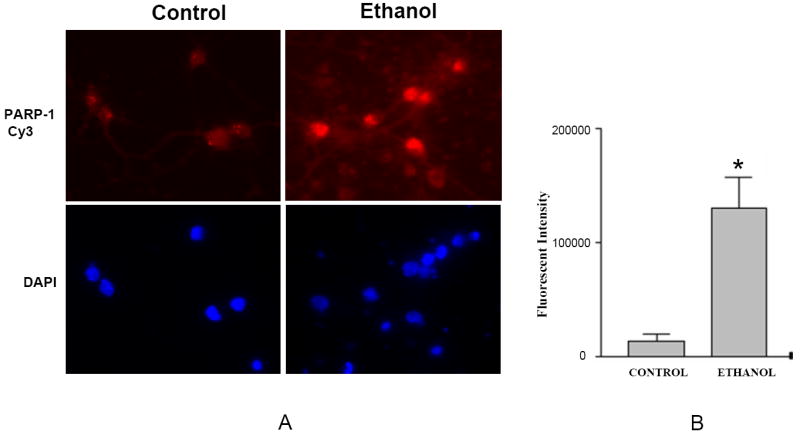
Neurons grown on cover slips were treated with 4mg/ml ethanol for 24 hours. Immunofluorescent staining was done using anti-PARP-1 antibody followed by Cy-3 conjugated rabbit secondary antibody. There is a robust increase in PARP-1 expression in ethanol exposed samples, primarily in the nuclei (Fig. 2A). Fig. 2B is a composite of five random fields. The intensity increase is approximately 10 fold (n=5, *= p<0.0002).
Nuclear and cytoplasmic proteins are covalently modified by PARP and poly(ADP-ribosyl)ation may play roles in PARP-1 -related cell death (Hong et al., 2004). The effects of ethanol on activity of PARP-1, as reflected by Poly(ADP-ribose) (PAR) polymer-protein formation in the nucleus, is illustrated in Figure 3. Since these are nuclear extracts, H2B was used as loading control. Increased PARylation of nuclear proteins was apparent as early as 15 minutes of ethanol exposure (4 mg/ml) and this peaked by 6 to 8 hours of continued treatment. This blot is a representation of n=3 experiments.
Fig 3. Ethanol elicits a time-dependent induction of nuclear protein PARylation.
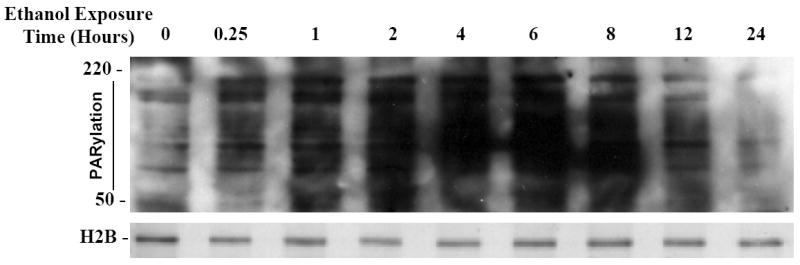
Increased PARylation is apparent within 0.25 hours of ethanol treatment and peaks between 6-8 hours of ethanol (4mg/ml) exposure as detected by western blot analysis with an antibody to PAR. H2B was used as loading control (Data represents n=3 experiments).
Prior studies have illustrated that ethanol rapidly activates caspase-3 –dependent apoptotic cascades in cultured fetal cortical neurons. Caspase-mediated inactivation of PARP-1 by cleavage from the full length 113kDa protein at the DEVD site, to 89- and 24kDa fragments is a feature of apoptosis. The antibody used to follow the cleavage of PARP-1 recognizes the N-terminal 24kDa fragment. Figure 4 demonstrates an ethanol-related increase in this 24kDa fragment within 4 hours of exposure. By 24 hours of ethanol exposure (4 mg/ml), PARP-1 cleavage was strikingly increased compared to respective controls (control data not shown). Fig 4 also illustrates that inhibition of caspase by a broad spectrum inhibitor, zVAD, dramatically reduced the ethanol-related PARP-1 cleavage. Fig. 4 is a representation of n=3 experiments.
Fig 4. PARP-1 is inactivated by caspase-3.
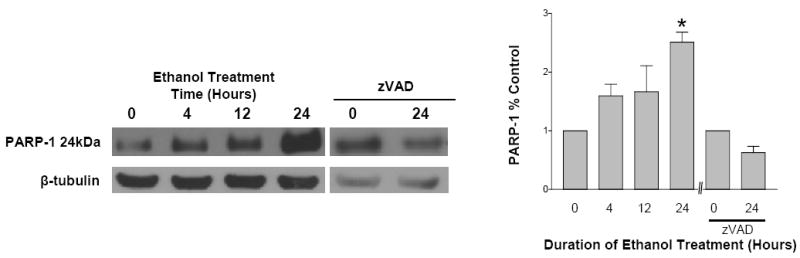
PARP-1 is cleaved from a full length 113kDa during apoptosis to generate an 89- and a 24kDa fragment. To detect the cleavage of PARP-1 an antibody was used that recognizes the N-terminal 24kDa fragment. With increased duration of ethanol exposure (4, 12 and 24 hours), there are increases in PARP-1 which reach statistical significance (p<0.05) by 24 hours of exposure. Caspase inhibition with zVAD during the 24 hour ethanol (4mg/ml) treatment reduced expression of the 24kDA band. Data represents n=3 experiments. (* = p< 0.05)
These experiments illustrate that in neurons exposed to ethanol 1). DNA damage occurs, 2). Initially, PARP-1 expression and activity are increased and, 3). With longer exposure, PARP-1 is inactivated, mostly by caspase cleavage.
Ethanol elicits rapid relocalization of AIF to the nucleus
In utero ethanol exposure can elicit release of AIF from mitochondria derived from fetal brains, an event potentially connected to PARP-1 activation (Hong et al., 2004; Ramachandran et al., 2003). Figure 5 illustrates this response in the cultured neurons showing AIF relocalization to the nucleus. On ethanol treatment (4.0 mg/ml), AIF translocates from mitochondria to the nuclei with a clear increase in nuclear content within 6 hours of exposure, by 24 hours of treatment, this translocation appeared complete. AIF together with EndoG (data not shown) can induce the chromatin condensation and DNA fragmentation, typical of apoptosis (Fig. 1).
Fig 5. Ethanol increases translocation of AIF from mitochondria to the nuclear compartment.
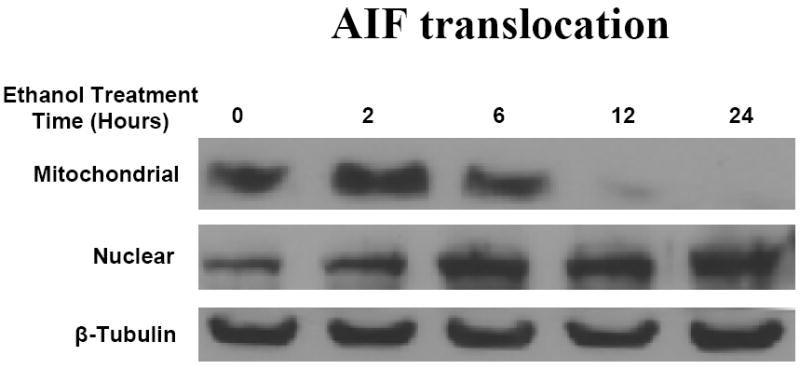
Neurons were treated with E (4 mg/ml) for 2, 6 and 24 hours. The expressions of AIF were detected by western blotting using anti-AIF primary antibodies. The primary antibodies were detected using HRP-conjugated secondary antibodies followed by use of a chemiluminescence reagent kit (ECL). Data represents n=3 experiments
DISCUSSION
During brain development juvenile neurons die in a normal physiological process that defines the adult number and functionality of neurons in the adult CNS. Programmed cell death plays a major role in this process (Yuan and Yankner, 2000) and numerous studies have documented that ethanol exposure increases this normal culling of neurons. A push by multiple laboratories has been directed towards elucidating specific apoptotic pathways activated by ethanol (Mooney and Miller, 2001; Ramachandran et al., 2003; Heaton et al., 2003; Young et al., 2005). Relatively unexplored areas are DNA damage responses to ethanol that would be expected to occur in the presence of oxidative stress (Ramachandran et al., 2003; Watts et al., 2005) and the related AIF components of neuron apoptosis pathways (Ramachandran et al., 2001).
Ethanol and DNA Damage
There are few reports detailing ethanol-related damage of neuron DNA of the sort that activates PARP-1. There is evidence that direct exposure to ethanol and acetaldehyde can elicit neuronal DNA damage, albeit with different profiles, and that ethanol-generated DNA alterations may not necessarily result in a measurable decline of neuron viability (Lamarche et al., 2004). In the liver, ethanol can elicit single-strand breaks/nicks which appear to be connected to free radical production (Navasumrit et al., 2000) and this is likely the case in the above studies. The occurrence of such rapid DNA damage in neurons is not surprising given the well documented ability of ethanol to generate reactive oxygen species in the developing brain and in cultured neurons (Heaton et al., 2003; Kotch et al., 1995; Maffi et al., 2008; Ramachandran et al., 2001, 2003). In these fetal rat cerebral cortical neuron cultures, we have previously observed a remarkably rapid onset of increased cell content of reactive oxygen species, within minutes of ethanol exposure (Ramachandran et al., 2003). However, measures of end-stage, apoptosis-related DNA fragmentation in these cultures did not reach significance until about 12 hours of treatment (Ramachandran et al., 2003, Watts et al, 2005). The data in Figure 1 illustrate that a relatively short exposure to ethanol can elicit DNA nick damage as evidenced by the increased tunnel staining and that this is concomitant with a relatively low abundance of overt nuclear condensation.
Ethanol and PARP-1 activation
The duality of PARP-1 function is well illustrated in the above studies with ethanol. DNA strand nicks and breaks (single and double strand) are triggers of PARP-1 activation, this damage being elicited by a variety of environmental stressors which include hydrogen peroxide, hydroxyl radicals and nitrogen centered radicals. With low levels of DNA damage, PARP-1 is a survival factor that is involved in DNA repair. However, at high levels of DNA damage, PARP-1 promotes cell death (Kim et al., 2005; Koh et al., 2005 for reviews). The products of PARP-1 are PAR polymers which covalently modify proteins (PARylation). Among nuclear target proteins for PARylation are a variety of transcription factors and core and linker histones, thus responses to alterations of these various activities and domains elicit multiple effects at the nuclear level (Kim et al., 2005). The rapid onset (15 minutes) of PARylation (Fig. 3) following ethanol exposure is a reflection of PARP-1 activity and, at this early stage, we are likely observing the nick sensor role of PARP-1 and commencement of related DNA repair (de Murcia and Menissier, 1994). The origin of this damage may be connected to the rapid onset of ethanol-related oxidative stress (Ramachandran et al., 2003, Watts et al., 2005) and many of the cells may not be committed to apoptotic death at these early time points. Recent studies have illustrated that the neurons which ultimately commit to ethanol-mediated apoptotic death are those least able to generate glutathione and counter the enhanced production of reactive oxygen species (Maffi et al., 2008). Thus, the increase in nuclear protein PARylation within 15 minutes of ethanol treatment is consistent with a chain of events comprising a rapid increase in neuron content of reactive oxygen species followed by DNA damage, and concomitant activation of PARP-1 –related DNA repair processes. However, with longer term ethanol exposure, the pro-death activities of PARP-1 may become significant.
There is abundant evidence supporting a connection between overactivation of PARP-1 and cell death, and this being a dominant process in CNS disorders (Kim et al., 2005; Koh et al., 2005). In a variety of pathological settings, inhibition of PARP-1 can provide cytoprotection e.g. ischemic injury, glutamate toxicity, and injury produced by reactive oxygen species (Hong et al., 2004, Pieper et al., 1999;Zhang et al., 1994). Details of the precise mechanisms underlying the cell death process are a work in progress, but among the multiple physiological roles for PARP-1, many are likely related to poly-ADP-ribosylations of proteins. In addition to ribosylation-related inhibition of an assortment of enzyme activities, PARP-1 is itself an aggressive enzyme that can rapidly deplete cellular NAD+ and ATP (Hong et al., 2004). Fig. 3 illustrates that ethanol strikingly increases PARylation of nuclear proteins with a peak at around 6 hours of exposure. There is a clear decrease in nuclear protein PARylation after 8 hours of ethanol treatment with it reverting to control levels by 24 hours of exposure. The data in Fig. 4 offer an explanation for this and illustrate a system by which the neuron can mitigate and ultimately block cytotoxic PARylation. PARP-1 is inactivated by caspase-3 cleavage into 89- and 24kDa fragments. This occurs during apoptosis due to caspase-3 activation (Kaufmann et al., 1993; Lazebenik et al., 1994). In these cultured cortical neurons, ethanol-mediated apoptosis is associated with caspase-3 activation as early as 2 hours after ethanol treatment but it only reaches statistical significance after 6 hours (Ramachandran et al., 2003). Fig 4 illustrates that within 4 hours of ethanol exposure, there is an increase (albeit not statistically significant) in the 24 kDa PARP-1 fragment and striking increases in the caspase-3 cleavage product within 24 hours of ethanol exposure. Also illustrated, is a substantial inhibition of PARP-1 cleavage at the 24 hour point by treatment of cells with a caspase inhibitor, zVAD. Taken together, Figures 3 and 4 illustrate that ethanol treatment generates a conspicuous increase in nuclear protein PARylation and that within 12 hours, PARP-1 cleavage/inactivation by caspase-3 reduces these PARylations. At this juncture, we do not know if PARylation plays significant roles in the ethanol-mediated apoptosis occurring in these cells, but it is highly likely that the apoptosis-related DNA fragmentation that occurs within 12 hours of ethanol treatment is connected to mitochondrial AIF release which, in turn, may be mediated by PARP-1 (Fig 6) (Hong et al., 2004).
Fig. 6. PARP-1 -related events in response to ethanol exposure.
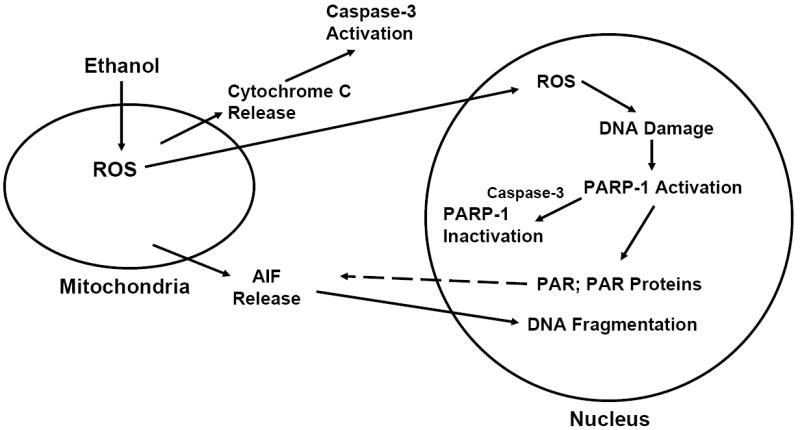
Within minutes of ethanol exposure, there is an enhanced expression of reactive oxygen species (ROS) in cultured fetal rat cerebral cortical neurons (Heaton et al., 2003; Kotch et al., 1995; Maffi et al., 2008; Ramachandran et al., 2001, 2003). This scheme presents the concept that PARP-1 is activated secondary to DNA damage, ultimately leading to PARylation of nuclear proteins. Within 2 hours of ethanol exposure there is an observable increase in translocation of AIF to the nuclear compartment, an event that may be mediated by PARP-1. Within 12 hours of ethanol treatment, there is caspase-3 -mediated inactivation of PARP-1. However, the DNA fragmentation process has already commenced in select neurons, in response to nuclear accumulation of AIF.
Ethanol and AIF
Prior studies have illustrated that in utero exposure to ethanol elicits release of AIF from mitochondria isolated from fetal brain (Ramachandran et al., 2001). When cells are exposed to cytotoxic insults that elicit apoptosis, AIF is released from mitochondria and its subsequent translocation to the nucleus is a typical feature of apoptotic death. We have also found that EndoG along with AIF (data not shown). Once translocated, these compounds play an apparently synergistic role in digestion of DNA (Modjtahedi et al., 2006 for a review). AIF induces chromatin condensation and DNA fragmentation of the sort that we have observed in these cells within 12 and 24 hours of ethanol exposure (Ramachandran et al., 2003; Susin et al., 1999; Watts et al., 2005; Ye et al., 2002). Figure 5 illustrates a time course for effects of ethanol on mitochondrial to nuclear translocation of AIF. Within 6 hours of exposure to ethanol, mitochondrial content of the protein is decreased and within 12 hours, depletion is extensive. This is accompanied by increases in nuclear AIF that is clearest at the 24 hour exposure point. The means by which AIF is released from mitochondria is controversial, but the composite of the literature supports there being caspase dependent and independent pathways, these depending on cell type and death stimuli (Dawson and Dawson, 2004; Yuste et al., 2005). Relevant to the current setting, is that an extensively documented function of PARP-1 is mediation of AIF translocation during cell death (Hong et al., 2004; Yu et al., 2002). One model has it that AIF release may be connected to actions of PAR polymers (Hong et al., 2004). It is tempting to speculate, based on the sequence of events documented in the above studies, that PARP-1 activation is a player in ethanol-mediated release of AIF. The time frames certainly support this role for PARP-1. Future studies will be needed to establish causality.
PARP-1 –related apoptotic pathways activated by ethanol
The ethanol-related DNA damage, PARP-1 activation, and AIF nuclear translocation documented in these studies are components of complex, interconnected events that ultimately generate apoptotic death of fetal cerebral cortical neurons. The activation of PARP-1, concomitant with increased cell ROS further supports previous studies illustrating that oxidative stress is a key initial player in the ethanol-mediated apoptotic death of these neurons (Maffi et al., 2008; Ramachandran et al., 2003; Rathinam et al., 2006; Watts et al., 2005). Figure 6 outlines key elements of apoptosis-related measures that we have found to be activated by ethanol in these cells and some of their interconnections. The earliest event in response to ethanol that we have detected is increased production of reactive oxygen species (ROS). Following this is increased expression of mitochondrial 4-hydroxynonenal (less than one hour), a lipid peroxidation product, and decreased cellular glutathione (Ramachandran et al., 2003). If cellular GSH is normalized either by added N-acetylcysteine or the presence of astrocytes, the increase in ROS is blocked as are the apoptotic responses (Ramachandran et al., 2003; Watts et al., 2005). Increased cytochrome C release follows within 2 hours and caspase-3 activation reaches statistical significance within 12 hours. We propose that the initial DNA damage which activates PARP-1 is caused by ROS and that the later DNA fragmentation and chromosomal condensation result from AIF (and EndoG) translocation to the nuclear compartment. The mechanisms underlying mitochondrial release of AIF remain to be firmly established, but PAR-related actions (dashed line) are strongly supported by studies from other laboratories.
Acknowledgments
Grant information: This work was supported by the National Institute of Health grant (RO1 AA010114)
References
- Akbar M, Baick J, Calderon F, Wen Z, Kim HY. Ethanol promotes neuronal apoptosis by inhibiting phosphatidylserine accumulation. J Neurosci Res. 2006;83:432–40. doi: 10.1002/jnr.20744. [DOI] [PubMed] [Google Scholar]
- Barnes DE, Walker DW. Prenatal ethanol exposure permanently reduces the number of pyramidal neurons in rat hippocampus. Brain Res Dev Brain Res. 1981;1:3–24. doi: 10.1016/0165-3806(81)90071-7. [DOI] [PubMed] [Google Scholar]
- Bhave SV, Snell LD, Tabakoff B, Hoffman PL. Chronic ethanol exposure attenuates the anti-apoptotic effect of NMDA in cerebellar granule neurons. J Neurochem. 2000;75:1035–1044. doi: 10.1046/j.1471-4159.2000.0751035.x. [DOI] [PubMed] [Google Scholar]
- Chandler JL, Sutton G, Norwiid D, Sumners C, Crews FT. Chronic alcohol increases N-methyl-D-aspartate-stimulated nitric oxide formation but not receptor density in cultured cortical neurons. Mol Pharmacol. 1997;51:733–740. doi: 10.1124/mol.51.5.733. [DOI] [PubMed] [Google Scholar]
- Cheema ZF, West JR, Miranda RC. Ethanol induces Fas/Apo [apoptosis]-1 mRNA and cell suicide in the developing cerebral cortex. Alcohol Clin Exp Res. 2000;24:535–43. [PubMed] [Google Scholar]
- Cohen-Armon M, Visochek L, Katzoff A, Levitan D, Susswein AJ, Klein R, Valbrun M, Schwartz JH. Long-term memory requires polyADP-ribosylation. Science. 2004;304:1820–1822. doi: 10.1126/science.1096775. [DOI] [PubMed] [Google Scholar]
- Crews FT, Waage HG, Wilkie MB, Lauder JM. Ethanol pretreatment enhances NMDA excitotoxicity in biogenic amine neurons: protection by brain derived neurotrophic factors. Alcohol Clin Exp Res. 1999;23:1834–1842. [PubMed] [Google Scholar]
- Dawson VL, Dawson TM. Deadly conversations: Nuclear-mitochondrial cross-talk. J Bioennerg and Biomemb. 2004;36:287–294. doi: 10.1023/B:JOBB.0000041755.22613.8d. [DOI] [PubMed] [Google Scholar]
- de la Monte SM, Wands JR. Chronic gestational exposure to ethanol impairs insulin-stimulated survival and mitochondrial function in cerebellar neurons. Cell Mol Life Sci. 2002;59:882–93. doi: 10.1007/s00018-002-8475-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Murcia G, Menissier-de Murcia J. Poly(ADP-ribose) polymerase: A molecular nick sensor. Trends Biochem Sci. 1994;19:172–176. doi: 10.1016/0968-0004(94)90280-1. [DOI] [PubMed] [Google Scholar]
- Devi BG, Henderson GI, Frosto RA, Schenker S. Effect of ethanol on rat fetal hepatocytes: studies on cell replication, lipid peroxidation and glutathione. Hepatology. 1993;18:648–659. [PubMed] [Google Scholar]
- Dikranian K, Qin YQ, Labruyere J, Nemmers B, Olney JW. Ethanol-induced neuroapoptosis in the developing rodent cerebellum and related brain stem structures. Brain Res Dev Brain Res. 2005;22(155(1)):1–13. doi: 10.1016/j.devbrainres.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Dutton GR. Isolation, culture and use of viable CNS perikarya. Meth Neurosci. 1990;2:87–102. [Google Scholar]
- Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, Dawson VL. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- Endres M, Wang ZQ, Namura S, Waeber C, Moskowitz MA. Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J Cereb Blood Flow Metab. 1997;17:1143–1151. doi: 10.1097/00004647-199711000-00002. [DOI] [PubMed] [Google Scholar]
- Goodlett CR, Thomas JD, West JR. Long-term deficits in cerebellar growth and rotorod performance of rats following “binge-like” alcohol exposure during the neonatal brain growth spurt. Neurotoxicol Teratol. 1991;13:69–74. doi: 10.1016/0892-0362(91)90029-v. [DOI] [PubMed] [Google Scholar]
- Heaton MB, Paiva M, Madoesky I, Shaw G. Ethanol effects on neonatal rat cortex: comparative analysis of neurotrophic factors, apoptosis-related proteins, and oxidative processes during vulnerable and resistant periods. Dev Brain Res. 2003;145:249–262. doi: 10.1016/j.devbrainres.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Hong SJ, Dawson TM, Dawson VL. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. TRENDS in Pharm Sci. 2004;25:259–272. doi: 10.1016/j.tips.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Jacobs JS, Miller MW. Proliferation and death of cultured fetal neocortical neurons: effects of ethanol on the dynamics of cell growth. J Neurocytology. 2001;30:391–401. doi: 10.1023/a:1015013609424. [DOI] [PubMed] [Google Scholar]
- Kameshita I, Matsuda Z, Taniguchi T, Shizuta Y. Poly (ADP-Ribose) synthetase. Separation and identification of three proteolytic fragments as the substrate-binding domain, the DNA-binding domain, and the automodification domain. J Biol Chem. 1984;25(259(8)):4770–6. [PubMed] [Google Scholar]
- Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE, Poirier GG. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res. 1993;53:3976–3985. [PubMed] [Google Scholar]
- Kauppinen TM. Multiple roles for poly(ADP-ribose)polymerase-1 in neurological disease. Neurochem International. 2007;50:954–958. doi: 10.1016/j.neuint.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Kim MY, Zhang T, Kraus WL. Poly(ADP-ribosyl)ation by PARP-1: PAR-laying’ NAD+into a nuclear signal. Genes and Devel. 2005;19:1951–1967. doi: 10.1101/gad.1331805. [DOI] [PubMed] [Google Scholar]
- Koh DW, Dawson TM, Dawson VL. Poly(ADP-ribosyl)ation regulation of life and death in the nervous system. Cell Mol Life Sci. 2005;62:760–768. doi: 10.1007/s00018-004-4508-y. [DOI] [PubMed] [Google Scholar]
- Kotch LE, Chen SY, Sulik KK. Ethanol-induced teratogenesis: Free radical damage as a possible mechanism. Teratol. 1995;52:128–136. doi: 10.1002/tera.1420520304. [DOI] [PubMed] [Google Scholar]
- Lamarche F, Gonthier B, Signorini B, Eysseric H, Barret L. Impact of ethanol and acetaldehyde on DNA and cell viability of cultured neurons. Cell Biol & Toxicol. 2004;20:361–374. doi: 10.1007/s10565-004-0087-9. [DOI] [PubMed] [Google Scholar]
- Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- Los M, Herr I, Friesen C, Fulda S, Schulze-Osthoff K, Debatin KM. Cross-resistance of CD95- and drug-induced apoptosis as a consequence of deficient activation of caspases (ICE/ Ced-3 proteases) Blood. 1997;90:3118–3129. [PubMed] [Google Scholar]
- Los M, Wesselborg S, Schulze-Osthoff K. The role of caspases in development, immunity, and apoptotic signal transduction: lessons from knockout mice. Immunity. 1999;10:629–639. doi: 10.1016/s1074-7613(00)80062-x. [DOI] [PubMed] [Google Scholar]
- Luo J, West JR, Pantazis NJ. Nerve growth factor and basic fibroblast growth factor protect rat cerebellar granule cells in culture against ethanol-induced cell death. Alcohol Clin Exp Res. 1997;12:1108–1120. [PubMed] [Google Scholar]
- Maffi SK, Rathinam ML, Cherian PP, Pate W, Hamby-Mason R, Schenker S, Henderson GI. Glutathione content as a potential mediator of the vulnerability of cultured fetal cortical neurons to ethanol-induced apoptosis. J NeuroSci Res. 2008;96:1289–1300. doi: 10.1002/jnr.21562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandir AS, Poitras MF, Berliner AR, Herring WJ, Guastella DB, Feldman A, Poirier GG, Wang ZQ, Dawson TM, Dawson VL. NMDA but not non-NMDA excitotoxicity is mediated by Poly(ADP-ribose) polymerase. J Neurosci. 2000;20:8005–8011. doi: 10.1523/JNEUROSCI.20-21-08005.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlhany RE, Jr, West JR, Miranda RC. Glial-derived neurotrophic factor (GDNF) prevents ethanol-induced apoptosis and JUN kinase phosphorylation. Dev Brain Res. 2000;119:209–16. doi: 10.1016/s0165-3806(99)00171-6. [DOI] [PubMed] [Google Scholar]
- Midrorikawa R, Takei Y, Hirokawa N. KIF4 motor regulates activity-dependent neuronal survival by suppressing PARP-1 enzymatic activity. Cell 2006. 2006 Apr 21;125(2):224–6. doi: 10.1016/j.cell.2006.02.039. [DOI] [PubMed] [Google Scholar]
- Miller MW. Limited ethanol exposure selectively alters the proliferation of precursor cells in the cerebral cortex. Alcohol Clin Exp Res. 1996;20:139–143. doi: 10.1111/j.1530-0277.1996.tb01056.x. [DOI] [PubMed] [Google Scholar]
- Miller MW, Potempa G. Numbers of neurons and glia in mature rat somatosensory cortex: effects of prenatal exposure to ethanol. J Comp Neurol. 1990;293:92–102. doi: 10.1002/cne.902930108. [DOI] [PubMed] [Google Scholar]
- Modjtahedi N, Giordanetto F, Madeo F, Kroemer G. Apoptosis-inducing factor: vital and Lethal. Trends in Cell Biol. 2006;16:264–272. doi: 10.1016/j.tcb.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Mooney SM, Miller MW. Effects of prenatal exposure to ethanol on the expression of bcl-2, bax and caspase 3 in the developing rat cerebral cortex and thalamus. Brain Res. 2001;911(1):71–81. doi: 10.1016/s0006-8993(01)02718-4. [DOI] [PubMed] [Google Scholar]
- Navasumrit P, Ward TH, Dodd NJ, O’Connor PJ. Ethanol-induced free radicals and hepatic DNA strand breaks are prevented in vivo by antioxidants: acute and chronic ethanol exposure. Carcinogenesis. 2000;21:93–99. doi: 10.1093/carcin/21.1.93. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;6(376(6535)):37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, Wang X. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes and Dev. 2003;17:1475–1486. doi: 10.1101/gad.1093903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowoslawski L, Klocke BJ, Roth KA. Molecular regulation of acute ethanol-induced neuron apoptosis. J Neuropathol Exp Neurol. 2005;64:490–7. doi: 10.1093/jnen/64.6.490. [DOI] [PubMed] [Google Scholar]
- Pieper AA, Verma A, Zhang J, Snyder SH. Poly(ADP-ribose) polymerase, nitric oxide and cell death. Trends Pharmacol Sci. 1999;20:171–181. doi: 10.1016/s0165-6147(99)01292-4. [DOI] [PubMed] [Google Scholar]
- Ramachandran V, Perez A, Chen J, Senthil D, Schenker S, Henderson GI. In utero ethanol exposure causes mitochondrial dysfunction, which can result in apoptotic cell death in fetal brain: a potential role for 4-hydroxynonenal. Alcohol Clin Exp Res. 2001;25(6):862–871. [PubMed] [Google Scholar]
- Ramachadran V, Watts LT, Maffi SK, Chen JJ, Schenker S, Henderson GI. Ethanol-induced oxidative stress precedes mitochondrially mediated apoptotic death of cultured fetal cortical neurons. J Neurosci Res. 2003;74:577–588. doi: 10.1002/jnr.10767. [DOI] [PubMed] [Google Scholar]
- Rathinam ML, Watts LT, Stark AA, Mahimainathan L, Stewart J, Schenker S, Henderson GI. Astrocyte control of fetal cortical neuron glutathione homeostasis: up-regulation by ethanol. J Neurochem. 2006;96:1289–1300. doi: 10.1111/j.1471-4159.2006.03674.x. [DOI] [PubMed] [Google Scholar]
- Sims JL, Berger SJ, Berger NA. Poly(ADP-ribose) Polymerase inhibitors preserve nicotinamide adenine dinucleotide and adenosine 5’-triphosphate pools in DNA-damaged cells: mechanism of stimulation of unscheduled DNA synthesis. Biochemistry. 1983;22:5188–5194. doi: 10.1021/bi00291a019. [DOI] [PubMed] [Google Scholar]
- Szabo SC, Dawson V. Role of poly(ADP-ribose) synthetase in inflammation and ischemia-reperfusion. Trends Pharmacol Sci. 1998;19:287–298. doi: 10.1016/s0165-6147(98)01193-6. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Marzo I, Snow BE, Brothers GM, Mangnion J, Jacotot E, Constantini P, Loeffler M. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Valles S, Pitarch J, Renau-Piqueras J, Guerri C. Ethanol exposure affects glial fibrillary acidic protein gene expression and transcription during rat brain development. J Neurochem. 1997;69(6):2484–93. doi: 10.1046/j.1471-4159.1997.69062484.x. [DOI] [PubMed] [Google Scholar]
- Vangipuram SD, Grever WE, Parker GC, Lyman WD. Ethanol increases fetal human neurosphere size and alters adhesion molecule gene expression. Alcohol Clin Exp Res. 2008;32(2):339–47. doi: 10.1111/j.1530-0277.2007.00568.x. [DOI] [PubMed] [Google Scholar]
- Vaudry D, Rousselle C, Basille M, Falluel-Morel A, Pamantung TF, Fontaine M, Fournier A, Vaudry H, Gonzalez BJ. Pituitary adenylate cyclase-activating polypeptide protects rat cerebellar granule neurons against ethanol-induced apoptotic cell death. Proc Natl Acad Sci U S A. 2002;99:6398–403. doi: 10.1073/pnas.082112699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virag L, Szabo C. The therapeutic potential of poly (ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- Visochek L, Steingart RA, Vulih-Shultzman I, Klein R, Priel E, Gozes I, Cohen-Armon M. PolyADP-ribosylation is involved in neurotrophic activity. J Neurosci. 2005;25:7420–7428. doi: 10.1523/JNEUROSCI.0333-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voehringer DW, McConkey DJ, McDonnell TJ, Brisbay S, Meyn RE. Bcl-2 expression causes redistribution of glutathione to the nucleus. PNAS. 1998;95(6):2956–2960. doi: 10.1073/pnas.95.6.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts LT, Rathinam ML, Schenker S, Henderson GI. Astrocytes protect neurons from ethanol-induced oxidative stress and apoptotic death. J Neurosci Res. 2005;80:655–666. doi: 10.1002/jnr.20502. [DOI] [PubMed] [Google Scholar]
- West JR, Dewey SL, Pierce DR, Black AC., Jr Prenatal and early postnatal exposure to ethanol permanently alters the rat hippocampus. Ciba Found Symp. 1984;105:8–25. doi: 10.1002/9780470720868.ch2. [DOI] [PubMed] [Google Scholar]
- West JR, Goodlett CR, Bonthius DJ, Hamre KM, Marcussen BL. Cell population depletion associated with fetal alcohol brain damage: mechanisms of BAC-dependent cell loss. Alcohol Clin Exp Res. 1990;14:813–818. doi: 10.1111/j.1530-0277.1990.tb01820.x. [DOI] [PubMed] [Google Scholar]
- Xu Y, Liu P, Li Y. Impaired development of mitochondria plays a role in the central nervous system defects of fetal alcohol syndrome. Birth Defects Res A Clin Mol Teratol. 2005;73:83–91. doi: 10.1002/bdra.20110. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Uchigata Y, Okamoto H. Streptozotocin and alloxan induce DNA strand breaks and poly(ADP-ribose) synthetase in pancreatic islets. Nature. 1981;294(5838):284–6. doi: 10.1038/294284a0. [DOI] [PubMed] [Google Scholar]
- Ye H, Cande C, Stephanou NC, Jiang S, Gurbuxani S, Larochette N, Daugas E, Garrido C, Kroemer G, Wu H. DNA binding is required for the apoptogenic action of apoptosis inducing factor. Nat Struct Biol. 2002;9(9):680–4. doi: 10.1038/nsb836. [DOI] [PubMed] [Google Scholar]
- Young C, Roth KA, Klocke BJ, West T, Holtzman DM, Labruyere J, Qin YQ, Dikranian K, Olney JW. Role of caspase-3 in ethanol-induced developmental neurodegeneration. Neurobiol Dis. 2005;20(2):608–614. doi: 10.1016/j.nbd.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, Dawson VL. Apoptosis Inducing Factor (AIF) mediates Poly (ADP-ribose) (PAR) polymer induced cell death. Proc Natl Acad Sci U S A. 2006;28(103(48)):18314–9. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;12(407(6805)):802–9. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- Yuste VJ, Moubarak RS, Delettre C, Bras M, Sancho P, Robert N, d’Alayer J, Susin SA. Cysteine proteases inhibition prevents mitochondrial apoptosis-inducing factor (AIF) release. Cell Death Differen. 2005;12:1445–1448. doi: 10.1038/sj.cdd.4401687. [DOI] [PubMed] [Google Scholar]
- Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]